
Histological Grade of Endometrioid Endometrial Cancer and Relapse Risk Can Be Predicted with Machine Learning from Gene Expression Data



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
- The current 3-tier grading system of endometrioid carcinoma is not ideal for patients included in the grade 2 group as the result could be either under- or overtreatment.
- An objective, reproducible gene expression based method could separate patients included currently in grade 2 class into low-risk and high risk subgroups.
- We have developped a machine learning classifier method that can stratify grade 2 patients in high-risk and low risk subgroups.
- For translational purposes we refined this method to perform the stratification of patients with simillar performance using as few as 12 biomarker genes.
- Based on recent developments in RNA-based diagnostics, translation into clinical practice of our proposed biomarkers could contribute to the development of a robust, binary grade classification system by which more reliable preoperative classification could be implemented.
2. Materials and Methods
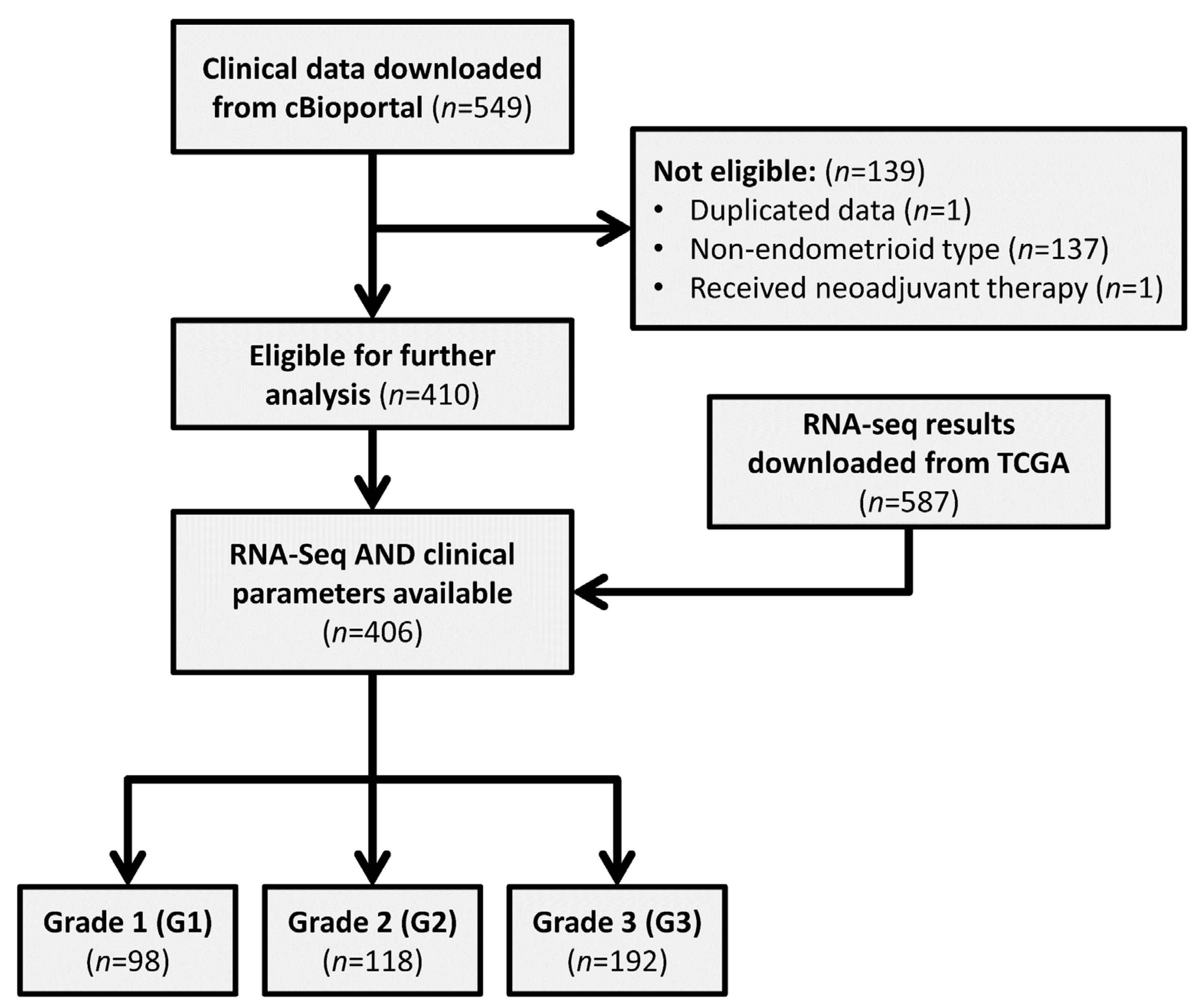
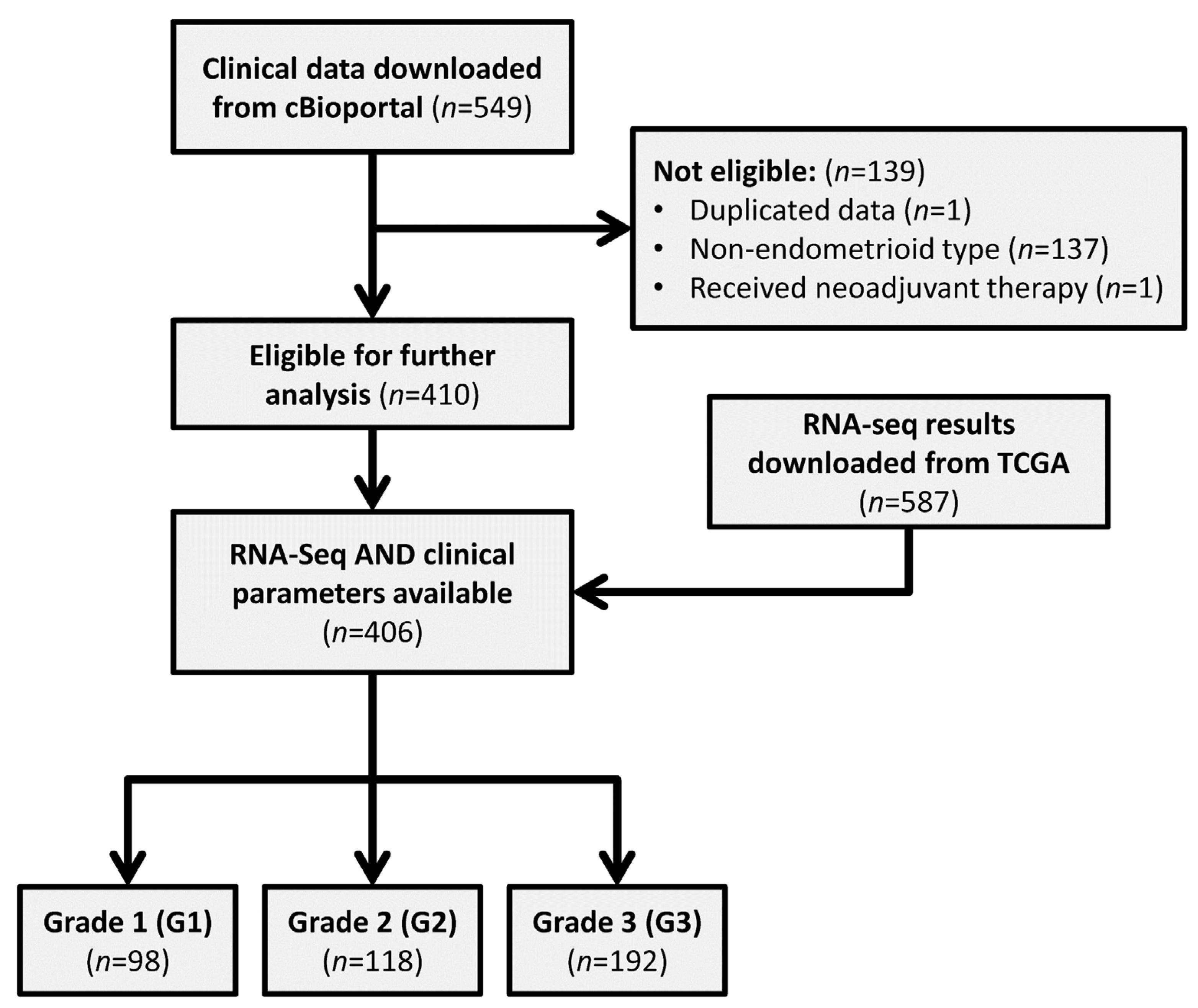
2.1. Downloading of Data
2.2. Screening of Data
2.3. Processing of RNA-Sequencing Data
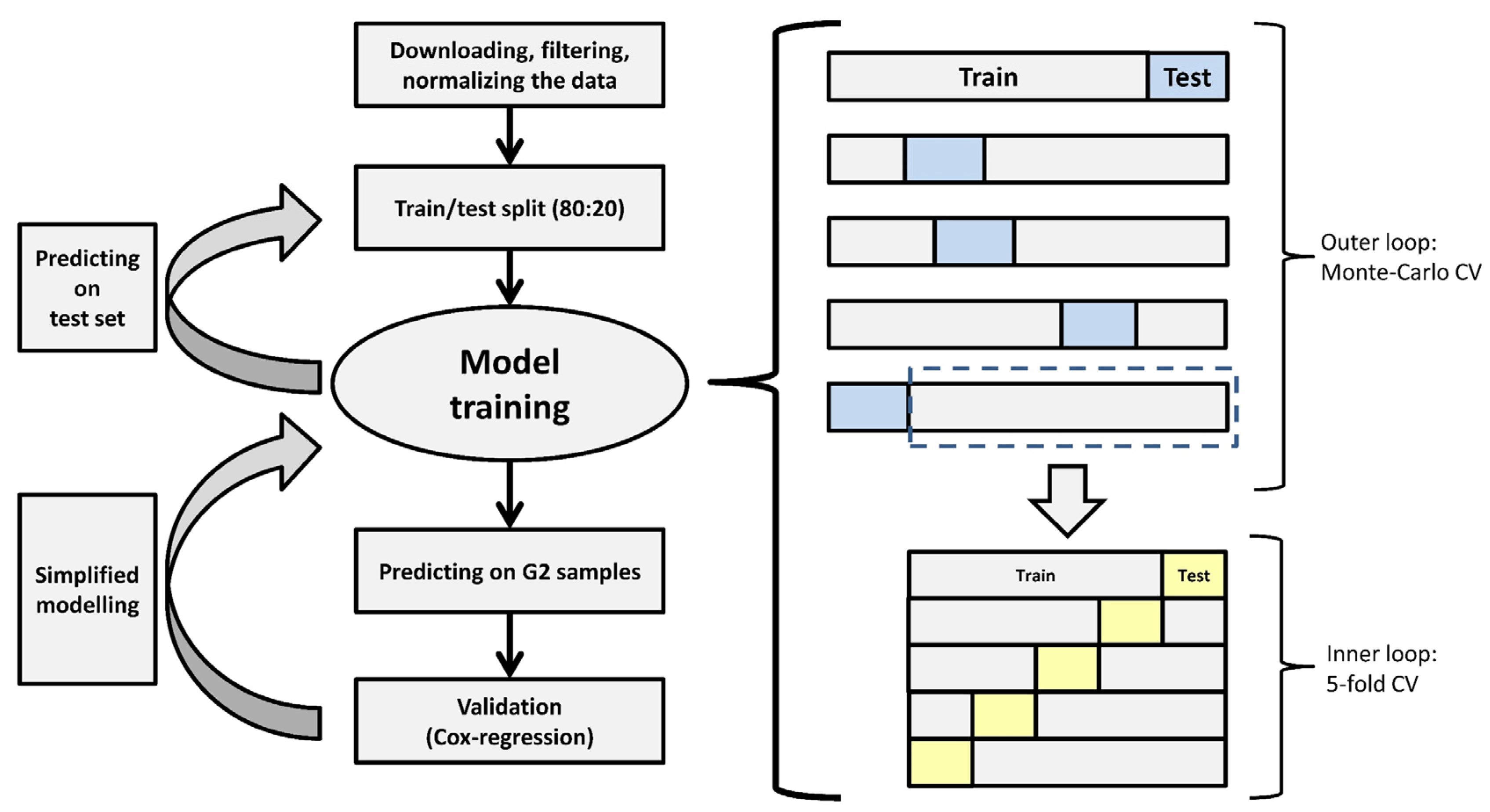
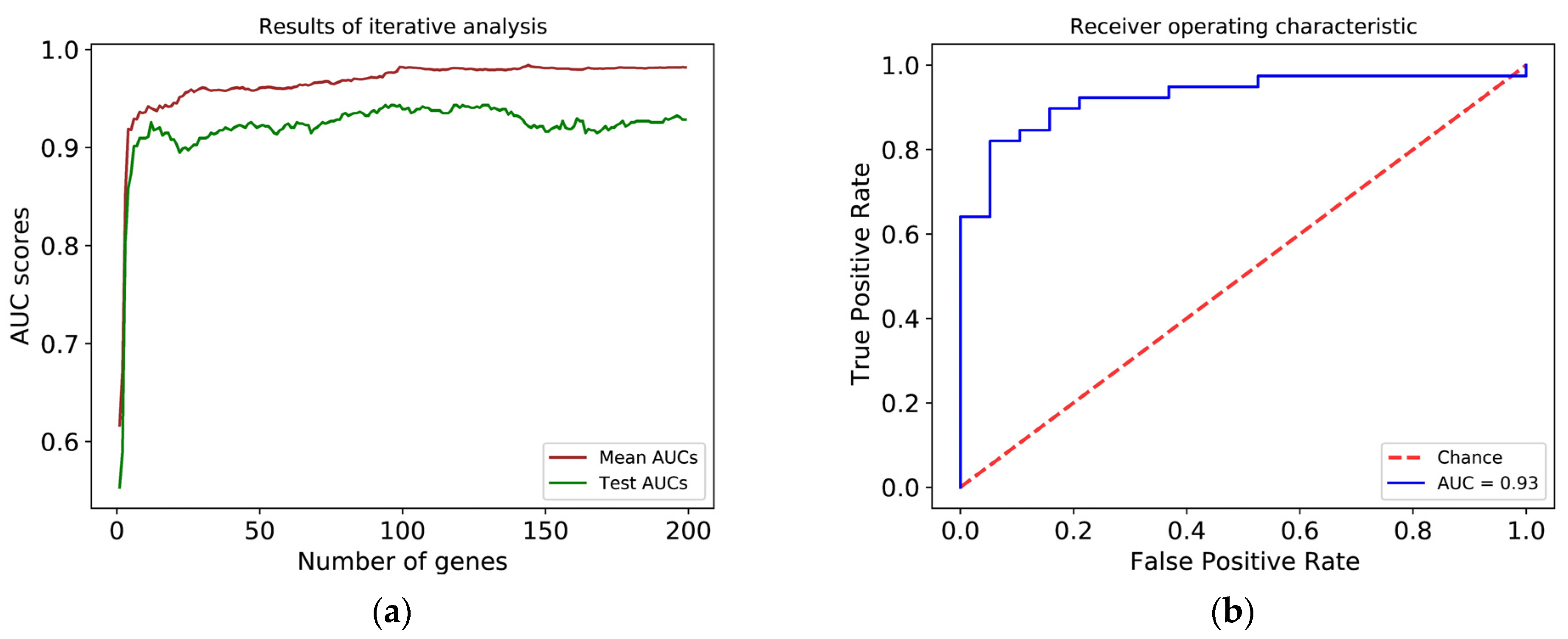
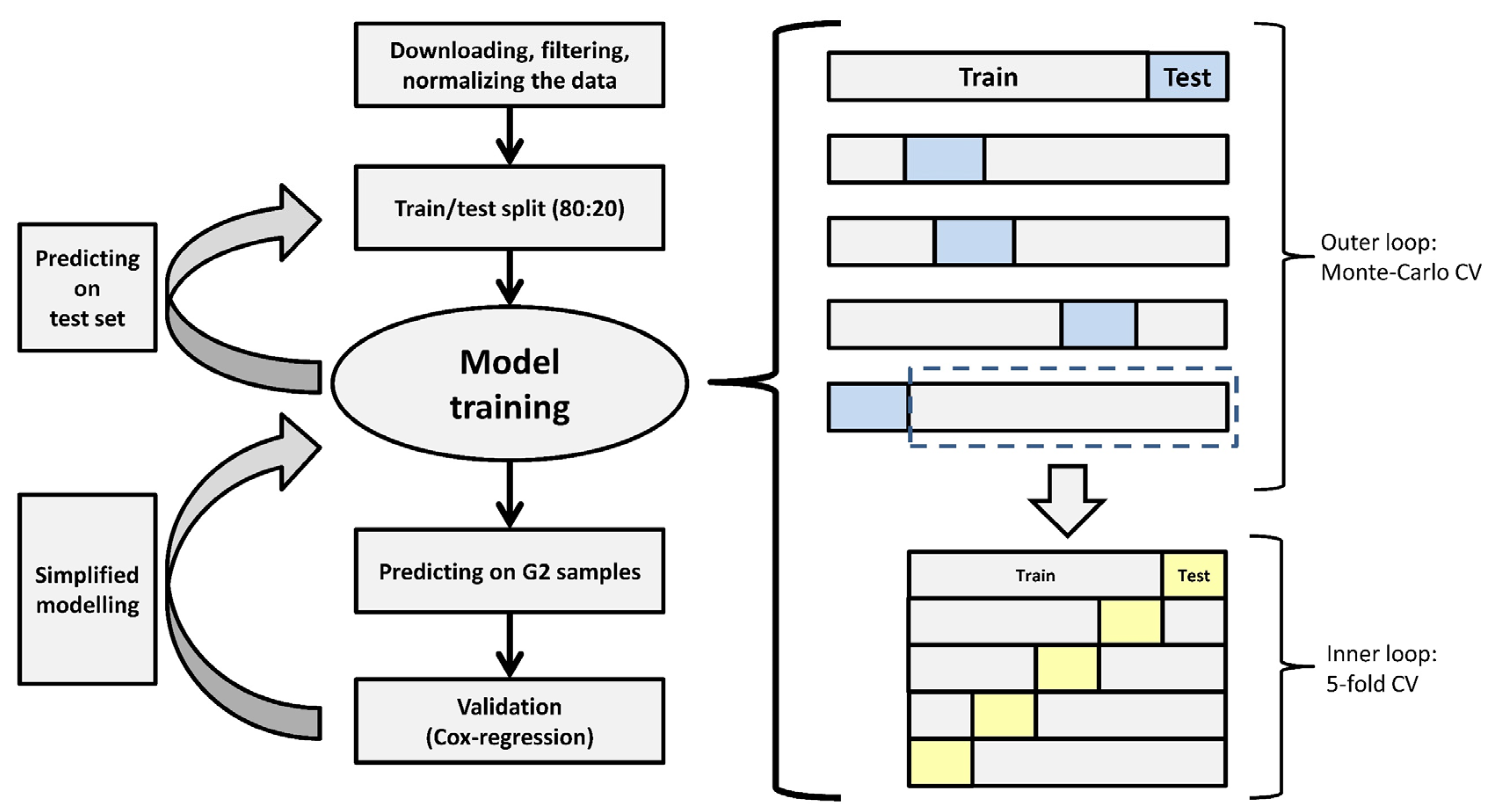
2.4. Machine-Learning Model Development
2.5. Analysis of Relapse-Free Survival
2.6. Model Development with a Reduced Number of Genes
3. Results
3.1. Patient Data Summary
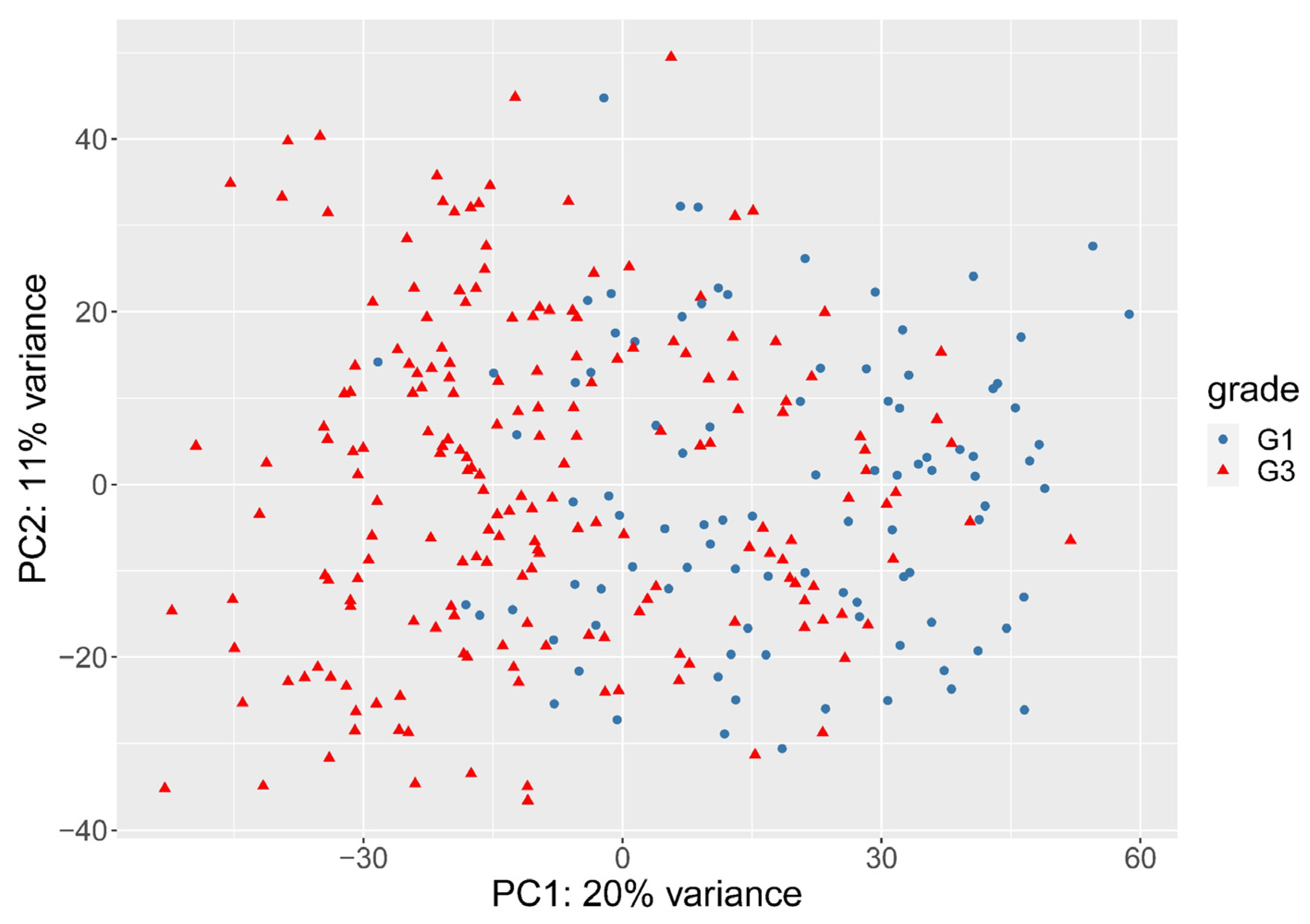
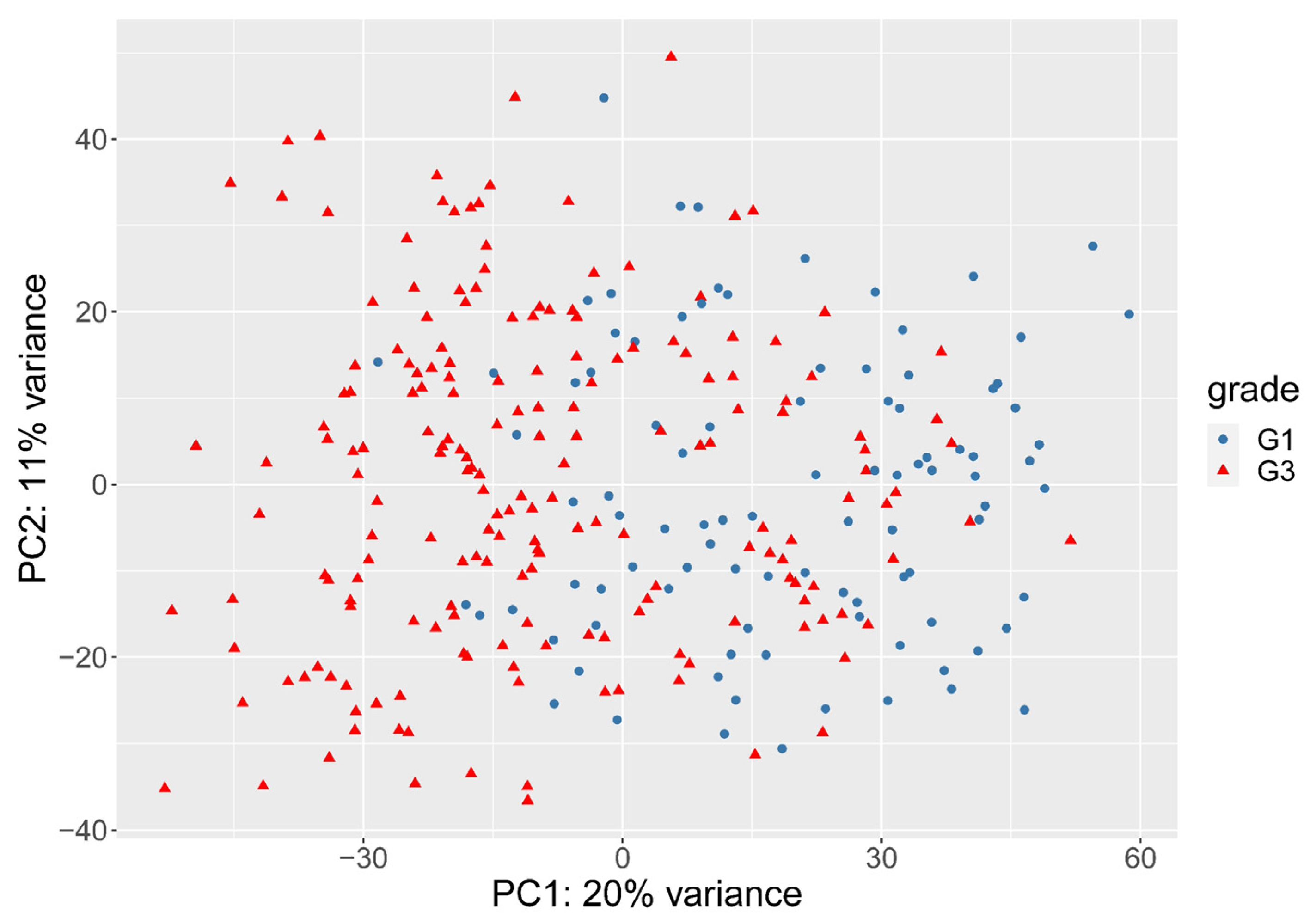
