
TYK2 in Cancer Metastases: Genomic and Proteomic Discovery



Abstract
Simple Summary
Abstract
1. Genomics, Transcriptomics and Proteomics in Target Discovery: TYK2 in Cancer
1.1. TYK2 in Carcinomas and Sarcomas
1.2. TYK2 in Hematological Cancers
1.3. Other Actions of TYK2 in Cancer
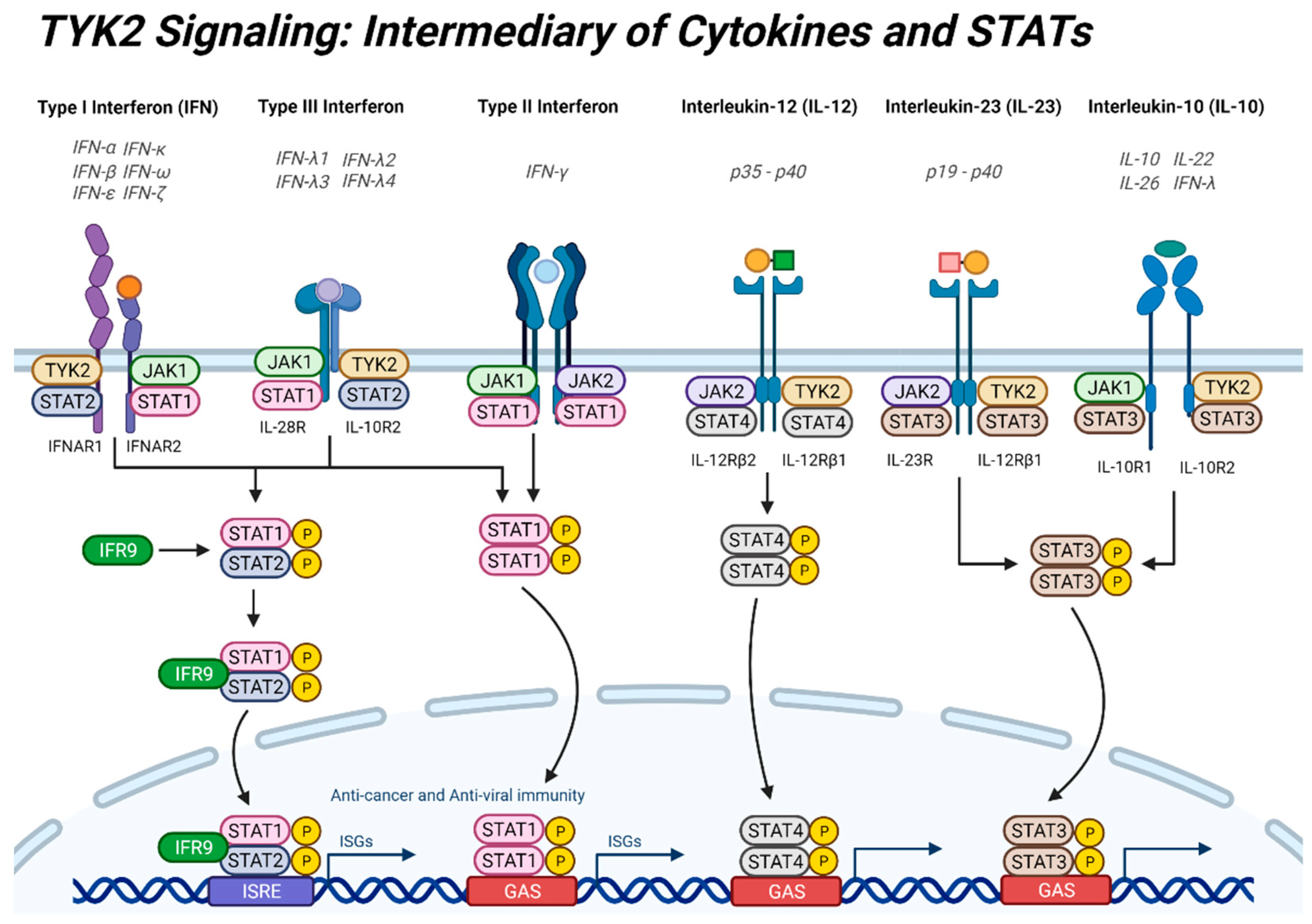
2. TYK2 Signaling: Intermediary of Cytokine Signaling and STATs
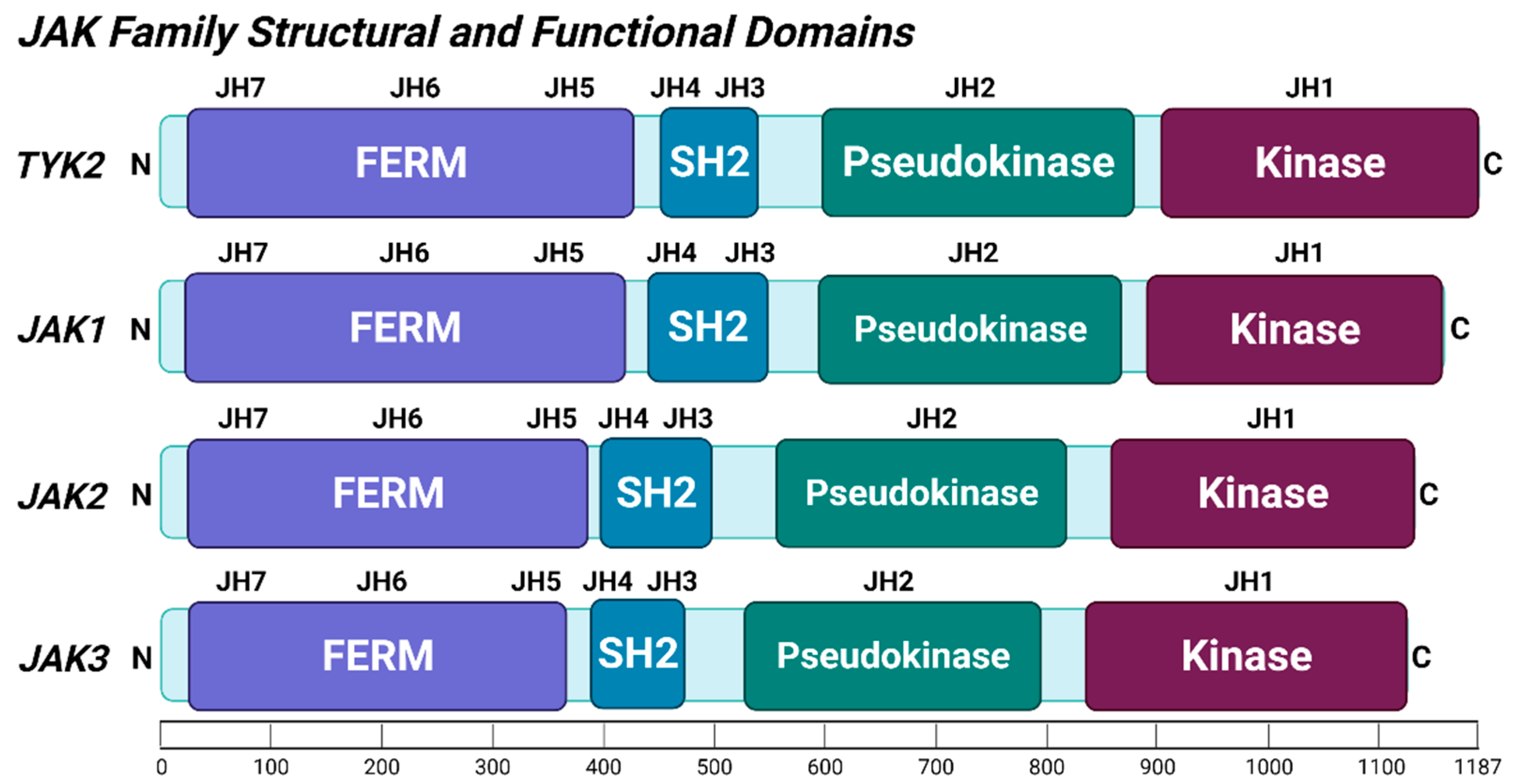
2.1. TYK2 Structure and Post-Translational Modifications
2.2. Interferons
2.3. Interleukins
3. Immune Modulatory and Inflammatory Effects of TYK2
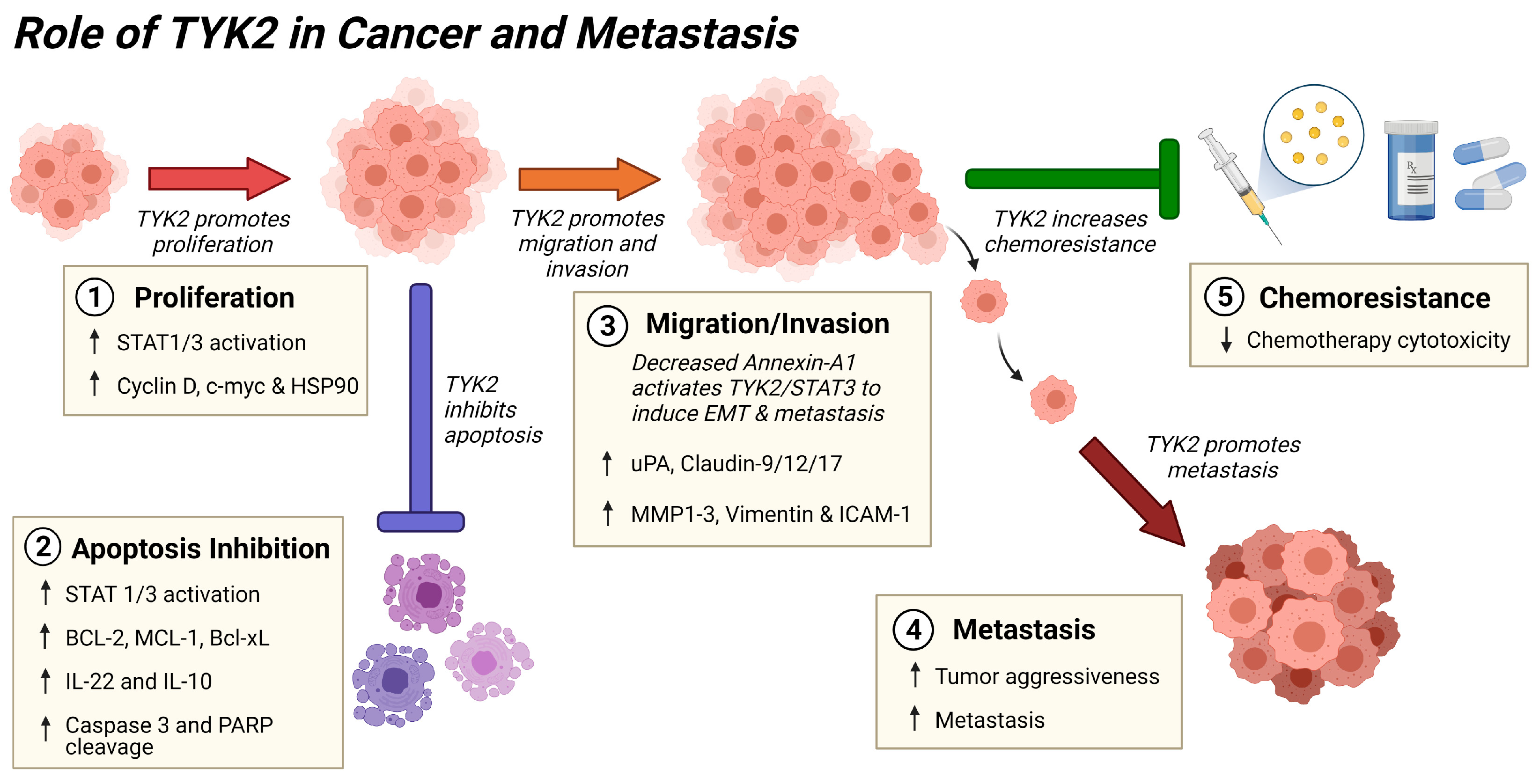
4. Pro-survival Actions of TYK2 and STATs in Cancer
5. Role of TYK2 in Metastasis
6. Pharmacologic Inhibition of TYK2 for Treatment of Cancer and Metastasis
7. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Berger, M.F.; Mardis, E.R. The emerging clinical relevance of genomics in cancer medicine. Nat. Rev. Clin. Oncol. 2018, 15, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Supplitt, S.; Karpinski, P.; Sasiadek, M.; Laczmanska, I. Current Achievements and Applications of Transcriptomics in Personalized Cancer Medicine. Int. J. Mol. Sci 2021, 22, 1422. [Google Scholar] [CrossRef] [PubMed]
- Shruthi, B.S.; Vinodhkumar, P. Selvamani. Proteomics: A new perspective for cancer. Adv. Biomed. Res. 2016, 5, 67. [Google Scholar] [CrossRef]
- Hammaren, H.M.; Virtanen, A.T.; Raivola, J.; Silvennoinen, O. The regulation of JAKs in cytokine signaling and its breakdown in disease. Cytokine 2019, 118, 48–63. [Google Scholar] [CrossRef] [PubMed]
- Firmbach-Kraft, I.; Byers, M.; Shows, T.; Dalla-Favera, R.; Krolewski, J.J. tyk2, prototype of a novel class of non-receptor tyrosine kinase genes. Oncogene 1990, 5, 1329–1336. [Google Scholar]
- Krolewski, J.J.; Lee, R.; Eddy, R.; Shows, T.B.; Dalla-Favera, R. Identification and chromosomal mapping of new human tyrosine kinase genes. Oncogene 1990, 5, 277–282. [Google Scholar] [PubMed]
- Ubel, C.; Mousset, S.; Trufa, D.; Sirbu, H.; Finotto, S. Establishing the role of tyrosine kinase 2 in cancer. Oncoimmunology 2013, 2, e22840. [Google Scholar] [CrossRef] [PubMed]
- Sohn, S.J.; Barrett, K.; Van Abbema, A.; Chang, C.; Kohli, P.B.; Kanda, H.; Smith, J.; Lai, Y.; Zhou, A.; Zhang, B.; et al. A Restricted Role for TYK2 Catalytic Activity in Human Cytokine Responses Revealed by Novel TYK2-Selective Inhibitors. J. Immunol. Res. 2013, 191, 2205–2216. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Holland, S.M.; Staudt, L.M. JAKs and STATs in immunity, immunodeficiency, and cancer. N. Engl. J. Med. 2013, 368, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Song, X.C.; Fu, G.; Yang, X.; Jiang, Z.; Wang, Y.; Zhou, G.W. Protein expression profiling of breast cancer cells by dissociable antibody microarray (DAMA) staining. Mol. Cell Proteomics 2008, 7, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Lv, J.; Yu, L.; Zhu, X.; Wu, J.; Zou, S.; Jiang, S. Proteomic identification of differentially-expressed proteins in squamous cervical cancer. Gynecol. Oncol. 2009, 112, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Organ, S.L.; Tong, J.; Taylor, P.; St-Germain, J.R.; Navab, R.; Moran, M.F.; Tsao, M.S. Quantitative phospho-proteomic profiling of hepatocyte growth factor (HGF)-MET signaling in colorectal cancer. J. Proteome Res. 2011, 10, 3200–3211. [Google Scholar] [CrossRef]
- Drake, J.M.; Graham, N.A.; Lee, J.K.; Stoyanova, T.; Faltermeier, C.M.; Sud, S.; Titz, B.; Huang, J.; Pienta, K.J.; Graeber, T.G.; et al. Metastatic castration-resistant prostate cancer reveals intrapatient similarity and interpatient heterogeneity of therapeutic kinase targets. Proc. Natl. Acad. Sci. USA 2013, 110, E4762–E4769. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Ding, L.; Yu, Y.; Li, W. JAK3 and TYK2 Serve as Prognostic Biomarkers and Are Associated with Immune Infiltration in Stomach Adenocarcinoma. Biomed. Res. Int. 2020, 2020, 7973568. [Google Scholar] [CrossRef]
- Ruhe, J.E.; Streit, S.; Hart, S.; Wong, C.H.; Specht, K.; Knyazev, P.; Knyazeva, T.; Tay, L.S.; Loo, H.L.; Foo, P.; et al. Genetic alterations in the tyrosine kinase transcriptome of human cancer cell lines. Cancer Res. 2007, 67, 11368–11376. [Google Scholar] [CrossRef]
