
Transcriptome of Male Breast Cancer Matched with Germline Profiling Reveals Novel Molecular Subtypes with Possible Clinical Relevance

 , , , , , , ,
, , , , , , ,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. RNA Isolation and Sequencing
2.3. Data Analysis
2.3.1. Differential Gene Expression Analysis
2.3.2. Gene Enrichment and Pathway-Based Analysis
2.3.3. Clustering Analysis
2.3.4. Statistical Analyses
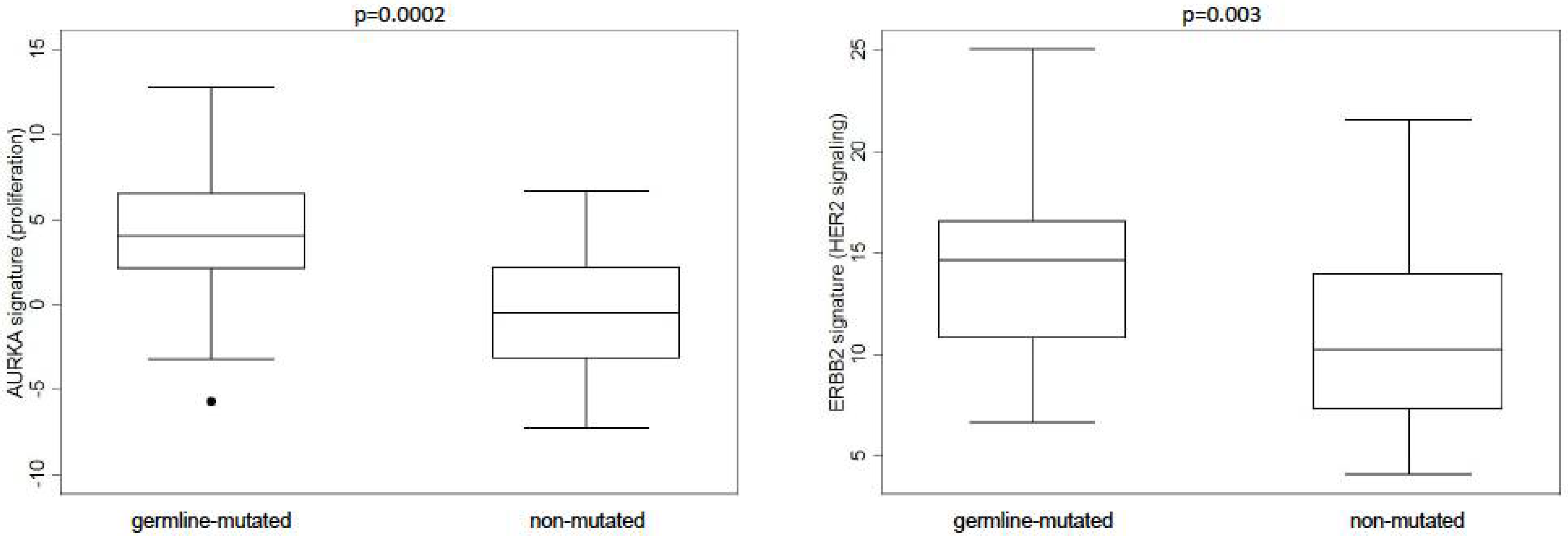
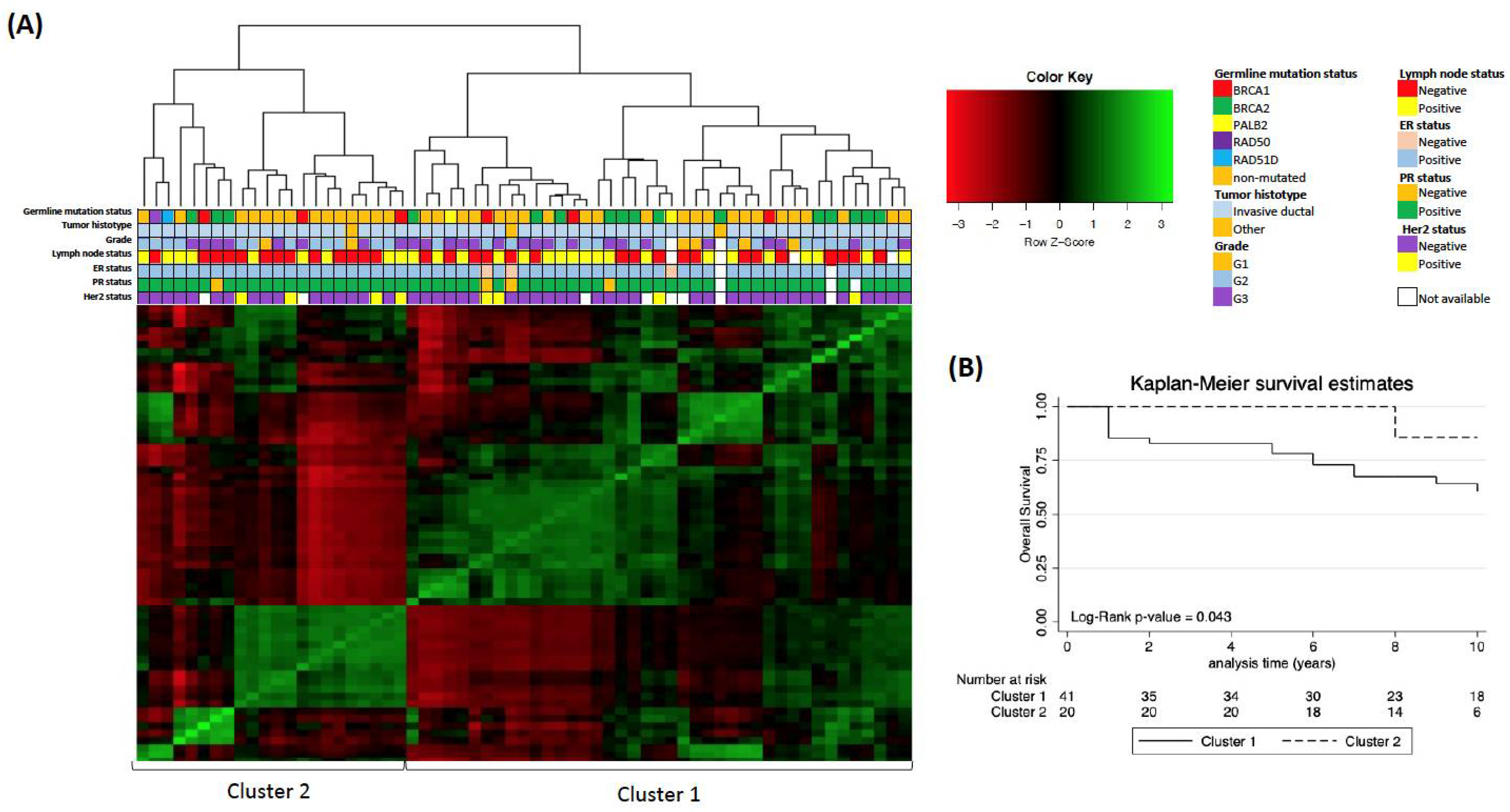
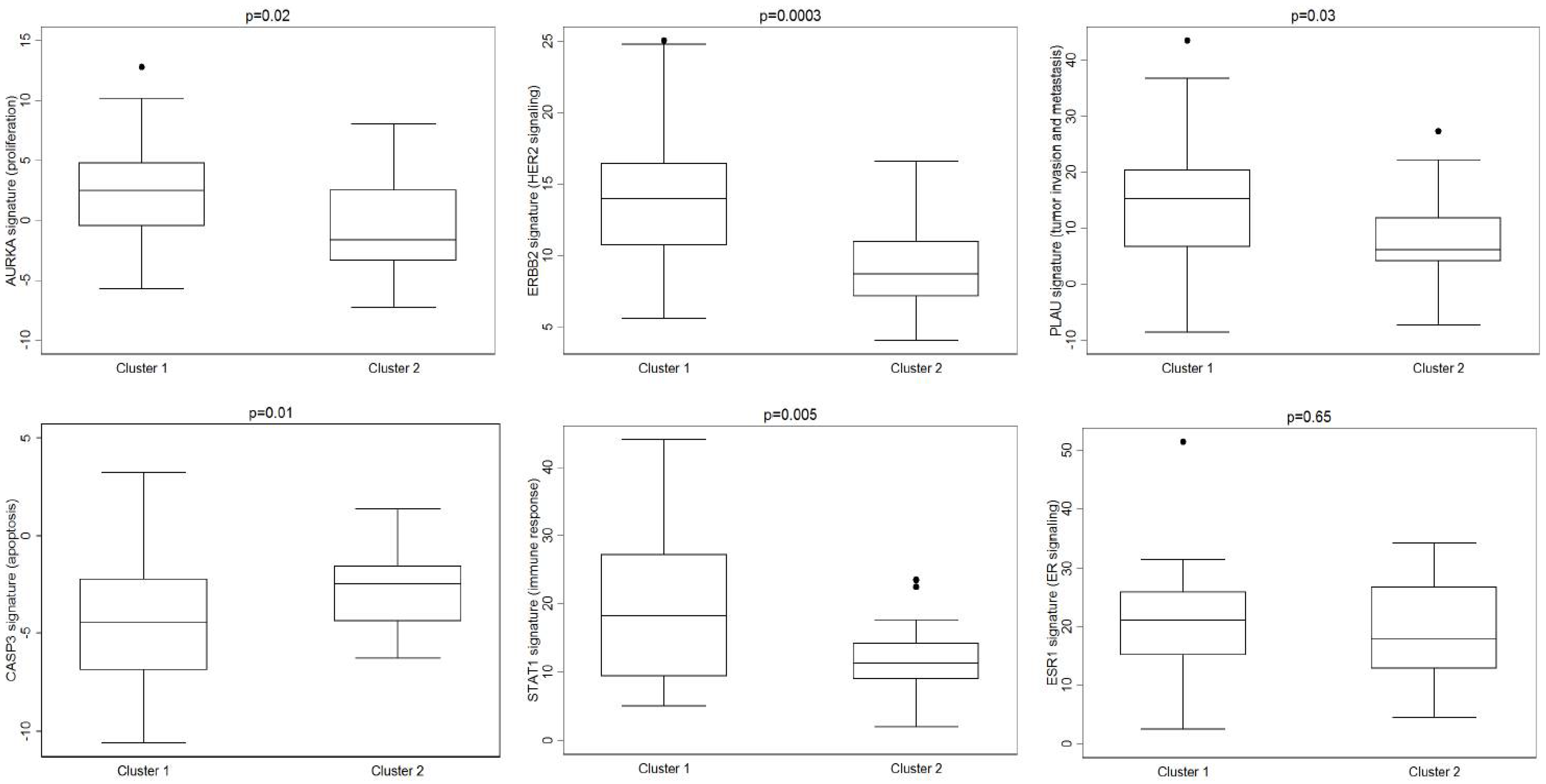
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rizzolo, P.; Silvestri, V.; Falchetti, M.; Ottini, L. Inherited and acquired alterations in development of breast cancer. Appl. Clin. Genet. 2011, 4, 145–158. [Google Scholar] [CrossRef]
- Silvestri, V.; Zelli, V.; Valentini, V.; Rizzolo, P.; Navazio, A.S.; Coppa, A.; Agata, S.; Oliani, C.; Barana, D.; Castrignanò, T.; et al. Whole-exome sequencing and targeted gene sequencing provide insights into the role of PALB2 as a male breast cancer susceptibility gene. Cancer 2017, 123, 210–218. [Google Scholar] [CrossRef]
- Rizzolo, P.; Zelli, V.; Silvestri, V.; Valentini, V.; Zanna, I.; Bianchi, S.; Masala, G.; Spinelli, A.M.; Tibiletti, M.G.; Russo, A.; et al. Insight into genetic susceptibility to male breast cancer by multigene panel testing: Results from a multicenter study in Italy. Int. J. Cancer 2019, 145, 390–400. [Google Scholar] [CrossRef]
- Pritzlaff, M.; Summerour, P.; McFarland, R.; Li, S.; Reineke, P.; Dolinsky, J.S.; Goldgar, D.E.; Shimelis, H.; Couch, F.J.; Chao, E.C.; et al. Male breast cancer in a multi-gene panel testing cohort: Insights and unexpected results. Breast Cancer Res. Treat. 2017, 161, 575–586. [Google Scholar] [CrossRef]