3.2. The Need for Weighting between Genes
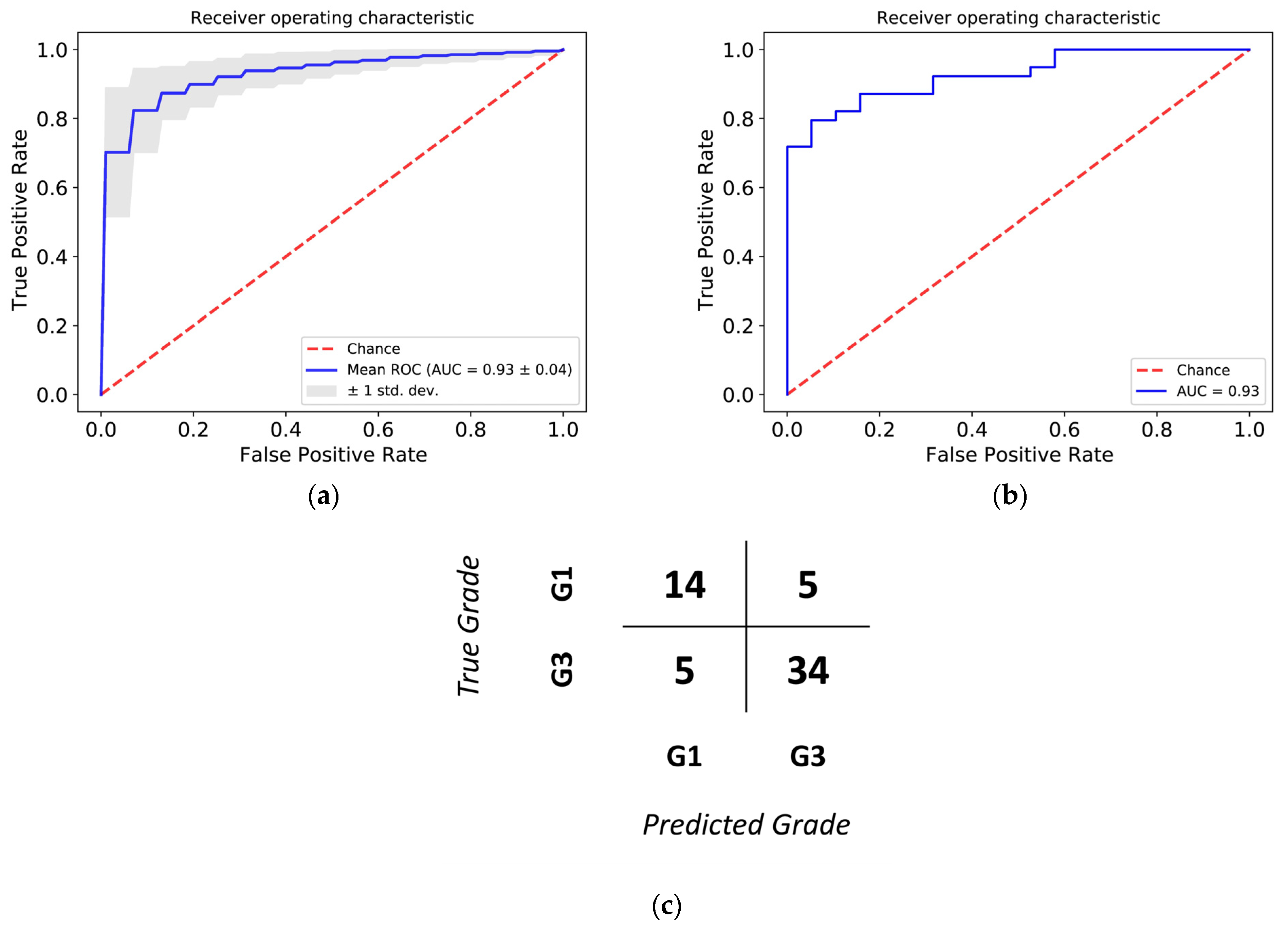
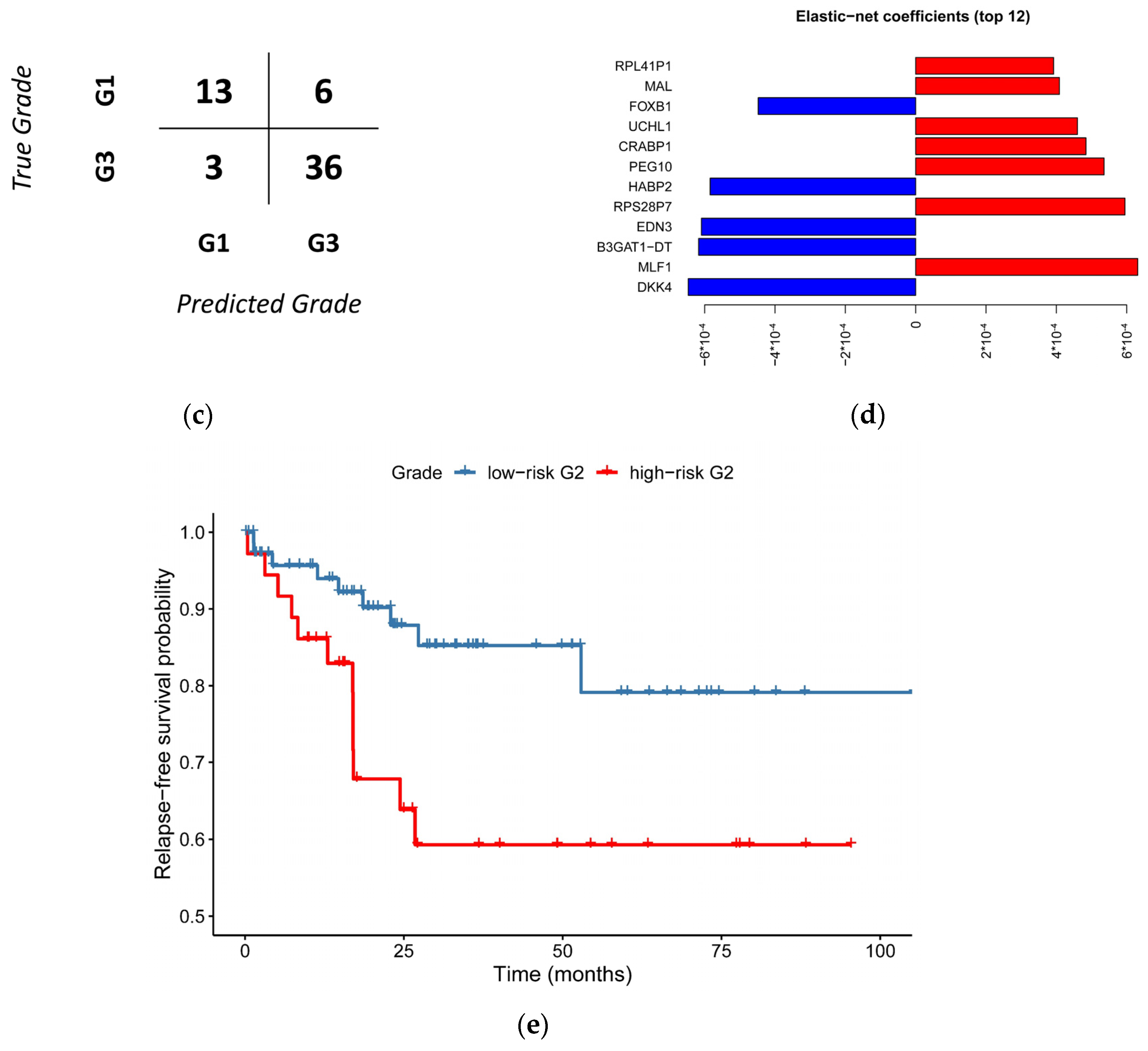
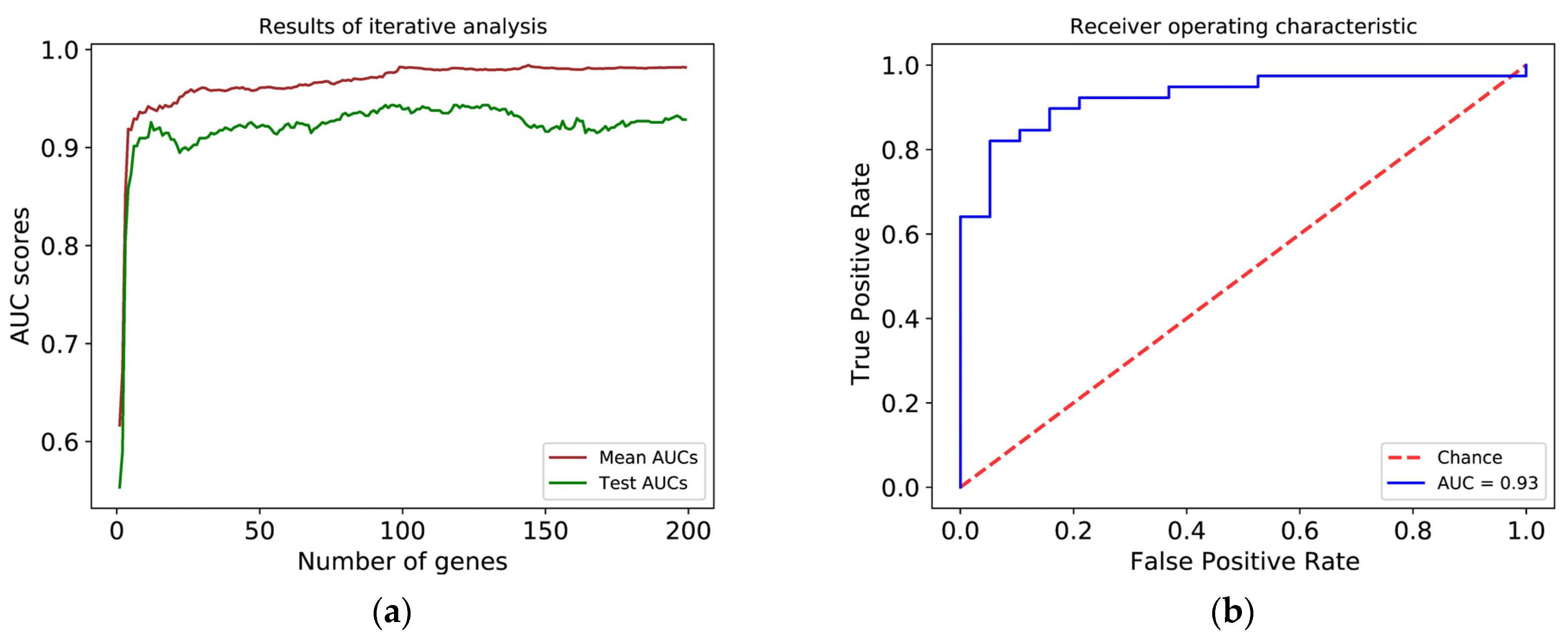
3.3. Histological Grade Can Be Predicted from Gene Expression
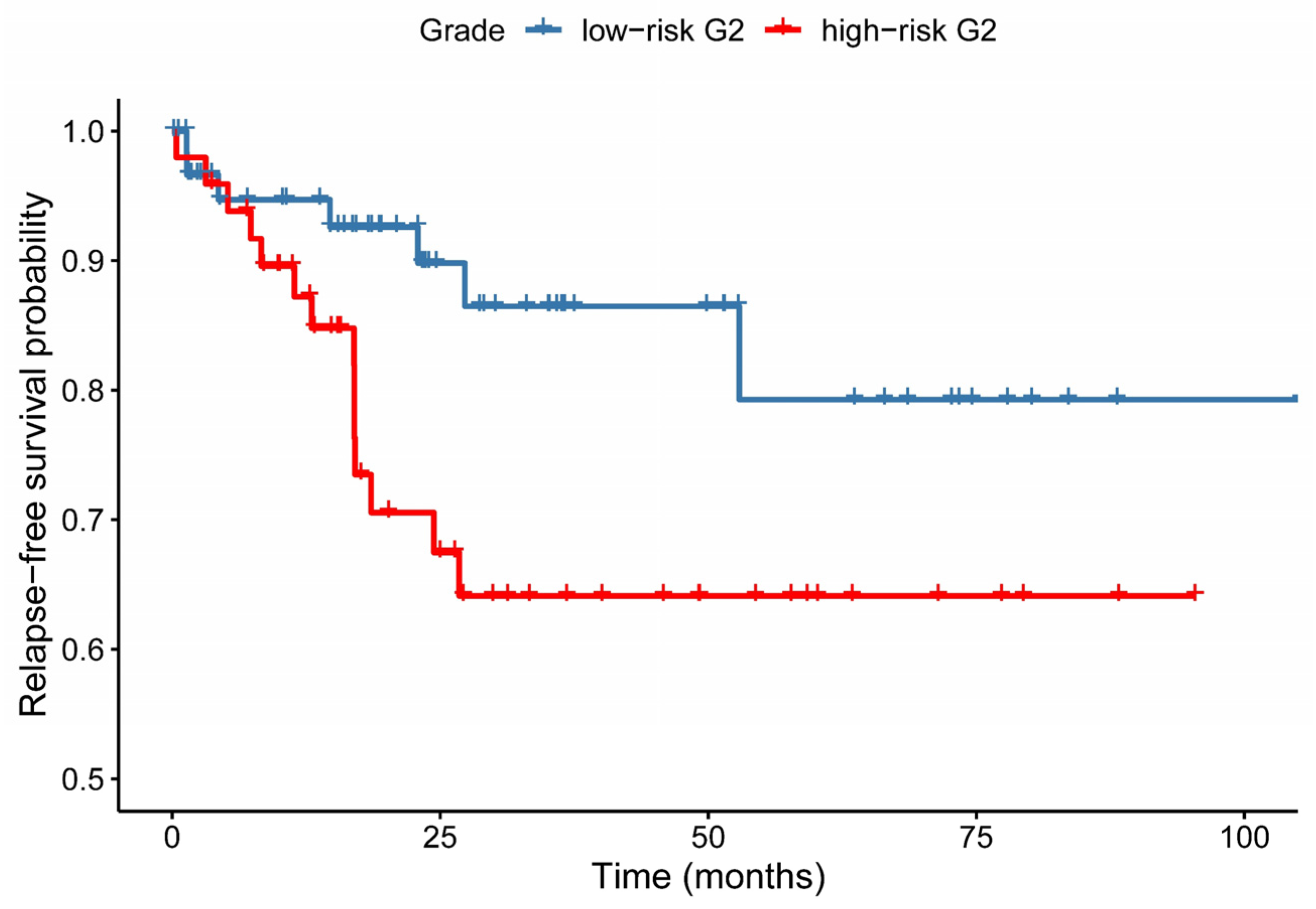
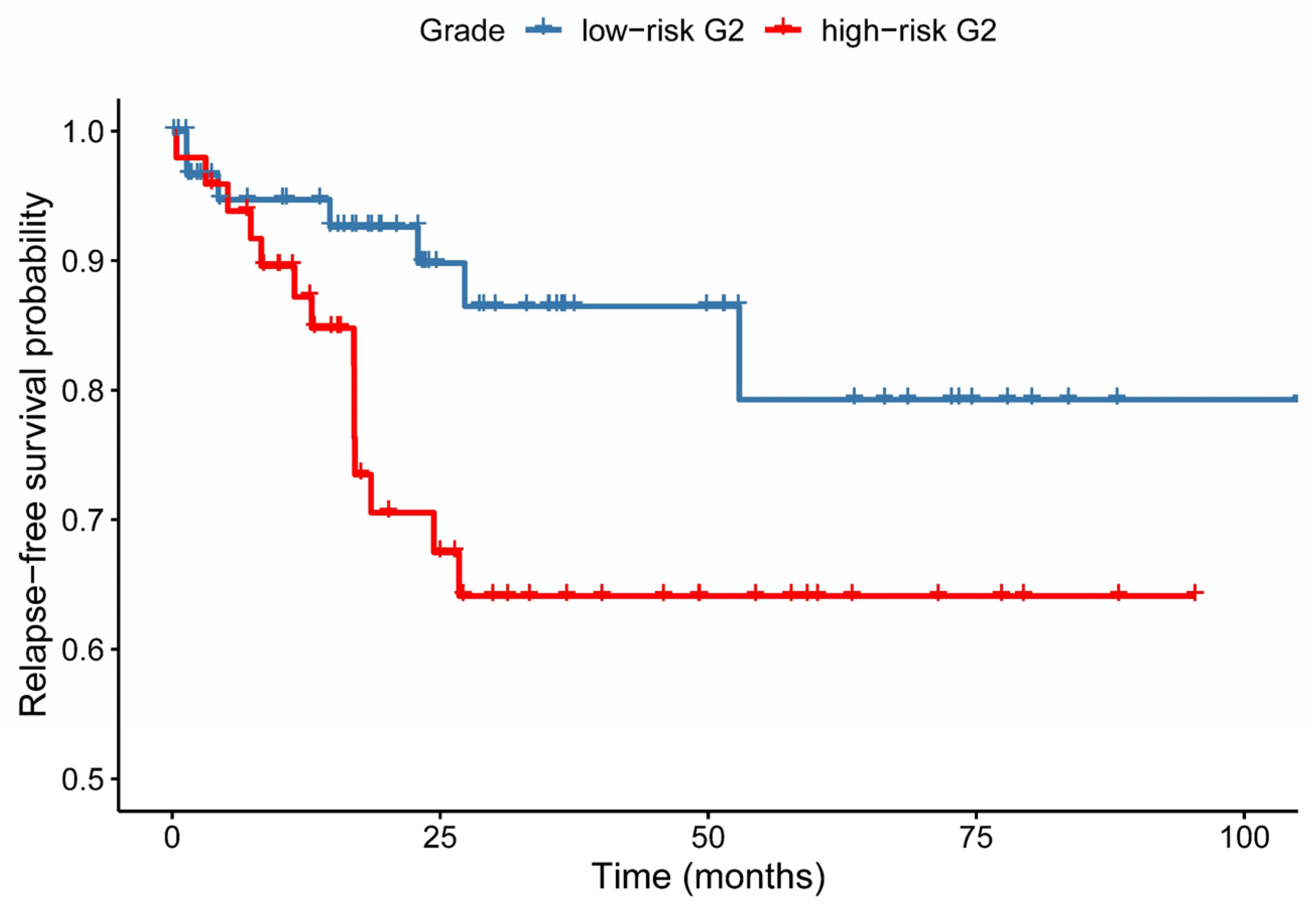
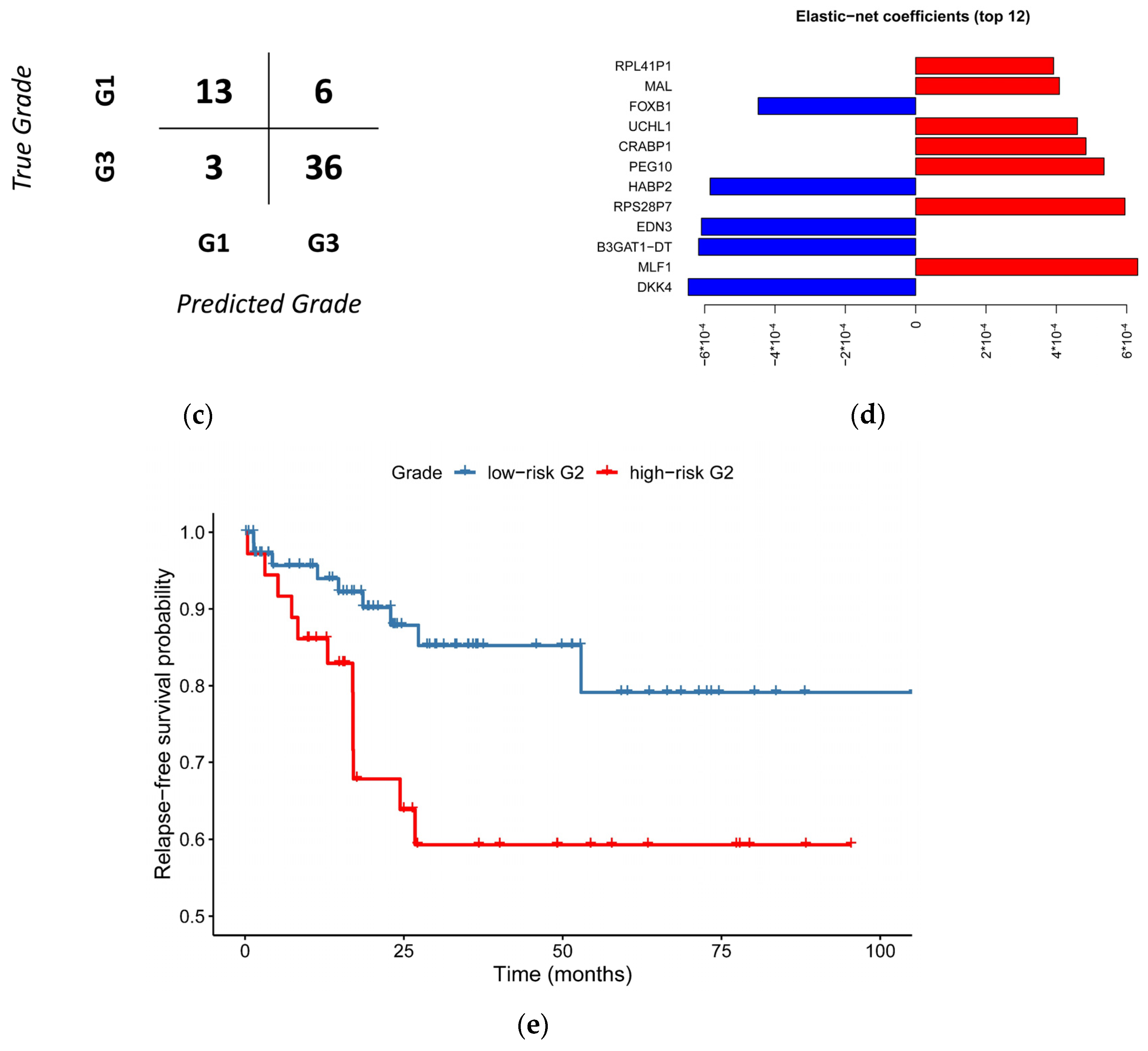
3.4. Relapse-Free Survival Varies across G2-Subgroups
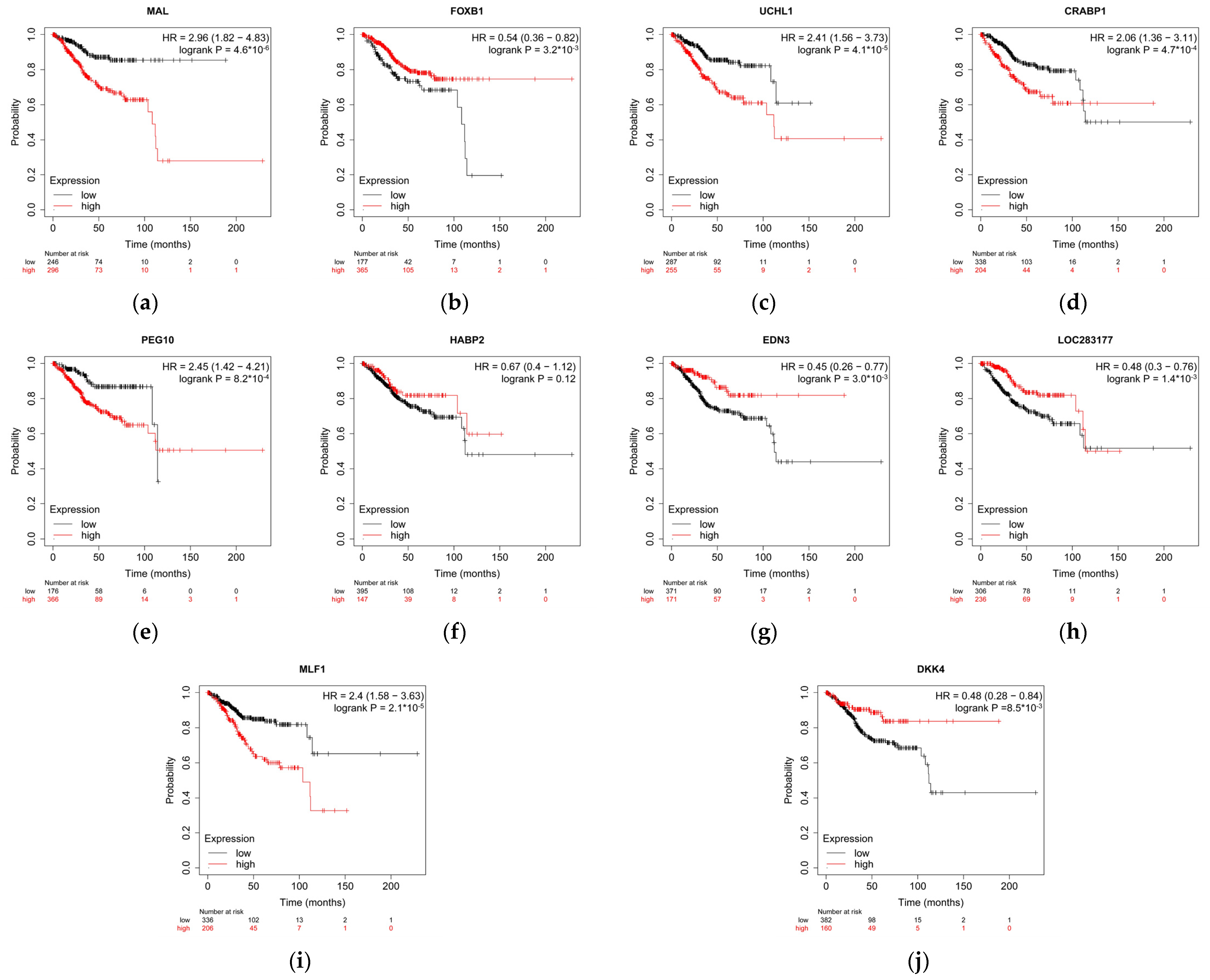
3.5. A Simplified Model Is Just as Accurate as of the Full Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Bokhman, J.V. Two pathogenetic types of endometrial carcinoma. Gynecol. Oncol. 1983, 15, 10–17. [Google Scholar] [CrossRef]
- Benedet, J.L.; Bender, H.; Jones, H.; Ngan, H.Y.; Pecorelli, S. FIGO staging classifications and clinical practice guidelines in the management of gynecologic cancers. FIGO Committee on Gynecologic Oncology. Int. J. Gynaecol. Obstet. 2000, 70, 209–262. [Google Scholar]
- Tejerizo-García, Á.; Jiménez-López, J.S.; Muñoz-González, J.L.; Bartolomé-Sotillos, S.; Marqueta-Marqués, L.; López-González, G.; Gómez, J.F.P.R. Overall survival and disease-free survival in endometrial cancer: Prognostic factors in 276 patients. Onco Targets Ther. 2013, 6, 1305–1313. [Google Scholar]
- Morice, P.; Leary, A.; Creutzberg, C.; Abu-Rustum, N.; Darai, E. Endometrial cancer. Lancet 2016, 387, 1094–1108. [Google Scholar] [CrossRef]