- Sang, Q.X.; Man, Y.G.; Sung, Y.M.; Khamis, Z.I.; Zhang, L.; Lee, M.H.; Byers, S.W.; Sahab, Z.J. Non-receptor tyrosine kinase 2 reaches its lowest expression levels in human breast cancer during regional nodal metastasis. Clin. Exp. Metastasis 2012, 29, 143–153. [Google Scholar] [CrossRef]
- Crescenzo, R.; Abate, F.; Lasorsa, E.; Tabbo’, F.; Gaudiano, M.; Chiesa, N.; Di Giacomo, F.; Spaccarotella, E.; Barbarossa, L.; Ercole, E.; et al. Convergent Mutations and Kinase Fusions Lead to Oncogenic STAT3 Activation in Anaplastic Large Cell Lymphoma. Cancer Cell 2015, 27, 516–532. [Google Scholar] [CrossRef]
- Sanda, T.; Tyner, J.W.; Gutierrez, A.; Ngo, V.N.; Glover, J.; Chang, B.H.; Yost, A.; Ma, W.; Fleischman, A.G.; Zhou, W.; et al. TYK2-STAT1-BCL2 pathway dependence in T-cell acute lymphoblastic leukemia. Cancer Discov. 2013, 3, 564–577. [Google Scholar] [CrossRef]
- Velusamy, T.; Kiel, M.J.; Sahasrabuddhe, A.A.; Rolland, D.; Dixon, C.A.; Bailey, N.G.; Betz, B.L.; Brown, N.A.; Hristov, A.C.; Wilcox, R.A.; et al. A novel recurrent NPM1-TYK2 gene fusion in cutaneous CD30-positive lymphoproliferative disorders. Blood 2014, 124, 3768–3771. [Google Scholar] [CrossRef]
- Qin, W.; Godec, A.; Zhang, X.; Zhu, C.; Shao, J.; Tao, Y.; Bu, X.; Hirbe, A.C. TYK2 promotes malignant peripheral nerve sheath tumor progression through inhibition of cell death. Cancer Med. 2019, 8, 5232–5241. [Google Scholar] [CrossRef] [PubMed]
- Groisberg, R.; Hong, D.S.; Holla, V.; Janku, F.; Piha-Paul, S.; Ravi, V.; Benjamin, R.; Kumar Patel, S.; Somaiah, N.; Conley, A.; et al. Clinical genomic profiling to identify actionable alterations for investigational therapies in patients with diverse sarcomas. Oncotarget 2017, 8, 39254–39267. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Ren, L.; Feng, Q.; Zhu, D.; Chang, W.; He, G.; Ji, M.; Jian, M.; Lin, Q.; Yi, T.; et al. Differences in clinical characteristics and mutational pattern between synchronous and metachronous colorectal liver metastases. Cancer Manag. Res. 2018, 10, 2871–2881. [Google Scholar] [CrossRef]
- Waanders, E.; Scheijen, B.; Jongmans, M.C.; Venselaar, H.; van Reijmersdal, S.V.; van Dijk, A.H.; Pastorczak, A.; Weren, R.D.; van der Schoot, C.E.; van de Vorst, M.; et al. Germline activating TYK2 mutations in pediatric patients with two primary acute lymphoblastic leukemia occurrences. Leukemia 2017, 31, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Diets, I.J.; Waanders, E.; Ligtenberg, M.J.; van Bladel, D.A.G.; Kamping, E.J.; Hoogerbrugge, P.M.; Hopman, S.; Olderode-Berends, M.J.; Gerkes, E.H.; Koolen, D.A.; et al. High Yield of Pathogenic Germline Mutations Causative or Likely Causative of the Cancer Phenotype in Selected Children with Cancer. Clin. Cancer Res. 2018, 24, 1594–1603. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.G.; Li, Y.; Payne-Turner, D.; Harvey, R.C.; Yang, Y.L.; Pei, D.; McCastlain, K.; Ding, L.; Lu, C.; Song, G.; et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N. Engl. J. Med. 2014, 371, 1005–1015. [Google Scholar] [CrossRef]
- Tasian, S.K.; Loh, M.L.; Hunger, S.P. Philadelphia chromosome-like acute lymphoblastic leukemia. Blood 2017, 130, 2064–2072. [Google Scholar] [CrossRef] [PubMed]
- Tomasson, M.H.; Xiang, Z.; Walgren, R.; Zhao, Y.; Kasai, Y.; Miner, T.; Ries, R.E.; Lubman, O.; Fremont, D.H.; McLellan, M.D.; et al. Somatic mutations and germline sequence variants in the expressed tyrosine kinase genes of patients with de novo acute myeloid leukemia. Blood 2008, 111, 4797–4808. [Google Scholar] [CrossRef] [PubMed]
- Turrubiartes-Martinez, E.; Bodega-Mayor, I.; Delgado-Wicke, P.; Molina-Jimenez, F.; Casique-Aguirre, D.; Gonzalez-Andrade, M.; Rapado, I.; Camos, M.; Diaz-de-Heredia, C.; Barragan, E.; et al. TYK2 Variants in B-Acute Lymphoblastic Leukaemia. Genes 2020, 11, 1434. [Google Scholar] [CrossRef]
- Pairo-Castineira, E.; Clohisey, S.; Klaric, L.; Bretherick, A.D.; Rawlik, K.; Pasko, D.; Walker, S.; Parkinson, N.; Fourman, M.H.; Russell, C.D.; et al. Genetic mechanisms of critical illness in COVID-19. Nature 2021, 591, 92. [Google Scholar] [CrossRef]
- Márquez, A.; Kerick, M.; Zhernakova, A.; Gutierrez-Achury, J.; Chen, W.M.; Onengut-Gumuscu, S.; González-Álvaro, I.; Rodriguez-Rodriguez, L.; Rios-Fernández, R.; González-Gay, M.A.; et al. Meta-analysis of Immunochip data of four autoimmune diseases reveals novel single-disease and cross-phenotype associations. Genome Med. 2018, 10, 97. [Google Scholar] [CrossRef]
