
Thyroid Hormone Receptor Beta as Tumor Suppressor: Untapped Potential in Treatment and Diagnostics in Solid Tumors



{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
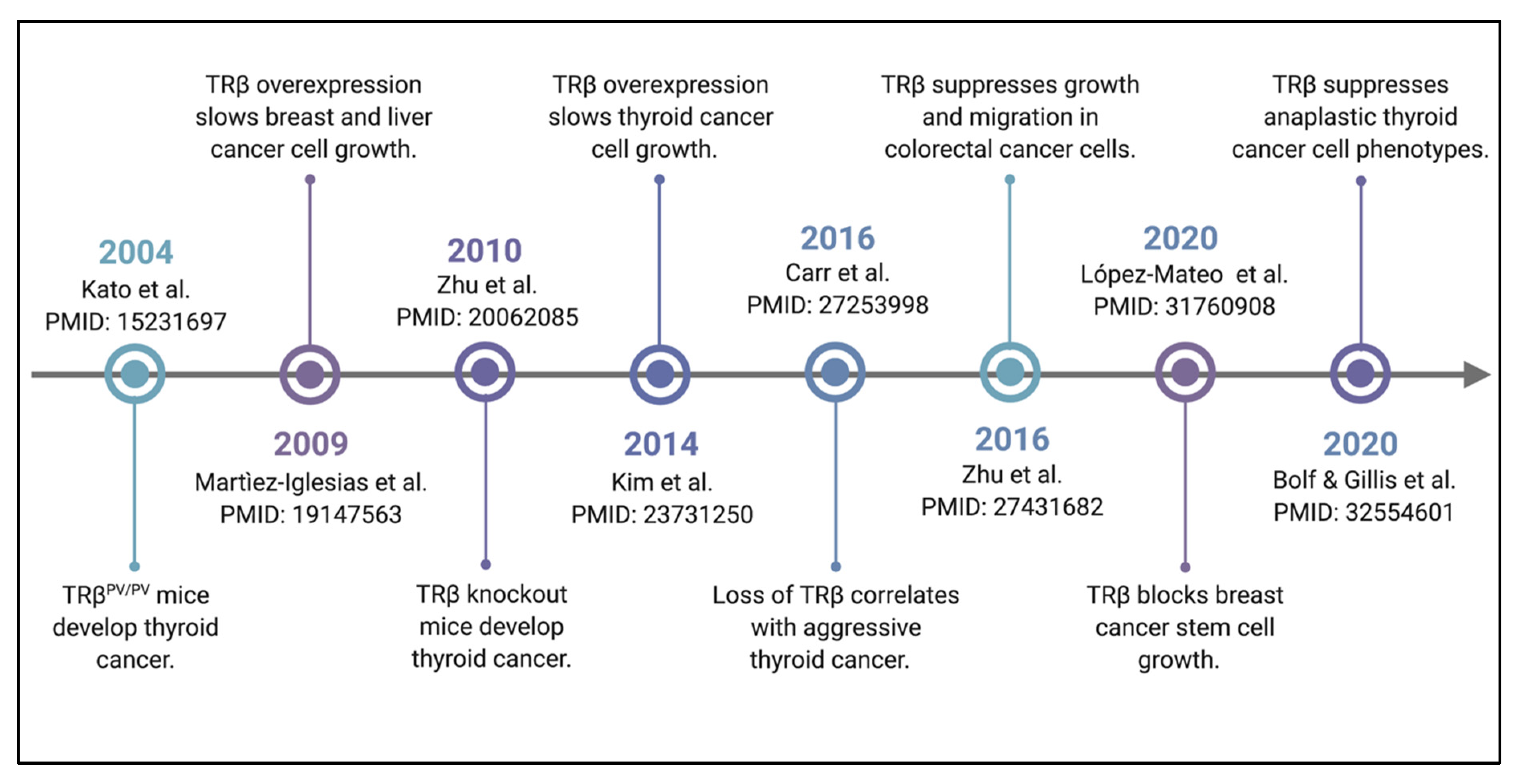
2. TRβ Satisfies the Criteria for a Tumor Suppressor
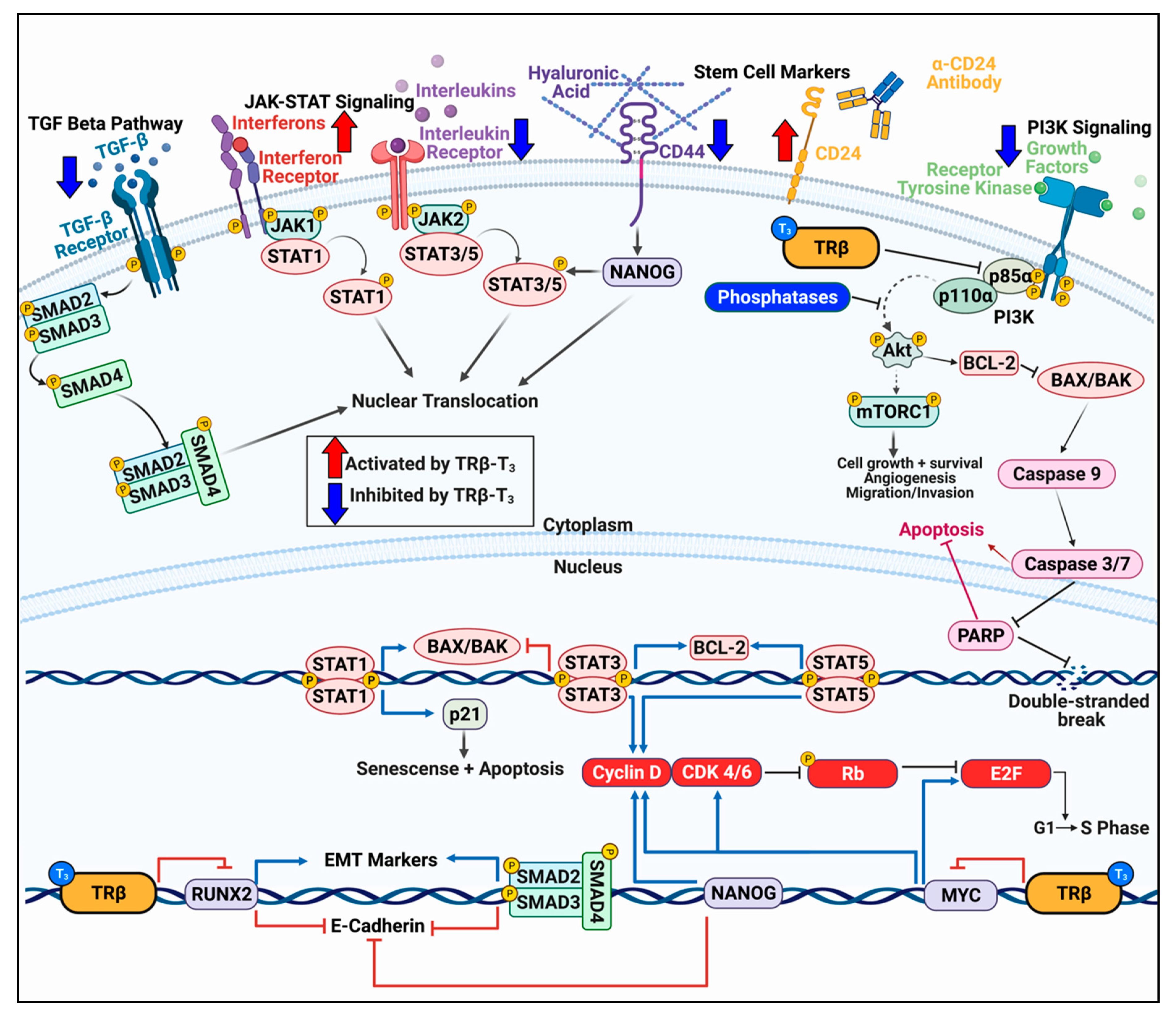
3. Mechanisms of TRβ-mediated Tumor Suppression
3.1. TRβ Attenuates the PI3K-Akt Signaling Pathway via Genomic and Nongenomic Mechanisms
3.2. TRβ Differentially Influences JAK-STAT Signaling
3.3. TRβ Regulation of Cell Cycle Progression
3.4. Impact of TRβ on TGF-β Signaling
3.5. TRβ Inhibits Epithelial–Mesenchymal Transition
3.6. TRβ Promotes Cancer Cell Re-Differentiation
3.7. TRβ Interactions with Epigenetic Modulators Are Key to Tumor Suppression
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kim, W.G.; Cheng, S.Y. Thyroid hormone receptors and cancer. Biochim. Biophys. Acta 2013, 1830, 3928–3936. [Google Scholar] [CrossRef]
- Aranda, A.; Martinez-Iglesias, O.; Ruiz-Llorente, L.; Garcia-Carpizo, V.; Zambrano, A. Thyroid receptor: Roles in cancer. Trends Endocrinol. Metab. TEM 2009, 20, 318–324. [Google Scholar] [CrossRef]
- Landa, I.; Ibrahimpasic, T.; Boucai, L.; Sinha, R.; Knauf, J.A.; Shah, R.H.; Dogan, S.; Ricarte-Filho, J.C.; Krishnamoorthy, G.P.; Xu, B.; et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J. Clin. Investig. 2016, 126, 1052–1066. [Google Scholar] [CrossRef]
- Joseph, B.; Ji, M.; Liu, D.; Hou, P.; Xing, M. Lack of mutations in the thyroid hormone receptor (TR) alpha and beta genes but frequent hypermethylation of the TRbeta gene in differentiated thyroid tumors. J. Clin. Endocrinol. Metab. 2007, 92, 4766–4770. [Google Scholar] [CrossRef] [PubMed]
- Puzianowska-Kuznicka, M.; Krystyniak, A.; Madej, A.; Cheng, S.Y.; Nauman, J. Functionally impaired TR mutants are present in thyroid papillary cancer. J. Clin. Endocrinol. Metab. 2002, 87, 1120–1128. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Iglesias, O.; Garcia-Silva, S.; Tenbaum, S.P.; Regadera, J.; Larcher, F.; Paramio, J.M.; Vennström, B.; Aranda, A. Thyroid hormone receptor beta1 acts as a potent suppressor of tumor invasiveness and metastasis. Cancer Res. 2009, 69, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Zhu, X.; Kim, D.; Zhang, L.; Kebebew, E.; Cheng, S. Reactivation of the silenced thyroid hormone receptor B gene expression delays thyroid tumor progression. Endocrinology 2013, 154, 25–35. [Google Scholar] [CrossRef]
- Park, J.W.; Zhao, L.; Cheng, S.Y. Inhibition of estrogen-dependent tumorigenesis by the thyroid hormone receptor beta in xenograft models. Am. J. Cancer Res. 2013, 3, 302–311. [Google Scholar]
- Kim, W.G.; Zhao, L.; Kim, D.W.; Willingham, M.C.; Cheng, S.Y. Inhibition of tumorigenesis by the thyroid hormone receptor beta in xenograft models. Thyroid 2014, 24, 260–269. [Google Scholar] [CrossRef]
- Kato, Y.; Ying, H.; Willingham, M.C.; Cheng, S.Y. A tumor suppressor role for thyroid hormone beta receptor in a mouse model of thyroid carcinogenesis. Endocrinology 2004, 145, 4430–4438. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Weinberg, R.A. Oncogenes and tumor suppressor genes. CA Cancer J. Clin. 1994, 44, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.G.; Zhao, L.; Willingham, M.C.; Cheng, S.Y. Thyroid hormone receptors are tumor suppressors in a mouse model of metastatic follicular thyroid carcinoma. Oncogene 2010, 29, 1909–1919. [Google Scholar] [CrossRef]
