
NF-κB and Pancreatic Cancer; Chapter and Verse


Simple Summary
Abstract
1. Introduction
2. NF-κB, a Potent and Versatile Transducer of Inflammatory Signals
2.1. The NF-κB Family
2.2. Downstream Signaling of Canonical and Non-Canonical NF-κB Activation
3. NF-κB and Pancreatic Inflammation
3.1. Pancreatitis Elevates the Risk for PDAC Development
3.2. NF-κB a Key Initiator of Inflammatory Mediators in the Pancreas
3.3. NF-κB Promotor or Inhibitor of Pancreatitis?
3.4. Beyond Inflammation; Multiple Roles for NF-κB in Pancreatic Tumorigenesis
4. The Role of NF-κB in Various Facets of PDAC Biology
4.1. Cell Death; Apoptosis and Autophagy
4.2. NF-κB and Angiogenesis
4.3. Metastasis and NF-κB
4.4. NF-κB and Chemotherapeutic Drug Resistance
5. An Emerging Role for NF-κB in the Tumor Microenvironment
5.1. NF-κB and Myeloid Subsets in PDAC
5.2. NF-κB and Pancreatic Stellate Cells (PSCs)
5.3. NF-κB and Tregs
6. Inhibiting NF-κB in PDAC; Is Therapy Possible without Unwanted Side-Effects?
6.1. Generalized Anti-Inflammatories
6.2. Curcumin, Flavinoids (Natural Phenolic Substances) and Proteasome Inhibitors
6.3. Direct Inhibition of NF-κB or NF-κB Subunits
6.4. Inhibition of NF-κB Signaling Components
6.4.1. IRAK4 Inhibitors
6.4.2. Transforming Growth Factor-β (TGF-β)-Activated Kinase 1 (TAK1) Inhibitors
6.4.3. TANK-Binding Kinase 1 (TBK1) Inhibitors
6.4.4. TNFR Pathway Inhibitors
6.5. Other Modes of NF-κB Inhibition and Combination Therapy
6.6. Smac Mimetics; The Future for NF-κB Inhibition and PDAC Therapy?
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- International Agency for Research on Cancer. Global Cancer Observatory. Available online: http://gco.iarc.fr/ (accessed on 28 June 2019).
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 2018, 24, 4846–4861. [Google Scholar] [CrossRef] [PubMed]
- Ilic, M.; Ilic, I. Epidemiology of pancreatic cancer. World J. Gastroenterol. 2016, 22, 9694–9705. [Google Scholar] [CrossRef]
- Chang, D.K.; Johns, A.L.; Merrett, N.D.; Gill, A.J.; Colvin, E.K.; Scarlett, C.J.; Nguyen, N.Q.; Leong, R.W.; Cosman, P.H.; Kelly, M.I.; et al. Margin clearance and outcome in resected pancreatic cancer. J. Clin. Oncol. 2009, 27, 2855–2862. [Google Scholar] [CrossRef]
- Luo, J.; Xiao, L.; Wu, C.; Zheng, Y.; Zhao, N. The incidence and survival rate of population-based pancreatic cancer patients: Shanghai Cancer Registry 2004–2009. PLoS ONE 2013, 8, e76052. [Google Scholar] [CrossRef]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef]
- Midha, S.; Chawla, S.; Garg, P.K. Modifiable and non-modifiable risk factors for pancreatic cancer: A review. Cancer Lett. 2016, 381, 269–277. [Google Scholar] [CrossRef]
- Becker, A.E.; Hernandez, Y.G.; Frucht, H.; Lucas, A.L. Pancreatic ductal adenocarcinoma: Risk factors, screening, and early detection. World J. Gastroenterol. 2014, 20, 11182–11198. [Google Scholar] [CrossRef] [PubMed]
- Hruban, R.H.; Canto, M.I.; Goggins, M.; Schulick, R.; Klein, A.P. Update on familial pancreatic cancer. Adv. Surg. 2010, 44, 293–311. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Liu, W.; Wu, J. Helicobacter pylori infection and pancreatic cancer risk: A meta-analysis. J. Cancer Res. Ther. 2016, 12, C229–C232. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, S.; Van Buren, G., 2nd; Fisher, W.E. Pancreatic cancer: Advances in treatment. World J. Gastroenterol. 2014, 20, 9354–9360. [Google Scholar] [CrossRef]
- Brune, K.; Abe, T.; Canto, M.; O’Malley, L.; Klein, A.P.; Maitra, A.; Volkan Adsay, N.; Fishman, E.K.; Cameron, J.L.; Yeo, C.J.; et al. Multifocal neoplastic precursor lesions associated with lobular atrophy of the pancreas in patients having a strong family history of pancreatic cancer. Am. J. Surg. Pathol. 2006, 30, 1067–1076. [Google Scholar]
- Notta, F.; Chan-Seng-Yue, M.; Lemire, M.; Li, Y.; Wilson, G.W.; Connor, A.A.; Denroche, R.E.; Liang, S.B.; Brown, A.M.; Kim, J.C.; et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 2016, 538, 378–382. [Google Scholar] [CrossRef]
- Basturk, O.; Hong, S.M.; Wood, L.D.; Adsay, N.V.; Albores-Saavedra, J.; Biankin, A.V.; Brosens, L.A.; Fukushima, N.; Goggins, M.; Hruban, R.H.; et al. A Revised Classification System and Recommendations From the Baltimore Consensus Meeting for Neoplastic Precursor Lesions in the Pancreas. Am. J. Surg. Pathol. 2015, 39, 1730–1741. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, Y.; Furukawa, T.; Yachida, S.; Nishimura, M.; Seki, A.; Nonaka, K.; Aida, J.; Takubo, K.; Ishiwata, T.; Kimura, W.; et al. The Prevalence and Clinicopathological Characteristics of High-Grade Pancreatic Intraepithelial Neoplasia: Autopsy Study Evaluating the Entire Pancreatic Parenchyma. Pancreas 2017, 46, 658–664. [Google Scholar] [CrossRef]
- Peters, M.L.B.; Eckel, A.; Mueller, P.P.; Tramontano, A.C.; Weaver, D.T.; Lietz, A.; Hur, C.; Kong, C.Y.; Pandharipande, P.V. Progression to pancreatic ductal adenocarcinoma from pancreatic intraepithelial neoplasia: Results of a simulation model. Pancreatology 2018, 18, 928–934. [Google Scholar] [CrossRef]
- Levy, P.; Dominguez-Munoz, E.; Imrie, C.; Lohr, M.; Maisonneuve, P. Epidemiology of chronic pancreatitis: Burden of the disease and consequences. United Eur. Gastroenterol. J. 2014, 2, 345–354. [Google Scholar] [CrossRef]
- Feldmann, G.; Beaty, R.; Hruban, R.H.; Maitra, A. Molecular genetics of pancreatic intraepithelial neoplasia. J. Hepatobiliary Pancreat. Surg. 2007, 14, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Hruban, R.H.; Maitra, A.; Goggins, M. Update on pancreatic intraepithelial neoplasia. Int. J. Clin. Exp. Pathol. 2008, 1, 306–316. [Google Scholar]
- Kanda, M.; Matthaei, H.; Wu, J.; Hong, S.M.; Yu, J.; Borges, M.; Hruban, R.H.; Maitra, A.; Kinzler, K.; Vogelstein, B.; et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology 2012, 142, 730–733.e9. [Google Scholar] [CrossRef] [PubMed]
- Hruban, R.H.; Goggins, M.; Parsons, J.; Kern, S.E. Progression model for pancreatic cancer. Clin. Cancer Res. 2000, 6, 2969–2972. [Google Scholar]
- Lohr, M.; Kloppel, G.; Maisonneuve, P.; Lowenfels, A.B.; Luttges, J. Frequency of K-ras mutations in pancreatic intraductal neoplasias associated with pancreatic ductal adenocarcinoma and chronic pancreatitis: A meta-analysis. Neoplasia 2005, 7, 17–23. [Google Scholar] [CrossRef]
- Cicenas, J.; Kvederaviciute, K.; Meskinyte, I.; Meskinyte-Kausiliene, E.; Skeberdyte, A.; Cicenas, J. KRAS, TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 Mutations in Pancreatic Cancer. Cancers 2017, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.e13. [Google Scholar] [CrossRef] [PubMed]
- Amundadottir, L.T. Pancreatic Cancer Genetics. Int. J. Biol. Sci. 2016, 12, 314–325. [Google Scholar] [CrossRef]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.L.; Chone, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Neoptolemos, J.P.; Palmer, D.H.; Ghaneh, P.; Psarelli, E.E.; Valle, J.W.; Halloran, C.M.; Faluyi, O.; O’Reilly, D.A.; Cunningham, D.; Wadsley, J.; et al. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): A multicentre, open-label, randomised, phase 3 trial. Lancet 2017, 389, 1011–1024. [Google Scholar] [CrossRef]
- Neoptolemos, J.P.; Kleeff, J.; Michl, P.; Costello, E.; Greenhalf, W.; Palmer, D.H. Therapeutic developments in pancreatic cancer: Current and future perspectives. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 333–348. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T. A multicenter international randomized phase III trial of adjuvant mFOLFIRINOX versus gemcitabine (gem) in patients with resected pancreatic ductal adenocarcinomas. J. Clin. Oncol. 2018, 36, LBA4001. [Google Scholar] [CrossRef]
- Ghosn, M.; Kourie, H.R.; El Rassy, E.; Haddad, F.G.; Hanna, C.; El Karak, F.; Nasr, D. Where does chemotherapy stands in the treatment of ampullary carcinoma? A review of literature. World J. Gastrointest. Oncol. 2016, 8, 745–750. [Google Scholar] [CrossRef]
- Kabacaoglu, D.; Ciecielski, K.J.; Ruess, D.A.; Algul, H. Immune Checkpoint Inhibition for Pancreatic Ductal Adenocarcinoma: Current Limitations and Future Options. Front. Immunol. 2018, 9, 1878. [Google Scholar] [CrossRef] [PubMed]
- Morrison, A.H.; Byrne, K.T.; Vonderheide, R.H. Immunotherapy and Prevention of Pancreatic Cancer. Trends Cancer 2018, 4, 418–428. [Google Scholar] [CrossRef]
- Thind, K.; Padrnos, L.J.; Ramanathan, R.K.; Borad, M.J. Immunotherapy in pancreatic cancer treatment: A new frontier. Ther. Adv. Gastroenterol. 2017, 10, 168–194. [Google Scholar] [CrossRef] [PubMed]
- Ben-Neriah, Y.; Karin, M. Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat. Immunol. 2011, 12, 715–723. [Google Scholar] [CrossRef]
- Karin, M. NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb. Perspect. Biol. 2009, 1, a000141. [Google Scholar] [CrossRef]
- Hoesel, B.; Schmid, J.A. The complexity of NF-kappaB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. NF-kappaB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Chen, X.; Ji, B.; Han, B.; Ernst, S.A.; Simeone, D.; Logsdon, C.D. NF-kappaB activation in pancreas induces pancreatic and systemic inflammatory response. Gastroenterology 2002, 122, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Stevens, R.J.; Roddam, A.W.; Beral, V. Pancreatic cancer in type 1 and young-onset diabetes: Systematic review and meta-analysis. Br. J. Cancer 2007, 96, 507–509. [Google Scholar] [CrossRef] [PubMed]
