
The Tumor Microenvironment-Dependent Transcription Factors AHR and HIF-1α Are Dispensable for Leukemogenesis in the Eµ-TCL1 Mouse Model of Chronic Lymphocytic Leukemia

 ,
,  ,
,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. B Cell Isolation
2.3. Validation of the Models at the gDNA, RNA, and Protein Levels
2.4. Sample Preparation for RNA Sequencing
2.5. RNA Sequencing Analysis
2.6. Murine B Cell-Specific Ahr-Induced Gene Signature
2.7. Gene Set Enrichment Analysis
2.8. Transcription Factor Enrichment Analysis
2.9. Statistical Analysis
3. Results
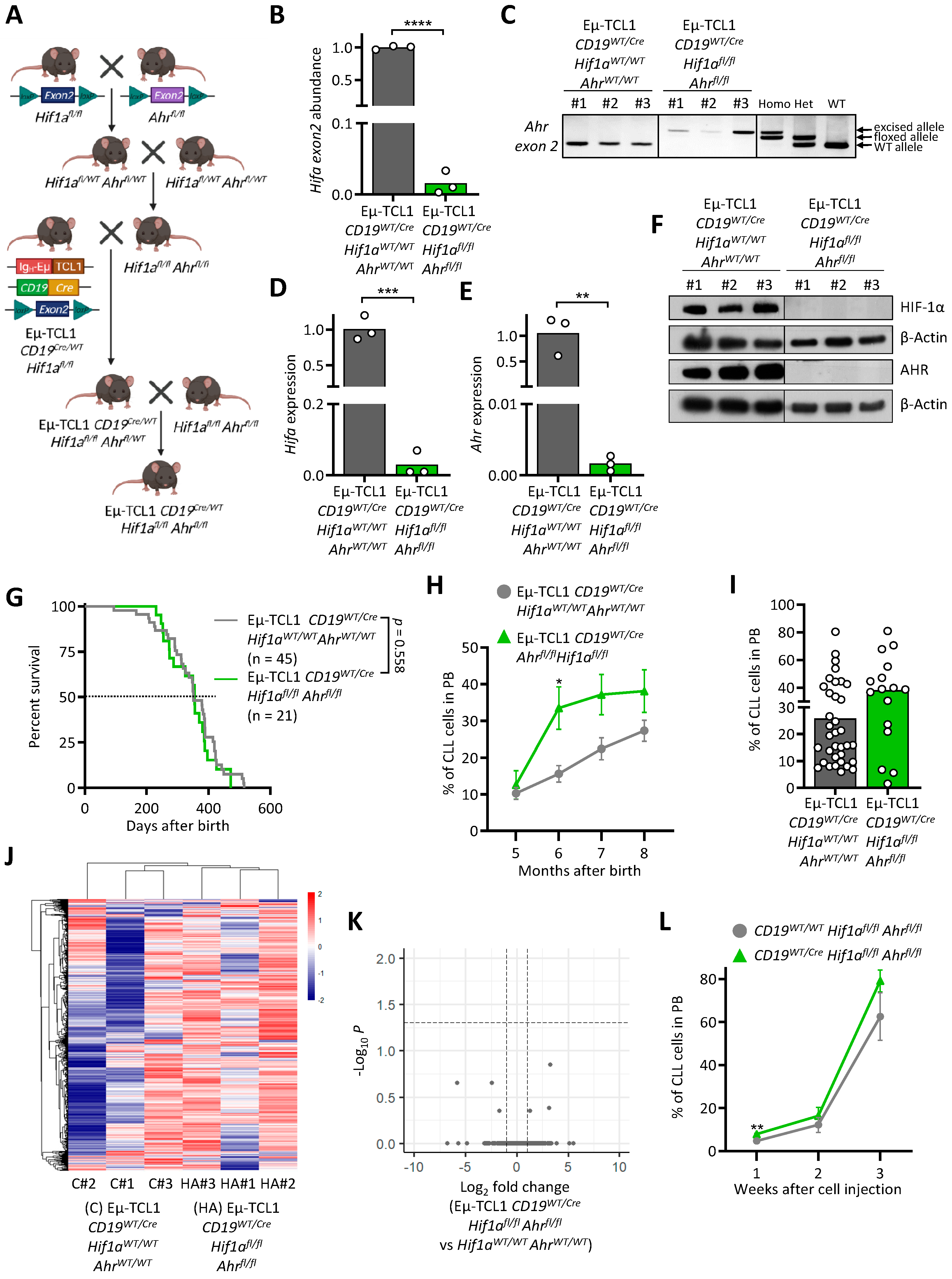
3.1. RNA Sequencing of CD5+CD19+ Leukemic B Cells from Eµ-TCL1 Mice
3.2. HIF-1α Knock-Out in Murine CLL Cells Does Not Impact Leukemogenesis
3.3. AHR Knock-Out in Murine CLL Cells Does Not Impact Leukemogenesis
3.4. AHR and HIF-1α Double Knock-Out Does Not Interfere with Murine CLL Development
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chiorazzi, N.; Chen, S.S.; Rai, K.R. Chronic Lymphocytic Leukemia. Cold Spring Harb. Perspect. Med. 2021, 11, a035220. [Google Scholar] [CrossRef] [PubMed]
- Herishanu, Y.; Katz, B.Z.; Lipsky, A.; Wiestner, A. Biology of chronic lymphocytic leukemia in different microenvironments: Clinical and therapeutic implications. Hematol. Oncol. Clin. 2013, 27, 173–206. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Belluti, S.; Rigillo, G.; Imbriano, C. Transcription Factors in Cancer: When Alternative Splicing Determines Opposite Cell Fates. Cells 2020, 9, 760. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.; et al. Mutational landscape and significance across 12 major cancer types. Nat. Cell Biol. 2013, 502, 333–339. [Google Scholar] [CrossRef]
- Venkatraman, S.; Meller, J.; Hongeng, S.; Tohtong, R.; Chutipongtanate, S. Transcriptional Regulation of Cancer Immune Checkpoints: Emerging Strategies for Immunotherapy. Vaccines 2020, 8, 735. [Google Scholar] [CrossRef] [PubMed]
- Moia, R.; Patriarca, A.; Deambrogi, C.; Rasi, S.; Favini, C.; Kodipad, A.A.; Schipani, M.; Gaidano, G. An update on: Molecular genetics of high-risk chronic lymphocytic leukemia. Expert Rev. Hematol. 2019, 13, 109–116. [Google Scholar] [CrossRef]
- Hallek, M. Chronic lymphocytic leukemia: 2020 update on diagnosis, risk stratification and treatment. Am. J. Hematol. 2019, 94, 1266–1287. [Google Scholar] [CrossRef]