- Carr, F.E.; Tai, P.W.; Barnum, M.S.; Gillis, N.E.; Evans, K.G.; Taber, T.H.; White, J.H.; Tomczak, J.A.; Jaworski, D.M.; Zaidi, S.K.; et al. Thyroid hormone receptor-β (TRβ) mediates runt-related transcription factor 2 (Runx2) expression in thyroid cancer cells: A novel signaling pathway in thyroid cancer. Endocrinology 2016, 157, 3278–3292. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Tian, G.; Yang, Q.; De, G.; Zhang, Z.; Wang, Y.; Nie, H.; Zhang, Y.; Yang, X.; Li, J. Thyroid hormone receptor β1 suppresses proliferation and migration by inhibiting PI3K/Akt signaling in human colorectal cancer cells. Oncol. Rep. 2016, 36, 1419–1426. [Google Scholar] [CrossRef]
- López-Mateo, I.; Alonso-Merino, E.; Suarez-Cabrera, C.; Park, J.W.; Cheng, S.Y.; Alemany, S.; Paramio, J.M.; Aranda, A. Thyroid hormone receptor β inhibits self-renewal capacity of breast cancer stem cells. Thyroid 2020, 30, 116–132. [Google Scholar] [CrossRef]
- Bolf, E.L.; Gillis, N.E.; Davidson, C.D.; Rodriguez, P.D.; Cozzens, L.; Tomczak, J.A.; Frietze, S.; Carr, F.E. Thyroid hormone receptor beta induces a tumor-suppressive program in anaplastic thyroid cancer. Mol. Cancer Res. 2020, 18, 1443–1452. [Google Scholar] [CrossRef]
- Carnero, A.; Blanco-Aparicio, C.; Renner, O.; Link, W.; Leal, J.F. The PTEN/PI3K/AKT signalling pathway in cancer, therapeutic implications. Curr. Cancer Drug Targets 2008, 8, 187–198. [Google Scholar] [CrossRef]
- Martini, M.; De Santis, M.C.; Braccini, L.; Gulluni, F.; Hirsch, E. PI3K/AKT signaling pathway and cancer: An updated review. Ann. Med. 2014, 46, 372–383. [Google Scholar] [CrossRef]
- Revathidevi, S.; Munirajan, A.K. Akt in cancer: Mediator and more. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Dai, Q.; Su, X.; Fu, J.; Feng, X.; Peng, J. Role of PI3K/AKT pathway in cancer: The framework of malignant behavior. Mol. Biol. Rep. 2020, 47, 4587–4629. [Google Scholar] [CrossRef]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Wang, Y.; Hou, P.; Yu, H.; Wang, W.; Ji, M.; Zhao, S.; Yan, S.; Sun, X.; Liu, D.; Shi, B.; et al. High prevalence and mutual exclusivity of genetic alterations in the phosphatidylinositol-3-kinase/akt pathway in thyroid tumors. J. Clin. Endocrinol. Metab. 2007, 92, 2387–2390. [Google Scholar] [CrossRef]
- Simoncini, T.; Hafezi-Moghadam, A.; Brazil, D.P.; Ley, K.; Chin, W.W.; Liao, J.K. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature 2000, 407, 538–541. [Google Scholar] [CrossRef]
- Cao, X.; Kambe, F.; Moeller, L.C.; Refetoff, S.; Seo, H. Thyroid hormone induces rapid activation of Akt/protein kinase B-mammalian target of rapamycin-p70S6K cascade through phosphatidylinositol 3-kinase in human fibroblasts. Mol. Endocrinol. 2005, 19, 102–112. [Google Scholar] [CrossRef]
- Storey, N.M.; Gentile, S.; Ullah, H.; Russo, A.; Muessel, M.; Erxleben, C.; Armstrong, D.L. Rapid signaling at the plasma membrane by a nuclear receptor for thyroid hormone. Proc. Natl. Acad. Sci. USA 2006, 103, 5197–5201. [Google Scholar] [CrossRef]