- Park, J.Y.; Kim, D.Y.; Kim, T.J.; Kim, J.W.; Kim, J.H.; Kim, Y.M.; Kim, Y.T.; Bae, D.S.; Nam, J.H. Hormonal therapy for women with stage IA endometrial cancer of all grades. Obstet. Gynecol. 2013, 122, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; Sidhu, D.; Duggan, M.A.; Arseneau, J.; Cesari, M.; Clement, P.B.; Ewanowich, C.A.; Kalloger, S.E.; Köbel, M. Reproducibility of histological cell type in high-grade endometrial carcinoma. Mod. Pathol. 2013, 26, 1594–1604. [Google Scholar] [CrossRef] [Green Version]
- Gilks, C.B.; Oliva, E.; Soslow, R.A. Poor interobserver reproducibility in the diagnosis of high-grade endometrial carcinoma. Am. J. Surg. Pathol. 2013, 37, 874–881. [Google Scholar] [CrossRef]
- Guan, H.; Semaan, A.; Bandyopadhyay, S.; Arabi, H.; Feng, J.; Fathallah, L.; Pansare, V.; Qazi, A.; Abdul-Karim, F.; Morris, R.T.; et al. Prognosis and reproducibility of new and existing binary grading systems for endometrial carcinoma comparedto FIGO grading in hysterectomy specimens. Int. J. Gynecol. Cancer 2011, 21, 654–660. [Google Scholar] [CrossRef]
- Nicole, C.M.V.; Casper, R.; Leon, F.A.G.M.; Iris, D.N.; Johan, B.; Johanna, M.A.P. Accuracy of Endometrial Sampling in Endometrial Carcinoma: A Systematic Review and Meta-analysis. Obstet. Gynecol. 2017, 130, 803–813. [Google Scholar]
- Getz, G.; Gabriel, S.B.; Cibulskis, K.; Lander, E.; Sivachenko, A.; Sougnez, C.; Lawrence, M.; Kandoth, C.; Dooling, D.; Fulton, R.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar]
- Abdulfatah, E.; Wakeling, E.; Sakr, S.; Al-Obaidy, K.; Bandyopadhyay, S.; Morris, R.; Feldman, G.; Ali-Fehmi, R. Molecular classification of endometrial carcinoma applied to endometrial biopsy specimens: Towards early personalized patient management. Gynecol. Oncol. 2019, 154, 467–474. [Google Scholar] [CrossRef]