- Huxley, R.; Ansary-Moghaddam, A.; Berrington de Gonzalez, A.; Barzi, F.; Woodward, M. Type-II diabetes and pancreatic cancer: A meta-analysis of 36 studies. Br. J. Cancer 2005, 92, 2076–2083. [Google Scholar] [CrossRef] [PubMed]
- Grote, V.A.; Rohrmann, S.; Nieters, A.; Dossus, L.; Tjonneland, A.; Halkjaer, J.; Overvad, K.; Fagherazzi, G.; Boutron-Ruault, M.C.; Morois, S.; et al. Diabetes mellitus, glycated haemoglobin and C-peptide levels in relation to pancreatic cancer risk: A study within the European Prospective Investigation into Cancer and Nutrition (EPIC) cohort. Diabetologia 2011, 54, 3037–3046. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. NF-kappaB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-kappaB: A Blossoming of Relevance to Human Pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef] [PubMed]
- Pramanik, K.C.; Makena, M.R.; Bhowmick, K.; Pandey, M.K. Advancement of NF-kappaB Signaling Pathway: A Novel Target in Pancreatic Cancer. Int. J. Mol. Sci. 2018, 19, 3890. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, L.; Mundade, R.; Korc, M.; Loehrer, P.J.; Lu, T. Critical role of NF-kappaB in pancreatic cancer. Oncotarget 2014, 5, 10969–10975. [Google Scholar] [CrossRef]
- Zhang, Z.; Rigas, B. NF-kappaB, inflammation and pancreatic carcinogenesis: NF-kappaB as a chemoprevention target (review). Int. J. Oncol. 2006, 29, 185–192. [Google Scholar]
- Kabacaoglu, D.; Ruess, D.A.; Ai, J.; Algul, H. NF-kappaB/Rel Transcription Factors in Pancreatic Cancer: Focusing on RelA, c-Rel, and RelB. Cancers 2019, 11, 937. [Google Scholar] [CrossRef]
- Sen, R.; Baltimore, D. Inducibility of kappa immunoglobulin enhancer-binding protein Nf-kappa B by a posttranslational mechanism. Cell 1986, 47, 921–928. [Google Scholar] [CrossRef]
- Sen, R.; Baltimore, D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 1986, 46, 705–716. [Google Scholar] [CrossRef]
- Gilmore, T.D. Introduction to NF-kappaB: Players, pathways, perspectives. Oncogene 2006, 25, 6680–6684. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-kappaB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Lin, A. NF-kB at the crossroads of life and death. Nat. Immunol. 2002, 3, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Vallabhapurapu, S.; Karin, M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef]
- Gerondakis, S.; Siebenlist, U. Roles of the NF-kappaB pathway in lymphocyte development and function. Cold Spring Harb. Perspect. Biol. 2010, 2, a000182. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal. Transduct Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Dolcet, X.; Llobet, D.; Pallares, J.; Matias-Guiu, X. NF-kB in development and progression of human cancer. Virchows Arch. 2005, 446, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Hong, J.T. Roles of NF-kappaB in Cancer and Inflammatory Diseases and Their Therapeutic Approaches. Cells 2016, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; May, M.J.; Jimi, E.; Ghosh, S. The phosphorylation status of nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1. Mol. Cell 2002, 9, 625–636. [Google Scholar] [CrossRef]
- Kitamura, H.; Kanehira, K.; Okita, K.; Morimatsu, M.; Saito, M. MAIL, a novel nuclear I kappa B protein that potentiates LPS-induced IL-6 production. FEBS Lett. 2000, 485, 53–56. [Google Scholar] [CrossRef]
- Haruta, H.; Kato, A.; Todokoro, K. Isolation of a novel interleukin-1-inducible nuclear protein bearing ankyrin-repeat motifs. J. Biol. Chem. 2001, 276, 12485–12488. [Google Scholar] [CrossRef]
- Yamazaki, S.; Muta, T.; Takeshige, K. A novel IkappaB protein, IkappaB-zeta, induced by proinflammatory stimuli, negatively regulates nuclear factor-kappaB in the nuclei. J. Biol. Chem. 2001, 276, 27657–27662. [Google Scholar] [CrossRef]
- Trinh, D.V.; Zhu, N.; Farhang, G.; Kim, B.J.; Huxford, T. The nuclear I kappaB protein I kappaB zeta specifically binds NF-kappaB p50 homodimers and forms a ternary complex on kappaB DNA. J. Mol. Biol. 2008, 379, 682–693. [Google Scholar] [CrossRef]
- Karin, M. How NF-kB is activated: The role of the IkB kinase (IKK) complex. Oncogene 1999, 18, 6867–6874. [Google Scholar] [CrossRef]
- Ghosh, S.; Karin, M. Missing pieces in the NF-kappaB puzzle. Cell 2002, 109, S81–S96. [Google Scholar] [CrossRef]
- Chen, F.E.; Huang, D.B.; Chen, Y.Q.; Ghosh, G. Crystal structure of p50/p65 heterodimer of transcription factor NF-kappaB bound to DNA. Nature 1998, 391, 410–413. [Google Scholar] [CrossRef]
- Huxford, T.; Huang, D.-B.; Malek, S.; Ghosh, G. The crystal structure of the IkBa/NF-kB complex reveals mechanisms of NF-kB inactivation. Cell 1998, 95, 759–770. [Google Scholar] [CrossRef]
- Page, A.; Navarro, M.; Suarez-Cabrera, C.; Bravo, A.; Ramirez, A. Context-Dependent Role of IKKbeta in Cancer. Genes 2017, 8, 376. [Google Scholar] [CrossRef] [PubMed]
- Mordmuller, B.; Krappmann, D.; Esen, M.; Wegener, E.; Scheidereit, C. Lymphotoxin and lipopolysaccharide induce NF-kappaB-p52 generation by a co-translational mechanism. EMBO Rep. 2003, 4, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Gerondakis, S.; Grumont, R.; Gugasyan, R.; Wong, L.; Isomura, I.; Ho, W.; Banerjee, A. Unravelling the complexities of the NF-kappaB signalling pathway using mouse knockout and transgenic models. Oncogene 2006, 25, 6781–6799. [Google Scholar] [CrossRef] [PubMed]
- Khurana, N.; Dodhiawala, P.B.; Bulle, A.; Lim, K.H. Deciphering the Role of Innate Immune NF-kB Pathway in Pancreatic Cancer. Cancers 2020, 12, 2675. [Google Scholar] [CrossRef] [PubMed]
- Padoan, A.; Plebani, M.; Basso, D. Inflammation and Pancreatic Cancer: Focus on Metabolism, Cytokines, and Immunity. Int. J. Mol. Sci. 2019, 20, 676. [Google Scholar] [CrossRef]
- Silke, J. The regulation of TNF signalling: What a tangled web we weave. Curr. Opin. Immunol. 2011, 23, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Rahighi, S.; Ikeda, F.; Kawasaki, M.; Akutsu, M.; Suzuki, N.; Kato, R.; Kensche, T.; Uejima, T.; Bloor, S.; Komander, D.; et al. Specific recognition of linear ubiquitin chains by NEMO is important for NF-kappaB activation. Cell 2009, 136, 1098–1109. [Google Scholar] [CrossRef]
- Haas, T.L.; Emmerich, C.H.; Gerlach, B.; Schmukle, A.C.; Cordier, S.M.; Rieser, E.; Feltham, R.; Vince, J.; Warnken, U.; Wenger, T.; et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol. Cell 2009, 36, 831–844. [Google Scholar] [CrossRef]
- Gerlach, B.; Cordier, S.M.; Schmukle, A.C.; Emmerich, C.H.; Rieser, E.; Haas, T.L.; Webb, A.I.; Rickard, J.A.; Anderton, H.; Wong, W.W.; et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 2011, 471, 591–596. [Google Scholar] [CrossRef]
- Israel, A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb. Perspect. Biol. 2010, 2, a000158. [Google Scholar] [CrossRef]
- Yang, Y.; Kelly, P.; Shaffer, A.L., 3rd; Schmitz, R.; Yoo, H.M.; Liu, X.; Huang, D.W.; Webster, D.; Young, R.M.; Nakagawa, M.; et al. Targeting Non-proteolytic Protein Ubiquitination for the Treatment of Diffuse Large B Cell Lymphoma. Cancer Cell 2016, 29, 494–507. [Google Scholar] [CrossRef] [PubMed]
- Damgaard, R.B.; Nachbur, U.; Yabal, M.; Wong, W.W.; Fiil, B.K.; Kastirr, M.; Rieser, E.; Rickard, J.A.; Bankovacki, A.; Peschel, C.; et al. The ubiquitin ligase XIAP recruits LUBAC for NOD2 signaling in inflammation and innate immunity. Mol. Cell 2012, 46, 746–758. [Google Scholar] [CrossRef]
- Basak, S.; Kim, H.; Kearns, J.D.; Tergaonkar, V.; O’Dea, E.; Werner, S.L.; Benedict, C.A.; Ware, C.F.; Ghosh, G.; Verma, I.M.; et al. A fourth IkappaB protein within the NF-kappaB signaling module. Cell 2007, 128, 369–381. [Google Scholar] [CrossRef] [PubMed]
- De Valle, E.; Grigoriadis, G.; O’Reilly, L.A.; Willis, S.N.; Maxwell, M.J.; Corcoran, L.M.; Tsantikos, E.; Cornish, J.K.; Fairfax, K.A.; Vasanthakumar, A.; et al. NFkappaB1 is essential to prevent the development of multiorgan autoimmunity by limiting IL-6 production in follicular B cells. J. Exp. Med. 2016, 213, 621–641. [Google Scholar] [CrossRef] [PubMed]
- Low, J.T.; Hughes, P.; Lin, A.; Siebenlist, U.; Jain, R.; Yaprianto, K.; Gray, D.H.; Gerondakis, S.; Strasser, A.; O’Reilly, L.A. Impact of loss of NF-kappaB1, NF-kappaB2 or c-REL on SLE-like autoimmune disease and lymphadenopathy in Fas(lpr/lpr) mutant mice. Immunol. Cell Biol. 2016, 94, 66–78. [Google Scholar] [CrossRef]
- O’Reilly, L.A.; Putoczki, T.L.; Mielke, L.A.; Low, J.T.; Lin, A.; Preaudet, A.; Herold, M.J.; Yaprianto, K.; Tai, L.; Kueh, A.; et al. Loss of NF-kappaB1 Causes Gastric Cancer with Aberrant Inflammation and Expression of Immune Checkpoint Regulators in a STAT-1-Dependent Manner. Immunity 2018, 48, 570.e8–583.e8. [Google Scholar] [CrossRef]
- Sun, S.C. The non-canonical NF-kappaB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Tegowski, M.; Baldwin, A. Noncanonical NF-κB in Cancer. Biomedicines 2018, 6, 66. [Google Scholar] [CrossRef]
- Zhang, H.; Sun, S.C. NF-kappaB in inflammation and renal diseases. Cell Biosci. 2015, 5, 63. [Google Scholar] [CrossRef]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [PubMed]
- Storz, P. Targeting the alternative NF-kappaB pathway in pancreatic cancer: A new direction for therapy? Expert Rev. Anticancer Ther. 2013, 13, 501–504. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lu, Z.; Li, Y.; Takwi, A.; Li, B.; Zhang, J.; Conklin, D.J.; Young, K.H.; Martin, R.; Li, Y. miR-301a as an NF-kappaB activator in pancreatic cancer cells. EMBO J. 2011, 30, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Arlt, A.; Schafer, H.; Kalthoff, H. The ‘N-factors’ in pancreatic cancer: Functional relevance of NF-kappaB, NFAT and Nrf2 in pancreatic cancer. Oncogenesis 2012, 1, e35. [Google Scholar] [CrossRef]