- Vito, A.; El-Sayes, N.; Mossman, K. Hypoxia-Driven Immune Escape in the Tumor Microenvironment. Cells 2020, 9, 992. [Google Scholar] [CrossRef] [PubMed]
- Campesato, L.F.; Budhu, S.; Tchaicha, J.; Weng, C.-H.; Gigoux, M.; Cohen, I.J.; Redmond, D.; Mangarin, L.; Pourpe, S.; Liu, C.; et al. Blockade of the AHR restricts a Treg-macrophage suppressive axis induced by L-Kynurenine. Nat. Commun. 2020, 11, 4011. [Google Scholar] [CrossRef] [PubMed]
- Irigoyen, M.; Garcia-Ruiz, J.C.; Berra, E. The hypoxia signalling pathway in haematological malignancies. Oncotarget 2017, 8, 36832–36844. [Google Scholar] [CrossRef]
- Paris, A.; Tardif, N.; Galibert, M.-D.; Corre, S. AhR and Cancer: From Gene Profiling to Targeted Therapy. Int. J. Mol. Sci. 2021, 22, 752. [Google Scholar] [CrossRef]
- Gentil, M.; Hugues, P.; Desterke, C.; Telliam, G.; Sloma, I.; Souza, L.E.B.; Baykal, S.; Artus, J.; Griscelli, F.; Guerci, A.; et al. Aryl hydrocarbon receptor (AHR) is a novel druggable pathway controlling malignant progenitor proliferation in chronic myeloid leukemia (CML). PLoS ONE 2018, 13, e0200923. [Google Scholar] [CrossRef]
- Cheong, J.E.; Sun, L. Targeting the IDO1/TDO2-KYN-AhR Pathway for Cancer Immunotherapy—Challenges and Opportunities. Trends Pharmacol. Sci. 2018, 39, 307–325. [Google Scholar] [CrossRef]
- Liu, Y.; Liang, X.; Dong, W.; Fang, Y.; Lv, J.; Zhang, T.; Fiskesund, R.; Xie, J.; Liu, J.; Yin, X.; et al. Tumor-Repopulating Cells Induce PD-1 Expression in CD8+ T Cells by Transferring Kynurenine and AhR Activation. Cancer Cell 2018, 33, 480–494. [Google Scholar] [CrossRef]
- Xue, P.; Fu, J.; Zhou, Y. The Aryl Hydrocarbon Receptor and Tumor Immunity. Front. Immunol. 2018, 9, 286. [Google Scholar] [CrossRef]
- Iommarini, L.; Porcelli, A.M.; Gasparre, G.; Kurelac, I. Non-Canonical Mechanisms Regulating Hypoxia-Inducible Factor 1 Alpha in Cancer. Front. Oncol. 2017, 7, 286. [Google Scholar] [CrossRef]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Kontos, C.; Papageorgiou, S.G.; Diamantopoulos, M.A.; Scorilas, A.; Bazani, E.; Vasilatou, D.; Gkontopoulos, K.; Glezou, E.; Stavroulaki, G.; Dimitriadis, G.; et al. mRNA overexpression of the hypoxia inducible factor 1 alpha subunit gene (HIF1A): An independent predictor of poor overall survival in chronic lymphocytic leukemia. Leuk. Res. 2016, 53, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Wagage, S.; Hunter, C.A. Interrelated Roles for the Aryl Hydrocarbon Receptor and Hypoxia Inducible Factor-1α in the Immune Response to Infection. Curr. Med. Chem. 2015, 11, 43–54. [Google Scholar] [CrossRef][Green Version]
