
Microdissected Tissue vs Tissue Slices—A Comparative Study of Tumor Explant Models Cultured On-Chip and Off-Chip

 , ,
, ,
Simple Summary
Abstract
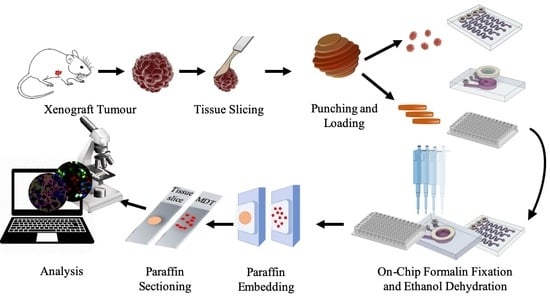
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Design and Fabrication of the Microfluidic Device
2.2. Finite Element Methodology
2.3. Ovarian and Prostate Cancer Cell Lines for Xenograft Production
2.4. MDT and Tissue Slice Production from Cell Line Xenograft Tumors
2.5. Tissue Loading, Trapping, and Culture of Tissue
2.6. Formalin Fixation and Paraffin Embedding Protocol and Tissue Staining
2.7. Quantification of Immunofluorescent Staining
2.8. Statistical Analysis
3. Results
3.1. Numerical Simulation Predicts Sufficient Oxygen and Glucose in MDTs and Deficiency in Tissue Slices
3.2. Tumor Models Preserve the Characteristics of the Primary Xenograft Tumor
3.3. Viability and Proliferation Activity in MDTs Are Higher Than Tissue Slices over the Culture Period
3.4. Elevated Levels of Hypoxia Are Found in Tissue Slices but Not in MDTs under Normoxic Culture Conditions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wong, C.H.; Siah, K.W.; Lo, A.W. Estimation of clinical trial success rates and related parameters. Biostatistics 2019, 20, 273–286. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Tycko, B. The history of cancer epigenetics. Nat. Rev. Cancer 2004, 4, 143–153. [Google Scholar] [CrossRef]
- Loeb, L.A.; Loeb, K.R.; Anderson, J.P. Multiple mutations and cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 776–781. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Ohlsson, R.; Henikoff, S. The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 2006, 7, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Nyga, A.; Cheema, U.; Loizidou, M. 3D tumour models: Novel in vitro approaches to cancer studies. J. Cell Commun. Signal. 2011, 5, 239. [Google Scholar] [CrossRef]
- Imamura, Y.; Mukohara, T.; Shimono, Y.; Funakoshi, Y.; Chayahara, N.; Toyoda, M.; Kiyota, N.; Takao, S.; Kono, S.; Nakatsura, T. Comparison of 2D-and 3D-culture models as drug-testing platforms in breast cancer. Oncol. Rep. 2015, 33, 1837–1843. [Google Scholar] [CrossRef] [PubMed]
- Zanoni, M.; Piccinini, F.; Arienti, C.; Zamagni, A.; Santi, S.; Polico, R.; Bevilacqua, A.; Tesei, A. 3D tumor spheroid models for in vitro therapeutic screening: A systematic approach to enhance the biological relevance of data obtained. Sci. Rep. 2016, 6, 19103. [Google Scholar] [CrossRef] [PubMed]
- Tung, Y.-C.; Hsiao, A.Y.; Allen, S.G.; Torisawa, Y.-s.; Ho, M.; Takayama, S. High-throughput 3D spheroid culture and drug testing using a 384 hanging drop array. Analyst 2011, 136, 473–478. [Google Scholar] [CrossRef]
- Anada, T.; Fukuda, J.; Sai, Y.; Suzuki, O. An oxygen-permeable spheroid culture system for the prevention of central hypoxia and necrosis of spheroids. Biomaterials 2012, 33, 8430–8441. [Google Scholar] [CrossRef]
- Jung, J.; Seol, H.S.; Chang, S. The generation and application of patient-derived xenograft model for cancer research. Cancer Res. Treat. 2018, 50, 1. [Google Scholar] [CrossRef]
- Scott, C.L.; Mackay, H.J.; Haluska Jr, P. Patient-derived xenograft models in gynecologic malignancies. Am. Soc. Clin. Oncol. Educ. Book 2014, 34, e258–e266. [Google Scholar] [CrossRef]
- Tuveson, D.; Clevers, H. Cancer modeling meets human organoid technology. Science 2019, 364, 952–955. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. Modeling development and disease with organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef] [PubMed]
- Van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef] [PubMed]