- Miraghazadeh, B.; Cook, M.C. Nuclear Factor-kappaB in Autoimmunity: Man and Mouse. Front. Immunol. 2018, 9, 613. [Google Scholar] [CrossRef]
- Courtois, G.; Gilmore, T.D. Mutations in the NF-kappaB signaling pathway: Implications for human disease. Oncogene 2006, 25, 6831–6843. [Google Scholar] [CrossRef] [PubMed]
- Thaventhiran, J.E.D.; Lango Allen, H.; Burren, O.S.; Rae, W.; Greene, D.; Staples, E.; Zhang, Z.; Farmery, J.H.R.; Simeoni, I.; Rivers, E.; et al. Whole-genome sequencing of a sporadic primary immunodeficiency cohort. Nature 2020, 583, 90–95. [Google Scholar] [CrossRef]
- Fliegauf, M.; Bryant, V.L.; Frede, N.; Slade, C.; Woon, S.T.; Lehnert, K.; Winzer, S.; Bulashevska, A.; Scerri, T.S.; Leung, E.; et al. Haploinsufficiency of the NF-κB1 Subunit p50 in Common Variable Immunodeficiency. Am. J. Hum. Genet. 2015, 97, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Low, J.T.; Christie, M.; Ernst, M.; Dumoutier, L.; Preaudet, A.; Ni, Y.; Griffin, M.D.W.; Mielke, L.A.; Strasser, A.; Putoczki, T.L.; et al. Loss of NFKB1 Results in Expression of Tumor Necrosis Factor and Activation of Signal Transducer and Activator of Transcription 1 to Promote Gastric Tumorigenesis in Mice. Gastroenterology 2020, 159, 1444–1458. [Google Scholar] [CrossRef] [PubMed]
- Concetti, J.; Wilson, C.L. NFKB1 and Cancer: Friend or Foe? Cells 2018, 7, 133. [Google Scholar] [CrossRef]
- Kolodecik, T.; Shugrue, C.; Ashat, M.; Thrower, E.C. Risk factors for pancreatic cancer: Underlying mechanisms and potential targets. Front. Physiol. 2013, 4, 415. [Google Scholar] [CrossRef]
- Raimondi, S.; Lowenfels, A.B.; Morselli-Labate, A.M.; Maisonneuve, P.; Pezzilli, R. Pancreatic cancer in chronic pancreatitis; Aetiology, incidence, and early detection. Best Pract. Research. Clin. Gastroenterol. 2010, 24, 349–358. [Google Scholar] [CrossRef]
- Sendler, M.; Weiss, F.U.; Golchert, J.; Homuth, G.; van den Brandt, C.; Mahajan, U.M.; Partecke, L.I.; Doring, P.; Gukovsky, I.; Gukovskaya, A.S.; et al. Cathepsin B-Mediated Activation of Trypsinogen in Endocytosing Macrophages Increases Severity of Pancreatitis in Mice. Gastroenterology 2018, 154, 704.e10–718.e10. [Google Scholar] [CrossRef] [PubMed]
- Gukovsky, I.; Li, N.; Todoric, J.; Gukovskaya, A.; Karin, M. Inflammation, autophagy, and obesity: Common features in the pathogenesis of pancreatitis and pancreatic cancer. Gastroenterology 2013, 144, 1199.e4–1209.e4. [Google Scholar] [CrossRef] [PubMed]
- Machicado, J.D.; Rebours, V.; Yadav, D. Epidemiology of chronic pancreatitis. In Pancreapedia; American Pancreatic Association: Prairie Village, KS, USA, 2016; pp. 1–15. Available online: https://www.pancreapedia.org/reviews/epidemiology-of-chronic-pancreatitis (accessed on 12 July 2021). [CrossRef]
- Howes, N.; Lerch, M.M.; Greenhalf, W.; Stocken, D.D.; Ellis, I.; Simon, P.; Truninger, K.; Ammann, R.; Cavallini, G.; Charnley, R.M.; et al. Clinical and genetic characteristics of hereditary pancreatitis in Europe. Clin. Gastroenterol. Hepatol. 2004, 2, 252–261. [Google Scholar] [CrossRef]
- DiDonato, J.A.; Mercurio, F.; Karin, M. NF-kappaB and the link between inflammation and cancer. Immunol. Rev. 2012, 246, 379–400. [Google Scholar] [CrossRef] [PubMed]
- Aghdassi, A.A.; Mayerle, J.; Christochowitz, S.; Weiss, F.U.; Sendler, M.; Lerch, M.M. Animal models for investigating chronic pancreatitis. Fibrogenesis Tissue Repair 2011, 4, 26. [Google Scholar] [CrossRef]
- Rakonczay, Z., Jr.; Hegyi, P.; Takacs, T.; McCarroll, J.; Saluja, A.K. The role of NF-kappaB activation in the pathogenesis of acute pancreatitis. Gut 2008, 57, 259–267. [Google Scholar] [CrossRef]
- Watanabe, T.; Kudo, M.; Strober, W. Immunopathogenesis of pancreatitis. Mucosal Immunol. 2017, 10, 283–298. [Google Scholar] [CrossRef]
- Huang, H.; Liu, Y.; Daniluk, J.; Gaiser, S.; Chu, J.; Wang, H.; Li, Z.S.; Logsdon, C.D.; Ji, B. Activation of nuclear factor-kappaB in acinar cells increases the severity of pancreatitis in mice. Gastroenterology 2013, 144, 202–210. [Google Scholar] [CrossRef]
- Gukovskaya, A.S.; Hosseini, S.; Satoh, A.; Cheng, J.H.; Nam, K.J.; Gukovsky, I.; Pandol, S.J. Ethanol differentially regulates NF-kappaB activation in pancreatic acinar cells through calcium and protein kinase C pathways. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G204–G213. [Google Scholar] [CrossRef][Green Version]
- Vonlaufen, A.; Phillips, P.A.; Xu, Z.; Zhang, X.; Yang, L.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Withdrawal of alcohol promotes regression while continued alcohol intake promotes persistence of LPS-induced pancreatic injury in alcohol-fed rats. Gut 2011, 60, 238–246. [Google Scholar] [CrossRef]
- Anastasi, A.; Erspamer, V.; Endean, R. Isolation and structure of caerulein, an active decapeptide from the skin of Hyla caerulea. Experientia 1967, 23, 699–700. [Google Scholar] [CrossRef]
- Sah, R.P.; Dudeja, V.; Dawra, R.K.; Saluja, A.K. Cerulein-induced chronic pancreatitis does not require intra-acinar activation of trypsinogen in mice. Gastroenterology 2013, 144, 1076–1085. [Google Scholar] [CrossRef]
- Kyttaris, V.C. Targeting cytokines to treat autoimmunity. Clin. Immunol. 2019, 206, 108251. [Google Scholar] [CrossRef]
- Sah, R.P.; Saluja, A. Molecular mechanisms of pancreatic injury. Curr. Opin. Gastroenterol. 2011, 27, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Steinle, A.U.; Weidenbach, H.; Wagner, M.; Adler, G.; Schmid, R.M. NF-kappaB/Rel activation in cerulein pancreatitis. Gastroenterology 1999, 116, 420–430. [Google Scholar] [CrossRef]
- Gukovsky, I.; Gukovskaya, A.S.; Blinman, T.A.; Zaninovic, V.; Pandol, S.J. Early NF-kappaB activation is associated with hormone-induced pancreatitis. Am. J. Physiol. 1998, 275, G1402–G1414. [Google Scholar] [CrossRef] [PubMed]
- Gukovskaya, A.S.; Gukovsky, I.; Zaninovic, V.; Song, M.; Sandoval, D.; Gukovsky, S.; Pandol, S.J. Pancreatic acinar cells produce, release, and respond to tumor necrosis factor-alpha. Role in regulating cell death and pancreatitis. J. Clin. Investig. 1997, 100, 1853–1862. [Google Scholar] [CrossRef]
- Grady, T.; Liang, P.; Ernst, S.A.; Logsdon, C.D. Chemokine gene expression in rat pancreatic acinar cells is an early event associated with acute pancreatitis. Gastroenterology 1997, 113, 1966–1975. [Google Scholar] [CrossRef]
- Altavilla, D.; Famulari, C.; Passaniti, M.; Galeano, M.; Macri, A.; Seminara, P.; Minutoli, L.; Marini, H.; Calo, M.; Venuti, F.S.; et al. Attenuated cerulein-induced pancreatitis in nuclear factor-kappaB-deficient mice. Lab. Investig. 2003, 83, 1723–1732. [Google Scholar] [CrossRef]
- Yang, H.T.; Wang, Y.; Zhao, X.; Demissie, E.; Papoutsopoulou, S.; Mambole, A.; O’Garra, A.; Tomczak, M.F.; Erdman, S.E.; Fox, J.G.; et al. NF-kappaB1 inhibits TLR-induced IFN-beta production in macrophages through TPL-2-dependent ERK activation. J. Immunol. 2011, 186, 1989–1996. [Google Scholar] [CrossRef] [PubMed]
- Dumitru, C.D.; Ceci, J.D.; Tsatsanis, C.; Kontoyiannis, D.; Stamatakis, K.; Lin, J.H.; Patriotis, C.; Jenkins, N.A.; Copeland, N.G.; Kollias, G.; et al. TNF-alpha induction by LPS is regulated posttranscriptionally via a Tpl2/ERK-dependent pathway. Cell 2000, 103, 1071–1083. [Google Scholar] [CrossRef]
- Gukovsky, I.; Gukovskaya, A. Nuclear factor-kappaB in pancreatitis: Jack-of-all-trades, but which one is more important? Gastroenterology 2013, 144, 26–29. [Google Scholar] [CrossRef] [PubMed]
- Baumann, B.; Wagner, M.; Aleksic, T.; von Wichert, G.; Weber, C.K.; Adler, G.; Wirth, T. Constitutive IKK2 activation in acinar cells is sufficient to induce pancreatitis in vivo. J. Clin. Investig. 2007, 117, 1502–1513. [Google Scholar] [CrossRef] [PubMed]
- Gaiser, S.; Daniluk, J.; Liu, Y.; Tsou, L.; Chu, J.; Lee, W.; Longnecker, D.S.; Logsdon, C.D.; Ji, B. Intracellular activation of trypsinogen in transgenic mice induces acute but not chronic pancreatitis. Gut 2011, 60, 1379–1388. [Google Scholar] [CrossRef] [PubMed]
- Algul, H.; Treiber, M.; Lesina, M.; Nakhai, H.; Saur, D.; Geisler, F.; Pfeifer, A.; Paxian, S.; Schmid, R.M. Pancreas-specific RelA/p65 truncation increases susceptibility of acini to inflammation-associated cell death following cerulein pancreatitis. J. Clin. Investig. 2007, 117, 1490–1501. [Google Scholar] [CrossRef] [PubMed]
- Neuhofer, P.; Liang, S.; Einwachter, H.; Schwerdtfeger, C.; Wartmann, T.; Treiber, M.; Zhang, H.; Schulz, H.U.; Dlubatz, K.; Lesina, M.; et al. Deletion of IkappaBalpha activates RelA to reduce acute pancreatitis in mice through up-regulation of Spi2A. Gastroenterology 2013, 144, 192–201. [Google Scholar] [CrossRef]
- Treiber, M.; Neuhofer, P.; Anetsberger, E.; Einwachter, H.; Lesina, M.; Rickmann, M.; Liang, S.; Kehl, T.; Nakhai, H.; Schmid, R.M.; et al. Myeloid, but not pancreatic, RelA/p65 is required for fibrosis in a mouse model of chronic pancreatitis. Gastroenterology 2011, 141, 1473.e7–1485.e7. [Google Scholar] [CrossRef]
- Qin, T.; Liu, C.J.; Zhang, H.W.; Pan, Y.F.; Tang, Q.; Liu, J.K.; Wang, Y.Z.; Hu, M.X.; Xue, F. Effect of the IkBalpha mutant gene delivery to mesenchymal stem cells on rat chronic pancreatitis. Genet. Mol. Res. 2014, 13, 371–385. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Shen, S.; Verma, I.M. NF-kappaB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef]
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a028936. [Google Scholar] [CrossRef]
