
Casein Kinase 1D Encodes a Novel Drug Target in Hedgehog—GLI-Driven Cancers and Tumor-Initiating Cells Resistant to SMO Inhibition

 , ,
, , 
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Genetic Perturbation of CSNK1D Interferes with Canonical, Oncogenic HH—GLI Signaling in Medulloblastoma Cells
2.2. Genetic Inhibition of CSNK1D Reduces HH—GLI Activity in SMOi-Resistant Tumor Entities Driven by Oncogenic GLI
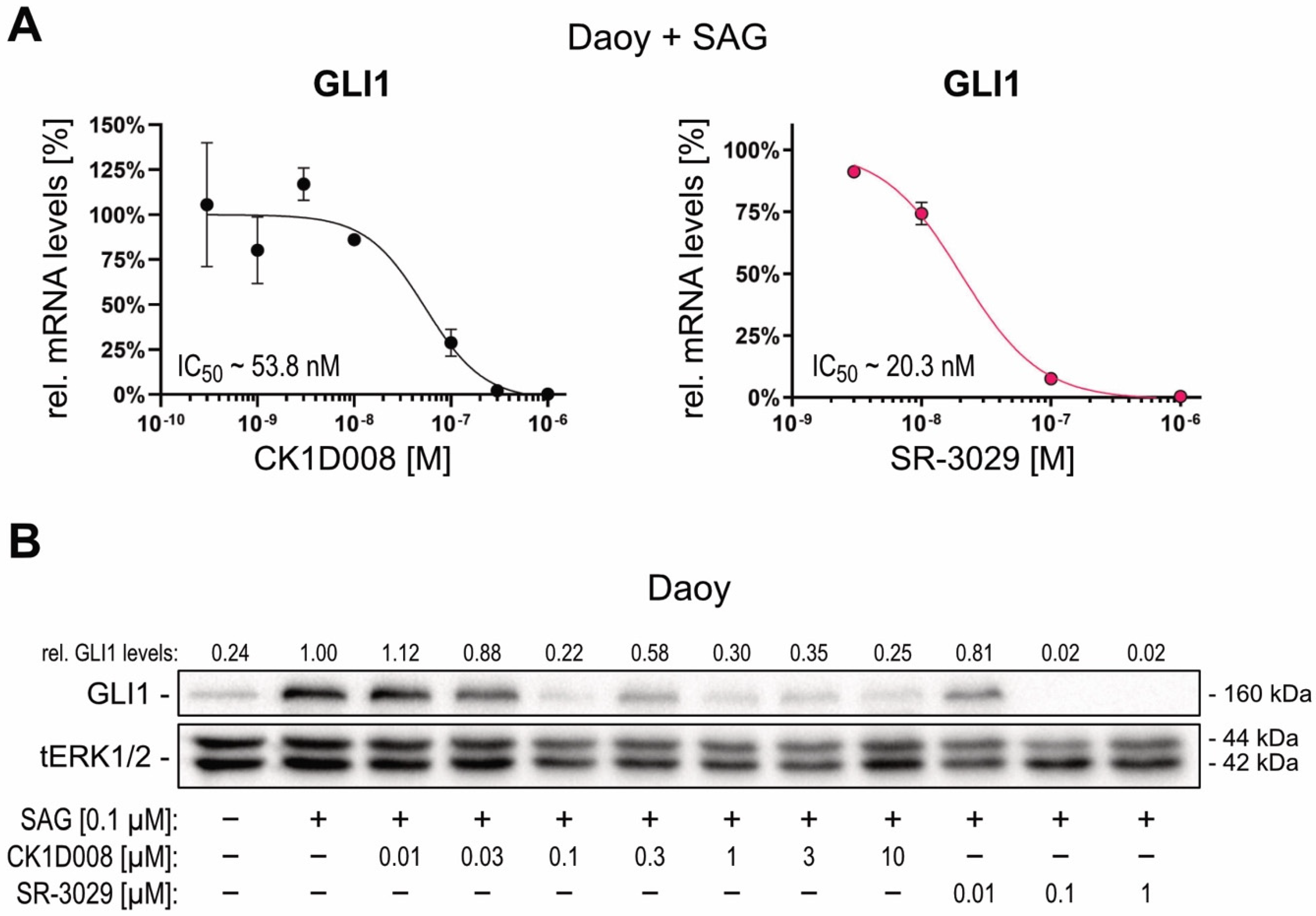
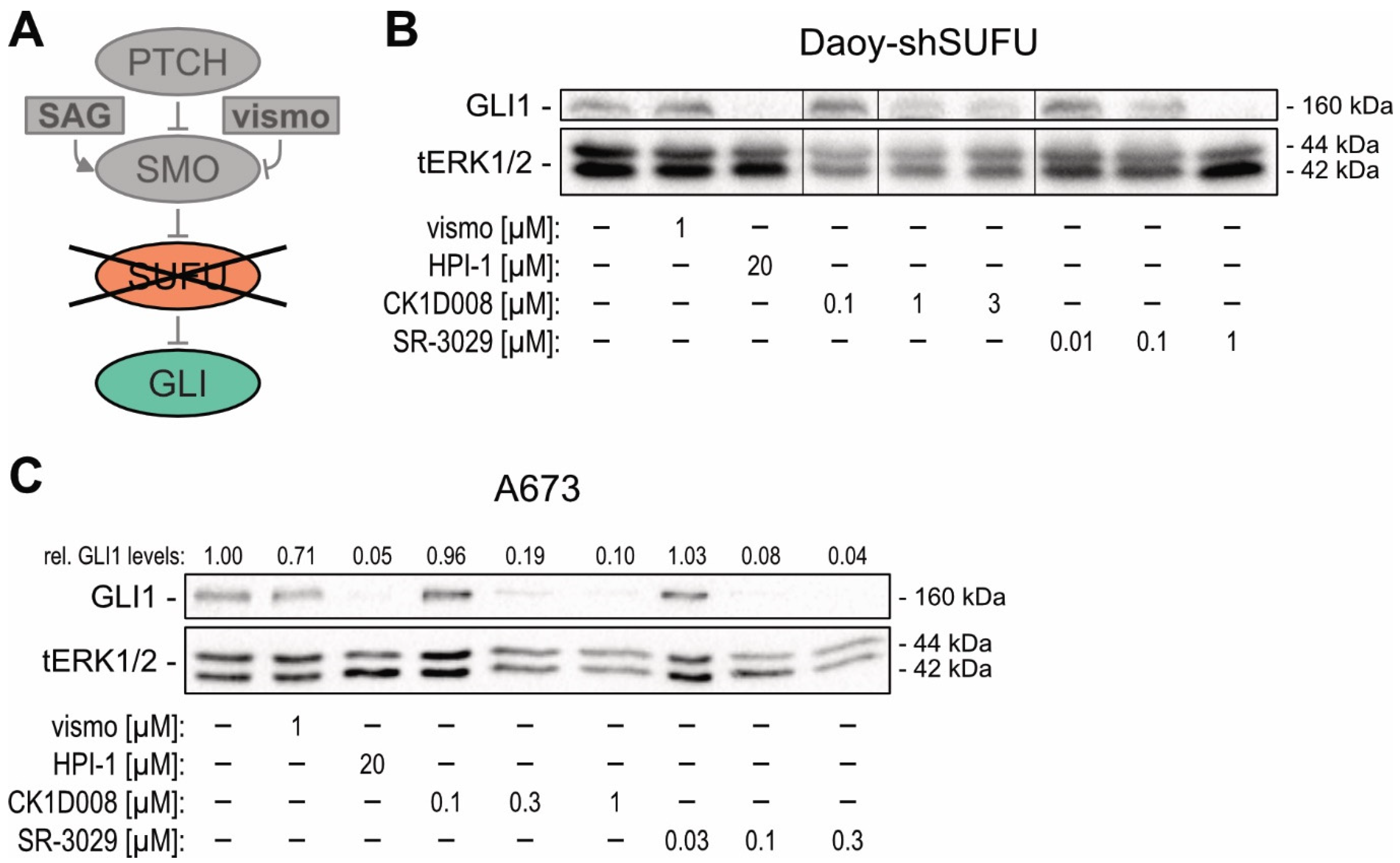
2.3. Pharmacological Targeting of CSNK1D Inhibits HH—GLI Signaling in Both Canonical and Non-Canonical Settings
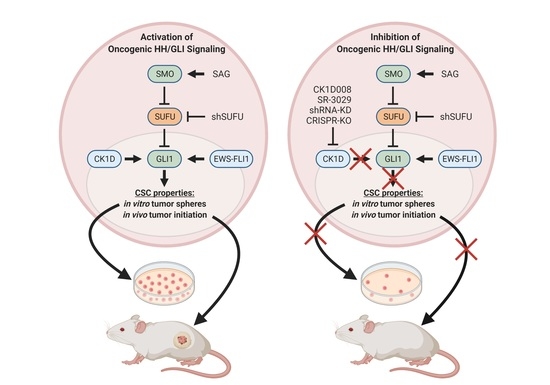
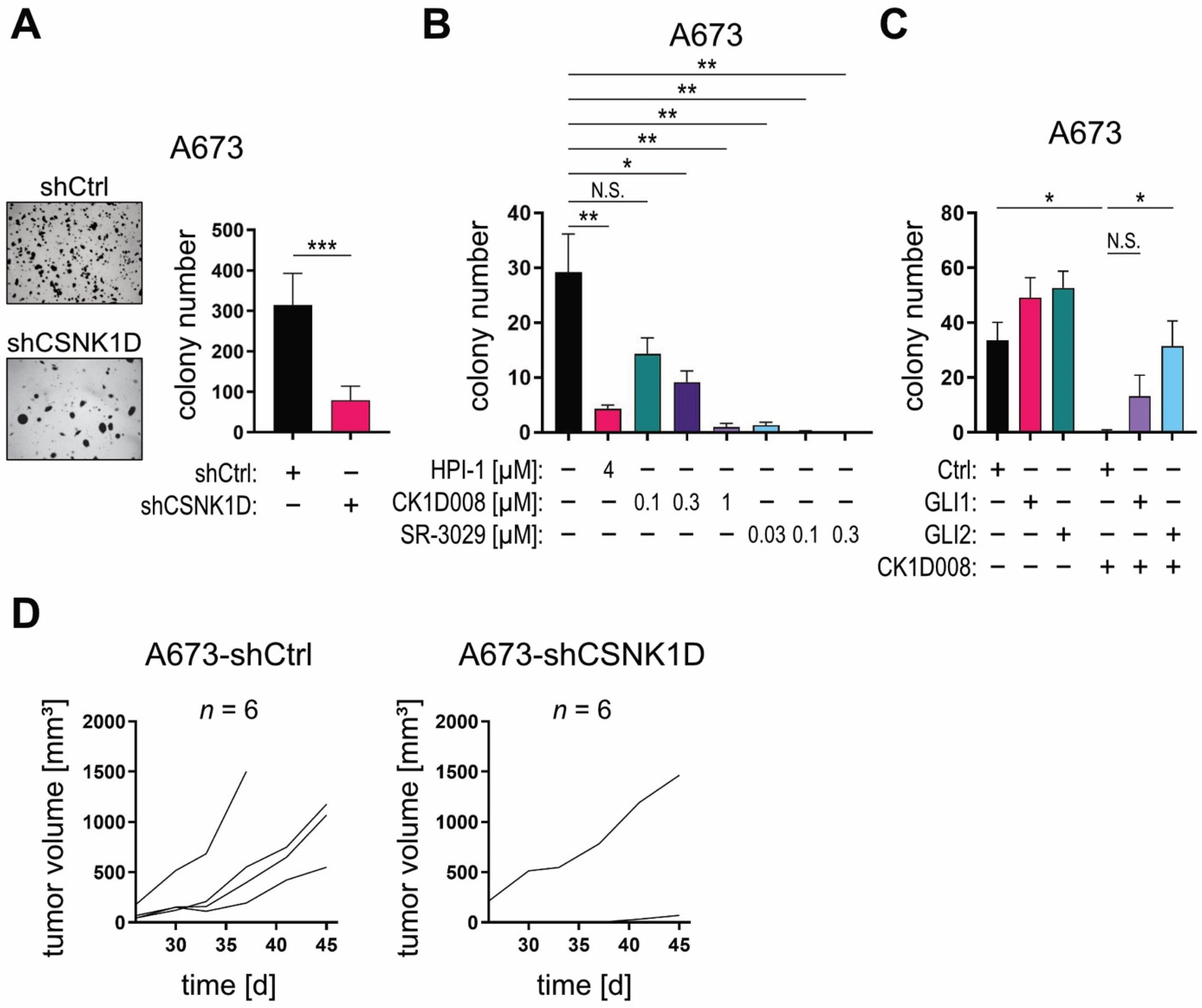
2.4. Targeting the CSNK1D-GLI Axis Inhibits CSC-Like Characteristics In Vitro and In Vivo
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Reagents
4.2. Identification and Characterization of CK1D008
4.3. Cell Proliferation and Anchorage-Dependent and -Independent Growth Assays
4.4. In Vivo Experiments
4.5. RNA Isolation and Quantitative PCR (qPCR)
4.6. Western Blot Analysis
4.7. RNA Interference and Overexpression Constructs
4.8. CRISPR-Mediated Knockout
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BCC | Basal cell carcinoma |
| CSNK1 | Casein kinase 1 |
| GLI | Glioma-associated oncogene homolog |
| HH | Hedgehog |
| HHIP | Hedgehog interacting protein |
| PTCH1 | Patched1 |
| SAG | Smoothened Agonist |
| SMO | Smoothened |
| SPOP | Speckle-type POZ protein |
| SUFU | Suppressor of Fused |
References
- Bajaj, J.; Diaz, E.; Reya, T. Stem cells in cancer initiation and progression. J. Cell Biol. 2020, 219, e201911053. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Bonnet, D.; De Maria, R.; Lapidot, T.; Copland, M.; Melo, J.V.; Chomienne, C.; Ishikawa, F.; Schuringa, J.J.; Stassi, G.; et al. Cancer stem cell definitions and terminology: The devil is in the details. Nat. Rev. Cancer 2012, 12, 767–775. [Google Scholar] [CrossRef]
- Ruiz i Altaba, A.; Mas, C.; Stecca, B. The Gli code: An information nexus regulating cell fate, stemness and cancer. Trends Cell Biol. 2007, 17, 438–447. [Google Scholar] [CrossRef] [Green Version]
- Stecca, B.; Ruiz i Altaba, A. Context-dependent regulation of the GLI code in cancer by HEDGEHOG and non-HEDGEHOG signals. J. Mol. Cell. Biol. 2010, 2, 84–95. [Google Scholar] [CrossRef] [Green Version]
- Zbinden, M.; Duquet, A.; Lorente-Trigos, A.; Ngwabyt, S.N.; Borges, I.; Ruiz i Altaba, A. NANOG regulates glioma stem cells and is essential in vivo acting in a cross-functional network with GLI1 and p53. EMBO J. 2010, 29, 2659–2674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varnat, F.; Duquet, A.; Malerba, M.; Zbinden, M.; Mas, C.; Gervaz, P.; Ruiz i Altaba, A. Human colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. EMBO Mol. Med. 2009, 1, 338–351. [Google Scholar] [CrossRef]