- Vaira, V.; Fedele, G.; Pyne, S.; Fasoli, E.; Zadra, G.; Bailey, D.; Snyder, E.; Faversani, A.; Coggi, G.; Flavin, R. Preclinical model of organotypic culture for pharmacodynamic profiling of human tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 8352–8356. [Google Scholar] [CrossRef]
- Meijer, T.G.; Naipal, K.A.; Jager, A.; van Gent, D.C. Ex vivo tumor culture systems for functional drug testing and therapy response prediction. Future Sci. 2017, 3, FSO190. [Google Scholar] [CrossRef]
- Belanger, M.C.; Ball, A.G.; Catterton, M.A.; Kinman, A.W.; Anbaei, P.; Groff, B.D.; Melchor, S.J.; Lukens, J.R.; Ross, A.E.; Pompano, R.R. Acute lymph node slices are a functional model system to study immunity ex vivo. ACS Pharmacol. Transl. Sci. 2021, 4, 128–142. [Google Scholar] [CrossRef]
- Powley, I.R.; Patel, M.; Miles, G.; Pringle, H.; Howells, L.; Thomas, A.; Kettleborough, C.; Bryans, J.; Hammonds, T.; MacFarlane, M. Patient-derived explants (PDEs) as a powerful preclinical platform for anti-cancer drug and biomarker discovery. Br. J. Cancer 2020, 122, 735–744. [Google Scholar] [CrossRef]
- Majumder, B.; Baraneedharan, U.; Thiyagarajan, S.; Radhakrishnan, P.; Narasimhan, H.; Dhandapani, M.; Brijwani, N.; Pinto, D.D.; Prasath, A.; Shanthappa, B.U. Predicting clinical response to anticancer drugs using an ex vivo platform that captures tumour heterogeneity. Nat. Commun. 2015, 6, 1–14. [Google Scholar]
- Van der Kuip, H.; Mürdter, T.E.; Sonnenberg, M.; McClellan, M.; Gutzeit, S.; Gerteis, A.; Simon, W.; Fritz, P.; Aulitzky, W.E. Short term culture of breast cancer tissues to study the activity of the anticancer drug taxol in an intact tumor environment. BMC Cancer 2006, 6, 86. [Google Scholar] [CrossRef]
- Vesci, L.; Carollo, V.; Roscilli, G.; Aurisicchio, L.; Ferrara, F.F.; Spagnoli, L.; De Santis, R. Trastuzumab and docetaxel in a preclinical organotypic breast cancer model using tissue slices from mammary fat pad: Translational relevance Corrigendum in/or/35/1/602. Oncol. Rep. 2015, 34, 1146–1152. [Google Scholar] [CrossRef]
- Van Midwoud, P.M.; Merema, M.T.; Verpoorte, E.; Groothuis, G.M. A microfluidic approach for in vitro assessment of interorgan interactions in drug metabolism using intestinal and liver slices. Lab Chip 2010, 10, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Van Midwoud, P.M.; Groothuis, G.M.; Merema, M.T.; Verpoorte, E. Microfluidic biochip for the perifusion of precision-cut rat liver slices for metabolism and toxicology studies. Biotechnol. Bioeng. 2010, 105, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Hattersley, S.M.; Dyer, C.E.; Greenman, J.; Haswell, S.J. Development of a microfluidic device for the maintenance and interrogation of viable tissue biopsies. Lab Chip 2008, 8, 1842–1846. [Google Scholar] [CrossRef]
- Shim, S.; Belanger, M.C.; Harris, A.R.; Munson, J.M.; Pompano, R.R. Two-way communication between ex vivo tissues on a microfluidic chip: Application to tumor–lymph node interaction. Lab Chip 2019, 19, 1013–1026. [Google Scholar] [CrossRef] [PubMed]
- Sontheimer-Phelps, A.; Hassell, B.A.; Ingber, D.E. Modelling cancer in microfluidic human organs-on-chips. Nat. Rev. Cancer 2019, 19, 65–81. [Google Scholar] [CrossRef]
- Holton, A.B.; Sinatra, F.L.; Kreahling, J.; Conway, A.J.; Landis, D.A.; Altiok, S. Microfluidic biopsy trapping device for the real-time monitoring of tumor microenvironment. PLoS ONE 2017, 12, e0169797. [Google Scholar] [CrossRef]
- Eduati, F.; Utharala, R.; Madhavan, D.; Neumann, U.P.; Longerich, T.; Cramer, T.; Saez-Rodriguez, J.; Merten, C.A. A microfluidics platform for combinatorial drug screening on cancer biopsies. Nat. Commun. 2018, 9, 1–13. [Google Scholar]