- Zhang, D.; Li, L.; Jiang, H.; Knolhoff, B.L.; Lockhart, A.C.; Wang-Gillam, A.; DeNardo, D.G.; Ruzinova, M.B.; Lim, K.-H. Constitutive IRAK4 Activation Underlies Poor Prognosis and Chemoresistance in Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2017, 23, 1748–1759. [Google Scholar] [CrossRef]
- Dodhiawala, P.B.; Khurana, N.; Zhang, D.; Cheng, Y.; Li, L.; Wei, Q.; Seehra, K.; Jiang, H.; Grierson, P.M.; Wang-Gillam, A.; et al. TPL2 enforces RAS-induced inflammatory signaling and is activated by point mutations. J. Clin. Investig. 2020, 130, 4771–4790. [Google Scholar] [CrossRef]
- Grimmig, T.; Moench, R.; Kreckel, J.; Haack, S.; Rueckert, F.; Rehder, R.; Tripathi, S.; Ribas, C.; Chandraker, A.; Germer, C.T.; et al. Toll Like Receptor 2, 4, and 9 Signaling Promotes Autoregulative Tumor Cell Growth and VEGF/PDGF Expression in Human Pancreatic Cancer. Int. J. Mol. Sci. 2016, 17, 2060. [Google Scholar] [CrossRef] [PubMed]
- Helgason, E.; Phung, Q.T.; Dueber, E.C. Recent insights into the complexity of Tank-binding kinase 1 signaling networks: The emerging role of cellular localization in the activation and substrate specificity of TBK1. FEBS Lett. 2013, 587, 1230–1237. [Google Scholar] [CrossRef] [PubMed]
- Shin, C.H.; Choi, D.S. Essential Roles for the Non-Canonical IκB Kinases in Linking Inflammation to Cancer, Obesity, and Diabetes. Cells 2019, 8, 178. [Google Scholar] [CrossRef] [PubMed]
- Cruz, V.H.; Brekken, R.A. Assessment of TANK-binding kinase 1 as a therapeutic target in cancer. Journal of cell communication and signaling. J. Cell Commun. Signal. 2018, 12, 83–90. [Google Scholar] [CrossRef]
- Herhaus, L. TBK1 (TANK-binding kinase 1)-mediated regulation of autophagy in health and disease. Matrix Biol. 2021, 100–101, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Durand, J.K.; Zhang, Q.; Baldwin, A.S. Roles for the IKK-Related Kinases TBK1 and IKKε in Cancer. Cells 2018, 7, 139. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef]
- Di Magliano, M.P.; Logsdon, C.D. Roles for KRAS in pancreatic tumor development and progression. Gastroenterology 2013, 144, 1220–1229. [Google Scholar] [CrossRef]
- Herreros-Villanueva, M.; Hijona, E.; Cosme, A.; Bujanda, L. Mouse models of pancreatic cancer. World J. Gastroenterol. 2012, 18, 1286–1294. [Google Scholar] [CrossRef]
- Steele, C.W.; Jamieson, N.B.; Evans, T.R.; McKay, C.J.; Sansom, O.J.; Morton, J.P.; Carter, C.R. Exploiting inflammation for therapeutic gain in pancreatic cancer. Br. J. Cancer 2013, 108, 997–1003. [Google Scholar] [CrossRef]
- Knudsen, E.S.; Vail, P.; Balaji, U.; Ngo, H.; Botros, I.W.; Makarov, V.; Riaz, N.; Balachandran, V.; Leach, S.; Thompson, D.M.; et al. Stratification of Pancreatic Ductal Adenocarcinoma: Combinatorial Genetic, Stromal, and Immunologic Markers. Clin. Cancer Res. 2017, 23, 4429–4440. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Imamura, Y.; Jenkins, R.W.; Cañadas, I.; Kitajima, S.; Aref, A.; Brannon, A.; Oki, E.; Castoreno, A.; Zhu, Z.; et al. Autophagy Inhibition Dysregulates TBK1 Signaling and Promotes Pancreatic Inflammation. Cancer Immunol. Res. 2016, 4, 520–530. [Google Scholar] [CrossRef]
- Chien, Y.; Kim, S.; Bumeister, R.; Loo, Y.M.; Kwon, S.W.; Johnson, C.L.; Balakireva, M.G.; Romeo, Y.; Kopelovich, L.; Gale, M.; et al. RalB GTPase-mediated activation of the IkappaB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell 2006, 127, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Nishina, T.; Yamaguchi, N.; Gohda, J.; Semba, K.; Inoue, J. NIK is involved in constitutive activation of the alternative NF-kappaB pathway and proliferation of pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2009, 388, 96–101. [Google Scholar] [CrossRef]
- Doppler, H.; Liou, G.Y.; Storz, P. Downregulation of TRAF2 mediates NIK-induced pancreatic cancer cell proliferation and tumorigenicity. PLoS ONE 2013, 8, e53676. [Google Scholar] [CrossRef] [PubMed]
- Wharry, C.E.; Haines, K.M.; Carroll, R.G.; May, M.J. Constitutive non-canonical NFkappaB signaling in pancreatic cancer cells. Cancer Biol. Ther. 2009, 8, 1567–1576. [Google Scholar] [CrossRef]
- Schneider, G.; Saur, D.; Siveke, J.T.; Fritsch, R.; Greten, F.R.; Schmid, R.M. IKKalpha controls p52/RelB at the skp2 gene promoter to regulate G1- to S-phase progression. EMBO J. 2006, 25, 3801–3812. [Google Scholar] [CrossRef] [PubMed]
- Geismann, C.; Schafer, H.; Gundlach, J.P.; Hauser, C.; Egberts, J.H.; Schneider, G.; Arlt, A. NF-kappaB Dependent Chemokine Signaling in Pancreatic Cancer. Cancers 2019, 11, 1445. [Google Scholar] [CrossRef] [PubMed]
- Bang, D.; Wilson, W.; Ryan, M.; Yeh, J.J.; Baldwin, A.S. GSK-3alpha promotes oncogenic KRAS function in pancreatic cancer via TAK1-TAB stabilization and regulation of noncanonical NF-kappaB. Cancer Discov. 2013, 3, 690–703. [Google Scholar] [CrossRef]
- Ling, J.; Kang, Y.; Zhao, R.; Xia, Q.; Lee, D.F.; Chang, Z.; Li, J.; Peng, B.; Fleming, J.B.; Wang, H.; et al. KrasG12D-induced IKK2/beta/NF-kappaB activation by IL-1alpha and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 105–120. [Google Scholar] [CrossRef]
- Maniati, E.; Bossard, M.; Cook, N.; Candido, J.B.; Emami-Shahri, N.; Nedospasov, S.A.; Balkwill, F.R.; Tuveson, D.A.; Hagemann, T. Crosstalk between the canonical NF-kappaB and Notch signaling pathways inhibits Ppargamma expression and promotes pancreatic cancer progression in mice. J. Clin. Investig. 2011, 121, 4685–4699. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Li, Z.; Peng, B.; Chiao, P.J. Identification of an autoregulatory feedback pathway involving interleukin-1alpha in induction of constitutive NF-kappaB activation in pancreatic cancer cells. J. Biol. Chem. 2004, 279, 16452–16462. [Google Scholar] [CrossRef]
- Doppler, H.; Panayiotou, R.; Reid, E.M.; Maimo, W.; Bastea, L.; Storz, P. The PRKD1 promoter is a target of the KRas-NF-kappaB pathway in pancreatic cancer. Sci. Rep. 2016, 6, 33758. [Google Scholar] [CrossRef]
- Bulle, A.; Lim, K.-H. Beyond just a tight fortress: Contribution of stroma to epithelial-mesenchymal transition in pancreatic cancer. Signal Transduct. Target. Ther. 2020, 5, 249. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Strasser, A.; Cory, S.; Adams, J.M. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J. 2011, 30, 3667–3683. [Google Scholar] [CrossRef]
- Baldwin, A.S. Regulation of cell death and autophagy by IKK and NF-kappaB: Critical mechanisms in immune function and cancer. Immunol. Rev. 2012, 246, 327–345. [Google Scholar] [CrossRef] [PubMed]
- Villunger, A.; Michalak, E.M.; Coultas, L.; Mullauer, F.; Bock, G.; Ausserlechner, M.J.; Adams, J.M.; Strasser, A. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science 2003, 302, 1036–1038. [Google Scholar] [CrossRef]
- Modi, S.; Kir, D.; Banerjee, S.; Saluja, A. Control of Apoptosis in Treatment and Biology of Pancreatic Cancer. J. Cell Biochem. 2016, 117, 279–288. [Google Scholar] [CrossRef]
- Song, S.; Wang, B.; Gu, S.; Li, X.; Sun, S. Expression of Beclin 1 and Bcl-2 in pancreatic neoplasms and its effect on pancreatic ductal adenocarcinoma prognosis. Oncology Lett. 2017, 14, 7849–7861. [Google Scholar] [CrossRef]
- Lopes, R.B.; Gangeswaran, R.; McNeish, I.A.; Wang, Y.; Lemoine, N.R. Expression of the IAP protein family is dysregulated in pancreatic cancer cells and is important for resistance to chemotherapy. Int. J. Cancer 2007, 120, 2344–2352. [Google Scholar] [CrossRef]
- Silke, J.; Meier, P. Inhibitor of apoptosis (IAP) proteins-modulators of cell death and inflammation. Cold Spring Harb. Perspect. Biol. 2013, 5, a008730. [Google Scholar] [CrossRef] [PubMed]
- New, M.; Van Acker, T.; Long, J.S.; Sakamaki, J.I.; Ryan, K.M.; Tooze, S.A. Molecular Pathways Controlling Autophagy in Pancreatic Cancer. Front. Oncol. 2017, 7, 28. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Trocoli, A.; Djavaheri-Mergny, M. The complex interplay between autophagy and NF-kappaB signaling pathways in cancer cells. Am. J. Cancer Res. 2011, 1, 629–649. [Google Scholar]
- Gui, X.; Yang, H.; Li, T.; Tan, X.; Shi, P.; Li, M.; Du, F.; Chen, Z.J. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 2019, 567, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Verzella, D.; Pescatore, A.; Capece, D.; Vecchiotti, D.; Ursini, M.V.; Franzoso, G.; Alesse, E.; Zazzeroni, F. Life, death, and autophagy in cancer: NF-kappaB turns up everywhere. Cell Death Dis. 2020, 11, 210. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Papademetrio, D.L.; Lompardia, S.L.; Simunovich, T.; Costantino, S.; Mihalez, C.Y.; Cavaliere, V.; Alvarez, E. Inhibition of Survival Pathways MAPK and NF-kB Triggers Apoptosis in Pancreatic Ductal Adenocarcinoma Cells via Suppression of Autophagy. Target. Oncol. 2016, 11, 183–195. [Google Scholar] [CrossRef]
- Gukovskaya, A.S.; Gukovsky, I.; Algul, H.; Habtezion, A. Autophagy, Inflammation, and Immune Dysfunction in the Pathogenesis of Pancreatitis. Gastroenterology 2017, 153, 1212–1226. [Google Scholar] [CrossRef] [PubMed]
- Criollo, A.; Senovilla, L.; Authier, H.; Maiuri, M.C.; Morselli, E.; Vitale, I.; Kepp, O.; Tasdemir, E.; Galluzzi, L.; Shen, S.; et al. IKK connects autophagy to major stress pathways. Autophagy 2010, 6, 189–191. [Google Scholar] [CrossRef]
- Criollo, A.; Senovilla, L.; Authier, H.; Maiuri, M.C.; Morselli, E.; Vitale, I.; Kepp, O.; Tasdemir, E.; Galluzzi, L.; Shen, S.; et al. The IKK complex contributes to the induction of autophagy. EMBO J. 2010, 29, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Bryant, K.L.; Stalnecker, C.A.; Zeitouni, D.; Klomp, J.E.; Peng, S.; Tikunov, A.P.; Gunda, V.; Pierobon, M.; Waters, A.M.; George, S.D.; et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med. 2019, 25, 628–640. [Google Scholar] [CrossRef]