- Kim, C.S.; Vasko, V.V.; Kato, Y.; Kruhlak, M.; Saji, M.; Cheng, S.Y.; Ringel, M.D. AKT activation promotes metastasis in a mouse model of follicular thyroid carcinoma. Endocrinology 2005, 146, 4456–4463. [Google Scholar] [CrossRef]
- Furuya, F.; Lu, C.; Willingham, M.C.; Cheng, S.Y. Inhibition of phosphatidylinositol 3-kinase delays tumor progression and blocks metastatic spread in a mouse model of thyroid cancer. Carcinogenesis 2007, 28, 2451–2458. [Google Scholar] [CrossRef] [PubMed]
- Moriggi, G.; Verga Falzacappa, C.; Mangialardo, C.; Michienzi, S.; Stigliano, A.; Brunetti, E.; Toscano, V.; Misiti, S. Thyroid hormones (T3 and T4): Dual effect on human cancer cell proliferation. Anticancer Res. 2011, 31, 89–96. [Google Scholar] [PubMed]
- Davidson, C.D.; Bolf, E.L.; Gillis, N.E.; Cozzens, L.M.; Tomczak, J.A.; Carr, F.E. Thyroid hormone receptor beta inhibits PI3K-Akt-mTOR signaling axis in anaplastic thyroid cancer via genomic mechanisms. J. Endocr. Soc. 2021, 5. [Google Scholar] [CrossRef] [PubMed]
- Li Chew, C.; Lunardi, A.; Gulluni, F.; Ruan, D.T.; Chen, M.; Salmena, L.; Nishino, M.; Papa, A.; Ng, C.; Fung, J.; et al. In vivo role of INPP4B in tumor and metastasis suppression through regulation of PI3K-AKT signaling at endosomes. Cancer Discov. 2015, 5, 740–751. [Google Scholar] [CrossRef]
- Pramfalk, C.; Pedrelli, M.; Parini, P. Role of thyroid receptor β in lipid metabolism. Biochim. Biophys. Acta 2011, 1812, 929–937. [Google Scholar] [CrossRef]
- Master, A.; Nauman, A. THRB (Thyroid Hormone Receptor, Beta). Atlas Genet. Cytogenet. Oncol. Haematol. 2014, 18, 400–433. [Google Scholar] [CrossRef]
- Evans, L.M.; Cowey, S.L.; Siegal, G.P.; Hardy, R.W. Stearate preferentially induces apoptosis in human breast cancer cells. Nutr. Cancer 2009, 61, 746–753. [Google Scholar] [CrossRef]
- Favaro, E.; Bensaad, K.; Chong, M.G.; Tennant, D.A.; Ferguson, D.J.; Snell, C.; Steers, G.; Turley, H.; Li, J.L.; Gunther, U.L.; et al. Glucose utilization via glycogen phosphorylase sustains proliferation and prevents premature senescence in cancer cells. Cell Metab. 2012, 16, 751–764. [Google Scholar] [CrossRef]
- Pelletier, J.; Bellot, G.; Gounon, P.; Lacas-Gervais, S.; Pouyssegur, J.; Mazure, N.M. Glycogen synthesis is induced in hypoxia by the hypoxia-inducible factor and promotes cancer cell survival. Front. Oncol. 2012, 2, 18. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.N.; Guo, P.; Lim, S.; Bassilian, S.; Lee, S.T.; Boren, J.; Cascante, M.; Go, V.L.; Boros, L.G. Metabolic sensitivity of pancreatic tumour cell apoptosis to glycogen phosphorylase inhibitor treatment. Br. J. Cancer 2004, 91, 2094–2100. [Google Scholar] [CrossRef]
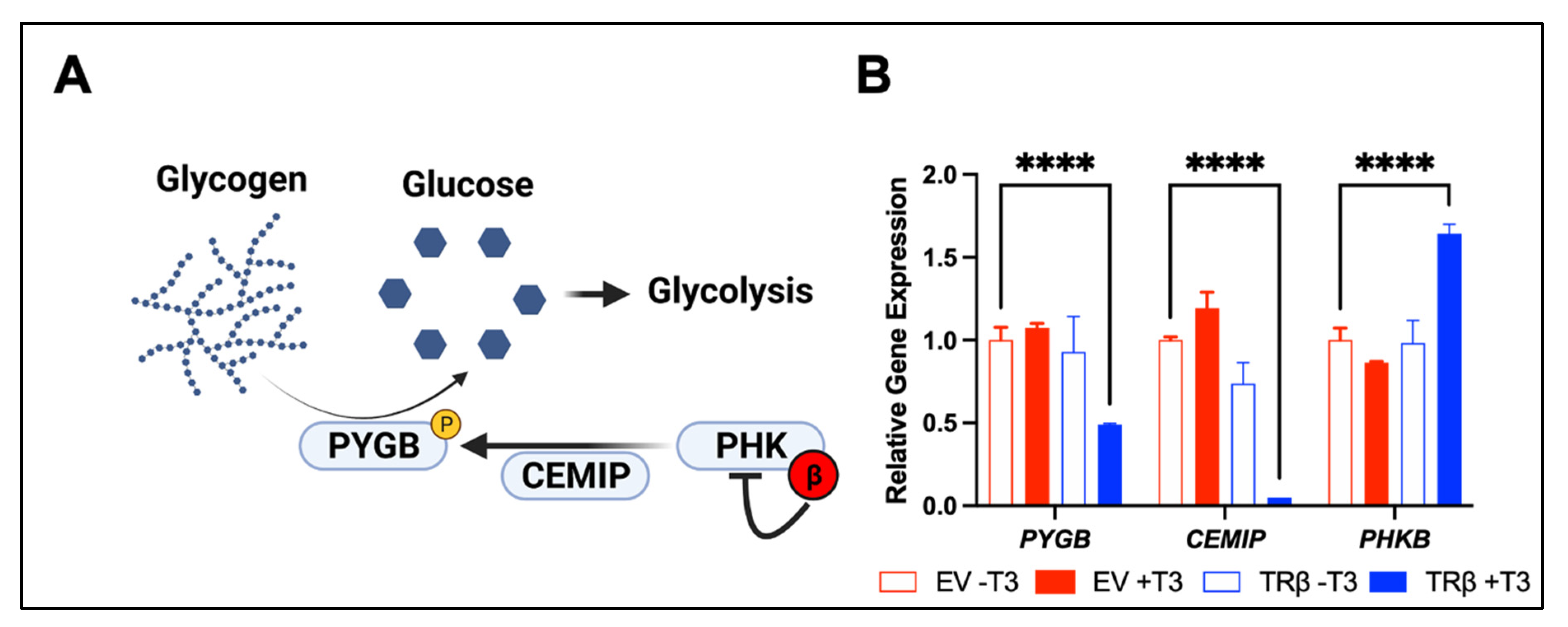
- Davidson CD, C.F. Review of pharmacological inhibition of thyroid cancer metabolism. J. Cancer Metastasis Treat. 2021, 7. [Google Scholar] [CrossRef]
- Dauer, P.; Lengyel, E. New roles for glycogen in tumor progression. Trends Cancer 2019, 5, 396–399. [Google Scholar] [CrossRef] [PubMed]