- Silvestri, V.; Rizzolo, P.; Zelli, V.; Valentini, V.; Zanna, I.; Bianchi, S.; Tibiletti, M.G.; Varesco, L.; Russo, A.; Tommasi, S.; et al. A possible role of FANCM mutations in male breast cancer susceptibility: Results from a multicenter study in Italy. Breast 2018, 38, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Fostira, F.; Saloustros, E.; Apostolou, P.; Vagena, A.; Kalfakakou, D.; Mauri, D.; Tryfonopoulos, D.; Georgoulias, V.; Yannoukakos, D.; Fountzilas, G.; et al. Germline deleterious mutations in genes other than BRCA2 are infrequent in male breast cancer. Breast Cancer Res. Treat. 2018, 169, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Ottini, L. Male breast cancer: A rare disease that might uncover underlying pathways of breast cancer. Nat. Rev. Cancer 2014, 14, 643. [Google Scholar] [CrossRef]
- Bayani, J.; Poncet, C.; Crozier, C.; Neven, A.; Piper, T.; Cunningham, C.; Sobol, M.; Aebi, S.; Benstead, K.; Bogler, O.; et al. Evaluation of multiple transcriptomic gene risk signatures in male breast cancer. NPJ Breast Cancer 2021, 7, 98. [Google Scholar] [CrossRef]
- Gucalp, A.; Traina, T.A.; Eisner, J.R.; Parker, J.S.; Selitsky, S.R.; Park, B.H. Male breast cancer: A disease distinct from female breast cancer. Breast Cancer Res. Treat. 2019, 173, 37–48. [Google Scholar] [CrossRef]
- Silvestri, V.; Barrowdale, D.; Mulligan, A.M.; Neuhausen, S.L.; Fox, S.; Karlan, B.Y.; Mitchell, G.; James, P.; Thull, D.L.; Zorn, K.K.; et al. Male breast cancer in BRCA1 and BRCA2 mutation carriers: Pathology data from the Consortium of Investigators of Modifiers of BRCA1/2. Breast Cancer Res. 2016, 18, 15. [Google Scholar] [CrossRef]
- Vermeulen, M.A.; Slaets, L.; Cardoso, F.; Giordano, S.H.; Tryfonidis, K.; van Diest, P.J.; Dijkstra, N.H.; Schröder, C.P.; van Asperen, C.J.; Linderholm, B.; et al. Pathological characterisation of male breast cancer: Results of the EORTC 10085/TBCRC/BIG/NABCG International Male Breast Cancer Program. Eur. J. Cancer 2017, 82, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.K.; Schultheis, A.M.; Bidard, F.C.; Weigelt, B.; Reis-Filho, J.S. Breast cancer genomics from microarrays to massively parallel sequencing: Paradigms and new insights. J. Natl. Cancer Inst. 2015, 107, djv015. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.J.; Thomassen, M.; Tan, Q.; Lænkholm, A.V.; Bak, M.; Sørensen, K.P.; Andersen, M.K.; Kruse, T.A.; Gerdes, A.N. RNA profiling reveals familial aggregation of molecular subtypes in non-BRCA breast cancer families. BMC Med. Genom. 2014, 7, 9. [Google Scholar] [CrossRef]
- Akbari, V.; Kallhor, M.; Akbari, M.T. Transcriptome mining of non-BRCA1/2 and BRCA1/2 familial breast cancer. J. Cell Biochem. 2019, 120, 575–583. [Google Scholar] [CrossRef]
- Callari, M.; Cappelletti, V.; De Cecco, L.; Musella, V.; Miodini, P.; Veneroni, S.; Gariboldi, M.; Pierotti, M.A.; Daidone, M.G. Gene expression analysis reveals a different transcriptomic landscape in female and male breast cancer. Breast Cancer Res. Treat. 2011, 127, 601–610. [Google Scholar] [CrossRef]
- Johansson, I.; Nilsson, C.; Berglund, P.; Lauss, M.; Ringnér, M.; Olsson, H.; Luts, L.; Sim, E.; Thorstensson, S.; Fjällskog, M.L.; et al. Gene expression profiling of primary male breast cancers reveals two unique subgroups and identifies N-acetyltransferase-1 (NAT1) as a novel prognostic biomarker. Breast Cancer Res. 2012, 14, R31. [Google Scholar] [CrossRef]
- Humphries, M.P.; Sundara Rajan, S.; Droop, A.; Suleman, C.A.B.; Carbone, C.; Nilsson, C.; Honarpisheh, H.; Cserni, G.; Dent, J.; Fulford, L.; et al. A Case-Matched Gender Comparison Transcriptomic Screen Identifies eIF4E and eIF5 as Potential Prognostic Markers in Male Breast Cancer. Clin. Cancer Res. 2017, 23, 2575–2583. [Google Scholar] [CrossRef][Green Version]
- André, S.; Pereira, T.; Silva, F.; Machado, P.; Vaz, F.; Aparício, M.; Silva, G.L.; Pinto, A.E. Male breast cancer: Specific biological characteristics and survival in a Portuguese cohort. Mol. Clin. Oncol. 2019, 10, 644–654. [Google Scholar] [CrossRef]