- Kommoss, S.; McConechy, M.K.; Kommoss, F.; Leung, S.; Bunz, A.; Magrill, J.; Britton, H.; Kommoss, F.; Grevenkamp, F.; Karnezis, A.; et al. Final validation of the ProMisE molecular classifier for endometrial carcinoma in a large population-based case series. Ann. Oncol. 2018, 29, 1180–1188. [Google Scholar] [CrossRef]
- Leon-Castillo, A.; De Boer, S.M.; Powell, M.E.; Mileshkin, L.R.; Mackay, H.J.; Leary, A.; Nijman, H.W.; Singh, N.; Pollock, P.M.; Bessette, P.; et al. Molecular classification of the PORTEC-3 trial for high-risk endometrial cancer: Impact on prognosis and benefit from adjuvant therapy. J. Clin. Oncol. 2020, 38, 3388–3397. [Google Scholar] [CrossRef]
- Yin, F.; Shao, X.; Zhao, L.; Li, X.; Zhou, J.; Cheng, Y.; He, X.; Lei, S.; Li, J.; Wang, J. Predicting prognosis of endometrioid endometrial adenocarcinoma on the basis of gene expression and clinical features using random forest. Oncol. Lett. 2019, 18, 1597–1606. [Google Scholar] [CrossRef]
- Besso, M.J.; Montivero, L.; Lacunza, E.; Argibay, M.C.; Abba, M.; Furlong, L.I.; Colas, E.; Gil-Moreno, A.; Reventos, J.; Bello, R.; et al. Identification of early stage recurrence endometrial cancer biomarkers using bioinformatics tools. Oncol. Rep. 2020, 44, 873–886. [Google Scholar] [CrossRef]
- Akazawa, M.; Hashimoto, K.; Noda, K.; Yoshida, K. The application of machine learning for predicting recurrence in patients with early-stage endometrial cancer: A pilot study. Obstet. Gynecol. Sci. 2021, 64, 266–273. [Google Scholar] [CrossRef]