- Terashima, M.; Fujita, Y.; Togashi, Y.; Sakai, K.; De Velasco, M.A.; Tomida, S.; Nishio, K. KIAA1199 interacts with glycogen phosphorylase kinase beta-subunit (PHKB) to promote glycogen breakdown and cancer cell survival. Oncotarget 2014, 5, 7040–7050. [Google Scholar] [CrossRef]
- Harrison, D.A. The Jak/STAT pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011205. [Google Scholar] [CrossRef]
- Seif, F.; Khoshmirsafa, M.; Aazami, H.; Mohsenzadegan, M.; Sedighi, G.; Bahar, M. The role of JAK-STAT signaling pathway and its regulators in the fate of T helper cells. Cell Commun. Signal. 2017, 15, 23. [Google Scholar] [CrossRef]
- Owen, K.L.; Brockwell, N.K.; Parker, B.S. JAK-STAT signaling: A double-edged sword of immune regulation and cancer progression. Cancers 2019, 11, 2002. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.J.; Putoczki, T. JAK-STAT signalling pathway in cancer. Cancers 2020, 12, 1971. [Google Scholar] [CrossRef] [PubMed]
- Kamran, M.Z.; Patil, P.; Gude, R.P. Role of STAT3 in cancer metastasis and translational advances. BioMed Res. Int. 2013, 2013, 421821. [Google Scholar] [CrossRef]
- Huynh, J.; Chand, A.; Gough, D.; Ernst, M. Therapeutically exploiting STAT3 activity in cancer—Using tissue repair as a road map. Nat. Rev. Cancer 2019, 19, 82–96. [Google Scholar] [CrossRef]
- Halim, C.E.; Deng, S.; Ong, M.S.; Yap, C.T. Involvement of STAT5 in Oncogenesis. Biomedicines 2020, 8, 316. [Google Scholar] [CrossRef] [PubMed]
- Chin, Y.E.; Kitagawa, M.; Kuida, K.; Flavell, R.A.; Fu, X.Y. Activation of the STAT signaling pathway can cause expression of caspase 1 and apoptosis. Mol. Cell Biol. 1997, 17, 5328–5337. [Google Scholar] [CrossRef]
- Stephanou, A.; Latchman, D.S. STAT-1: A novel regulator of apoptosis. Int. J. Exp. Pathol. 2003, 84, 239–244. [Google Scholar] [CrossRef]
- Sironi, J.J.; Ouchi, T. STAT1-induced apoptosis is mediated by caspases 2, 3, and 7. J. Biol. Chem. 2004, 279, 4066–4074. [Google Scholar] [CrossRef]
- Su, Q.; Wang, F.; Dong, Z.; Chen, M.; Cao, R. IFN-γ induces apoptosis in human melanocytes by activating the JAK1/STAT1 signaling pathway. Mol. Med. Rep. 2020, 22, 3111–3116. [Google Scholar] [CrossRef] [PubMed]
- Guigon, C.J.; Kim, D.W.; Willingham, M.C.; Cheng, S.Y. Mutation of thyroid hormone receptor-β in mice predisposes to the development of mammary tumors. Oncogene 2011, 30, 3381–3390. [Google Scholar] [CrossRef] [PubMed]
- Debierre-Grockiego, F. Anti-apoptotic role of STAT5 in haematopoietic cells and in the pathogenesis of malignancies. Apoptosis 2004, 9, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Perez-Juste, G.; Aranda, A. The cyclin-dependent kinase inhibitor p27(Kip1) is involved in thyroid hormone-mediated neuronal differentiation. J. Biol. Chem. 1999, 274, 5026–5031. [Google Scholar] [CrossRef]
- Pérez-Juste, G.; García-Silva, S.; Aranda, A. An element in the region responsible for premature termination of transcription mediates repression of c-myc gene expression by thyroid hormone in neuroblastoma cells. J. Biol. Chem. 2000, 275, 1307–1314. [Google Scholar] [CrossRef]
- Porlan, E.; Vega, S.; Iglesias, T.; Rodríguez-Peña, A. Unliganded thyroid hormone receptor beta1 inhibits proliferation of murine fibroblasts by delaying the onset of the G1 cell-cycle signals. Oncogene 2004, 23, 8756–8765. [Google Scholar] [CrossRef]