- Aberger, F.; Ruiz i Altaba, A. Context-dependent signal integration by the GLI code: The oncogenic load, pathways, modifiers and implications for cancer therapy. Semin. Cell Dev. Biol. 2014, 33, 93–104. [Google Scholar] [CrossRef]
- Teglund, S.; Toftgard, R. Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim. Biophys. Acta 2010, 1805, 181–208. [Google Scholar] [CrossRef]
- Hui, C.C.; Angers, S. Gli proteins in development and disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 513–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.; Hui, C.C. Hedgehog signaling in development and cancer. Dev. Cell 2008, 15, 801–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieuwenhuis, E.; Hui, C.C. Hedgehog signaling and congenital malformations. Clin. Genet. 2005, 67, 193–208. [Google Scholar] [CrossRef]
- Rohatgi, R.; Milenkovic, L.; Scott, M.P. Patched1 regulates hedgehog signaling at the primary cilium. Science 2007, 317, 372–376. [Google Scholar] [CrossRef] [Green Version]
- Kasper, M.; Regl, G.; Frischauf, A.M.; Aberger, F. GLI transcription factors: Mediators of oncogenic Hedgehog signalling. Eur. J. Cancer 2006, 42, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Briscoe, J.; Therond, P. Hedgehog signaling: From the Drosophila cuticle to anti-cancer drugs. Dev. Cell 2005, 8, 143–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooper, J.E.; Scott, M.P. Communicating with Hedgehogs. Nat. Rev. Mol. Cell Biol. 2005, 6, 306–317. [Google Scholar] [CrossRef]
- Goetz, S.C.; Anderson, K.V. The primary cilium: A signalling centre during vertebrate development. Nat Rev Genet. 2010, 11, 331–344. [Google Scholar] [CrossRef]
- He, M.; Agbu, S.; Anderson, K.V. Microtubule motors drive hedgehog signaling in primary cilia. Trends Cell Biol. 2017, 27, 110–125. [Google Scholar] [CrossRef] [Green Version]
- Atwood, S.X.; Whitson, R.J.; Oro, A.E. Advanced treatment for basal cell carcinomas. Cold Spring Harb. Perspect. Med. 2014, 4, a013581. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.Y.; Ally, M.S.; Chanana, A.M.; Mackay-Wiggan, J.M.; Aszterbaum, M.; Lindgren, J.A.; Ulerio, G.; Rezaee, M.R.; Gildengorin, G.; Marji, J.; et al. Inhibition of the hedgehog pathway in patients with basal-cell nevus syndrome: Final results from the multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2016, 17, 1720–1731. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; LoRusso, P.M.; Rudin, C.M.; Reddy, J.C.; Yauch, R.L.; Tibes, R.; Weiss, G.J.; Borad, M.J.; Hann, C.L.; Brahmer, J.R.; et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N. Engl. J. Med. 2009, 361, 1164–1172. [Google Scholar] [CrossRef] [Green Version]
- Migden, M.R.; Guminski, A.; Gutzmer, R.; Dirix, L.; Lewis, K.D.; Combemale, P.; Herd, R.M.; Kudchadkar, R.; Trefzer, U.; Gogov, S.; et al. Treatment with two different doses of sonidegib in patients with locally advanced or metastatic basal cell carcinoma (BOLT): A multicentre, randomised, double-blind phase 2 trial. Lancet Oncol. 2015, 16, 716–728. [Google Scholar] [CrossRef]