- Ottini, L.; Silvestri, V.; Rizzolo, P.; Falchetti, M.; Zanna, I.; Saieva, C.; Masala, G.; Bianchi, S.; Manoukian, S.; Barile, M.; et al. Clinical and pathologic characteristics of BRCA-positive and BRCA-negative male breast cancer patients: Results from a collaborative multicenter study in Italy. Breast Cancer Res. Treat. 2012, 134, 411–418. [Google Scholar] [CrossRef]
- Thorne, H.; Mitchell, G.; Fox, S.; kConFab Consortium. kConFab: A familial breast cancer consortium facilitating research and translational oncology. J. Natl. Cancer Inst. Monogr. 2011, 2011, 79–81. [Google Scholar] [CrossRef]
- Desmedt, C.; Haibe-Kains, B.; Wirapati, P.; Buyse, M.; Larsimont, D.; Bontempi, G.; Delorenzi, M.; Piccart, M.; Sotiriou, C. Biological processes associated with breast cancer clinical outcome depend on the molecular subtypes. Clin. Cancer Res. 2008, 14, 5158–5165. [Google Scholar] [CrossRef]
- Suzuki, R.; Shimodaira, H. Pvclust: An R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 2006, 22, 1540–1542. [Google Scholar] [CrossRef]
- Zanna, I.; Silvestri, V.; Palli, D.; Magrini, A.; Rizzolo, P.; Saieva, C.; Zelli, V.; Bendinelli, B.; Vezzosi, V.; Valentini, V.; et al. Smoking and FGFR2 rs2981582 variant independently modulate male breast cancer survival: A population-based study in Tuscany, Italy. Breast 2018, 40, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhang, Y.; Williams, J.; Antoniou, E.; McCombie, W.R.; Wu, S.; Zhu, W.; Davidson, N.O.; Denoya, P.; Li, E. Parallel comparison of Illumina RNA-Seq and Affymetrix microarray platforms on transcriptomic profiles generated from 5-aza-deoxy-cytidine treated HT-29 colon cancer cells and simulated datasets. BMC Bioinform. 2013, 14 (Suppl. 9), S1. [Google Scholar] [CrossRef]
- Zhao, S.; Fung-Leung, W.P.; Bittner, A.; Ngo, K.; Liu, X. Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. PLoS ONE 2014, 9, e78644. [Google Scholar] [CrossRef]