- Dendrou, C.A.; Cortes, A.; Shipman, L.; Evans, H.G.; Attfield, K.E.; Jostins, L.; Barber, T.; Kaur, G.; Kuttikkatte, S.B.; Leach, O.A.; et al. Resolving TYK2 locus genotype-to-phenotype differences in autoimmunity. Sci. Transl. Med. 2016, 8, 363ra149. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.R.; Cheng, L.; Gillooly, K.M.; Strnad, J.; Zupa-Fernandez, A.; Catlett, I.M.; Zhang, Y.; Heimrich, E.M.; McIntyre, K.W.; Cunningham, M.D.; et al. Autoimmune pathways in mice and humans are blocked by pharmacological stabilization of the TYK2 pseudokinase domain. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Couturier, N.; Bucciarelli, F.; Nurtdinov, R.N.; Debouverie, M.; Lebrun-Frenay, C.; Defer, G.; Moreau, T.; Confavreux, C.; Vukusic, S.; Cournu-Rebeix, I.; et al. Tyrosine kinase 2 variant influences T lymphocyte polarization and multiple sclerosis susceptibility. Brain 2011, 134, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Rotival, M.; Patin, E.; Michel, F.; Pellegrini, S. Two common disease-associated TYK2 variants impact exon splicing and TYK2 dosage. PLoS ONE 2020, 15, e0225289. [Google Scholar] [CrossRef] [PubMed]
- Diogo, D.; Bastarache, L.; Liao, K.P.; Graham, R.R.; Fulton, R.S.; Greenberg, J.D.; Eyre, S.; Bowes, J.; Cui, J.; Lee, A.; et al. TYK2 protein-coding variants protect against rheumatoid arthritis and autoimmunity, with no evidence of major pleiotropic effects on non-autoimmune complex traits. PLoS ONE 2015, 10, e0122271. [Google Scholar] [CrossRef]
- Hirbe, A.C.; Kaushal, M.; Sharma, M.K.; Dahiya, S.; Pekmezci, M.; Perry, A.; Gutmann, D.H. Clinical genomic profiling identifies TYK2 mutation and overexpression in patients with neurofibromatosis type 1-associated malignant peripheral nerve sheath tumors. Cancer 2017, 123, 1194–1201. [Google Scholar] [CrossRef]
- Silvennoinen, O.; Hubbard, S.R. Molecular insights into regulation of JAK2 in myeloproliferative neoplasms. Blood 2015, 125, 3388–3392. [Google Scholar] [CrossRef]
- Vainchenker, W.; Constantinescu, S.N. JAK/STAT signaling in hematological malignancies. Oncogene 2013, 32, 2601–2613. [Google Scholar] [CrossRef]
- Li, Z.; Gakovic, M.; Ragimbeau, J.; Eloranta, M.L.; Ronnblom, L.; Michel, F.; Pellegrini, S. Two rare disease-associated Tyk2 variants are catalytically impaired but signaling competent. J. Immunol. 2013, 190, 2335–2344. [Google Scholar] [CrossRef]
- Yokota, T.; Kanakura, Y. Genetic abnormalities associated with acute lymphoblastic leukemia. Cancer Sci. 2016, 107, 721–725. [Google Scholar] [CrossRef]
- Prutsch, N.; Gurnhofer, E.; Suske, T.; Liang, H.C.; Schlederer, M.; Roos, S.; Wu, L.C.; Simonitsch-Klupp, I.; Alvarez-Hernandez, A.; Kornauth, C.; et al. Dependency on the TYK2/STAT1/MCL1 axis in anaplastic large cell lymphoma. Leukemia 2019, 33, 696–709. [Google Scholar] [CrossRef]
- Zhang, Q.; Sturgill, J.L.; Kmieciak, M.; Szczepanek, K.; Derecka, M.; Koebel, C.; Graham, L.J.; Dai, Y.; Chen, S.; Grant, S.; et al. The role of Tyk2 in regulation of breast cancer growth. J. Interferon Cytokine Res. 2011, 31, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Stoiber, D.; Kovacic, B.; Schuster, C.; Schellack, C.; Karaghiosoff, M.; Kreibich, R.; Weisz, E.; Artwohl, M.; Kleine, O.C.; Muller, M.; et al. TYK2 is a key regulator of the surveillance of B lymphoid tumors. J. Clin. Investig. 2004, 114, 1650–1658. [Google Scholar] [CrossRef]
- Karjalainen, A.; Shoebridge, S.; Krunic, M.; Simonovic, N.; Tebb, G.; Macho-Maschler, S.; Strobl, B.; Muller, M. TYK2 in Tumor Immunosurveillance. Cancers 2020, 12, 150. [Google Scholar] [CrossRef] [PubMed]
- Kaminker, J.S.; Zhang, Y.; Waugh, A.; Haverty, P.M.; Peters, B.; Sebisanovic, D.; Stinson, J.; Forrest, W.F.; Bazan, J.F.; Seshagiri, S.; et al. Distinguishing cancer-associated missense mutations from common polymorphisms. Cancer Res. 2007, 67, 465–473. [Google Scholar] [CrossRef]
- Ghoreschi, K.; Laurence, A.; O’Shea, J.J. Janus kinases in immune cell signaling. Immunol. Rev. 2009, 228, 273–287. [Google Scholar] [CrossRef]
- Strobl, B.; Stoiber, D.; Sexl, V.; Mueller, M. Tyrosine kinase 2 (TYK2) in cytokine signalling and host immunity. Front. Biosci. 2011, 16, 3214–3232. [Google Scholar] [CrossRef]
- Garrido-Trigo, A.; Salas, A. Molecular Structure and Function of Janus Kinases: Implications for the Development of Inhibitors. J. Crohn’s Colitis 2019, 14, S713–S724. [Google Scholar] [CrossRef]