- Sekulic, A.; Migden, M.R.; Oro, A.E.; Dirix, L.; Lewis, K.D.; Hainsworth, J.D.; Solomon, J.A.; Yoo, S.; Arron, S.T.; Friedlander, P.A.; et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N. Engl. J. Med. 2012, 366, 2171–2179. [Google Scholar] [CrossRef] [Green Version]
- Norsworthy, K.J.; By, K.; Subramaniam, S.; Zhuang, L.; Del Valle, P.L.; Przepiorka, D.; Shen, Y.L.; Sheth, C.M.; Liu, C.; Leong, R.; et al. FDA approval summary: Glasdegib for newly diagnosed acute myeloid leukemia. Clin. Cancer Res. 2019, 25, 6021–6025. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.Y.; Mackay-Wiggan, J.M.; Aszterbaum, M.; Yauch, R.L.; Lindgren, J.; Chang, K.; Coppola, C.; Chanana, A.M.; Marji, J.; Bickers, D.R.; et al. Inhibiting the hedgehog pathway in patients with the basal-cell nevus syndrome. N. Engl. J. Med. 2012, 366, 2180–2188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atwood, S.X.; Sarin, K.Y.; Whitson, R.J.; Li, J.R.; Kim, G.; Rezaee, M.; Ally, M.S.; Kim, J.; Yao, C.; Chang, A.L.; et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell 2015, 27, 342–353. [Google Scholar] [CrossRef] [Green Version]
- Sharpe, H.J.; Pau, G.; Dijkgraaf, G.J.; Basset-Seguin, N.; Modrusan, Z.; Januario, T.; Tsui, V.; Durham, A.B.; Dlugosz, A.A.; Haverty, P.M.; et al. Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell 2015, 27, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Yauch, R.L.; Dijkgraaf, G.J.; Alicke, B.; Januario, T.; Ahn, C.P.; Holcomb, T.; Pujara, K.; Stinson, J.; Callahan, C.A.; Tang, T.; et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science 2009, 326, 572–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buonamici, S.; Williams, J.; Morrissey, M.; Wang, A.; Guo, R.; Vattay, A.; Hsiao, K.; Yuan, J.; Green, J.; Ospina, B.; et al. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci. Transl. Med. 2010, 2, 51ra70. [Google Scholar] [CrossRef] [Green Version]
- Bonilla, X.; Parmentier, L.; King, B.; Bezrukov, F.; Kaya, G.; Zoete, V.; Seplyarskiy, V.B.; Sharpe, H.J.; McKee, T.; Letourneau, A.; et al. Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nat. Genet. 2016, 48, 398–406. [Google Scholar] [CrossRef]
- Atwood, S.X.; Li, M.; Lee, A.; Tang, J.Y.; Oro, A.E. GLI activation by atypical protein kinase C iota/lambda regulates the growth of basal cell carcinomas. Nature 2013, 494, 484–488. [Google Scholar] [CrossRef] [Green Version]
- Whitson, R.J.; Lee, A.; Urman, N.M.; Mirza, A.; Yao, C.Y.; Brown, A.S.; Li, J.R.; Shankar, G.; Fry, M.A.; Atwood, S.X.; et al. Noncanonical hedgehog pathway activation through SRF-MKL1 promotes drug resistance in basal cell carcinomas. Nat. Med. 2018, 24, 271–281. [Google Scholar] [CrossRef]