- Yen, C.C.; Huang, Y.H.; Liao, C.Y.; Liao, C.J.; Cheng, W.L.; Chen, W.J.; Lin, K.H. Mediation of the inhibitory effect of thyroid hormone on proliferation of hepatoma cells by transforming growth factor-beta. J. Mol. Endocrinol. 2006, 36, 9–21. [Google Scholar] [CrossRef]
- Michienzi, S.; Bucci, B.; Verga Falzacappa, C.; Patriarca, V.; Stigliano, A.; Panacchia, L.; Brunetti, E.; Toscano, V.; Misiti, S. 3,3′,5-Triiodo-L-thyronine inhibits ductal pancreatic adenocarcinoma proliferation improving the cytotoxic effect of chemotherapy. J. Endocrinol. 2007, 193, 209–223. [Google Scholar] [CrossRef]
- Martinez-Iglesias, O.; Garcia-Silva, S.; Regadera, J.; Aranda, A. Hypothyroidism enhances tumor invasiveness and metastasis development. PLoS ONE 2009, 4, e6428. [Google Scholar] [CrossRef]
- Lin, Y.H.; Huang, Y.H.; Wu, M.H.; Wu, S.M.; Chi, H.C.; Liao, C.J.; Chen, C.Y.; Tseng, Y.H.; Tsai, C.Y.; Tsai, M.M.; et al. Thyroid hormone suppresses cell proliferation through endoglin-mediated promotion of p21 stability. Oncogene 2013, 32, 3904–3914. [Google Scholar] [CrossRef]
- Turner, N.C.; Ro, J.; André, F.; Loi, S.; Verma, S.; Iwata, H.; Harbeck, N.; Loibl, S.; Huang Bartlett, C.; Zhang, K.; et al. Palbociclib in hormone-receptor-positive advanced breast cancer. N. Engl. J. Med. 2015, 373, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Karasic, T.B.; O’Hara, M.H.; Teitelbaum, U.R.; Damjanov, N.; Giantonio, B.J.; d’Entremont, T.S.; Gallagher, M.; Zhang, P.J.; O’Dwyer, P.J. Phase II trial of palbociclib in patients with advanced esophageal or gastric cancer. Oncologist 2020, 25, e1864–e1868. [Google Scholar] [CrossRef] [PubMed]
- Sepúlveda-Sánchez, J.M.; Gil-Gil, M.; Alonso-García, M.; Vaz Salgado, M.; Vicente, E.; Mesía Barroso, C.; Rodríguez Sánchez, Á.; Durán, G.; De Las Peñas, R.; Muñoz-Langa, J.; et al. Phase II trial of palbociclib in recurrent retinoblastoma-positive anaplastic oligodendroglioma: A study from the spanish group for research in neuro-oncology (GEINO). Target. Oncol. 2020, 15, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Serra, F.; Lapidari, P.; Quaquarini, E.; Tagliaferri, B.; Sottotetti, F.; Palumbo, R. Palbociclib in metastatic breast cancer: Current evidence and real-life data. Drugs Context 2019, 8, 212579. [Google Scholar] [CrossRef] [PubMed]
- Hata, A.; Chen, Y.G. TGF-β Signaling from receptors to smads. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-β and the TGF-β Family: Context-dependent roles in cell and tissue physiology. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef]
- Vander Ark, A.; Cao, J.; Li, X. TGF-β receptors: In and beyond TGF-β signaling. Cell. Signal. 2018, 52, 112–120. [Google Scholar] [CrossRef]
- Alonso-Merino, E.; Martín Orozco, R.; Ruíz-Llorente, L.; Martínez-Iglesias, O.A.; Velasco-Martín, J.P.; Montero-Pedrazuela, A.; Fanjul-Rodríguez, L.; Contreras-Jurado, C.; Regadera, J.; Aranda, A. Thyroid hormones inhibit TGF-β signaling and attenuate fibrotic responses. Proc. Natl. Acad. Sci. USA 2016, 113, E3451–E3460. [Google Scholar] [CrossRef]