- Ragimbeau, J.; Dondi, E.; Vasserot, A.; Romero, P.; Uze, G.; Pellegrini, S. The receptor interaction region of Tyk2 contains a motif required for its nuclear localization. J. Biol. Chem. 2001, 276, 30812–30818. [Google Scholar] [CrossRef]
- Chrencik, J.E.; Patny, A.; Leung, I.K.; Korniski, B.; Emmons, T.L.; Hall, T.; Weinberg, R.A.; Gormley, J.A.; Williams, J.M.; Day, J.E.; et al. Structural and thermodynamic characterization of the TYK2 and JAK3 kinase domains in complex with CP-690550 and CMP-6. J. Mol. Biol. 2010, 400, 413–433. [Google Scholar] [CrossRef]
- Nicholas, C.; Lesinski, B.G. The Jak-STAT Signal Transduction Pathway in Melanoma. In Breakthroughs in Melanoma Research; IntechOpen: London, UK, 2011. [Google Scholar]
- Vainchenker, W.; Leroy, E.; Gilles, L.; Marty, C.; Plo, I.; Constantinescu, S.N. JAK inhibitors for the treatment of myeloproliferative neoplasms and other disorders. F1000Research 2018, 7, 82. [Google Scholar] [CrossRef]
- Leitner, N.R.; Witalisz-Siepracka, A.; Strobl, B.; Muller, M. Tyrosine kinase 2—Surveillant of tumours and bona fide oncogene. Cytokine 2017, 89, 209–218. [Google Scholar] [CrossRef][Green Version]
- Wallweber, H.J.; Tam, C.; Franke, Y.; Starovasnik, M.A.; Lupardus, P.J. Structural basis of recognition of interferon-alpha receptor by tyrosine kinase 2. Nat. Struct. Mol. Biol. 2014, 21, 443–448. [Google Scholar] [CrossRef]
- Ferrao, R.; Lupardus, P.J. The Janus Kinase (JAK) FERM and SH2 Domains: Bringing Specificity to JAK-Receptor Interactions. Front. Endocrinol. 2017, 8. [Google Scholar] [CrossRef]
- Woss, K.; Simonovic, N.; Strobl, B.; Macho-Maschler, S.; Muller, M. TYK2: An Upstream Kinase of STATs in Cancer. Cancers 2019, 11, 1728. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Hu, P.; Quinn, D.F.; Wang, Y.K. Phosphotyrosine proteomic study of interferon alpha signaling pathway using a combination of immunoprecipitation and immobilized metal affinity chromatography. Mol. Cell Proteomics 2005, 4, 721–730. [Google Scholar] [CrossRef]
- Babon, J.J.; Nicola, N.A. The biology and mechanism of action of suppressor of cytokine signaling 3. Growth Factors 2012, 30, 207–219. [Google Scholar] [CrossRef]
- Yoshimura, A.; Naka, T.; Kubo, M. SOCS proteins, cytokine signalling and immune regulation. Nat. Rev. Immunol. 2007, 7, 454–465. [Google Scholar] [CrossRef]
- Linossi, E.M.; Nicholson, S.E. Kinase inhibition, competitive binding and proteasomal degradation: Resolving the molecular function of the suppressor of cytokine signaling (SOCS) proteins. Immunol. Rev. 2015, 266, 123–133. [Google Scholar] [CrossRef]
- Babon, J.J.; Kershaw, N.J.; Murphy, J.M.; Varghese, L.N.; Laktyushin, A.; Young, S.N.; Lucet, I.S.; Norton, R.S.; Nicola, N.A. Suppression of Cytokine Signaling by SOCS3: Characterization of the Mode of Inhibition and the Basis of Its Specificity. Immunity 2012, 36, 239–250. [Google Scholar] [CrossRef]
- David, M.; Chen, H.E.; Goelz, S.; Larner, A.C.; Neel, B.G. Differential regulation of the alpha/beta interferon-stimulated Jak/Stat pathway by the SH2 domain-containing tyrosine phosphatase SHPTP1. Mol. Cell. Biol. 1995, 15, 7050–7058. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Han, X.; Tang, Y.; Wu, Y.; Qu, B.; Shen, N. miR-744 enhances type I interferon signaling pathway by targeting PTP1B in primary human renal mesangial cells. Sci. Rep. 2015, 5, 12987. [Google Scholar] [CrossRef] [PubMed]
- Irie-Sasaki, J.; Sasaki, T.; Matsumoto, W.; Opavsky, A.; Cheng, M.; Welstead, G.; Griffiths, E.; Krawczyk, C.; Richardson, C.D.; Aitken, K.; et al. CD45 is a JAK phosphatase and negatively regulates cytokine receptor signalling. Nature 2001, 409, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Akahane, K.; Sanda, T.; Mansour, M.R.; Radimerski, T.; DeAngelo, D.J.; Weinstock, D.M.; Look, A.T. HSP90 inhibition leads to degradation of the TYK2 kinase and apoptotic cell death in T-cell acute lymphoblastic leukemia. Leukemia 2016, 30, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Velazquez, L.; Fellous, M.; Stark, G.R.; Pellegrini, S. A protein tyrosine kinase in the interferon alpha/beta signaling pathway. Cell 1992, 70, 313–322. [Google Scholar] [CrossRef]
- Hervas-Stubbs, S.; Perez-Gracia, J.L.; Rouzaut, A.; Sanmamed, M.F.; Le Bon, A.; Melero, I. Direct effects of type I interferons on cells of the immune system. Clin. Cancer Res. 2011, 17, 2619–2627. [Google Scholar] [CrossRef] [PubMed]