- Sherr, D.H.; Monti, S. The role of the aryl hydrocarbon receptor in normal and malignant B cell development. Semin. Immunopathol. 2013, 35, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Burrows, N.; Maxwell, P.H. Hypoxia and B cells. Exp. Cell Res. 2017, 356, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Villa, M.; Gialitakis, M.; Tolaini, M.; Ahlfors, H.; Henderson, C.J.; Wolf, C.R.; Brink, R.; Stockinger, B. Aryl hydrocarbon receptor is required for optimal B-cell proliferation. EMBO J. 2017, 36, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Piper, C.J.; Rosser, E.C.; Oleinika, K.; Nistala, K.; Krausgruber, T.; Rendeiro, A.; Banos, A.; Drozdov, I.; Villa, M.; Thomson, S.; et al. Aryl Hydrocarbon Receptor Contributes to the Transcriptional Program of IL-10-Producing Regulatory B Cells. Cell Rep. 2019, 29, 1878–1892. [Google Scholar] [CrossRef]
- DiLillo, D.J.; Weinberg, J.B.; Yoshizaki, A.; Horikawa, M.; Bryant, J.M.; Iwata, Y.; Matsushita, T.; Matta, K.M.; Chen, Y.; Venturi, G.M.; et al. Chronic lymphocytic leukemia and regulatory B cells share IL-10 competence and immunosuppressive function. Leukemia 2013, 27, 170–182. [Google Scholar] [CrossRef]
- Valsecchi, R.; Coltella, N.; Belloni, D.; Ponente, M.; Hacken, E.T.; Scielzo, C.; Scarfo’, L.; Bertilaccio, M.T.S.; Brambilla, P.; Lenti, E.; et al. HIF-1α regulates the interaction of chronic lymphocytic leukemia cells with the tumor microenvironment. Blood 2016, 127, 1987–1997. [Google Scholar] [CrossRef]
- Griggio, V.; Vitale, C.; Todaro, M.; Riganti, C.; Kopecka, J.; Salvetti, C.; Bomben, R.; Bo, M.D.; Magliulo, D.; Rossi, D.; et al. HIF-1α is over-expressed in leukemic cells from TP53-disrupted patients and is a promising therapeutic target in chronic lymphocytic leukemia. Haematologica 2019, 105, 1042–1054. [Google Scholar] [CrossRef] [PubMed]
- Bichi, R.; Shinton, S.A.; Martin, E.S.; Koval, A.; Calin, G.; Cesari, R.; Russo, G.; Hardy, R.R.; Croce, C.M. Human chronic lymphocytic leukemia modeled in mouse by targeted TCL1 expression. Proc. Natl. Acad. Sci. USA 2002, 99, 6955–6960. [Google Scholar] [CrossRef]