- Tabruyn, S.P.; Griffioen, A.W. NF-kappa B: A new player in angiostatic therapy. Angiogenesis 2008, 11, 101–106. [Google Scholar] [CrossRef]
- Huang, S.; Pettaway, C.A.; Uehara, H.; Bucana, C.D.; Fidler, I.J. Blockade of NF-kappaB activity in human prostate cancer cells is associated with suppression of angiogenesis, invasion, and metastasis. Oncogene 2001, 20, 4188–4197. [Google Scholar] [CrossRef]
- Fujioka, S.; Sclabas, G.M.; Schmidt, C.; Frederick, W.A.; Dong, Q.G.; Abbruzzese, J.L.; Evans, D.B.; Baker, C.; Chiao, P.J. Function of nuclear factor kappaB in pancreatic cancer metastasis. Clin. Cancer Res. 2003, 9, 346–354. [Google Scholar] [PubMed]
- Shamoto, T.; Matsuo, Y.; Shibata, T.; Tsuboi, K.; Nagasaki, T.; Takahashi, H.; Funahashi, H.; Okada, Y.; Takeyama, H. Zerumbone inhibits angiogenesis by blocking NF-kappaB activity in pancreatic cancer. Pancreas 2014, 43, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, Z.A.; Yang, J.; Wang, Q.; Kowalski, J.; Freed, I.; Murter, C.; Hong, S.M.; Koorstra, J.B.; Rajeshkumar, N.V.; He, X.; et al. Prognostic significance of tumorigenic cells with mesenchymal features in pancreatic adenocarcinoma. J. Natl. Cancer Inst. 2010, 102, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [PubMed]
- Maier, H.J.; Schmidt-Strassburger, U.; Huber, M.A.; Wiedemann, E.M.; Beug, H.; Wirth, T. NF-kappaB promotes epithelial-mesenchymal transition, migration and invasion of pancreatic carcinoma cells. Cancer Lett. 2010, 295, 214–228. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Liu, J.; Zhang, L.; Xiao, X.; Li, W. Curcumin inhibits H2O2-induced invasion and migration of human pancreatic cancer via suppression of the ERK/NF-kappaB pathway. Oncol. Rep. 2016, 36, 2245–2251. [Google Scholar] [CrossRef]
- Huber, M.A.; Azoitei, N.; Baumann, B.; Grunert, S.; Sommer, A.; Pehamberger, H.; Kraut, N.; Beug, H.; Wirth, T. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J. Clin. Investig. 2004, 114, 569–581. [Google Scholar] [CrossRef]
- Pires, B.R.; Mencalha, A.L.; Ferreira, G.M.; de Souza, W.F.; Morgado-Diaz, J.A.; Maia, A.M.; Correa, S.; Abdelhay, E.S. NF-kappaB Is Involved in the Regulation of EMT Genes in Breast Cancer Cells. PLoS ONE 2017, 12, e0169622. [Google Scholar] [CrossRef]
- Li, C.W.; Xia, W.; Huo, L.; Lim, S.O.; Wu, Y.; Hsu, J.L.; Chao, C.H.; Yamaguchi, H.; Yang, N.K.; Ding, Q.; et al. Epithelial-mesenchymal transition induced by TNF-alpha requires NF-kappaB-mediated transcriptional upregulation of Twist1. Cancer Res. 2012, 72, 1290–1300. [Google Scholar] [CrossRef]
- D’Ignazio, L.; Bandarra, D.; Rocha, S. NF-κB and HIF crosstalk in immune responses. FEBS J. 2016, 283, 413–424. [Google Scholar] [CrossRef]
- Zhang, Z.; Duan, Q.; Zhao, H.; Liu, T.; Wu, H.; Shen, Q.; Wang, C.; Yin, T. Gemcitabine treatment promotes pancreatic cancer stemness through the Nox/ROS/NF-kappaB/STAT3 signaling cascade. Cancer Lett. 2016, 382, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Amrutkar, M.; Gladhaug, I.P. Pancreatic Cancer Chemoresistance to Gemcitabine. Cancers 2017, 9, 157. [Google Scholar] [CrossRef]
- Vasseur, R.; Skrypek, N.; Duchene, B.; Renaud, F.; Martinez-Maqueda, D.; Vincent, A.; Porchet, N.; Van Seuningen, I.; Jonckheere, N. The mucin MUC4 is a transcriptional and post-transcriptional target of K-ras oncogene in pancreatic cancer. Implication of MAPK/AP-1, NF-kappaB and RalB signaling pathways. Biochim. Biophys. Acta 2015, 1849, 1375–1384. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Arumugam, T.; Yamamoto, T.; Levin, P.A.; Ramachandran, V.; Ji, B.; Lopez-Berestein, G.; Vivas-Mejia, P.E.; Sood, A.K.; McConkey, D.J.; et al. Nuclear factor-kappaB p65/relA silencing induces apoptosis and increases gemcitabine effectiveness in a subset of pancreatic cancer cells. Clin. Cancer Res. 2008, 14, 8143–8151. [Google Scholar] [CrossRef] [PubMed]
- Kong, R.; Sun, B.; Jiang, H.; Pan, S.; Chen, H.; Wang, S.; Krissansen, G.W.; Sun, X. Downregulation of nuclear factor-kappaB p65 subunit by small interfering RNA synergizes with gemcitabine to inhibit the growth of pancreatic cancer. Cancer Lett. 2010, 291, 90–98. [Google Scholar] [CrossRef]
- Gong, J.; Munoz, A.R.; Pingali, S.; Payton-Stewart, F.; Chan, D.E.; Freeman, J.W.; Ghosh, R.; Kumar, A.P. Downregulation of STAT3/NF-kappaB potentiates gemcitabine activity in pancreatic cancer cells. Mol. Carcinog. 2017, 56, 402–411. [Google Scholar] [CrossRef]
- Yu, C.; Chen, S.; Guo, Y.; Sun, C. Oncogenic TRIM31 confers gemcitabine resistance in pancreatic cancer via activating the NF-kappaB signaling pathway. Theranostics 2018, 8, 3224–3236. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, J.S. Tumor stroma as targets for cancer therapy. Pharmacol. Ther. 2013, 137, 200–215. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef]
- Lankadasari, M.B.; Mukhopadhyay, P.; Mohammed, S.; Harikumar, K.B. TAMing pancreatic cancer: Combat with a double edged sword. Mol. Cancer 2019, 18, 48. [Google Scholar] [CrossRef]
- Neesse, A.; Bauer, C.A.; Ohlund, D.; Lauth, M.; Buchholz, M.; Michl, P.; Tuveson, D.A.; Gress, T.M. Stromal biology and therapy in pancreatic cancer: Ready for clinical translation? Gut 2019, 68, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A. Macrophages, innate immunity and cancer: Balance, tolerance, and diversity. Curr. Opin. Immunol. 2010, 22, 231–237. [Google Scholar] [CrossRef]
- Ino, Y.; Yamazaki-Itoh, R.; Shimada, K.; Iwasaki, M.; Kosuge, T.; Kanai, Y.; Hiraoka, N. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br. J. Cancer 2013, 108, 914–923. [Google Scholar] [CrossRef] [PubMed]
- Kurahara, H.; Shinchi, H.; Mataki, Y.; Maemura, K.; Noma, H.; Kubo, F.; Sakoda, M.; Ueno, S.; Natsugoe, S.; Takao, S. Significance of M2-polarized tumor-associated macrophage in pancreatic cancer. J. Surg. Res. 2011, 167, e211–e219. [Google Scholar] [CrossRef] [PubMed]
- Saccani, A.; Schioppa, T.; Porta, C.; Biswas, S.K.; Nebuloni, M.; Vago, L.; Bottazzi, B.; Colombo, M.P.; Mantovani, A.; Sica, A. p50 nuclear factor-kappaB overexpression in tumor-associated macrophages inhibits M1 inflammatory responses and antitumor resistance. Cancer Res. 2006, 66, 11432–11440. [Google Scholar] [CrossRef]
- Porta, C.; Rimoldi, M.; Raes, G.; Brys, L.; Ghezzi, P.; Di Liberto, D.; Dieli, F.; Ghisletti, S.; Natoli, G.; De Baetselier, P.; et al. Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor kappaB. Proc. Natl. Acad. Sci. USA 2009, 106, 14978–14983. [Google Scholar] [CrossRef] [PubMed]
- Suresh, R.; Barakat, D.J.; Barberi, T.; Zheng, L.; Jaffee, E.; Pienta, K.J.; Friedman, A.D. NF-kappaB p50-deficient immature myeloid cell (p50-IMC) adoptive transfer slows the growth of murine prostate and pancreatic ductal carcinoma. J. Immunother. Cancer 2020, 8, e000244. [Google Scholar] [CrossRef]
- Zhang, Y.; Velez-Delgado, A.; Mathew, E.; Li, D.; Mendez, F.M.; Flannagan, K.; Rhim, A.D.; Simeone, D.M.; Beatty, G.L.; Pasca di Magliano, M. Myeloid cells are required for PD-1/PD-L1 checkpoint activation and the establishment of an immunosuppressive environment in pancreatic cancer. Gut 2017, 66, 124–136. [Google Scholar] [CrossRef]
- Xue, R.; Jia, K.; Wang, J.; Yang, L.; Wang, Y.; Gao, L.; Hao, J. A Rising Star in Pancreatic Diseases: Pancreatic Stellate Cells. Front. Physiol. 2018, 9, 754. [Google Scholar] [CrossRef]
- Apte, M.V.; Wilson, J.S. Dangerous liaisons: Pancreatic stellate cells and pancreatic cancer cells. J. Gastroenterol. Hepatol. 2012, 27 (Suppl. 2), 69–74. [Google Scholar] [CrossRef]
- Masamune, A.; Shimosegawa, T. Signal transduction in pancreatic stellate cells. J. Gastroenterol. 2009, 44, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFbeta to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef] [PubMed]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef]
- Lenggenhager, D.; Amrutkar, M.; Santha, P.; Aasrum, M.; Lohr, J.M.; Gladhaug, I.P.; Verbeke, C.S. Commonly Used Pancreatic Stellate Cell Cultures Differ Phenotypically and in Their Interactions with Pancreatic Cancer Cells. Cells 2019, 8, 23. [Google Scholar] [CrossRef]
- Garg, B.; Giri, B.; Modi, S.; Sethi, V.; Castro, I.; Umland, O.; Ban, Y.; Lavania, S.; Dawra, R.; Banerjee, S.; et al. NFkappaB in Pancreatic Stellate Cells Reduces Infiltration of Tumors by Cytotoxic T Cells and Killing of Cancer Cells, via Up-regulation of CXCL12. Gastroenterology 2018, 155, 880.e8–891.e8. [Google Scholar] [CrossRef]
- Kobayashi, H.; Enomoto, A.; Woods, S.L.; Burt, A.D.; Takahashi, M.; Worthley, D.L. Cancer-associated fibroblasts in gastrointestinal cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 282–295. [Google Scholar] [CrossRef]
- Martinez-Bosch, N.; Vinaixa, J.; Navarro, P. Immune Evasion in Pancreatic Cancer: From Mechanisms to Therapy. Cancers 2018, 10, 6. [Google Scholar] [CrossRef]
- Isomura, I.; Palmer, S.; Grumont, R.J.; Bunting, K.; Hoyne, G.; Wilkinson, N.; Banerjee, A.; Proietto, A.; Gugasyan, R.; Li, W.; et al. c-Rel is required for the development of thymic Foxp3+ CD4 regulatory T cells. J. Exp. Med. 2009, 206, 3001–3014. [Google Scholar] [CrossRef]
- Ruan, Q.; Kameswaran, V.; Tone, Y.; Li, L.; Liou, H.C.; Greene, M.I.; Tone, M.; Chen, Y.H. Development of Foxp3(+) regulatory t cells is driven by the c-Rel enhanceosome. Immunity 2009, 31, 932–940. [Google Scholar] [CrossRef]