- Karaghiosoff, M.; Neubauer, H.; Lassnig, C.; Kovarik, P.; Schindler, H.; Pircher, H.; McCoy, B.; Bogdan, C.; Decker, T.; Brem, G.; et al. Partial impairment of cytokine responses in Tyk2-deficient mice. Immunity 2000, 13, 549–560. [Google Scholar] [CrossRef]
- Alspach, E.; Lussier, D.M.; Schreiber, R.D. Interferon gamma and Its Important Roles in Promoting and Inhibiting Spontaneous and Therapeutic Cancer Immunity. Csh. Perspect. Biol. 2019, 11. [Google Scholar] [CrossRef]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immun. 2015, 15, 87–103. [Google Scholar] [CrossRef]
- Dunn, G.P.; Koebel, C.M.; Schreiber, R.D. Interferons, immunity and cancer immunoediting. Nat. Rev. Immun. 2006, 6, 836–848. [Google Scholar] [CrossRef]
- Barrat, F.J.; Lu, T.T. Role of type I interferons and innate immunity in systemic sclerosis: Unbalanced activities on distinct cell types? Curr. Opin. Rheumatol. 2019, 31, 569–575. [Google Scholar] [CrossRef]
- Kretschmer, S.; Lee-Kirsch, M.A. Type I interferon-mediated autoinflammation and autoimmunity. Curr. Opin. Immunol. 2017, 49, 96–102. [Google Scholar] [CrossRef]
- Crow, M.K.; Ronnblom, L. Type I interferons in host defence and inflammatory diseases. Lupus Sci. Med. 2019, 6. [Google Scholar] [CrossRef]
- Saraiva, M.; O’Garra, A. The regulation of IL-10 production by immune cells. Nat. Rev. Immun. 2010, 10, 170–181. [Google Scholar] [CrossRef]
- Ouyang, W.; O’Garra, A. IL-10 Family Cytokines IL-10 and IL-22: From Basic Science to Clinical Translation. Immunity 2019, 50, 871–891. [Google Scholar] [CrossRef] [PubMed]
- Wack, A.; Terczynska-Dyla, E.; Hartmann, R. Guarding the frontiers: The biology of type III interferons. Nat. Immunol. 2015, 16, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Zanoni, I.; Granucci, F.; Broggi, A. Interferon (IFN)-lambda Takes the Helm: Immunomodulatory Roles of Type III IFNs. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the Eye of the Cytokine Storm. Microbiol. Mol. Biol. R. 2012, 76, 16–32. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Darnell, J.E. STATs: Transcriptional control and biological impact. Nat. Rev. Mol. Cell. Biol. 2002, 3, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Kortylewski, M.; Xin, H.; Kujawski, M.; Lee, H.; Liu, Y.; Harris, T.; Drake, C.; Pardoll, D.; Yu, H. Regulation of the IL-23 and IL-12 Balance by Stat3 Signaling in the Tumor Microenvironment. Cancer Cell 2009, 15, 114–123. [Google Scholar] [CrossRef]
- Tait Wojno, E.D.; Hunter, C.A.; Stumhofer, J.S. The Immunobiology of the Interleukin-12 Family: Room for Discovery. Immunity 2019, 50, 851–870. [Google Scholar] [CrossRef]
- Walter, M.R. The molecular basis of IL-10 function: From receptor structure to the onset of signaling. Curr. Top. Microbiol. Immunol. 2014, 380, 191–212. [Google Scholar] [CrossRef]
- Donnelly, R.P.; Sheikh, F.; Kotenko, S.V.; Dickensheets, H. The expanded family of class II cytokines that share the IL-10 receptor-2 (IL-10R2) chain. J. Leukoc. Biol. 2004, 76, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef]
- Simma, O.; Zebedin, E.; Neugebauer, N.; Schellack, C.; Pilz, A.; Chang-Rodriguez, S.; Lingnau, K.; Weisz, E.; Putz, E.M.; Pickl, W.F.; et al. Identification of an Indispensable Role for Tyrosine Kinase 2 in CTL-Mediated Tumor Surveillance. Cancer Res. 2009, 69, 203–211. [Google Scholar] [CrossRef][Green Version]
- Prchal-Murphy, M.; Witalisz-Siepracka, A.; Bednarik, K.T.; Putz, E.M.; Gotthardt, D.; Meissl, K.; Sexl, V.; Muller, M.; Strobl, B. In vivo tumor surveillance by NK cells requires TYK2 but not TYK2 kinase activity. Oncoimmunology 2015, 4. [Google Scholar] [CrossRef]
- Kreins, A.Y.; Ciancanelli, M.J.; Okada, S.; Kong, X.F.; Ramirez-Alejo, N.; Kilic, S.S.; El Baghdadi, J.; Nonoyama, S.; Mahdaviani, S.A.; Ailal, F.; et al. Human TYK2 deficiency: Mycobacterial and viral infections without hyper-IgE syndrome. J. Exp. Med. 2015, 212, 1641–1662. [Google Scholar] [CrossRef]
- Holland, S.M.; DeLeo, F.R.; Elloumi, H.Z.; Hsu, A.P.; Uzel, G.; Brodsky, N.; Freeman, A.F.; Demidowich, A.; Davis, J.; Turner, M.L.; et al. STAT3 mutations in the hyper-IgE syndrome. N. Engl. J. Med. 2007, 357, 1608–1619. [Google Scholar] [CrossRef]
- Freeman, A.F.; Holland, S.M. The hyper-IgE syndromes. Immunol. Allergy Clin. 2008, 28, 277–291. [Google Scholar] [CrossRef]