- Huang, C.Y.; Liao, K.W.; Chou, C.H.; Shrestha, S.; Yang, C.D.; Chiew, M.Y.; Huang, H.T.; Hong, H.C.; Huang, S.H.; Chang, T.H.; et al. Pilot Study to Establish a Novel Five-Gene Biomarker Panel for Predicting Lymph Node Metastasis in Patients with Early Stage Endometrial Cancer. Front. Oncol. 2020, 9, 1508. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Klevebring, D.; Lindberg, J.; Czene, K.; Grönberg, H.; Rantalainen, M. Determining breast cancer histological grade from RNA-sequencing data. Breast Cancer Res. 2016, 18, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Wong, M.L.; Medrano, J.F. One-Step Versus Two-Step Real-Time PCR. BioTechniques 2005, 39, 75–85. [Google Scholar] [CrossRef]
- Soslow, R.A.; Tornos, C.; Park, K.J.; Malpica, A.; Matias-Guiu, X.; Oliva, E.; Parkash, V.; Carlson, J.; McCluggage, W.G.; Gilks, C.B. Endometrial Carcinoma Diagnosis: Use of FIGO Grading and Genomic Subcategories in Clinical Practice: Recommendations of the International Society of Gynecological Pathologists. Int. J. Gynecol. Pathol. 2019, 38, S64. [Google Scholar] [CrossRef] [Green Version]
- Patten, D.K.; Corleone, G.; Győrffy, B.; Perone, Y.; Slaven, N.; Barozzi, I.; Erdős, E.; Saiakhova, A.; Goddard, K.; Vingiani, A.; et al. Enhancer mapping uncovers phenotypic heterogeneity and evolution in patients with luminal breast cancer. Nat. Med. 2018, 24, 1469–1480. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Zou, H.; Hastie, T. Erratum: Regularization and variable selection via the elastic net. J. R. Stat. Soc. Ser. B Stat. Methodol. 2005, 67, 768. [Google Scholar] [CrossRef]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Nagy, Á.; Munkácsy, G.; Győrffy, B. Pancancer survival analysis of cancer hallmark genes. Sci. Rep. 2021, 11, 6047. [Google Scholar] [CrossRef] [PubMed]