- Gonder, S.; Botana, I.F.; Wierz, M.; Pagano, G.; Gargiulo, E.; Cosma, A.; Moussay, E.; Paggetti, J.; Largeot, A. Method for the Analysis of the Tumor Microenvironment by Mass Cytometry: Application to Chronic Lymphocytic Leukemia. Front. Immunol. 2020, 11, 578176. [Google Scholar] [CrossRef]
- Wierz, M.; Pierson, S.; Chouha, N.; Désaubry, L.; François, J.-H.; Berchem, G.; Paggetti, J.; Moussay, E. The prohibitin-binding compound fluorizoline induces apoptosis in chronic lymphocytic leukemia cells ex vivo but fails to prevent leukemia development in a murine model. Haematologica 2018, 103, e154–e157. [Google Scholar] [CrossRef]
- Wierz, M.; Janji, B.; Berchem, G.; Moussay, E.; Paggetti, J. High-dimensional mass cytometry analysis revealed microenvironment complexity in chronic lymphocytic leukemia. OncoImmunology 2018, 7, e1465167. [Google Scholar] [CrossRef]
- Wierz, M.; Pierson, S.; Guyonnet, L.; Viry, E.; Lequeux, A.; Oudin, A.; Niclou, S.P.; Ollert, M.; Berchem, G.; Janji, B.; et al. Dual PD1/LAG3 immune checkpoint blockade limits tumor development in a murine model of chronic lymphocytic leukemia. Blood 2018, 131, 1617–1621. [Google Scholar] [CrossRef]
- Meng, X.; Grötsch, B.; Luo, Y.; Knaup, K.X.; Wiesener, M.S.; Chen, X.-X.; Jantsch, J.; Fillatreau, S.; Schett, G.; Bozec, A. Hypoxia-inducible factor-1α is a critical transcription factor for IL-10-producing B cells in autoimmune disease. Nat. Commun. 2018, 9, 251. [Google Scholar] [CrossRef]
- Kovalova, N.; Nault, R.; Crawford, R.; Zacharewski, T.R.; Kaminski, N.E. Comparative analysis of TCDD-induced AhR-mediated gene expression in human, mouse and rat primary B cells. Toxicol. Appl. Pharmacol. 2017, 316, 95–106. [Google Scholar] [CrossRef]
- Wang, G.L.; Jiang, B.-H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed]
- Bacsi, S.G.; Reisz-Porszasz, S.; Hankinson, O. Orientation of the heterodimeric aryl hydrocarbon (dioxin) receptor complex on its asymmetric DNA recognition sequence. Mol. Pharmacol. 1995, 47, 432–438. [Google Scholar] [PubMed]
- Vaidyanathan, B.; Chaudhry, A.; Yewdell, W.T.; Angeletti, D.; Yen, W.-F.; Wheatley, A.; Bradfield, C.A.; McDermott, A.B.; Yewdell, J.W.; Rudensky, A.Y.; et al. The aryl hydrocarbon receptor controls cell-fate decisions in B cells. J. Exp. Med. 2017, 214, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Vakili-Samiani, S.; Jalil, A.T.; Abdelbasset, W.K.; Yumashev, A.V.; Karpisheh, V.; Jalali, P.; Adibfar, S.; Ahmadi, M.; Feizi, A.A.H.; Jadidi-Niaragh, F. Targeting Wee1 kinase as a therapeutic approach in Hematological Malignancies. DNA Repair 2021, 107, 103203. [Google Scholar] [CrossRef]