- Dentice, M.; Luongo, C.; Ambrosio, R.; Sibilio, A.; Casillo, A.; Iaccarino, A.; Troncone, G.; Fenzi, G.; Larsen, P.R.; Salvatore, D. β-Catenin regulates deiodinase levels and thyroid hormone signaling in colon cancer cells. Gastroenterology 2012, 143, 1037–1047. [Google Scholar] [CrossRef]
- Bolf, E.L.; Gillis, N.E.; Barnum, M.S.; Beaudet, C.M.; Yu, G.Y.; Tomczak, J.A.; Stein, J.L.; Lian, J.B.; Stein, G.S.; Carr, F.E. The thyroid hormone receptor-RUNX2 Axis: A novel tumor suppressive pathway in breast cancer. Horm. Cancer 2020, 11, 34–41. [Google Scholar] [CrossRef]
- Nassar, D.; Blanpain, C. Cancer stem cells: Basic concepts and therapeutic implications. Annu. Rev. Pathol. 2016, 11, 47–76. [Google Scholar] [CrossRef] [PubMed]
- Liu, J. The dualistic origin of human tumors. Semin. Cancer Biol. 2018, 53, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Unternaehrer, J.J. Epithelial-mesenchymal transition and cancer stem cells: At the Crossroads of differentiation and dedifferentiation. Dev. Dyn. 2019, 248, 10–20. [Google Scholar] [CrossRef]
- Vora, H.H.; Patel, N.A.; Rajvik, K.N.; Mehta, S.V.; Brahmbhatt, B.V.; Shah, M.J.; Shukla, S.N.; Shah, P.M. Cytokeratin and vimentin expression in breast cancer. Int. J. Biol. Markers 2009, 24, 38–46. [Google Scholar] [CrossRef]
- Alshareeda, A.T.; Soria, D.; Garibaldi, J.M.; Rakha, E.; Nolan, C.; Ellis, I.O.; Green, A.R. Characteristics of basal cytokeratin expression in breast cancer. Breast Cancer Res. Treat. 2013, 139, 23–37. [Google Scholar] [CrossRef]
- Karantza, V. Keratins in health and cancer: More than mere epithelial cell markers. Oncogene 2011, 30, 127–138. [Google Scholar] [CrossRef]
- Mathisen, P.M.; Miller, L. Thyroid hormone induces constitutive keratin gene expression during Xenopus laevis development. Mol. Cell Biol. 1989, 9, 1823–1831. [Google Scholar] [CrossRef]
- Perra, A.; Kowalik, M.A.; Pibiri, M.; Ledda-Columbano, G.M.; Columbano, A. Thyroid hormone receptor ligands induce regression of rat preneoplastic liver lesions causing their reversion to a differentiated phenotype. Hepatology 2009, 49, 1287–1296. [Google Scholar] [CrossRef]
- Gillis, N.E.; Davidson, C.D.; Cozzens, L.M.; Wilson, E.; Bolf, E.L.; Tomczak, J.A.; Carr, F.E. A thyroid hormone receptor beta specific agonist suppresses anaplastic thyroid cancer cell phenotype and increases efficacy of therapeutic agents. bioRxiv 2021. [Google Scholar] [CrossRef]
- Salnikov, A.V.; Bretz, N.P.; Perne, C.; Hazin, J.; Keller, S.; Fogel, M.; Herr, I.; Schlange, T.; Moldenhauer, G.; Altevogt, P. Antibody targeting of CD24 efficiently retards growth and influences cytokine milieu in experimental carcinomas. Br. J. Cancer 2013, 108, 1449–1459. [Google Scholar] [CrossRef]
- Barkal, A.A.; Brewer, R.E.; Markovic, M.; Kowarsky, M.; Barkal, S.A.; Zaro, B.W.; Krishnan, V.; Hatakeyama, J.; Dorigo, O.; Barkal, L.J.; et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature 2019, 572, 392–396. [Google Scholar] [CrossRef]
- Hahm, J.B.; Schroeder, A.C.; Privalsky, M.L. The two major isoforms of thyroid hormone receptor, TRα1 and TRβ1, preferentially partner with distinct panels of auxiliary proteins. Mol. Cell Endocrinol. 2014, 383, 80–95. [Google Scholar] [CrossRef]