- Győrffy, B. Survival analysis across the entire transcriptome identifies biomarkers with the highest prognostic power in breast cancer. Comput. Struct. Biotechnol. J. 2021, 19, 4101–4109. [Google Scholar] [CrossRef]
- Cai, X.; Yao, Z.; Li, L.; Huang, J. Role of DKK4 in tumorigenesis and tumor progression. Int. J. Biol. Sci. 2018, 14, 616–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamae, I.; Kato, J.Y.; Yokoyama, T.; Ito, H.; Yoneda-Kato, N. Myeloid leukemia factor 1 stabilizes tumor suppressor C/EBPa to prevent Trib1-driven acute myeloid leukemia. Blood Adv. 2017, 1, 1682–1693. [Google Scholar] [CrossRef]
- Nobili, L.; Ronchetti, D.; Taiana, E.; Neri, A. Long non-coding RNAs in B-cell malignancies: A comprehensive overview. Oncotarget 2017, 8, 60605–60623. [Google Scholar] [CrossRef] [Green Version]
- Baldini, F.; Calderoni, M.; Vergani, L.; Modesto, P.; Florio, T.; Pagano, A. An overview of long non-coding (Lnc)rnas in neuroblastoma. Int. J. Mol. Sci. 2021, 22, 4234. [Google Scholar] [CrossRef] [PubMed]
- Piergentili, R.; Zaami, S.; Cavaliere, A.F.; Signore, F.; Scambia, G.; Mattei, A.; Marinelli, E.; Gulia, C.; Perelli, F. Non-coding rnas as prognostic markers for endometrial cancer. Int. J. Mol. Sci. 2021, 22, 3151. [Google Scholar] [CrossRef]
- Fridrichova, I.; Kalinkova, L.; Karhanek, M.; Smolkova, B.; Machalekova, K.; Wachsmannova, L.; Nikolaieva, N.; Kajo, K. Mir-497-5p decreased expression associated with high-risk endometrial cancer. Int. J. Mol. Sci. 2021, 22, 127. [Google Scholar] [CrossRef]
- Dy, J.G.; Brodley, C.E. Feature selection for unsupervised learning. J. Mach. Learn. Res. 2004, 5, 845–889. [Google Scholar]