- Lambert, M.; Jambon, S.; Depauw, S.; David-Cordonnier, M.-H. Targeting Transcription Factors for Cancer Treatment. Molecules 2018, 23, 1479. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, G.; Cantrell, D. Environmental and metabolic sensors that control T cell biology. Front. Immunol. 2015, 6, 99. [Google Scholar] [CrossRef] [PubMed]
- Magliulo, D.; Bernardi, R. HIF-α factors as potential therapeutic targets in leukemia. Expert Opin. Ther. Targets 2018, 22, 917–928. [Google Scholar] [CrossRef]
- Bianchi-Smiraglia, A.; Bagati, A.; Fink, E.E.; Affronti, H.C.; Lipchick, B.C.; Moparthy, S.; Long, M.D.; Rosario, S.R.; Lightman, S.M.; Moparthy, K.; et al. Inhibition of the aryl hydrocarbon receptor/polyamine biosynthesis axis suppresses multiple myeloma. J. Clin. Investig. 2018, 128, 4682–4696. [Google Scholar] [CrossRef]
- Largeot, A.; Pagano, G.; Gonder, S.; Moussay, E.; Paggetti, J. The B-side of Cancer Immunity: The Underrated Tune. Cells 2019, 8, 449. [Google Scholar] [CrossRef] [PubMed]
- Alhakeem, S.; McKenna, M.K.; Oben, K.Z.; Noothi, S.K.; Rivas, J.R.; Hildebrandt, G.C.; Fleischman, R.A.; Rangnekar, V.M.; Muthusamy, N.; Bondada, S. Chronic Lymphocytic Leukemia–Derived IL-10 Suppresses Antitumor Immunity. J. Immunol. 2018, 200, 4180–4189. [Google Scholar] [CrossRef] [PubMed]
- Vitale, C.; Griggio, V.; Riganti, C.; Todaro, M.; Kopecka, J.; Jones, R.; Salvetti, C.; Boccellato, E.; Perutelli, F.; Voena, C.; et al. Targeting HIF-1α Regulatory Pathways as a Strategy to Hamper Tumor-Microenvironment Interactions in CLL. Cancers 2021, 13, 2883. [Google Scholar] [CrossRef]
- Serra, S.; Vaisitti, T.; Audrito, V.; Bologna, C.; Buonincontri, R.; Chen, S.-S.; Arruga, F.; Brusa, D.; Coscia, M.; Jaksic, O.; et al. Adenosine signaling mediates hypoxic responses in the chronic lymphocytic leukemia microenvironment. Blood Adv. 2016, 1, 47–61. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Yue, L.; Shi, J.; Shao, M.; Wu, T. Role of IDO and TDO in Cancers and Related Diseases and the Therapeutic Implications. J. Cancer 2019, 10, 2771–2782. [Google Scholar] [CrossRef]