- Freeman, A.F.; Holland, S.M. Clinical Manifestations, Etiology, and Pathogenesis of the Hyper-IgE Syndromes. Pediatr. Res. 2009, 65, 32r–37r. [Google Scholar] [CrossRef]
- Rael, E.L.; Marshall, R.T.; McClain, J.J. The Hyper-IgE Syndromes: Lessons in Nature, From Bench to Bedside. World Allergy Organ. J. 2012, 5, 79–87. [Google Scholar] [CrossRef]
- Woellner, C.; Schaffer, A.A.; Pluck, J.M.; Renner, E.D.; Knebel, C.; Holland, S.M.; Plebani, A.; Grimbacher, B. The hyper IgE syndrome and mutations in TYK2. Immunity 2007, 26, 535. [Google Scholar] [CrossRef]
- Minegishi, Y.; Saito, M.; Morio, T.; Watanabe, K.; Agematsu, K.; Tsuchiya, S.; Takada, H.; Hara, T.; Kawamura, N.; Ariga, T.; et al. Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved in innate and acquired immunity. Immunity 2006, 25, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, S.; Kaiser-Labusch, P.; Bank, J.; Ammann, S.; Kolb-Kokocinski, A.; Edelbusch, C.; Omran, H.; Ehl, S. Tyrosine kinase 2 is not limiting human antiviral type III interferon responses. Eur. J. Immunol. 2016, 46, 2639–2649. [Google Scholar] [CrossRef]
- Sarrafzadeh, S.A.; Mahloojirad, M.; Casanova, J.L.; Badalzadeh, M.; Bustamante, J.; Boisson-Dupuis, S.; Pourpak, Z.; Nourizadeh, M.; Moin, M. A New Patient with Inherited TYK2 Deficiency. J. Clin. Immunol 2020, 40, 232–235. [Google Scholar] [CrossRef]
- Shaw, M.H.; Boyartchuk, V.; Wong, S.; Karaghiosoff, M.; Ragimbeau, J.; Pellegrini, S.; Muller, M.; Dietrich, W.F.; Yap, G.S. A natural mutation in the Tyk2 pseudokinase domain underlies altered susceptibility of B10.Q/J mice to infection and autoimmunity. Proc. Natl. Acad. Sci. USA 2003, 100, 11594–11599. [Google Scholar] [CrossRef]
- Shimoda, K.; Kato, K.; Aoki, K.; Matsuda, T.; Miyamoto, A.; Shibamori, M.; Yamashita, M.; Numata, A.; Takase, K.; Kobayashi, S.; et al. Tyk2 plays a restricted role in IFN alpha signaling, although it is required for IL-12-mediated T cell function. Immunity 2000, 13, 561–571. [Google Scholar] [CrossRef]
- Luo, W.; Li, Y.X.; Jiang, L.J.; Chen, Q.; Wang, T.; Ye, D.W. Targeting JAK-STAT Signaling to Control Cytokine Release Syndrome in COVID-19. Trends Pharmacol. Sci. 2020, 41, 531–543. [Google Scholar] [CrossRef]
- Kalil, A.C.; Patterson, T.F.; Mehta, A.K.; Tomashek, K.M.; Wolfe, C.R.; Ghazaryan, V.; Marconi, V.C.; Ruiz-Palacios, G.M.; Hsieh, L.; Kline, S.; et al. Baricitinib plus Remdesivir for Hospitalized Adults with COVID-19. N. Engl. J. Med. 2021, 384, 795–807. [Google Scholar] [CrossRef]
- Berg, J.; Zscheppang, K.; Fatykhova, D.; Tönnies, M.; Bauer, T.T.; Schneider, P.; Neudecker, J.; Rückert, J.C.; Eggeling, S.; Schimek, M.; et al. Tyk2 as a target for immune regulation in human viral/bacterial pneumonia. Eur. Respir. J. 2017, 50. [Google Scholar] [CrossRef] [PubMed]
- Subbarao, S.A. Cancer vs. SARS-CoV-2 induced inflammation, overlapping functions, and pharmacological targeting. Inflammopharmacology 2021, 29, 343–366. [Google Scholar] [CrossRef] [PubMed]
- Gorman, J.A.; Hundhausen, C.; Kinsman, M.; Arkatkar, T.; Allenspach, E.J.; Clough, C.; West, S.E.; Thomas, K.; Eken, A.; Khim, S.; et al. The TYK2-P1104A Autoimmune Protective Variant Limits Coordinate Signals Required to Generate Specialized T Cell Subsets. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Owen, K.L.; Brockwell, N.K.; Parker, B.S. JAK-STAT Signaling: A Double-Edged Sword of Immune Regulation and Cancer Progression. Cancers 2019, 11, 2002. [Google Scholar] [CrossRef]
- Jia, X.; Huang, C.; Hu, Y.; Wu, Q.; Liu, F.; Nie, W.; Chen, H.; Li, X.; Dong, Z.; Liu, K. Cirsiliol targets tyrosine kinase 2 to inhibit esophageal squamous cell carcinoma growth in vitro and in vivo. J. Exp. Clin. Cancer Res. 2021, 40, 105. [Google Scholar] [CrossRef]
- Carmo, C.R.; Lyons-Lewis, J.; Seckl, M.J.; Costa-Pereira, A.P. A novel requirement for Janus kinases as mediators of drug resistance induced by fibroblast growth factor-2 in human cancer cells. PLoS ONE 2011, 6, e19861. [Google Scholar] [CrossRef]
- Herrmann, A.; Lahtz, C.; Nagao, T.; Song, J.Y.; Chan, W.C.; Lee, H.; Yue, C.; Look, T.; Mulfarth, R.; Li, W.; et al. CTLA4 Promotes Tyk2-STAT3-Dependent B-cell Oncogenicity. Cancer Res. 2017, 77, 5118–5128. [Google Scholar] [CrossRef]