- Zhao, X.; Ponomaryov, T.; Ornell, K.J.; Zhou, P.; Dabral, S.K.; Pak, E.; Li, W.; Atwood, S.X.; Whitson, R.J.; Chang, A.L.; et al. RAS/MAPK activation drives resistance to smo inhibition, metastasis, and tumor evolution in shh pathway-dependent tumors. Cancer Res. 2015, 75, 3623–3635. [Google Scholar] [CrossRef] [Green Version]
- Kuonen, F.; Huskey, N.E.; Shankar, G.; Jaju, P.; Whitson, R.J.; Rieger, K.E.; Atwood, S.X.; Sarin, K.Y.; Oro, A.E. Loss of primary cilia drives switching from hedgehog to Ras/MAPK pathway in resistant basal cell carcinoma. J. Investig. Dermatol. 2019, 139, 1439–1448. [Google Scholar] [CrossRef]
- Brewster, R.; Mullor, J.L.; Ruiz i Altaba, A. Gli2 functions in FGF signaling during antero-posterior patterning. Development 2000, 127, 4395–4405. [Google Scholar] [CrossRef]
- Gruber, W.; Hutzinger, M.; Elmer, D.P.; Parigger, T.; Sternberg, C.; Cegielkowski, L.; Zaja, M.; Leban, J.; Michel, S.; Hamm, S.; et al. DYRK1B as therapeutic target in Hedgehog/GLI-dependent cancer cells with Smoothened inhibitor resistance. Oncotarget 2016, 7, 7134–7148. [Google Scholar] [CrossRef] [Green Version]
- Kasper, M.; Schnidar, H.; Neill, G.W.; Hanneder, M.; Klingler, S.; Blaas, L.; Schmid, C.; Hauser-Kronberger, C.; Regl, G.; Philpott, M.P.; et al. Selective modulation of Hedgehog/GLI target gene expression by epidermal growth factor signaling in human keratinocytes. Mol. Cell. Biol. 2006, 26, 6283–6298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauth, M.; Bergstrom, A.; Shimokawa, T.; Tostar, U.; Jin, Q.; Fendrich, V.; Guerra, C.; Barbacid, M.; Toftgard, R. DYRK1B-dependent autocrine-to-paracrine shift of Hedgehog signaling by mutant RAS. Nat. Struct. Mol. Biol. 2010, 17, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Neill, G.W.; Harrison, W.J.; Ikram, M.S.; Williams, T.D.; Bianchi, L.S.; Nadendla, S.K.; Green, J.L.; Ghali, L.; Frischauf, A.M.; O’Toole, E.A.; et al. GLI1 repression of ERK activity correlates with colony formation and impaired migration in human epidermal keratinocytes. Carcinogenesis 2008, 29, 738–746. [Google Scholar] [CrossRef] [Green Version]
- Pelczar, P.; Zibat, A.; van Dop, W.A.; Heijmans, J.; Bleckmann, A.; Gruber, W.; Nitzki, F.; Uhmann, A.; Guijarro, M.V.; Hernando, E.; et al. Inactivation of Patched1 in mice leads to development of gastrointestinal stromal-like tumors that express Pdgfralpha but not kit. Gastroenterology 2013, 144, 134–144.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sternberg, C.; Gruber, W.; Eberl, M.; Tesanovic, S.; Stadler, M.; Elmer, D.P.; Schlederer, M.; Grund, S.; Roos, S.; Wolff, F.; et al. Synergistic cross-talk of hedgehog and interleukin-6 signaling drives growth of basal cell carcinoma. Int. J. Cancer 2018, 143, 2943–2954. [Google Scholar] [CrossRef] [Green Version]