- Lindström, V.; Aittoniemi, J.; Jylhävä, J.; Eklund, C.; Hurme, M.; Paavonen, T.; Oja, S.S.; Itälä-Remes, M.; Sinisalo, M. Indoleamine 2,3-Dioxygenase Activity and Expression in Patients with Chronic Lymphocytic Leukemia. Clin. Lymphoma Myeloma Leuk. 2012, 12, 363–365. [Google Scholar] [CrossRef] [PubMed]
- Jitschin, R.; Braun, M.; Büttner, M.; Dettmer-Wilde, K.; Bricks, J.; Berger, J.; Eckart, M.J.; Krause, S.; Oefner, P.J.; Le Blanc, K.; et al. CLL-cells induce IDOhi CD14+HLA-DRlo myeloid-derived suppressor cells that inhibit T-cell responses and promote TRegs. Blood 2014, 124, 750–760. [Google Scholar] [CrossRef]
- Sadik, A.; Patterson, L.F.S.; Öztürk, S.; Mohapatra, S.R.; Panitz, V.; Secker, P.F.; Pfänder, P.; Loth, S.; Salem, H.; Prentzell, M.T.; et al. IL4I1 Is a Metabolic Immune Checkpoint that Activates the AHR and Promotes Tumor Progression. Cell 2020, 182, 1252–1270. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rank | GeneSets | SIZE | NES | NOM p-Val |
|---|---|---|---|---|
| 1 | HALLMARK_MYC_TARGETS_V1 | 200 | 1.46 | 0.000 |
| 2 | HALLMARK_CHOLESTEROL_HOMEOSTASIS | 73 | 1.45 | 0.000 |
| 3 | HALLMARK_GLYCOLYSIS | 200 | 1.4 | 0.000 |
| 4 | HALLMARK_E2F_TARGETS | 200 | 1.4 | 0.185 |
| 6 | HALLMARK_MYC_TARGETS_V2 | 57 | 1.38 | 0.000 |
| 7 | HALLMARK_OXIDATIVE_PHOSPHORYLATION | 197 | 1.35 | 0.000 |
| 8 | HALLMARK_REACTIVE_OXYGEN_SPECIES_PATHWAY | 49 | 1.34 | 0.000 |
| 9 | HALLMARK_FATTY_ACID_METABOLISM | 157 | 1.34 | 0.000 |
| 10 | HALLMARK_G2M_CHECKPOINT | 196 | 1.29 | 0.185 |
| 11 | HALLMARK_UV_RESPONSE_UP | 156 | 1.28 | 0.000 |
| 15 | HALLMARK_HYPOXIA | 198 | 1.22 | 0.000 |
| 16 | HALLMARK_P53_PATHWAY | 200 | 1.22 | 0.084 |
| 18 | HALLMARK_IL2_STAT5_SIGNALING | 199 | 1.2 | 0.094 |
| 20 | HALLMARK_MTORC1_SIGNALING | 199 | 1.18 | 0.287 |
| 21 | HALLMARK_UNFOLDED_PROTEIN_RESPONSE | 112 | 1.15 | 0.185 |
| 24 | HALLMARK_MITOTIC_SPINDLE | 199 | 1.13 | 0.269 |
| 25 | HALLMARK_DNA_REPAIR | 148 | 1.13 | 0.084 |
| 28 | HALLMARK_XENOBIOTIC_METABOLISM | 196 | 1.09 | 0.299 |
| 30 | HALLMARK_APOPTOSIS | 161 | 1.01 | 0.376 |
| 31 | HALLMARK_IL6_JAK_STAT3_SIGNALING | 87 | 1.01 | 0.381 |
| 32 | HALLMARK_PROTEIN_SECRETION | 96 | 1.01 | 0.472 |
| 33 | HALLMARK_WNT_BETA_CATENIN_SIGNALING | 41 | 1 | 0.362 |
| Rank | TF | Score | Rank | TF | Score |
|---|---|---|---|---|---|
| 1 | FOXM1 | 4.333 | 26 | CEBPB | 88.83 |
| 2 | CENPA | 5.0 | 27 | NFE2L2 | 89.4 |
| 3 | ZNF367 | 8.333 | 28 | ARNTL2 | 94.67 |
| 4 | PA2G4 | 15.5 | 29 | JDP2 | 95.67 |