- Hung, J.H.; Weng, Z. Analysis of microarray and RNA-seq expression profiling data. Cold Spring Harb. Protoc. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Abdeljaoued, S.; Bettaieb, I.; Nasri, M.; Adouni, O.; Goucha, A.; El Amine, O.; Boussen, H.; Rahal, K.; Gamoudi, A. Overexpression of FOXM1 Is a Potential Prognostic Marker in Male Breast Cancer. Oncol. Res. Treat. 2017, 40, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Tang, A.; Gao, K.; Chu, L.; Zhang, R.; Yang, J.; Zheng, J. Aurora kinases: Novel therapy targets in cancers. Oncotarget 2017, 8, 23937–23954. [Google Scholar] [CrossRef]
- Davar, D.; Beumer, J.H.; Hamieh, L.; Tawbi, H. Role of PARP inhibitors in cancer biology and therapy. Curr. Med. Chem. 2012, 19, 3907–3921. [Google Scholar] [CrossRef]
- Giordano, S.H. Breast Cancer in Men. N. Engl. J. Med. 2018, 378, 2311–2320. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Bieche, I.; Girault, I.; Urbain, E.; Tozlu, S.; Lidereau, R. Relationship between intratumoral expression of genes coding for xenobiotic-metabolizing enzymes and benefit from adjuvant tamoxifen in estrogen receptor alpha-positive postmenopausal breast carcinoma. Breast Cancer Res. 2004, 6, R252–R263. [Google Scholar] [CrossRef] [PubMed]
- Hassett, M.J.; Somerfield, M.R.; Baker, E.R.; Cardoso, F.; Kansal, K.J.; Kwait, D.C.; Plichta, J.K.; Ricker, C.; Roshal, A.; Ruddy, K.J.; et al. Management of Male Breast Cancer: ASCO Guideline. J. Clin. Oncol. 2020, 38, JCO1903120. [Google Scholar] [CrossRef]
- US Department of Health and Human Services, Food and Drug Administration, Oncology Cener of Excellence, Center for Drug Evaluation and Research and Center for Biologics Evaluation and Research. Male Breast Cancer: Developing Drugs for Treatment Draft Guidance for Industry. August 2019. Available online: www.fda.gov/media/130061/download (accessed on 23 September 2019).