- Mirzapoiazova, T.; Mambetsariev, N.; Lennon, F.E.; Mambetsariev, B.; Berlind, J.E.; Salgia, R.; Singleton, P.A. HABP2 is a novel regulator of hyaluronan-mediated human lung cancer progression. Front. Oncol. 2015, 5, 164. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wan, Y.; Zhang, Z.; Jiang, Y.; Gu, Z.; Ma, X.; Nie, S.; Yang, J.; Lang, J.; Cheng, W.; et al. IGF2BP1 overexpression stabilizes PEG10 mRNA in an m6A-dependent manner and promotes endometrial cancer progression. Theranostics 2021, 11, 1100–1114. [Google Scholar] [CrossRef]
- Siddiqui, N.A.; Thomas, E.J.; Dunlop, W.; Redfern, C.P.F. Retinoic acid receptors and retinoid binding proteins in endometrial adenocarcinoma: Differential expression of cellular retinoid binding proteins in endometrioid tumours. Int. J. Cancer 1995, 64, 253–263. [Google Scholar] [CrossRef]
- Nakao, K.; Hirakawa, T.; Suwa, H.; Kogure, K.; Ikeda, S.; Yamashita, S.; Minegishi, T.; Kishi, H. High Expression of Ubiquitin C-terminal Hydrolase L1 Is Associated With Poor Prognosis in Endometrial Cancer Patients. Int. J. Gynecol. Cancer 2018, 28, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Iny Stein, T.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards suite: From gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinform. 2016, 2016, 1.30.1–1.30.33. [Google Scholar] [CrossRef]
- Lara-Lemus, R. On the role of myelin and lymphocyte protein (MAL) in cancer: A puzzle with two faces. J. Cancer 2019, 10, 2312–2318. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.A.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Wheeler, D.L. GenBank: Update. Nucleic Acids Res. 2004, 32, D23–D26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Yang, L.; Mo, Y.Y. Role of pseudogenes in tumorigenesis. Cancers 2018, 10, 256. [Google Scholar] [CrossRef] [Green Version]