- Varjosalo, M.; Bjorklund, M.; Cheng, F.; Syvanen, H.; Kivioja, T.; Kilpinen, S.; Sun, Z.; Kallioniemi, O.; Stunnenberg, H.G.; He, W.W.; et al. Application of active and kinase-deficient kinome collection for identification of kinases regulating hedgehog signaling. Cell 2008, 133, 537–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javelaud, D.; Pierrat, M.J.; Mauviel, A. Crosstalk between TGF-beta and hedgehog signaling in cancer. FEBS Lett. 2012, 586, 2016–2025. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.H.; Andl, T.; Grachtchouk, V.; Wang, A.; Liu, J.; Syu, L.J.; Ferris, J.; Wang, T.S.; Glick, A.B.; Millar, S.E.; et al. Pathological responses to oncogenic Hedgehog signaling in skin are dependent on canonical Wnt/beta3-catenin signaling. Nat. Genet. 2008, 40, 1130–1135. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J. CK1 in developmental signaling: Hedgehog and Wnt. Curr. Top. Dev. Biol. 2017, 123, 303–329. [Google Scholar] [CrossRef] [Green Version]
- Price, M.A.; Kalderon, D. Proteolysis of the Hedgehog signaling effector Cubitus interruptus requires phosphorylation by Glycogen Synthase Kinase 3 and Casein Kinase 1. Cell 2002, 108, 823–835. [Google Scholar] [CrossRef] [Green Version]
- Price, M.A. CKI, there’s more than one: Casein kinase I family members in Wnt and Hedgehog signaling. Genes Dev. 2006, 20, 399–410. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Sasai, N.; Ma, G.; Yue, T.; Jia, J.; Briscoe, J.; Jiang, J. Sonic Hedgehog dependent phosphorylation by CK1alpha and GRK2 is required for ciliary accumulation and activation of smoothened. PLoS Biol. 2011, 9, e1001083. [Google Scholar] [CrossRef] [Green Version]
- Purzner, T.; Purzner, J.; Buckstaff, T.; Cozza, G.; Gholamin, S.; Rusert, J.M.; Hartl, T.A.; Sanders, J.; Conley, N.; Ge, X.; et al. Developmental phosphoproteomics identifies the kinase CK2 as a driver of Hedgehog signaling and a therapeutic target in medulloblastoma. Sci. Signal 2018, 11, eaau5147. [Google Scholar] [CrossRef] [Green Version]
- Jia, J.; Zhang, L.; Zhang, Q.; Tong, C.; Wang, B.; Hou, F.; Amanai, K.; Jiang, J. Phosphorylation by double-time/CKIepsilon and CKIalpha targets cubitus interruptus for Slimb/beta-TRCP-mediated proteolytic processing. Dev. Cell 2005, 9, 819–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Q.; Li, S.; Li, S.; Jiang, A.; Chen, Y.; Jiang, J. Hedgehog-induced phosphorylation by CK1 sustains the activity of Ci/Gli activator. Proc. Natl. Acad. Sci. USA 2014, 111, E5651–E5660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank-Kamenetsky, M.; Zhang, X.M.; Bottega, S.; Guicherit, O.; Wichterle, H.; Dudek, H.; Bumcrot, D.; Wang, F.Y.; Jones, S.; Shulok, J.; et al. Small-molecule modulators of Hedgehog signaling: Identification and characterization of Smoothened agonists and antagonists. J. Biol. 2002, 1, 10. [Google Scholar] [CrossRef] [Green Version]