- Hansra, D.; Jackson, S.; Sequeira, J.; Vazirani, R.; Alvarez, R. Male patient with metastatic stage IV breast cancer achieves complete remission on second line Abemaciclib, Fulvestrant and Leuprolide: A case report. Mol. Clin. Oncol. 2020, 12, 120–125. [Google Scholar] [CrossRef]
- Paik, S.; Kim, C.; Wolmark, N. HER2 status and benefit from adjuvant trastuzumab in breast cancer. N. Engl. J. Med. 2008, 358, 1409–1411. [Google Scholar] [CrossRef] [PubMed]
- Perez, E.A.; Reinholz, M.M.; Hillman, D.W.; Tenner, K.S.; Schroeder, M.J.; Davidson, N.E.; Martino, S.; Sledge, G.W.; Harris, L.N.; Gralow, J.R.; et al. HER2 and chromosome 17 effect on patient outcome in the N9831 adjuvant trastuzumab trial. J. Clin. Oncol. 2010, 28, 4307–4315. [Google Scholar] [CrossRef] [PubMed]
- Vikas, P.; Borcherding, N.; Chennamadhavuni, A.; Garje, R. Therapeutic Potential of Combining PARP Inhibitor and Immunotherapy in Solid Tumors. Front. Oncol. 2020, 10, 570. [Google Scholar] [CrossRef]
- Moelans, C.B.; de Ligt, J.; van der Groep, P.; Prins, P.; Besselink, N.J.M.; Hoogstraat, M.; Ter Hoeve, N.D.; Lacle, M.M.; Kornegoor, R.; van der Pol, C.C.; et al. The molecular genetic make-up of male breast cancer. Endocr. Relat. Cancer 2019, 26, 779–794. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Characteristic 1 | MBCs (N. 63) | Germline- Mutated MBCs (N. 26) | Non-Mutated MBCs (N. 37) | p-Value 2 | |||
|---|---|---|---|---|---|---|---|
| N | % | N | % | N | % | ||
| Mean age at diagnosis ± SD (range) | 65.5 ± 11.0 (40–91) | 65.0 ± 12.3 (43–85) | 65.7 ± 10.1 (40–91) | 0.8 | |||
| Mean follow-up, years ± SD (range) | 7.3 ± 3.1 (1–10) | 6.8 ± 3.2 (1–10) | 7.6 ± 3.2 (1–10) | 0.3 | |||
| Tumor histotype | |||||||
| Invasive ductal carcinoma | 60 | 95.2 | 25 | 100.0 | 35 | 92.1 | |
| Other | 3 | 4.8 | 0 | 0.0 | 3 | 7.9 | 0.3 |
| Histologic grade | |||||||
| 1 | 6 | 9.8 | 0 | 0.0 | 6 | 16.2 | |
| 2 | 29 | 47.6 | 10 | 41.7 | 19 | 51.4 | |
| 3 | 26 | 42.6 | 14 | 58.3 | 12 | 32.4 | 0.03 |
| Lymph node status | |||||||
| Negative | 31 | 52.5 | 13 | 54.2 | 18 | 51.4 | |
| Positive | 28 | 47.5 | 11 | 45.8 | 17 | 48.6 | 1.0 |
| ER status | |||||||
| Negative | 3 | 4.9 | 2 | 8.3 | 1 | 2.7 | |
| Positive | 58 | 95.1 | 22 | 91.7 | 36 | 97.3 | 0.6 |
| PR status | |||||||
| Negative | 4 | 6.7 | 3 | 13.0 | 1 | 2.7 | |
| Positive | 56 | 93.3 | 20 | 87.0 | 36 | 97.3 | 0.2 |
| HER2 status | |||||||
| Negative | 47 | 85.5 | 17 | 81.0 | 30 | 88.2 | |
| Positive | 8 | 14.5 | 4 | 19.0 | 4 | 11.8 | 0.5 |
| Category | Term | Expression 1 | Count | Genes | p-Value |
|---|---|---|---|---|---|