| 5 | HMGA2 | 24.67 | 30 | ZNF888 | 97.0 |
| 6 | AHRR | 26.5 | 31 | BHLHE40 | 97.75 |
| 7 | ZNF695 | 29.0 | 32 | ZNF670 | 98.5 |
| 8 | E2F7 | 29.2 | 33 | OSR1 | 99.5 |
| 9 | E2F1 | 33.83 | 34 | GLMP | 101.5 |
| 10 | FOSL1 | 35.4 | 35 | HMGN3 | 105.0 |
| 11 | ELK3 | 49.0 | 36 | ZNF93 | 106.7 |
| 12 | FOXD1 | 51.5 | 37 | AHR | 107.4 |
| 13 | MYBL2 | 55.17 | 38 | PPARG | 108.0 |
| 14 | TFDP1 | 56.0 | 39 | TAL1 | 109.0 |
| 15 | E2F2 | 65.33 | 40 | ATF3 | 110.3 |
| 16 | RFX8 | 68.0 | 41 | TEAD4 | 119.8 |
| 17 | CENPX | 68.0 | 42 | NR1H3 | 120.0 |
| 18 | KLF6 | 72.0 | 43 | HIF1A | 120.2 |
| 19 | CREB3L2 | 75.33 | 44 | VDR | 120.8 |
| 20 | ETV4 | 76.5 | 45 | KLF9 | 123.8 |
| 21 | ZNF878 | 78.0 | 46 | GLIS2 | 126.3 |
| 22 | E2F8 | 80.0 | 47 | RELB | 128.3 |
| 23 | DNMT1 | 81.0 | 48 | PLSCR1 | 131.0 |
| 24 | NR2F6 | 85.5 | 49 | TEAD3 | 132.7 |
| 25 | KLF4 | 87.0 | 50 | HLX | 136.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonder, S.; Largeot, A.; Gargiulo, E.; Pierson, S.; Fernandez Botana, I.; Pagano, G.; Paggetti, J.; Moussay, E. The Tumor Microenvironment-Dependent Transcription Factors AHR and HIF-1α Are Dispensable for Leukemogenesis in the Eµ-TCL1 Mouse Model of Chronic Lymphocytic Leukemia. Cancers 2021, 13, 4518. https://doi.org/10.3390/cancers13184518
Gonder S, Largeot A, Gargiulo E, Pierson S, Fernandez Botana I, Pagano G, Paggetti J, Moussay E. The Tumor Microenvironment-Dependent Transcription Factors AHR and HIF-1α Are Dispensable for Leukemogenesis in the Eµ-TCL1 Mouse Model of Chronic Lymphocytic Leukemia. Cancers. 2021; 13(18):4518. https://doi.org/10.3390/cancers13184518
Chicago/Turabian StyleGonder, Susanne, Anne Largeot, Ernesto Gargiulo, Sandrine Pierson, Iria Fernandez Botana, Giulia Pagano, Jerome Paggetti, and Etienne Moussay. 2021. "The Tumor Microenvironment-Dependent Transcription Factors AHR and HIF-1α Are Dispensable for Leukemogenesis in the Eµ-TCL1 Mouse Model of Chronic Lymphocytic Leukemia" Cancers 13, no. 18: 4518. https://doi.org/10.3390/cancers13184518
APA StyleGonder, S., Largeot, A., Gargiulo, E., Pierson, S., Fernandez Botana, I., Pagano, G., Paggetti, J., & Moussay, E. (2021). The Tumor Microenvironment-Dependent Transcription Factors AHR and HIF-1α Are Dispensable for Leukemogenesis in the Eµ-TCL1 Mouse Model of Chronic Lymphocytic Leukemia. Cancers, 13(18), 4518. https://doi.org/10.3390/cancers13184518