- Horowitz, L.F.; Rodriguez, A.D.; Dereli-Korkut, Z.; Lin, R.; Castro, K.; Mikheev, A.; Monnat, R.J.; Folch, A.; Rostomily, R.C. Multiplexed drug testing of tumor slices using a microfluidic platform. NPJ Precis. Oncol. 2020, 4, 1–15. [Google Scholar] [CrossRef]
- Astolfi, M.; Péant, B.; Lateef, M.; Rousset, N.; Kendall-Dupont, J.; Carmona, E.; Monet, F.; Saad, F.; Provencher, D.; Mes-Masson, A.-M. Micro-dissected tumor tissues on chip: An ex vivo method for drug testing and personalized therapy. Lab Chip 2016, 16, 312–325. [Google Scholar] [CrossRef]
- Simeone, K.; Guay-Lord, R.; Lateef, M.A.; Péant, B.; Kendall-Dupont, J.; Orimoto, A.M.; Carmona, E.; Provencher, D.; Saad, F.; Gervais, T. Paraffin-embedding lithography and micro-dissected tissue micro-arrays: Tools for biological and pharmacological analysis of ex vivo solid tumors. Lab Chip 2019, 19, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-C.; Lam, R.H.; Thorsen, T.; Asada, H.H. Mathematical analysis of oxygen transfer through polydimethylsiloxane membrane between double layers of cell culture channel and gas chamber in microfluidic oxygenator. Microfluid. Nanofluid. 2013, 15, 285–296. [Google Scholar] [CrossRef]
- Casciari, J.J.; Sotirchos, S.V.; Sutherland, R.M. Variations in tumor cell growth rates and metabolism with oxygen concentration, glucose concentration, and extracellular pH. J. Cell. Physiol. 1992, 151, 386–394. [Google Scholar] [CrossRef]
- Bertuzzi, A.; Fasano, A.; Gandolfi, A.; Sinisgalli, C. Necrotic core in EMT6/Ro tumour spheroids: Is it caused by an ATP deficit? J. Theor. Biol. 2010, 262, 142–150. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sorensen, R.; Novak, N. The use of Michaelis-Menten kinetics in cell biology and physiology teaching laboratories. Biochem. Educ. 1996, 24, 26–28. [Google Scholar] [CrossRef]
- Al-Ani, A.; Toms, D.; Kondro, D.; Thundathil, J.; Yu, Y.; Ungrin, M. Oxygenation in cell culture: Critical parameters for reproducibility are routinely not reported. PLoS ONE 2018, 13, e0204269. [Google Scholar] [CrossRef] [PubMed]
- Wenger, R.H.; Kurtcuoglu, V.; Scholz, C.C.; Marti, H.H.; Hoogewijs, D. Frequently asked questions in hypoxia research. Hypoxia 2015, 3, 35. [Google Scholar] [CrossRef]
- Hockel, M.; Vaupel, P. Tumor hypoxia: Definitions and current clinical, biologic, and molecular aspects. J. Natl. Cancer Inst. 2001, 93, 266–276. [Google Scholar] [CrossRef]
- Rousset, N.; Monet, F.; Gervais, T. Simulation-assisted design of microfluidic sample traps for optimal trapping and culture of non-adherent single cells, tissues, and spheroids. Sci. Rep. 2017, 7, 245. [Google Scholar] [CrossRef]
- Labouba, I.; Le Page, C.; Communal, L.; Kristessen, T.; You, X.; Péant, B.; Barrès, V.; Gannon, P.O.; Mes-Masson, A.-M.; Saad, F. Potential Cross-Talk between Alternative and Classical NF-κB Pathways in Prostate Cancer Tissues as Measured by a Multi-Staining Immunofluorescence Co-Localization Assay. PLoS ONE 2015, 10, e0131024. [Google Scholar] [CrossRef]
- Molina, M.F.; Fabre, T.; Cleret-Buhot, A.; Soucy, G.; Meunier, L.; Abdelnabi, M.N.; Belforte, N.; Turcotte, S.; Shoukry, N.H. Visualization, Quantification, and Mapping of Immune Cell Populations in the Tumor Microenvironment. JoVE J. Vis. Exp. 2020, 157, e60740. [Google Scholar]
- Adigun, R.; Basit, H.; Murray, J. Necrosis, cell (liquefactive, coagulative, caseous, fat, fibrinoid, and gangrenous). StatPearls. Available online: https://www.ncbi.nlm.nih.gov/books/NBK430935/ (accessed on 10 June 2021).