- Sen, B.; Saigal, B.; Parikh, N.; Gallick, G.; Johnson, F.M. Sustained Src inhibition results in signal transducer and activator of transcription 3 (STAT3) activation and cancer cell survival via altered Janus-activated kinase-STAT3 binding. Cancer Res. 2009, 69, 1958–1965. [Google Scholar] [CrossRef]
- Shahmarvand, N.; Nagy, A.; Shahryari, J.; Ohgami, R.S. Mutations in the signal transducer and activator of transcription family of genes in cancer. Cancer Sci. 2018, 109, 926–933. [Google Scholar] [CrossRef]
- Verhoeven, Y.; Tilborghs, S.; Jacobs, J.; De Waele, J.; Quatannens, D.; Deben, C.; Prenen, H.; Pauwels, P.; Trinh, X.B.; Wouters, A.; et al. The potential and controversy of targeting STAT family members in cancer. Semin. Cancer Biol. 2020, 60, 41–56. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Z.Y. STAT1 in Cancer: Friend or Foe? Discov. Med. 2017, 24, 19–29. [Google Scholar]
- Carpenter, R.L.; Lo, H.W. STAT3 Target Genes Relevant to Human Cancers. Cancers 2014, 6, 897–925. [Google Scholar] [CrossRef] [PubMed]
- Ide, H.; Nakagawa, T.; Terado, Y.; Kamiyama, Y.; Muto, S.; Horie, S. Tyk2 expression and its signaling enhances the invasiveness of prostate cancer cells. Biochem. Biophys. Res. Commun. 2008, 369, 292–296. [Google Scholar] [CrossRef]
- Sun, L.; Feng, L.; Cui, J. Increased expression of claudin-17 promotes a malignant phenotype in hepatocyte via Tyk2/Stat3 signaling and is associated with poor prognosis in patients with hepatocellular carcinoma. Diagn. Pathol. 2018, 13, 72. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Feng, L.; Cui, J. Increased expression of claudin-12 promotes the metastatic phenotype of human bronchial epithelial cells and is associated with poor prognosis in lung squamous cell carcinoma. Exp. Ther. Med. 2019, 17, 165–174. [Google Scholar] [CrossRef]
- Liu, H.; Wang, M.; Liang, N.; Guan, L. Claudin-9 enhances the metastatic potential of hepatocytes via Tyk2/Stat3 signaling. Turk. J. Gastroenterol. 2019, 30, 722–731. [Google Scholar] [CrossRef]
- Maschler, S.; Gebeshuber, C.A.; Wiedemann, E.M.; Alacakaptan, M.; Schreiber, M.; Custic, I.; Beug, H. Annexin A1 attenuates EMT and metastatic potential in breast cancer. EMBO Mol. Med. 2010, 2, 401–414. [Google Scholar] [CrossRef]
- Marroqui, L.; Dos Santos, R.S.; Floyel, T.; Grieco, F.A.; Santin, I.; Op de Beeck, A.; Marselli, L.; Marchetti, P.; Pociot, F.; Eizirik, D.L. TYK2, a Candidate Gene for Type 1 Diabetes, Modulates Apoptosis and the Innate Immune Response in Human Pancreatic beta-Cells. Diabetes 2015, 64, 3808–3817. [Google Scholar] [CrossRef]
- Gamero, A.M.; Potla, R.; Wegrzyn, J.; Szelag, M.; Edling, A.E.; Shimoda, K.; Link, D.C.; Dulak, J.; Baker, D.P.; Tanabe, Y.; et al. Activation of Tyk2 and Stat3 is required for the apoptotic actions of interferon-beta in primary pro-B cells. J. Biol. Chem. 2006, 281, 16238–16244. [Google Scholar] [CrossRef]
- Shimoda, H.K.; Shide, K.; Kameda, T.; Matsunaga, T.; Shimoda, K. Tyrosine kinase 2 interacts with the proapoptotic protein Siva-1 and augments its apoptotic functions. Biochem. Biophys. Res. Commun. 2010, 400, 252–257. [Google Scholar] [CrossRef]
- Rani, M.R.; Pandalai, S.; Shrock, J.; Almasan, A.; Ransohoff, R.M. Requirement of catalytically active Tyk2 and accessory signals for the induction of TRAIL mRNA by IFN-beta. J. Interferon Cytokine Res. 2007, 27, 767–779. [Google Scholar] [CrossRef]
- Wan, J.; Fu, A.K.; Ip, F.C.; Ng, H.K.; Hugon, J.; Page, G.; Wang, J.H.; Lai, K.O.; Wu, Z.; Ip, N.Y. Tyk2/STAT3 signaling mediates beta-amyloid-induced neuronal cell death: Implications in Alzheimer’s disease. J. Neurosci. 2010, 30, 6873–6881. [Google Scholar] [CrossRef]
- Mittal, V. Epithelial Mesenchymal Transition in Tumor Metastasis. Annu. Rev. Pathol. 2018, 13, 395–412. [Google Scholar] [CrossRef]
- Dave, B.; Mittal, V.; Tan, N.M.; Chang, J.C. Epithelial-mesenchymal transition, cancer stem cells and treatment resistance. Breast Cancer Res. 2012, 14, 202. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Nagase, H.; Visse, R.; Murphy, G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res. 2006, 69, 562–573. [Google Scholar] [CrossRef]
- He, A.; Zhang, R.; Wang, J.; Huang, Z.; Liao, W.; Li, Y.; Wang, C.; Yang, J.; Feng, Q.; Wu, L. TYK2 is a prognostic biomarker and associated with immune infiltration in the lung adenocarcinoma microenvironment. Asia Pac. J. Clin. Oncol. 2021. [Google Scholar] [CrossRef]