- Long, M.; Park, S.G.; Strickland, I.; Hayden, M.S.; Ghosh, S. Nuclear factor-kappaB modulates regulatory T cell development by directly regulating expression of Foxp3 transcription factor. Immunity 2009, 31, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.; Grinberg-Bleyer, Y.; Liao, W.; Maloney, D.; Wang, P.; Wu, Z.; Wang, J.; Bhatt, D.M.; Heise, N.; Schmid, R.M.; et al. An NF-kappaB Transcription-Factor-Dependent Lineage-Specific Transcriptional Program Promotes Regulatory T Cell Identity and Function. Immunity 2017, 47, 450.e5–465.e5. [Google Scholar] [CrossRef]
- Jang, J.E.; Hajdu, C.H.; Liot, C.; Miller, G.; Dustin, M.L.; Bar-Sagi, D. Crosstalk between Regulatory T Cells and Tumor-Associated Dendritic Cells Negates Anti-tumor Immunity in Pancreatic Cancer. Cell Rep. 2017, 20, 558–571. [Google Scholar] [CrossRef]
- Zhang, Y.; Lazarus, J.; Steele, N.G.; Yan, W.; Lee, H.J.; Nwosu, Z.C.; Halbrook, C.J.; Menjivar, R.E.; Kemp, S.B.; Sirihorachai, V.R.; et al. Regulatory T-cell Depletion Alters the Tumor Microenvironment and Accelerates Pancreatic Carcinogenesis. Cancer Discov. 2020, 10, 422–439. [Google Scholar] [CrossRef]
- Grinberg-Bleyer, Y.; Oh, H.; Desrichard, A.; Bhatt, D.M.; Caron, R.; Chan, T.A.; Schmid, R.M.; Klein, U.; Hayden, M.S.; Ghosh, S. NF-kappaB c-Rel Is Crucial for the Regulatory T Cell Immune Checkpoint in Cancer. Cell 2017, 170, 1096–1108.e13. [Google Scholar] [CrossRef]
- Wang, D.J.; Ratnam, N.M.; Byrd, J.C.; Guttridge, D.C. NF-kappaB functions in tumor initiation by suppressing the surveillance of both innate and adaptive immune cells. Cell Rep. 2014, 9, 90–103. [Google Scholar] [CrossRef]
- Pandey, V.; Storz, P. Targeting the tumor microenvironment in pancreatic ductal adenocarcinoma. Expert Rev. Anticancer Ther. 2019, 19, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef]
- Holcomb, B.; Yip-Schneider, M.; Schmidt, C.M. The role of nuclear factor kappaB in pancreatic cancer and the clinical applications of targeted therapy. Pancreas 2008, 36, 225–235. [Google Scholar] [CrossRef]
- Geismann, C.; Erhart, W.; Grohmann, F.; Schreiber, S.; Schneider, G.; Schafer, H.; Arlt, A. TRAIL/NF-kappaB/CX3CL1 Mediated Onco-Immuno Crosstalk Leading to TRAIL Resistance of Pancreatic Cancer Cell Lines. Int. J. Mol. Sci. 2018, 19, 1661. [Google Scholar] [CrossRef] [PubMed]
- Surh, Y.J.; Bode, A.M.; Dong, Z. Breaking the NF-kappaB and STAT3 alliance inhibits inflammation and pancreatic tumorigenesis. Cancer Prev. Res. 2010, 3, 1379–1381. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Weber, C.K.; Greten, T.F.; Schneider, G.; Wagner, M.; Adler, G.; Schmid, R.M. Stat3 and NF-kappaB activation prevents apoptosis in pancreatic carcinogenesis. Gastroenterology 2002, 123, 2052–2063. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, M.; Jing, Y.; Yin, X.; Ma, P.; Zhang, Z.; Wang, X.; Di, W.; Zhuang, G. Deubiquitinase USP13 dictates MCL1 stability and sensitivity to BH3 mimetic inhibitors. Nat. Commun. 2018, 9, 215. [Google Scholar] [CrossRef] [PubMed]
- Ranson, J. The roles of Hedgehog signalling and NF-κB activity in pancreatic cancer and opportunities for treatment. Biosci. Horiz. Int. J. Stud. Res. 2014, 7, hzu004. [Google Scholar] [CrossRef]
- Gilmore, T.D.; Herscovitch, M. Inhibitors of NF-kappaB signaling: 785 and counting. Oncogene 2006, 25, 6887–6899. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.C.; Sundaram, C.; Reuter, S.; Aggarwal, B.B. Inhibiting NF-kappaB activation by small molecules as a therapeutic strategy. Biochim. Biophys. Acta 2010, 1799, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Herrington, F.D.; Carmody, R.J.; Goodyear, C.S. Modulation of NF-kappaB Signaling as a Therapeutic Target in Autoimmunity. J. Biomol. Screen 2016, 21, 223–242. [Google Scholar] [CrossRef]
- O’Reilly, E.; Santocanale, C.; Szegezdi, É. Molecular targets for novel drug development in pancreatic cancer. Ann. Cancer Res. 2015, 2, 1–11. [Google Scholar]
- Freitas, R.H.C.N.; Fraga, C.A.M. NF-κB-IKKβ Pathway as a Target for Drug Development: Realities, Challenges and Perspectives. Curr. Drug Targets 2018, 19, 1933–1942. [Google Scholar] [CrossRef]
- Yin, M.-J.; Yamamoto, Y.; Gaynor, R.B. The anti-inflammatory agents aspirin and salicylate inhibit the activity of IkB kinase-b. Nature 1998, 396, 77–80. [Google Scholar] [CrossRef]
- Smith, C.E.; Soti, S.; Jones, T.A.; Nakagawa, A.; Xue, D.; Yin, H. Non-steroidal Anti-inflammatory Drugs Are Caspase Inhibitors. Cell Chem. Biol. 2017, 24, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Muerkoster, S.; Arlt, A.; Witt, M.; Gehrz, A.; Haye, S.; March, C.; Grohmann, F.; Wegehenkel, K.; Kalthoff, H.; Folsch, U.R.; et al. Usage of the NF-kappaB inhibitor sulfasalazine as sensitizing agent in combined chemotherapy of pancreatic cancer. Int. J. Cancer 2003, 104, 469–476. [Google Scholar] [CrossRef]
- Yip-Schneider, M.T.; Nakshatri, H.; Sweeney, C.J.; Marshall, M.S.; Wiebke, E.A.; Schmidt, C.M. Parthenolide and sulindac cooperate to mediate growth suppression and inhibit the nuclear factor-kappa B pathway in pancreatic carcinoma cells. Mol. Cancer Ther. 2005, 4, 587–594. [Google Scholar] [CrossRef]
- El-Rayes, B.F.; Ali, S.; Sarkar, F.H.; Philip, P.A. Cyclooxygenase-2-dependent and -independent effects of celecoxib in pancreatic cancer cell lines. Mol. Cancer Ther. 2004, 3, 1421–1426. [Google Scholar]
- Tak, P.P.; Firestein, G.S. NF-kappaB: A key role in inflammatory diseases. J. Clin. Investig. 2001, 107, 7–11. [Google Scholar] [CrossRef]
- Surh, Y.J.; Chun, K.S.; Cha, H.H.; Han, S.S.; Keum, Y.S.; Park, K.K.; Lee, S.S. Molecular mechanisms underlying chemopreventive activities of anti-inflammatory phytochemicals: Down-regulation of COX-2 and iNOS through suppression of NF-kappa B activation. Mutat. Res. 2001, 480–481, 243–268. [Google Scholar] [CrossRef]
- Singh, S.; Aggarwal, B.B. Activation of transcription factor NF-kappa B is suppressed by curcumin (diferuloylmethane). J. Biol. Chem. 1995, 270, 24995–25000. [Google Scholar] [CrossRef] [PubMed]
- Bharti, A.C.; Donato, N.; Singh, S.; Aggarwal, B.B. Curcumin (diferuloylmethane) down-regulates the constitutive activation of nuclear factor-kappa B and IkappaBalpha kinase in human multiple myeloma cells, leading to suppression of proliferation and induction of apoptosis. Blood 2003, 101, 1053–1062. [Google Scholar] [CrossRef]
- Dhillon, N.; Aggarwal, B.B.; Newman, R.A.; Wolff, R.A.; Kunnumakkara, A.B.; Abbruzzese, J.L.; Ng, C.S.; Badmaev, V.; Kurzrock, R. Phase II trial of curcumin in patients with advanced pancreatic cancer. Clin. Cancer Res. 2008, 14, 4491–4499. [Google Scholar] [CrossRef]
- Bimonte, S.; Barbieri, A.; Leongito, M.; Piccirillo, M.; Giudice, A.; Pivonello, C.; de Angelis, C.; Granata, V.; Palaia, R.; Izzo, F. Curcumin Anti Cancer Studies in Pancreatic Cancer. Nutrients 2016, 8, 433. [Google Scholar] [CrossRef]
- Miasari, M.; Puthalakath, H.; Silke, J. Ubiquitylation and cancer development. Cancer Drug Targets 2008, 8, 118–123. [Google Scholar] [CrossRef]
- Wente, M.N.; Eibl, G.; Reber, H.A.; Friess, H.; Buchler, M.W.; Hines, O.J. The proteasome inhibitor MG132 induces apoptosis in human pancreatic cancer cells. Oncol. Rep. 2005, 14, 1635–1638. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, Y.; Sawai, H.; Ochi, N.; Yasuda, A.; Sakamoto, M.; Takahashi, H.; Funahashi, H.; Takeyama, H.; Guha, S. Proteasome inhibitor MG132 inhibits angiogenesis in pancreatic cancer by blocking NF-kappaB activity. Dig. Dis. Sci. 2010, 55, 1167–1176. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, S.T.; Sweeney-Gotsch, B.; Takamori, R.; McConkey, D.J. The proteasome inhibitor bortezomib enhances the activity of docetaxel in orthotopic human pancreatic tumor xenografts. Mol. Cancer Ther. 2004, 3, 59–70. [Google Scholar]
- Alberts, S.R.; Foster, N.R.; Morton, R.F.; Kugler, J.; Schaefer, P.; Wiesenfeld, M.; Fitch, T.R.; Steen, P.; Kim, G.P.; Gill, S. PS-341 and gemcitabine in patients with metastatic pancreatic adenocarcinoma: A North Central Cancer Treatment Group (NCCTG) randomized phase II study. Ann. Oncol. 2005, 16, 1654–1661. [Google Scholar] [CrossRef] [PubMed]
- Catley, M.C.; Chivers, J.E.; Holden, N.S.; Barnes, P.J.; Newton, R. Validation of IKK beta as therapeutic target in airway inflammatory disease by adenoviral-mediated delivery of dominant-negative IKK beta to pulmonary epithelial cells. Br. J. Pharmacol. 2005, 145, 114–122. [Google Scholar] [CrossRef]
- May, M.J.; D’Acquisto, F.; Madge, L.A.; Glockner, J.; Pober, J.S.; Ghosh, S. Selective inhibition of NF-kappaB activation by a peptide that blocks the interaction of NEMO with the IkappaB kinase complex. Science 2000, 289, 1550–1554. [Google Scholar] [CrossRef]
- Greten, F.R.; Arkan, M.C.; Bollrath, J.; Hsu, L.C.; Goode, J.; Miething, C.; Goktuna, S.I.; Neuenhahn, M.; Fierer, J.; Paxian, S.; et al. NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell 2007, 130, 918–931. [Google Scholar] [CrossRef]
- Prescott, J.A.; Cook, S.J. Targeting IKKβ in Cancer: Challenges and Opportunities for the Therapeutic Utilisation of IKKβ Inhibitors. Cells 2018, 7, 115. [Google Scholar] [CrossRef]
- Ramadass, V.; Vaiyapuri, T.; Tergaonkar, V. Small Molecule NF-κB Pathway Inhibitors in Clinic. Int. J. Mol. Sci. 2020, 21, 5164. [Google Scholar] [CrossRef]
- Ochiai, T.; Saito, Y.; Saitoh, T.; Dewan, M.Z.; Shioya, A.; Kobayashi, M.; Kawachi, H.; Muto, S.; Itai, A.; Uota, S.; et al. Inhibition of IkappaB kinase beta restrains oncogenic proliferation of pancreatic cancer cells. J. Med Dent. Sci. 2008, 55, 49–59. [Google Scholar]