| UP_KEYWORDS | Cell cycle | ↑ | 22 | KIFC1, PARD6B, DBF4B, TICRR, DLGAP5, FOXM1, RBL1, KNTC1, KIF18B, AURKA, CENPE, UBE2C, SYCP2, CDKN3, CCNE2, CDC45, NCAPG, HJURP, CIT, CCNA2, ASPM, MELK | 4.70 × 10−7 |
| UP_KEYWORDS | Cell division | ↑ | 13 | KIFC1, PARD6B, KNTC1, KIF18B, CENPE, AURKA, UBE2C, SYCP2, CCNE2, NCAPG, CIT, CCNA2, ASPM | 8.00 × 10−4 |
| UP_KEYWORDS | Mitosis | ↑ | 10 | KIFC1, NCAPG, KNTC1, KIF18B, CENPE, AURKA, UBE2C, CIT, CCNA2, ASPM | 0.002 |
| KEGG_PATHWAY | hsa03010:Ribosome | ↓ | 9 | RPS25, RPS17, RPL34, RPLP1, RPL26, RPS9, RPL24, RSL24D1, RPL4 | 0.003 |
| GOTERM_BP_DIRECT | GO:0051301—cell division | ↑ | 13 | KIF14, KIFC1, PARD6B, KNTC1, KIF18B, CENPE, AURKA, UBE2C, SYCP2, CCNE2, NCAPG, CCNA2, TUBA1C | 0.005 |
| GOTERM_BP_DIRECT | GO:0006413~translational initiation | ↓ | 9 | RPS25, RPS17, RPL34, RPLP1, RPL26, EIF3L, RPS9, RPL24, RPL4 | 0.009 |
| UP_KEYWORDS | DNA damage | ↑ | 9 | XRCC2, TICRR, FOXM1, BRIP1, ATAD5, RAD54B, POLQ, RAD54L, BARD1 | 0.032 |
| GOTERM_CC_DIRECT | GO:0070062—extracellular exosome | ↓ | 40 | KRT6C, NAMPT, C3, CSF1, SORL1, HEXB, CLU, ITM2B, RPS25, AZGP1, GPM6A, SERINC1, RPL34, RPLP1, FAT2, HSPA6, IGKV1D-12, LTF, RPL4, PRKACB, PDGFD, ARL6IP5, MUC13, RHOBTB3, PLAT, FLOT2, TMC5, RPL26, RPS9, RPL24, ENDOD1, SERPINI1, NUCB1, GNB2, RPS17, NUCB2, CYBRD1, CYFIP2, CA2, LRP2 | 0.039 |
| GOTERM_CC_DIRECT | GO:0005783—endoplasmic reticulum | ↓ | 18 | CAST, EEF1B2, CAMLG, CLU, SORL1, APH1B, VASH1, LRPAP1, FMO5, EPHA4, BRINP1, NUCB2, BCAP29, RAB38, ANO7, LRP2, KCNQ1, ARL6IP5 | 0.040 |
| UP_KEYWORDS | DNA repair | ↑ | 8 | XRCC2, TICRR, FOXM1, BRIP1, RAD54B, POLQ, RAD54L, BARD1 | 0.041 |
| KEGG_PATHWAY | hsa04110:Cell cycle | ↑ | 6 | CCNE2, CDC45, RBL1, TTK, ESPL1, CCNA2 | 0.044 |
| Category | Term | Expression 1 | Count | Genes | p-Value |
|---|---|---|---|---|---|
| UP_KEYWORDS | Immunity | ↑ | 16 | MICB, S100A8, IFITM2, GSDMD, IDO2, C4BPB, LY9, APOBEC3H, IGKV1-12, TLR9, IGHV3-11, NUDCD1, IGHV3-23, IGKV1D-39, IGKV3-20, IGHV3-13 | 0.002 |
| UP_KEYWORDS | Immunoglobulin domain | ↑ | 16 | IGHG1, MICB, IL18RAP, MPZL2, LY9, PIGR, SIRPB1, SIGIRR, IGHV3-11, LINGO1, IGSF5, IGHV3-23, IGKV1D-39, IGKV3-20, IGHV3-13, LAG3 | 0.003 |
| GOTERM_MF_DIRECT | GO:0003823~antigen binding | ↑ | 8 | IGHG1, IGHV3-11, MICB, IGHV3-23, IGKV1D-39, IGKV3-20, IGHV3-13, LAG3 | 0.005 |
| REACTOME_PATHWAY | R-HSA-166663:R-HSA-166663 Initial triggering of complement | ↑ | 6 | IGHG1, IGHV3-11, IGHV3-23, IGKV1D-39, IGKV3-20, IGHV3-13 | 0.015 |
| REACTOME_PATHWAY | R-HSA-2029481:R-HSA-2029481 FCGR activation | ↑ | 6 | IGHG1, IGHV3-11, IGHV3-23, IGKV1D-39, IGKV3-20, IGHV3-13 | 0.017 |