- Caruso, R.A.; Branca, G.; Fedele, F.; Irato, E.; Finocchiaro, G.; Parisi, A.; Ieni, A. Mechanisms of coagulative necrosis in malignant epithelial tumors. Oncol. Lett. 2014, 8, 1397–1402. [Google Scholar] [CrossRef]
- Tafreshi, N.K.; Lloyd, M.C.; Proemsey, J.B.; Bui, M.M.; Kim, J.; Gillies, R.J.; Morse, D.L. Evaluation of CAIX and CAXII expression in breast cancer at varied O 2 levels: CAIX is the superior surrogate imaging biomarker of tumor hypoxia. Mol. Imaging Biol. 2016, 18, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Tostain, J.; Li, G.; Gentil-Perret, A.; Gigante, M. Carbonic anhydrase 9 in clear cell renal cell carcinoma: A marker for diagnosis, prognosis and treatment. Eur. J. Cancer 2010, 46, 3141–3148. [Google Scholar] [CrossRef]
- Moon, E.J.; Brizel, D.M.; Chi, J.T.A.; Dewhirst, M.W. The potential role of intrinsic hypoxia markers as prognostic variables in cancer. Antioxid. Redox Signal. 2007, 9, 1237–1294. [Google Scholar] [CrossRef] [PubMed]
- Schornack, P.A.; Gillies, R.J. Contributions of cell metabolism and H+ diffusion to the acidic pH of tumors. Neoplasia 2003, 5, 135–145. [Google Scholar] [CrossRef]
- Rademakers, S.E.; Lok, J.; van der Kogel, A.J.; Bussink, J.; Kaanders, J.H. Metabolic markers in relation to hypoxia; staining patterns and colocalization of pimonidazole, HIF-1α, CAIX, LDH-5, GLUT-1, MCT1 and MCT4. BMC Cancer 2011, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.H.; Diaz-Gonzalez, J.A.; Russell, J.; Chen, Q.; Burgman, P.; Li, X.-F.; Ling, C.C. Detecting changes in tumor hypoxia with carbonic anhydrase IX and pimonidazole. Cancer Biol. Ther. 2007, 6, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.J.; Parker, C.A.; Stratford, I.J. Exogenous and Endogenous Markers of Tumour Oxygenation Status. In Oxygen Transport to Tissue XXVI; Advances in Experimental Medicine and biology; Springer: Boston, MA, USA, 2005; Volume 566, pp. 285–294. [Google Scholar]
- Horvath, P.; Aulner, N.; Bickle, M.; Davies, A.M.; Del Nery, E.; Ebner, D.; Montoya, M.C.; Östling, P.; Pietiäinen, V.; Price, L.S. Screening out irrelevant cell-based models of disease. Nat. Rev. Drug Discov. 2016, 15, 751–769. [Google Scholar] [CrossRef]
- Riley, A.; Green, V.; Cheah, R.; McKenzie, G.; Karsai, L.; England, J.; Greenman, J. A novel microfluidic device capable of maintaining functional thyroid carcinoma specimens ex vivo provides a new drug screening platform. BMC Cancer 2019, 19, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Iyer, N.G.; Tay, H.T.u.; Wu, Y.; Lim, T.K.; Zheng, L.; Song, I.C.; Kwoh, C.K.; Huynh, H.; Tan, P.O. Microarray profiling shows distinct differences between primary tumors and commonly used preclinical models in hepatocellular carcinoma. BMC Cancer 2015, 15, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Mak, I.W.; Evaniew, N.; Ghert, M. Lost in translation: Animal models and clinical trials in cancer treatment. Am. J. Transl. Res. 2014, 6, 114. [Google Scholar]
- Begley, C.G.; Ellis, L.M. Raise standards for preclinical cancer research. Nature 2012, 483, 531–533. [Google Scholar] [CrossRef] [PubMed]