- Cooper, L.A.; Demicco, E.G.; Saltz, J.H.; Powell, R.T.; Rao, A.; Lazar, A.J. PanCancer insights from The Cancer Genome Atlas: The pathologist’s perspective. J. Pathol. 2018, 244, 512–524. [Google Scholar] [CrossRef]
- Lazar, A.J.; McLellan, M.D.; Bailey, M.H.; Miller, C.A.; Appelbaum, E.L.; Cordes, M.G.; Fronick, C.C.; Fulton, L.A.; Fulton, R.S.; Mardis, E.R.; et al. Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas. Cell 2017, 171, 950–965.e28. [Google Scholar]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gargya, P.; Bálint, B.L. Histological Grade of Endometrioid Endometrial Cancer and Relapse Risk Can Be Predicted with Machine Learning from Gene Expression Data. Cancers 2021, 13, 4348. https://doi.org/10.3390/cancers13174348
Gargya P, Bálint BL. Histological Grade of Endometrioid Endometrial Cancer and Relapse Risk Can Be Predicted with Machine Learning from Gene Expression Data. Cancers. 2021; 13(17):4348. https://doi.org/10.3390/cancers13174348
Chicago/Turabian StyleGargya, Péter, and Bálint László Bálint. 2021. "Histological Grade of Endometrioid Endometrial Cancer and Relapse Risk Can Be Predicted with Machine Learning from Gene Expression Data" Cancers 13, no. 17: 4348. https://doi.org/10.3390/cancers13174348
APA StyleGargya, P., & Bálint, B. L. (2021). Histological Grade of Endometrioid Endometrial Cancer and Relapse Risk Can Be Predicted with Machine Learning from Gene Expression Data. Cancers, 13(17), 4348. https://doi.org/10.3390/cancers13174348