| REACTOME_PATHWAY | R-HSA-2029485:R-HSA-2029485 Role of phospholipids in phagocytosis | ↑ | 6 | IGHG1, IGHV3-11, IGHV3-23, IGKV1D-39, IGKV3-20, IGHV3-13 | 0.019 |
| REACTOME_PATHWAY | R-HSA-198933:R-HSA-198933 Immunoregulatory interactions between a Lymphoid and a non-Lymphoid cell | ↑ | 8 | KLRB1, IGHV3-11, MICB, IGHV3-23, IGKV1D-39, IGKV3-20, IGHV3-13, HCST | 0.019 |
| REACTOME_PATHWAY | R-HSA-173623:R-HSA-173623 Classical antibody-mediated complement activation | ↑ | 6 | IGHG1, IGHV3-11, IGHV3-23, IGKV1D-39, IGKV3-20, IGHV3-13 | 0.022 |
| UP_KEYWORDS | Immunoglobulin V region | ↑ | 5 | IGHV3-11, IGHV3-23, IGKV1D-39, IGKV3-20, IGHV3-13 | 0.022 |
| UP_KEYWORDS | Adaptive immunity | ↑ | 8 | IGHV3-11, MICB, IGHV3-23, IGKV1D-39, IGKV3-20, LY9, IGKV1-12, IGHV3-13 | 0.025 |
| REACTOME_PATHWAY | R-HSA-5690714:R-HSA-5690714 CD22 mediated BCR regulation | ↑ | 5 | IGHV3-11, IGHV3-23, IGKV1D-39, IGKV3-20, IGHV3-13 | 0.035 |
| REACTOME_PATHWAY | R-HSA-2454202:R-HSA-2454202 Fc epsilon receptor (FCERI) signaling | ↑ | 5 | IGHV3-11, IGHV3-23, IGKV1D-39, IGKV3-20, IGHV3-13 | 0.035 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zelli, V.; Silvestri, V.; Valentini, V.; Bucalo, A.; Rizzolo, P.; Zanna, I.; Bianchi, S.; Coppa, A.; Giannini, G.; Cortesi, L.; et al. Transcriptome of Male Breast Cancer Matched with Germline Profiling Reveals Novel Molecular Subtypes with Possible Clinical Relevance. Cancers 2021, 13, 4515. https://doi.org/10.3390/cancers13184515
Zelli V, Silvestri V, Valentini V, Bucalo A, Rizzolo P, Zanna I, Bianchi S, Coppa A, Giannini G, Cortesi L, et al. Transcriptome of Male Breast Cancer Matched with Germline Profiling Reveals Novel Molecular Subtypes with Possible Clinical Relevance. Cancers. 2021; 13(18):4515. https://doi.org/10.3390/cancers13184515
Chicago/Turabian StyleZelli, Veronica, Valentina Silvestri, Virginia Valentini, Agostino Bucalo, Piera Rizzolo, Ines Zanna, Simonetta Bianchi, Anna Coppa, Giuseppe Giannini, Laura Cortesi, and et al. 2021. "Transcriptome of Male Breast Cancer Matched with Germline Profiling Reveals Novel Molecular Subtypes with Possible Clinical Relevance" Cancers 13, no. 18: 4515. https://doi.org/10.3390/cancers13184515
APA StyleZelli, V., Silvestri, V., Valentini, V., Bucalo, A., Rizzolo, P., Zanna, I., Bianchi, S., Coppa, A., Giannini, G., Cortesi, L., Calistri, D., Tibiletti, M. G., Fox, S. B., Fab, k., Palli, D., & Ottini, L. (2021). Transcriptome of Male Breast Cancer Matched with Germline Profiling Reveals Novel Molecular Subtypes with Possible Clinical Relevance. Cancers, 13(18), 4515. https://doi.org/10.3390/cancers13184515