- Weichert, W.; Boehm, M.; Gekeler, V.; Bahra, M.; Langrehr, J.; Neuhaus, P.; Denkert, C.; Imre, G.; Weller, C.; Hofmann, H.P.; et al. High expression of RelA/p65 is associated with activation of nuclear factor-kappaB-dependent signaling in pancreatic cancer and marks a patient population with poor prognosis. Br. J. Cancer 2007, 97, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Abbruzzese, J.L.; Evans, D.B.; Larry, L.; Cleary, K.R.; Chiao, P.J. The nuclear factor-kappa B RelA transcription factor is constitutively activated in human pancreatic adenocarcinoma cells. Clin. Cancer Res. 1999, 5, 119–127. [Google Scholar]
- Yang, X.D.; Huang, B.; Li, M.; Lamb, A.; Kelleher, N.L.; Chen, L.F. Negative regulation of NF-kappaB action by Set9-mediated lysine methylation of the RelA subunit. EMBO J. 2009, 28, 1055–1066. [Google Scholar] [CrossRef] [PubMed]
- Grumont, R.J.; Gerondakis, S. Murine c-rel transcription is rapidly induced in T-cells and fibroblasts by mitogenic agents and the phorbol ester 12-O-tetradecanoylphorbol-13-acetate. Cell Growth Differ. 1990, 1, 345–350. [Google Scholar]
- Gilmore, T.D.; Gerondakis, S. The c-Rel Transcription Factor in Development and Disease. Genes Cancer 2011, 2, 695–711. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.E.; Leslie, J.; Perkins, N.D. c-Rel and its many roles in cancer: An old story with new twists. Br. J. Cancer 2016, 114, 1–6. [Google Scholar] [CrossRef]
- Ouk, S.; Liou, M.L.; Liou, H.C. Direct Rel/NF-kappaB inhibitors: Structural basis for mechanism of action. Future Med. Chem. 2009, 1, 1683–1707. [Google Scholar] [CrossRef]
- Shono, Y.; Tuckett, A.Z.; Liou, H.C.; Doubrovina, E.; Derenzini, E.; Ouk, S.; Tsai, J.J.; Smith, O.M.; Levy, E.R.; Kreines, F.M.; et al. Characterization of a c-Rel Inhibitor That Mediates Anticancer Properties in Hematologic Malignancies by Blocking NF-kappaB-Controlled Oxidative Stress Responses. Cancer Res. 2016, 76, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Shono, Y.; Tuckett, A.Z.; Liou, H.C.; Ouk, S.; Doubrovina, E.; Doubrovin, M.; Derenzini, E.; Scallion, M.I.; Tsai, J.J.; Smith, O.M.; et al. Targeted Therapy of B Cell Lymphoma with a Direct Inhibitor of the NF-κB Subunit c-Rel. Blood 2014, 124, 507. [Google Scholar] [CrossRef]
- Geismann, C.; Grohmann, F.; Sebens, S.; Wirths, G.; Dreher, A.; Hasler, R.; Rosenstiel, P.; Hauser, C.; Egberts, J.H.; Trauzold, A.; et al. c-Rel is a critical mediator of NF-kappaB-dependent TRAIL resistance of pancreatic cancer cells. Cell Death Dis. 2014, 5, e1455. [Google Scholar] [CrossRef] [PubMed]
- Rhyasen, G.; Starczynowski, D. IRAK signalling in cancer. Br. J. Cancer 2015, 112, 232–237. [Google Scholar] [CrossRef]
- Zhan, D.; Park, C.Y. Stem Cells in the Myelodysplastic Syndromes. Front. Aging 2021, 2, 27. [Google Scholar] [CrossRef]
- Zarrin, A.A.; Bao, K.; Lupardus, P.; Vucic, D. Kinase inhibition in autoimmunity and inflammation. Nat. Rev. Drug Discov. 2021, 20, 39–63. [Google Scholar] [CrossRef]
- Nowakowski, G.S.; Leslie, L.A.; Younes, A.; Rosenthal, A.C.; Lunning, M.A.; Patel, K.; Landsburg, D.J.; Martinez, E.; von Roemeling, R.; Lieberman, C.; et al. Safety, Pharmacokinetics and Activity of CA-4948, an IRAK4 Inhibitor, for Treatment of Patients with Relapsed or Refractory Hematologic Malignancies: Results from the Phase 1 Study. Blood 2020, 136, 44–45. [Google Scholar] [CrossRef]
- Xu, Y.-R.; Lei, C.-Q. TAK1-TABs Complex: A Central Signalosome in Inflammatory Responses. Front. Immunol. 2021, 11, 608976. [Google Scholar] [CrossRef] [PubMed]
- Malireddi, R.K.S.; Sannula, K.; Kanneganti, T.-D. ZBP1 and TAK1: Master Regulators of NLRP3 Inflammasome/Pyroptosis, Apoptosis, and Necroptosis (PAN-optosis). Front. Cell. Infect. Microbiol. 2021, 9, 406. [Google Scholar] [CrossRef]
- Scarneo, S.A.; Mansourati, A.; Eibschutz, L.S.; Totzke, J.; Roques, J.; Loiselle, D.; Carlson, D.; Hughes, P.; Haystead, T. Genetic and pharmacological validation of TAK1 inhibition in macrophages as a therapeutic strategy to effectively inhibit TNF secretion. Sci. Rep. 2018, 8, 17058. [Google Scholar] [CrossRef] [PubMed]
- Totzke, J.; Gurbani, D.; Raphemot, R.; Hughes, P.F.; Bodoor, K.; Carlson, D.A.; Loiselle, D.R.; Bera, A.K.; Eibschutz, L.S.; Perkins, M.M.; et al. Takinib, a Selective TAK1 Inhibitor, Broadens the Therapeutic Efficacy of TNF-α Inhibition for Cancer and Autoimmune Disease. Cell Chem. Biol. 2017, 24, 1029.e7–1039.e7. [Google Scholar] [CrossRef]
- Zhou, J.; Zheng, B.; Ji, J.; Shen, F.; Min, H.; Liu, B.; Wu, J.; Zhang, S. LYTAK1, a novel TAK1 inhibitor, suppresses KRAS mutant colorectal cancer cell growth in vitro and in vivo. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2015, 36, 3301–3308. [Google Scholar] [CrossRef] [PubMed]
- Louis, C.; Burns, C.; Wicks, I.P. TANK-Binding Kinase 1-Dependent Responses in Health and Autoimmunity. Front. Immunol. 2018, 9, 434. [Google Scholar] [CrossRef]
- Alam, M.; Hasan, G.M.; Hassan, I. A review on the role of TANK-binding kinase 1 signaling in cancer. Int. J. Biol. Macromol. 2021, 183, 2364–2375. [Google Scholar] [CrossRef] [PubMed]
- Adjuto-Saccone, M.; Soubeyran, P.; Garcia, J.; Audebert, S.; Camoin, L.; Rubis, M.; Roques, J.; Binétruy, B.; Iovanna, J.L.; Tournaire, R. TNF-α induces endothelial–mesenchymal transition promoting stromal development of pancreatic adenocarcinoma. Cell Death Dis. 2021, 12, 649. [Google Scholar] [CrossRef]
- Hasan, M.; Yan, N. Therapeutic potential of targeting TBK1 in autoimmune diseases and interferonopathies. Pharmacol. Res. 2016, 111, 336–342. [Google Scholar] [CrossRef]
- Zhu, Z.; Aref, A.R.; Cohoon, T.J.; Barbie, T.U.; Imamura, Y.; Yang, S.; Moody, S.E.; Shen, R.R.; Schinzel, A.C.; Thai, T.C.; et al. Inhibition of KRAS-driven tumorigenicity by interruption of an autocrine cytokine circuit. Cancer Discov. 2014, 4, 452–465. [Google Scholar] [CrossRef] [PubMed]
- Burton, V.H.; Ou, Y.-H.; Toombs, J.E.; White, M.A.; Brekken, R.A. TBK1 as a novel mediator of K-Ras driven pancreatic cancer. Mol. Cancer Res. 2014, 12. [Google Scholar] [CrossRef]
- Li, P.; Zheng, Y.; Chen, X. Drugs for Autoimmune Inflammatory Diseases: From Small Molecule Compounds to Anti-TNF Biologics. Front. Pharmacol. 2017, 8, 460. [Google Scholar] [CrossRef] [PubMed]
- Egberts, J.H.; Cloosters, V.; Noack, A.; Schniewind, B.; Thon, L.; Klose, S.; Kettler, B.; von Forstner, C.; Kneitz, C.; Tepel, J.; et al. Anti–Tumor Necrosis Factor Therapy Inhibits Pancreatic Tumor Growth and Metastasis. Cancer Res. 2008, 68, 1443–1450. [Google Scholar] [CrossRef]
- Zhao, X.; Fan, W.; Xu, Z.; Chen, H.; He, Y.; Yang, G.; Yang, G.; Hu, H.; Tang, S.; Wang, P.; et al. Inhibiting tumor necrosis factor-alpha diminishes desmoplasia and inflammation to overcome chemoresistance in pancreatic ductal adenocarcinoma. Oncotarget 2016, 7, 81110–81122. [Google Scholar] [CrossRef] [PubMed]
- Otto, L.; Rahn, S.; Daunke, T.; Walter, F.; Winter, E.; Möller, J.L.; Rose-John, S.; Wesch, D.; Schäfer, H.; Sebens, S. Initiation of Pancreatic Cancer: The Interplay of Hyperglycemia and Macrophages Promotes the Acquisition of Malignancy-Associated Properties in Pancreatic Ductal Epithelial Cells. Int. J. Mol. Sci. 2021, 22, 5086. [Google Scholar] [CrossRef] [PubMed]
- Wiedenmann, B.; Malfertheiner, P.; Friess, H.; Ritch, P.; Arseneau, J.; Mantovani, G.; Caprioni, F.; Van Cutsem, E.; Richel, D.; DeWitte, M.; et al. A multicenter, phase II study of infliximab plus gemcitabine in pancreatic cancer cachexia. J. Supportive Oncol. 2008, 6, 18–25. [Google Scholar]
- Wu, C.; Fernandez, S.A.; Criswell, T.; Chidiac, T.A.; Guttridge, D.; Villalona-Calero, M.; Bekaii-Saab, T.S. Disrupting cytokine signaling in pancreatic cancer: A phase I/II study of etanercept in combination with gemcitabine in patients with advanced disease. Pancreas 2013, 42, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Van Mackelenbergh, M.G.; Stroes, C.I.; Spijker, R.; van Eijck, C.; Wilmink, J.W.; Bijlsma, M.F.; van Laarhoven, H. Clinical Trials Targeting the Stroma in Pancreatic Cancer: A Systematic Review and Meta-Analysis. Cancers 2019, 11, 588. [Google Scholar] [CrossRef]
- Elsayed, M.; Abdelrahim, M. The Latest Advancement in Pancreatic Ductal Adenocarcinoma Therapy: A Review Article for the Latest Guidelines and Novel Therapies. Biomedicines 2021, 9, 389. [Google Scholar] [CrossRef]
- Martens, S.; Hofmans, S.; Declercq, W.; Augustyns, K.; Vandenabeele, P. Inhibitors Targeting RIPK1/RIPK3: Old and New Drugs. Trends Pharmacol. Sci. 2020, 41, 209–224. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Webster, J.D.; Varfolomeev, E.; Kwon, Y.C.; Cheng, J.H.; Zhang, J.; Dugger, D.L.; Wickliffe, K.E.; Maltzman, A.; Sujatha-Bhaskar, S.; et al. RIP1 inhibition blocks inflammatory diseases but not tumor growth or metastases. Cell Death Differ. 2020, 27, 161–175. [Google Scholar] [CrossRef]
- Mifflin, L.; Ofengeim, D.; Yuan, J. Receptor-interacting protein kinase 1 (RIPK1) as a therapeutic target. Nat. Rev. Drug Discov. 2020, 19, 553–571. [Google Scholar] [CrossRef]
- Jensen, S.; Seidelin, J.B.; LaCasse, E.C.; Nielsen, O.H. SMAC mimetics and RIPK inhibitors as therapeutics for chronic inflammatory diseases. Sci. Signal. 2020, 13, eaax8295. [Google Scholar] [CrossRef]
- Degterev, A.; Ofengeim, D.; Yuan, J. Targeting RIPK1 for the treatment of human diseases. Proc. Natl. Acad. Sci. USA 2019, 116, 9714–9722. [Google Scholar] [CrossRef]