- Gotschel, F.; Berg, D.; Gruber, W.; Bender, C.; Eberl, M.; Friedel, M.; Sonntag, J.; Rungeler, E.; Hache, H.; Wierling, C.; et al. Synergism between Hedgehog-GLI and EGFR signaling in Hedgehog-responsive human medulloblastoma cells induces downregulation of canonical Hedgehog-target genes and stabilized expression of GLI1. PLoS ONE 2013, 8, e65403. [Google Scholar] [CrossRef]
- Eggenschwiler, J.T.; Anderson, K.V. Cilia and developmental signaling. Annu. Rev. Cell Dev. Biol. 2007, 23, 345–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauchamp, E.; Bulut, G.; Abaan, O.; Chen, K.; Merchant, A.; Matsui, W.; Endo, Y.; Rubin, J.S.; Toretsky, J.; Uren, A. GLI1 is a direct transcriptional target of EWS-FLI1 oncoprotein. J. Biol. Chem. 2009, 284, 9074–9082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauchamp, E.M.; Ringer, L.; Bulut, G.; Sajwan, K.P.; Hall, M.D.; Lee, Y.C.; Peaceman, D.; Ozdemirli, M.; Rodriguez, O.; Macdonald, T.J.; et al. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathway. J. Clin. Investig. 2011, 121, 148–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyman, J.M.; Firestone, A.J.; Heine, V.M.; Zhao, Y.; Ocasio, C.A.; Han, K.; Sun, M.; Rack, P.G.; Sinha, S.; Wu, J.J.; et al. Small-molecule inhibitors reveal multiple strategies for Hedgehog pathway blockade. Proc. Natl. Acad. Sci. USA 2009, 106, 14132–14137. [Google Scholar] [CrossRef] [Green Version]
- Sekulic, A.; Von Hoff, D. Hedgehog pathway inhibition. Cell 2016, 164, 831. [Google Scholar] [CrossRef]
- Rosenberg, L.H.; Lafitte, M.; Quereda, V.; Grant, W.; Chen, W.; Bibian, M.; Noguchi, Y.; Fallahi, M.; Yang, C.; Chang, J.C.; et al. Therapeutic targeting of casein kinase 1delta in breast cancer. Sci. Transl. Med. 2015, 7, 318ra202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kogerman, P.; Grimm, T.; Kogerman, L.; Krause, D.; Unden, A.B.; Sandstedt, B.; Toftgard, R.; Zaphiropoulos, P.G. Mammalian suppressor-of-fused modulates nuclear-cytoplasmic shuttling of Gli-1. Nat. Cell Biol. 1999, 1, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Eberl, M.; Klingler, S.; Mangelberger, D.; Loipetzberger, A.; Damhofer, H.; Zoidl, K.; Schnidar, H.; Hache, H.; Bauer, H.C.; Solca, F.; et al. Hedgehog-EGFR cooperation response genes determine the oncogenic phenotype of basal cell carcinoma and tumour-initiating pancreatic cancer cells. EMBO Mol. Med. 2012, 4, 218–233. [Google Scholar] [CrossRef]
- Rodriguez-Blanco, J.; Schilling, N.S.; Tokhunts, R.; Giambelli, C.; Long, J.; Liang Fei, D.; Singh, S.; Black, K.E.; Wang, Z.; Galimberti, F.; et al. The hedgehog processing pathway is required for NSCLC growth and survival. Oncogene 2013, 32, 2335–2345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clement, V.; Sanchez, P.; de Tribolet, N.; Radovanovic, I.; Ruiz i Altaba, A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef]