- Gould, S.E.; Junttila, M.R.; de Sauvage, F.J. Translational value of mouse models in oncology drug development. Nat. Med. 2015, 21, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Ibarrola-Villava, M.; Cervantes, A.; Bardelli, A. Preclinical models for precision oncology. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2018, 1870, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Grimes, D.R.; Fletcher, A.G.; Partridge, M. Oxygen consumption dynamics in steady-state tumour models. R. Soc. Open Sci. 2014, 1, 140080. [Google Scholar] [CrossRef]
- Shannon, A.M.; Bouchier-Hayes, D.J.; Condron, C.M.; Toomey, D. Tumour hypoxia, chemotherapeutic resistance and hypoxia-related therapies. Cancer Treat. Rev. 2003, 29, 297–307. [Google Scholar] [CrossRef]
- Brown, J.M.; Wilson, W.R. Exploiting tumour hypoxia in cancer treatment. Nat. Rev. Cancer 2004, 4, 437. [Google Scholar] [CrossRef]
- Minchinton, A.I.; Tannock, I.F. Drug penetration in solid tumours. Nat. Rev. Cancer 2006, 6, 583–592. [Google Scholar] [CrossRef]
- Wijeratne, P.A.; Vavourakis, V. A quantitative in silico platform for simulating cytotoxic and nanoparticle drug delivery to solid tumours. Interface Focus 2019, 9, 20180063. [Google Scholar] [CrossRef]
- Muraro, M.G.; Muenst, S.; Mele, V.; Quagliata, L.; Iezzi, G.; Tzankov, A.; Weber, W.P.; Spagnoli, G.C.; Soysal, S.D. Ex-vivo assessment of drug response on breast cancer primary tissue with preserved microenvironments. Oncoimmunology 2017, 6, e1331798. [Google Scholar] [CrossRef] [PubMed]
- Richardson, A.; Schwerdtfeger, L.A.; Eaton, D.; Mclean, I.; Henry, C.S.; Tobet, S.A. A microfluidic organotypic device for culture of mammalian intestines ex vivo. Anal. Methods 2020, 12, 297–303. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dorrigiv, D.; Simeone, K.; Communal, L.; Kendall-Dupont, J.; St-Georges-Robillard, A.; Péant, B.; Carmona, E.; Mes-Masson, A.-M.; Gervais, T. Microdissected Tissue vs Tissue Slices—A Comparative Study of Tumor Explant Models Cultured On-Chip and Off-Chip. Cancers 2021, 13, 4208. https://doi.org/10.3390/cancers13164208
Dorrigiv D, Simeone K, Communal L, Kendall-Dupont J, St-Georges-Robillard A, Péant B, Carmona E, Mes-Masson A-M, Gervais T. Microdissected Tissue vs Tissue Slices—A Comparative Study of Tumor Explant Models Cultured On-Chip and Off-Chip. Cancers. 2021; 13(16):4208. https://doi.org/10.3390/cancers13164208
Chicago/Turabian StyleDorrigiv, Dina, Kayla Simeone, Laudine Communal, Jennifer Kendall-Dupont, Amélie St-Georges-Robillard, Benjamin Péant, Euridice Carmona, Anne-Marie Mes-Masson, and Thomas Gervais. 2021. "Microdissected Tissue vs Tissue Slices—A Comparative Study of Tumor Explant Models Cultured On-Chip and Off-Chip" Cancers 13, no. 16: 4208. https://doi.org/10.3390/cancers13164208
APA StyleDorrigiv, D., Simeone, K., Communal, L., Kendall-Dupont, J., St-Georges-Robillard, A., Péant, B., Carmona, E., Mes-Masson, A.-M., & Gervais, T. (2021). Microdissected Tissue vs Tissue Slices—A Comparative Study of Tumor Explant Models Cultured On-Chip and Off-Chip. Cancers, 13(16), 4208. https://doi.org/10.3390/cancers13164208