- Fozzatti, L.; Lu, C.; Kim, D.-W.; Cheng, S.-Y. Differential recruitment of nuclear coregulators directs the isoform-dependent action of mutant thyroid hormone receptors. Mol. Endocrinol. 2011, 25, 908–921. [Google Scholar] [CrossRef]
- Gillis, N.E.; Boyd, J.R.; Tomczak, J.A.; Frietze, S.; Carr, F.E. Thyroid hormone dependent transcriptional programming by TRβ requires SWI/SNF chromatin remodelers. bioRxiv 2021. [Google Scholar] [CrossRef]
- Lee, K.C.; Li, J.; Cole, P.A.; Wong, J.; Kraus, W.L. Transcriptional activation by thyroid hormone receptor-β involves chromatin remodeling, histone acetylation, and synergistic stimulation by p300 and steroid receptor coactivators. Mol. Endocrinol. 2003, 17, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Fondell, J.D. Ordered recruitment of histone acetyltransferases and the TRAP/Mediator complex to thyroid hormone-responsive promoters in vivo. Proc. Natl. Acad. Sci. USA 2002, 99, 7934–7939. [Google Scholar] [CrossRef] [PubMed]
- Skowron, K.J.; Booker, K.; Cheng, C.; Creed, S.; David, B.P.; Lazzara, P.R.; Lian, A.; Siddiqui, Z.; Speltz, T.E.; Moore, T.W. Steroid receptor/coactivator binding inhibitors: An update. Mol. Cell Endocrinol. 2019, 493, 110471. [Google Scholar] [CrossRef] [PubMed]
- Astapova, I.; Hollenberg, A.N. The in vivo role of nuclear receptor corepressors in thyroid hormone action. Biochim. Biophys. Acta 2013, 1830, 3876–3881. [Google Scholar] [CrossRef]
- Yen, P.M. Physiological and molecular basis of thyroid hormone action. Physiol. Rev. 2001, 81, 1097–1142. [Google Scholar] [CrossRef]
- Astapova, I. Role of co-regulators in metabolic and transcriptional actions of thyroid hormone. J. Mol. Endocrinol. 2016, 56, 73–97. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Lazar, M.A. The CoRNR motif controls the recruitment of corepressors by nuclear hormone receptors. Nature 1999, 402, 93–96. [Google Scholar] [CrossRef]
- Horlein, A.J.; Naar, A.M.; Heinzel, T.; Torchia, J.; Gloss, B.; Kurokawa, R.; Ryan, A.; Kamei, Y.; Soderstrom, M.; Glass, C.K.; et al. Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature 1995, 377, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Aranda, A.; Pascual, A. Nuclear hormone receptors and gene expression. Physiol. Rev. 2001, 81, 1269–1304. [Google Scholar] [CrossRef]
- Zhang, Y.; Dufau, M.L. Dual mechanisms of regulation of transcription of luteinizing hormone receptor gene by nuclear orphan receptors and histone deacetylase complexes. J. Steroid Biochem. Mol. Biol. 2003, 85, 401–414. [Google Scholar] [CrossRef]
- Yao, Y.L.; Yang, W.M. The metastasis-associated proteins 1 and 2 form distinct protein complexes with histone deacetylase activity. J. Biol. Chem. 2003, 278, 42560–42568. [Google Scholar] [CrossRef]
- Fleischer, T.C.; Yun, U.J.; Ayer, D.E. Identification and characterization of three new components of the mSin3A corepressor complex. Mol. Cell Biol. 2003, 23, 3456–3467. [Google Scholar] [CrossRef]
- Martínez-Iglesias, O.; Olmeda, D.; Alonso-Merino, E.; Gómez-Rey, S.; González-López, A.M.; Luengo, E.; Soengas, M.S.; Palacios, J.; Regadera, J.; Aranda, A. The nuclear corepressor 1 and the thyroid hormone receptor β suppress breast tumor lymphangiogenesis. Oncotarget 2016, 7, 78971–78984. [Google Scholar] [CrossRef]