- Regl, G.; Neill, G.W.; Eichberger, T.; Kasper, M.; Ikram, M.S.; Koller, J.; Hintner, H.; Quinn, A.G.; Frischauf, A.M.; Aberger, F. Human GLI2 and GLI1 are part of a positive feedback mechanism in Basal Cell Carcinoma. Oncogene 2002, 21, 5529–5539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peer, E.; Tesanovic, S.; Aberger, F. Next-generation hedgehog/GLI pathway inhibitors for cancer therapy. Cancers 2019, 11, 538. [Google Scholar] [CrossRef] [Green Version]
- Schnidar, H.; Eberl, M.; Klingler, S.; Mangelberger, D.; Kasper, M.; Hauser-Kronberger, C.; Regl, G.; Kroismayr, R.; Moriggl, R.; Sibilia, M.; et al. Epidermal growth factor receptor signaling synergizes with Hedgehog/GLI in oncogenic transformation via activation of the MEK/ERK/JUN pathway. Cancer Res. 2009, 69, 1284–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauth, M.; Bergstrom, A.; Shimokawa, T.; Toftgard, R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl. Acad. Sci. USA 2007, 104, 8455–8460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Infante, P.; Mori, M.; Alfonsi, R.; Ghirga, F.; Aiello, F.; Toscano, S.; Ingallina, C.; Siler, M.; Cucchi, D.; Po, A.; et al. Gli1/DNA interaction is a druggable target for Hedgehog-dependent tumors. EMBO J. 2015, 34, 200–217. [Google Scholar] [CrossRef]
- Bibian, M.; Rahaim, R.J.; Choi, J.Y.; Noguchi, Y.; Schurer, S.; Chen, W.; Nakanishi, S.; Licht, K.; Rosenberg, L.H.; Li, L.; et al. Development of highly selective casein kinase 1delta/1epsilon (CK1delta/epsilon) inhibitors with potent antiproliferative properties. Bioorg. Med. Chem. Lett. 2013, 23, 4374–4380. [Google Scholar] [CrossRef] [Green Version]
- Bayik, D.; Lathia, J.D. Cancer stem cell-immune cell crosstalk in tumour progression. Nat. Rev. Cancer 2021. [Google Scholar] [CrossRef]
- Grund-Groschke, S.; Ortner, D.; Szenes-Nagy, A.B.; Zaborsky, N.; Weiss, R.; Neureiter, D.; Wipplinger, M.; Risch, A.; Hammerl, P.; Greil, R.; et al. Epidermal activation of Hedgehog signaling establishes an immunosuppressive microenvironment in basal cell carcinoma by modulating skin immunity. Mol. Oncol. 2020, 14, 1930–1946. [Google Scholar] [CrossRef]
- Grund-Groschke, S.; Stockmaier, G.; Aberger, F. Hedgehog/GLI signaling in tumor immunity-new therapeutic opportunities and clinical implications. Cell Commun Signal 2019, 17, 172. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, J.; Holokai, L.; Syu, L.; Steele, N.G.; Chang, J.; Wang, J.; Ahmed, S.; Dlugosz, A.; Zavros, Y. Hedgehog signaling induces PD-L1 expression and tumor cell proliferation in gastric cancer. Oncotarget 2018, 9, 37439–37457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holokai, L.; Chakrabarti, J.; Broda, T.; Chang, J.; Hawkins, J.A.; Sundaram, N.; Wroblewski, L.E.; Peek, R.M., Jr.; Wang, J.; Helmrath, M.; et al. Increased Programmed death-ligand 1 is an early epithelial cell response to helicobacter pylori infection. PLoS Pathog. 2019, 15, e1007468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schittek, B.; Sinnberg, T. Biological functions of casein kinase 1 isoforms and putative roles in tumorigenesis. Mol. Cancer 2014, 13, 231. [Google Scholar] [CrossRef] [Green Version]