- Shirai, Y.; Saito, N.; Uwagawa, T.; Shiba, H.; Horiuchi, T.; Iwase, R.; Haruki, K.; Ohashi, T.; Yanaga, K. Pomalidomide promotes chemosensitization of pancreatic cancer by inhibition of NF-kappaB. Oncotarget 2018, 9, 15292–15301. [Google Scholar] [CrossRef] [PubMed]
- Santoro, R.; Zanotto, M.; Piro, G.; Carbone, C.; Tortora, G.; Melisi, D. Modulating TAK1 expression through the inhibition of GSK3 impairs YAP/TAZ oncogenic functions in pancreatic cancer. Cancer Res. 2018, 78, 3339. [Google Scholar]
- Kazi, A.; Xiang, S.; Yang, H.; Delitto, D.; Trevino, J.; Jiang, R.H.Y.; Ayaz, M.; Lawrence, H.R.; Kennedy, P.; Sebti, S.M. GSK3 suppression upregulates beta-catenin and c-Myc to abrogate KRas-dependent tumors. Nat. Commun. 2018, 9, 5154. [Google Scholar] [CrossRef] [PubMed]
- Fabre, C.; Mimura, N.; Bobb, K.; Kong, S.Y.; Gorgun, G.; Cirstea, D.; Hu, Y.; Minami, J.; Ohguchi, H.; Zhang, J.; et al. Dual inhibition of canonical and noncanonical NF-kappaB pathways demonstrates significant antitumor activities in multiple myeloma. Clin. Cancer Res. 2012, 18, 4669–4681. [Google Scholar] [CrossRef] [PubMed]
- Vince, J.E.; Wong, W.W.; Khan, N.; Feltham, R.; Chau, D.; Ahmed, A.U.; Benetatos, C.A.; Chunduru, S.K.; Condon, S.M.; McKinlay, M.; et al. IAP Antagonists Target cIAP1 to Induce TNFalpha-Dependent Apoptosis. Cell 2007, 131, 682–693. [Google Scholar] [CrossRef] [PubMed]
- Varfolomeev, E.; Blankenship, J.W.; Wayson, S.M.; Fedorova, A.V.; Kayagaki, N.; Garg, P.; Zobel, K.; Dynek, J.N.; Elliott, L.O.; Wallweber, H.J.; et al. IAP Antagonists Induce Autoubiquitination of c-IAPs, NF-kappaB Activation, and TNFalpha-Dependent Apoptosis. Cell 2007, 131, 669–681. [Google Scholar] [CrossRef]
- Gaither, A.; Porter, D.; Yao, Y.; Borawski, J.; Yang, G.; Donovan, J.; Sage, D.; Slisz, J.; Tran, M.; Straub, C.; et al. A Smac mimetic rescue screen reveals roles for inhibitor of apoptosis proteins in tumor necrosis factor-alpha signaling. Cancer Res. 2007, 67, 11493–11498. [Google Scholar] [CrossRef]
- Morrish, E.; Brumatti, G.; Silke, J. Future Therapeutic Directions for Smac-Mimetics. Cells 2020, 9, 406. [Google Scholar] [CrossRef]
- Zhao, X.Y.; Wang, X.Y.; Wei, Q.Y.; Xu, Y.M.; Lau, A.T.Y. Potency and Selectivity of SMAC/DIABLO Mimetics in Solid Tumor Therapy. Cells 2020, 9, 1012. [Google Scholar] [CrossRef]
- Liang, W.; Liao, Y.; Zhang, J.; Huang, Q.; Luo, W.; Yu, J.; Gong, J.; Zhou, Y.; Li, X.; Tang, B.; et al. Heat shock factor 1 inhibits the mitochondrial apoptosis pathway by regulating second mitochondria-derived activator of caspase to promote pancreatic tumorigenesis. J. Exp. Clin. Cancer Res. 2017, 36, 64. [Google Scholar] [CrossRef]
- Giagkousiklidis, S.; Vellanki, S.H.; Debatin, K.M.; Fulda, S. Sensitization of pancreatic carcinoma cells for gamma-irradiation-induced apoptosis by XIAP inhibition. Oncogene 2007, 26, 7006–7016. [Google Scholar] [CrossRef][Green Version]
- Vogler, M.; Walczak, H.; Stadel, D.; Haas, T.L.; Genze, F.; Jovanovic, M.; Gschwend, J.E.; Simmet, T.; Debatin, K.M.; Fulda, S. Targeting XIAP bypasses Bcl-2-mediated resistance to TRAIL and cooperates with TRAIL to suppress pancreatic cancer growth in vitro and in vivo. Cancer Res. 2008, 68, 7956–7965. [Google Scholar] [CrossRef]
- Feltham, R.; Bettjeman, B.; Budhidarmo, R.; Mace, P.D.; Shirley, S.; Condon, S.M.; Chunduru, S.K.; McKinlay, M.A.; Vaux, D.L.; Silke, J.; et al. Smac Mimetics Activate the E3 Ligase Activity of cIAP1 Protein by Promoting RING Domain Dimerization. J. Biol. Chem. 2011, 286, 17015–17028. [Google Scholar] [CrossRef] [PubMed]
- Dueber, E.C.; Schoeffler, A.J.; Lingel, A.; Elliott, J.M.; Fedorova, A.V.; Giannetti, A.M.; Zobel, K.; Maurer, B.; Varfolomeev, E.; Wu, P.; et al. Antagonists induce a conformational change in cIAP1 that promotes autoubiquitination. Science 2011, 334, 376–380. [Google Scholar] [CrossRef]
- Feoktistova, M.; Geserick, P.; Kellert, B.; Dimitrova, D.P.; Langlais, C.; Hupe, M.; Cain, K.; MacFarlane, M.; Häcker, G.; Leverkus, M. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell 2011, 43, 449–463. [Google Scholar] [CrossRef]
- Tenev, T.; Bianchi, K.; Darding, M.; Broemer, M.; Langlais, C.; Wallberg, F.; Zachariou, A.; Lopez, J.; MacFarlane, M.; Cain, K.; et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol. Cell 2011, 43, 432–448. [Google Scholar] [CrossRef] [PubMed]
- Condon, S.M.; Mitsuuchi, Y.; Deng, Y.; LaPorte, M.G.; Rippin, S.R.; Haimowitz, T.; Alexander, M.D.; Kumar, P.T.; Hendi, M.S.; Lee, Y.H.; et al. Birinapant, a smac-mimetic with improved tolerability for the treatment of solid tumors and hematological malignancies. J. Med. Chem. 2014, 57, 3666–3677. [Google Scholar] [CrossRef]
- Brumatti, G.; Ma, C.; Lalaoui, N.; Nguyen, N.Y.; Navarro, M.; Tanzer, M.C.; Richmond, J.; Ghisi, M.; Salmon, J.M.; Silke, N.; et al. The caspase-8 inhibitor emricasan combines with the SMAC mimetic birinapant to induce necroptosis and treat acute myeloid leukemia. Sci. Transl. Med. 2016, 8, 339ra369. [Google Scholar] [CrossRef] [PubMed]
- Lalaoui, N.; Hanggi, K.; Brumatti, G.; Chau, D.; Nguyen, N.Y.; Vasilikos, L.; Spilgies, L.M.; Heckmann, D.A.; Ma, C.; Ghisi, M.; et al. Targeting p38 or MK2 Enhances the Anti-Leukemic Activity of Smac-Mimetics. Cancer Cell 2016, 29, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Kearney, C.J.; Lalaoui, N.; Freeman, A.J.; Ramsbottom, K.M.; Silke, J.; Oliaro, J. PD-L1 and IAPs co-operate to protect tumors from cytotoxic lymphocyte-derived TNF. Cell Death Differ. 2017, 24, 1705–1716. [Google Scholar] [CrossRef]
- Vredevoogd, D.W.; Kuilman, T.; Ligtenberg, M.A.; Boshuizen, J.; Stecker, K.E.; de Bruijn, B.; Krijgsman, O.; Huang, X.; Kenski, J.C.N.; Lacroix, R.; et al. Augmenting Immunotherapy Impact by Lowering Tumor TNF Cytotoxicity Threshold. Cell 2019, 178, 585–599. [Google Scholar] [CrossRef]
- Michie, J.; Beavis, P.A.; Freeman, A.J.; Vervoort, S.J.; Ramsbottom, K.M.; Narasimhan, V.; Lelliott, E.J.; Lalaoui, N.; Ramsay, R.G.; Johnstone, R.W.; et al. Antagonism of IAPs Enhances CAR T-cell Efficacy. Cancer Immunol. Res. 2019, 7, 183–192. [Google Scholar] [CrossRef]
- Dufva, O.; Koski, J.; Maliniemi, P.; Ianevski, A.; Klievink, J.; Leitner, J.; Pölönen, P.; Hohtari, H.; Saeed, K.; Hannunen, T.; et al. Integrated drug profiling and CRISPR screening identify essential pathways for CAR T-cell cytotoxicity. Blood 2020, 135, 597–609. [Google Scholar] [CrossRef]
- Zhu, X.; Shen, X.; Qu, J.; Straubinger, R.M.; Jusko, W.J. Proteomic Analysis of Combined Gemcitabine and Birinapant in Pancreatic Cancer Cells. Front. Pharmacol. 2018, 9, 84. [Google Scholar] [CrossRef]
- Awasthi, N.; Kirane, A.; Schwarz, M.A.; Toombs, J.E.; Brekken, R.A.; Schwarz, R.E. Smac mimetic-derived augmentation of chemotherapeutic response in experimental pancreatic cancer. BMC Cancer 2011, 11, 15. [Google Scholar] [CrossRef] [PubMed]
- Hashim, Y.M.; Vangveravong, S.; Sankpal, N.V.; Binder, P.S.; Liu, J.S.; Goedegebuure, P.S.; Mach, R.H.; Spitzer, D.; Hawkins, W.G. The Targeted SMAC Mimetic SW IV-134 is a strong enhancer of standard chemotherapy in pancreatic canceR. J. Exp. Clin. Cancer Res. 2017, 36, 14. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.H.; Shin, J.S.; Hong, S.W.; Jung, S.A.; Hwang, I.Y.; Kim, J.H.; Choi, E.K.; Ha, S.H.; Kim, J.S.; Kim, K.M.; et al. A novel small-molecule IAP antagonist, AZD5582, draws Mcl-1 down-regulation for induction of apoptosis through targeting of cIAP1 and XIAP in human pancreatic cancer. Oncotarget 2015, 6, 26895–26908. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Straubinger, R.M.; Jusko, W.J. Mechanism-based mathematical modeling of combined gemcitabine and birinapant in pancreatic cancer cells. J. Pharmacokinet. Pharmacodyn. 2015, 42, 477–496. [Google Scholar] [CrossRef] [PubMed]
- West, A.C.; Martin, B.P.; Andrews, D.A.; Hogg, S.J.; Banerjee, A.; Grigoriadis, G.; Johnstone, R.W.; Shortt, J. The SMAC mimetic, LCL-161, reduces survival in aggressive MYC-driven lymphoma while promoting susceptibility to endotoxic shock. Oncogenesis 2016, 5, e216. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
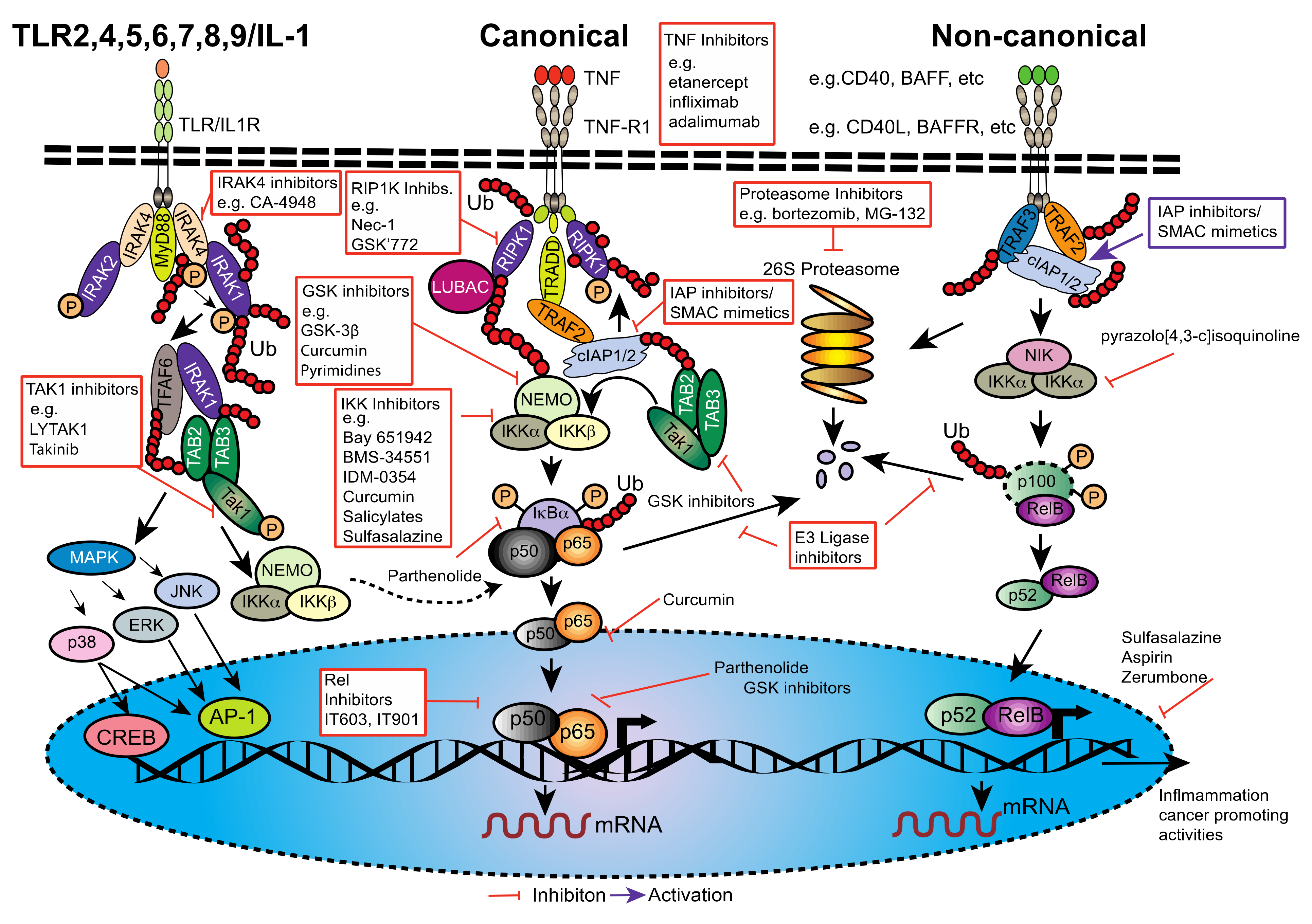{kind=link}
| Smac-Mimetic | Adjuvant Therapy | Phase | Clinical Trial/Start Date | Status (12 February 2021) | Trial Centres |
|---|---|---|---|---|---|
| Debio 1143 a | Pembrolizumab | I | NCT03871959/Sep-19 | Recruiting | France |
| Debio 1143 b | Nivolumab | I/II | NCT04122625/Apr-19 | Recruiting | USA, France, Spain |
| LCL161 | Gemcitabine/Nab-Paclitaxel | I | NCT01934634/Mar-14 | Unknown | USA |
| APG1387 c | None | I/II | NCT03386526/Nov-17 | Recruiting | USA |
| APG1387 b | None | Ib | NCT04284488/Dec-17 | Recruiting | USA |
| APG1387 | Gemcitabine/Nab-Paclitaxel | I/II | NCT04643405/Nov-20 | Not yet recruiting | China |
| BI 891065 b | Immunotherapy (BI 754091) | I | NCT04138823/Oct-19 | Active, not recruiting | Japan |
| ASTX660 d | None | I/II | NCT02503423/July-15 | Recruiting | USA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silke, J.; O’Reilly, L.A. NF-κB and Pancreatic Cancer; Chapter and Verse. Cancers 2021, 13, 4510. https://doi.org/10.3390/cancers13184510
Silke J, O’Reilly LA. NF-κB and Pancreatic Cancer; Chapter and Verse. Cancers. 2021; 13(18):4510. https://doi.org/10.3390/cancers13184510
Chicago/Turabian StyleSilke, John, and Lorraine Ann O’Reilly. 2021. "NF-κB and Pancreatic Cancer; Chapter and Verse" Cancers 13, no. 18: 4510. https://doi.org/10.3390/cancers13184510
APA StyleSilke, J., & O’Reilly, L. A. (2021). NF-κB and Pancreatic Cancer; Chapter and Verse. Cancers, 13(18), 4510. https://doi.org/10.3390/cancers13184510