- Kaushik, D.; Vashistha, V.; Isharwal, S.; Sediqe, S.A.; Lin, M.F. Histone deacetylase inhibitors in castration-resistant prostate cancer: Molecular mechanism of action and recent clinical trials. Adv. Urol. 2015, 7, 388–395. [Google Scholar] [CrossRef]
- Hodges-Gallagher, L.; Valentine, C.D.; Bader, S.E.; Kushner, P.J. Inhibition of histone deacetylase enhances the anti-proliferative action of antiestrogens on breast cancer cells and blocks tamoxifen-induced proliferation of uterine cells. Breast Cancer Res. Treat. 2007, 105, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Elbers, L.P.; Kastelein, J.J.; Sjouke, B. Thyroid hormone mimetics: The past, current status and future challenges. Curr. Atheroscler. Rep. 2016, 18, 14. [Google Scholar] [CrossRef] [PubMed]
- Saponaro, F.; Sestito, S.; Runfola, M.; Rapposelli, S.; Chiellini, G. Selective thyroid hormone receptor-beta (TRβ) agonists: New perspectives for the treatment of metabolic and neurodegenerative disorders. Front. Med. 2020, 7, 331. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, S.J.; Chaudhary, P.; DeBell, M.J.; Marracci, G.; Miller, H.; Calkins, E.; Pocius, E.; Napier, B.A.; Emery, B.; Bourdette, D.; et al. TREM2 is thyroid hormone regulated making the TREM2 pathway druggable with ligands for thyroid hormone receptor. bioRxiv 2021. [Google Scholar] [CrossRef]
- Scanlan, T.S. Sobetirome: A case history of bench-to-clinic drug discovery and development. Heart Fail. Rev. 2010, 15, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Lin, J.Z.; Sieglaff, D.H.; Ayers, S.D.; Denoto-Reynolds, F.; Baxter, J.D.; Webb, P. Identical gene regulation patterns of T3 and selective thyroid hormone receptor modulator GC-1. Endocrinology 2012, 153, 501–511. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Grover, G.J.; Mellström, K.; Ye, L.; Malm, J.; Li, Y.L.; Bladh, L.G.; Sleph, P.G.; Smith, M.A.; George, R.; Vennström, B.; et al. Selective thyroid hormone receptor-beta activation: A strategy for reduction of weight, cholesterol, and lipoprotein (a) with reduced cardiovascular liability. Proc. Natl. Acad. Sci. USA 2003, 100, 10067–10072. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davidson, C.D.; Gillis, N.E.; Carr, F.E. Thyroid Hormone Receptor Beta as Tumor Suppressor: Untapped Potential in Treatment and Diagnostics in Solid Tumors. Cancers 2021, 13, 4254. https://doi.org/10.3390/cancers13174254
Davidson CD, Gillis NE, Carr FE. Thyroid Hormone Receptor Beta as Tumor Suppressor: Untapped Potential in Treatment and Diagnostics in Solid Tumors. Cancers. 2021; 13(17):4254. https://doi.org/10.3390/cancers13174254
Chicago/Turabian StyleDavidson, Cole D., Noelle E. Gillis, and Frances E. Carr. 2021. "Thyroid Hormone Receptor Beta as Tumor Suppressor: Untapped Potential in Treatment and Diagnostics in Solid Tumors" Cancers 13, no. 17: 4254. https://doi.org/10.3390/cancers13174254
APA StyleDavidson, C. D., Gillis, N. E., & Carr, F. E. (2021). Thyroid Hormone Receptor Beta as Tumor Suppressor: Untapped Potential in Treatment and Diagnostics in Solid Tumors. Cancers, 13(17), 4254. https://doi.org/10.3390/cancers13174254