- Fiaschi, M.; Rozell, B.; Bergstrom, A.; Toftgard, R.; Kleman, M.I. Targeted expression of GLI1 in the mammary gland disrupts pregnancy-induced maturation and causes lactation failure. J. Biol. Chem. 2007, 282, 36090–36101. [Google Scholar] [CrossRef] [Green Version]
- Fiaschi, M.; Rozell, B.; Bergstrom, A.; Toftgard, R. Development of mammary tumors by conditional expression of GLI1. Cancer Res. 2009, 69, 4810–4817. [Google Scholar] [CrossRef] [Green Version]
- Norum, J.H.; Frings, O.; Kasper, M.; Bergholtz, H.; Zell Thime, H.; Bergstrom, A.; Andersson, A.; Kuiper, R.; Fredlund, E.; Sorlie, T.; et al. GLI1-induced mammary gland tumours are transplantable and maintain major molecular features. Int. J. Cancer 2020, 146, 1125–1138. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Kasper, M.; Regl, G.; Eichberger, T.; Frischauf, A.M.; Aberger, F. Efficient manipulation of Hedgehog/GLI signaling using retroviral expression systems. Methods Mol. Biol. 2007, 397, 67–78. [Google Scholar] [CrossRef]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinkman, E.K.; Chen, T.; Amendola, M.; van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014, 42, e168. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peer, E.; Aichberger, S.K.; Vilotic, F.; Gruber, W.; Parigger, T.; Grund-Gröschke, S.; Elmer, D.P.; Rathje, F.; Ramspacher, A.; Zaja, M.; et al. Casein Kinase 1D Encodes a Novel Drug Target in Hedgehog—GLI-Driven Cancers and Tumor-Initiating Cells Resistant to SMO Inhibition. Cancers 2021, 13, 4227. https://doi.org/10.3390/cancers13164227
Peer E, Aichberger SK, Vilotic F, Gruber W, Parigger T, Grund-Gröschke S, Elmer DP, Rathje F, Ramspacher A, Zaja M, et al. Casein Kinase 1D Encodes a Novel Drug Target in Hedgehog—GLI-Driven Cancers and Tumor-Initiating Cells Resistant to SMO Inhibition. Cancers. 2021; 13(16):4227. https://doi.org/10.3390/cancers13164227
Chicago/Turabian StylePeer, Elisabeth, Sophie Karoline Aichberger, Filip Vilotic, Wolfgang Gruber, Thomas Parigger, Sandra Grund-Gröschke, Dominik Patrick Elmer, Florian Rathje, Andrea Ramspacher, Mirko Zaja, and et al. 2021. "Casein Kinase 1D Encodes a Novel Drug Target in Hedgehog—GLI-Driven Cancers and Tumor-Initiating Cells Resistant to SMO Inhibition" Cancers 13, no. 16: 4227. https://doi.org/10.3390/cancers13164227
APA StylePeer, E., Aichberger, S. K., Vilotic, F., Gruber, W., Parigger, T., Grund-Gröschke, S., Elmer, D. P., Rathje, F., Ramspacher, A., Zaja, M., Michel, S., Hamm, S., & Aberger, F. (2021). Casein Kinase 1D Encodes a Novel Drug Target in Hedgehog—GLI-Driven Cancers and Tumor-Initiating Cells Resistant to SMO Inhibition. Cancers, 13(16), 4227. https://doi.org/10.3390/cancers13164227