- Taylor, P.C. Clinical efficacy of launched JAK inhibitors in rheumatoid arthritis. Rheumatology (Oxford) 2019, 58, i17–i26. [Google Scholar] [CrossRef]
- Agrawal, M.; Kim, E.S.; Colombel, J.F. JAK Inhibitors Safety in Ulcerative Colitis: Practical Implications. J. Crohns Colitis 2020, 14, S755–S760. [Google Scholar] [CrossRef]
- Strand, V.; Kremer, J.M.; Li, Z.G.; Hall, S.; Fleischmann, R.M.; Genovese, M.C.; Martin-Mola, E.; Isaacs, J.; Gruben, D.; Wallenstein, G.; et al. Tofacitinib (CP-690,550) in Combination with Traditional Disease-Modifying Anti-Rheumatic Drugs: Phase 3 Study Patient-Reported Outcomes in Patients with Active Rheumatoid Arthritis and An Inadequate Response to Disease-Modifying Anti-Rheumatic Drugs. Arthritis Rheum. 2011, 63, S1032. [Google Scholar]
- Van der Heijde, D.; Tanaka, Y.; Fleischmann, R.; Keystone, E.C.; Kremer, J.M.; Zerbini, C.A.F.; Cardiel, M.; Cohen, S.B.; Nash, P.T.; Song, Y.W.; et al. Tofacitinib (CP-690,550), An Oral Janus Kinase Inhibitor, in Combination with Methotrexate Reduced the Progression of Structural Damage in Patients with Rheumatoid Arthritis: A 24-Month Phase 3 Study. Arthritis Rheum. 2011, 63, S1017–S1018. [Google Scholar]
- Harrison, C.; Kiladjian, J.-J.; Al-Ali, H.K.; Gisslinger, H.; Waltzman, R.; Stalbovskaya, V.; McQuitty, M.; Hunter, D.S.; Levy, R.; Knoops, L.; et al. JAK Inhibition with Ruxolitinib versus Best Available Therapy for Myelofibrosis. N. Engl. J. Med. 2012, 366, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Georgeon, S.; Moser, R.; Moore, D.J.; Caflisch, A.; Hantschel, O. Specificity and mechanism-of-action of the JAK2 tyrosine kinase inhibitors ruxolitinib and SAR302503 (TG101348). Leukemia 2014, 28, 404–407. [Google Scholar] [CrossRef]
- Xue, E.; Lorentino, F.; Pavesi, F.; Assanelli, A.; Peccatori, J.; Bernardi, M.; Corti, C.; Ciceri, F.; Lupo Stanghellini, M.T. Ruxolitinib for chronic steroid-refractory graft versus host disease: A single center experience. Leuk. Res. 2021, 109, 106642. [Google Scholar] [CrossRef] [PubMed]
- Sarmiento, M.; Rojas, P.; Jerez, J.; Bertín, P.; Campbell, J.; García, M.J.; Pereira, J.; Triantafilo, N.; Ocqueteau, M. Ruxolitinib for Severe COVID-19-Related Hyperinflammation in Nonresponders to Steroids. Acta Haematol. 2021, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Vannucchi, A.M.; Griesshammer, M.; Masszi, T.; Durrant, S.; Passamonti, F.; Harrison, C.N.; Pane, F.; Zachee, P.; Hino, M.; et al. Ruxolitinib in polycythemia vera: Follow-up from the RESPONSE trial. J. Clin. Oncol. 2015, 33. [Google Scholar] [CrossRef]
- Zhang, X.H.; Zhang, Y.; Qiao, W.Z.; Zhang, J.; Qi, Z.G. Baricitinib, a drug with potential effect to prevent SARS-CoV-2 from entering target cells and control cytokine storm induced by COVID-19. Int. Immunopharmacol. 2020, 86. [Google Scholar] [CrossRef]
- Biggioggero, M.; Becciolini, A.; Crotti, C.; Agape, E.; Favalli, E.G. Upadacitinib and filgotinib: The role of JAK1 selective inhibition in the treatment of rheumatoid arthritis. Drugs Context 2019, 8, 212595. [Google Scholar] [CrossRef]
- Harrison, C.N.; Schaap, N.; Vannucchi, A.M.; Kiladjian, J.-J.; Jourdan, E.; Silver, R.T.; Schouten, H.C.; Passamonti, F.; Zweegman, S.; Talpaz, M.; et al. Fedratinib in patients with myelofibrosis previously treated with ruxolitinib: An updated analysis of the JAKARTA2 study using stringent criteria for ruxolitinib failure. Am. J. Hematol. 2020, 95, 594–603. [Google Scholar] [CrossRef]
- Liu, C.; Lin, J.; Langevine, C.; Smith, D.; Li, J.; Tokarski, J.S.; Khan, J.; Ruzanov, M.; Strnad, J.; Zupa-Fernandez, A.; et al. Discovery of BMS-986202: A Clinical Tyk2 Inhibitor that Binds to Tyk2 JH2. J. Med. Chem. 2021, 64, 677–694. [Google Scholar] [CrossRef]
- Harrison, C.N.; Vannucchi, A.M.; Platzbecker, U.; Cervantes, F.; Gupta, V.; Lavie, D.; Passamonti, F.; Winton, E.F.; Dong, H.; Kawashima, J.; et al. Momelotinib versus best available therapy in patients with myelofibrosis previously treated with ruxolitinib (SIMPLIFY 2): A randomised, open-label, phase 3 trial. Lancet Haematol. 2018, 5, E73–E81. [Google Scholar] [CrossRef]
- Coffey, G.; Betz, A.; DeGuzman, F.; Pak, Y.; Inagaki, M.; Baker, D.C.; Hollenbach, S.J.; Pandey, A.; Sinha, U. The Novel Kinase Inhibitor PRT062070 (Cerdulatinib) Demonstrates Efficacy in Models of Autoimmunity and B-Cell Cancer. J. Pharmacol. Exp. Ther. 2014, 351, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Hamlin, P.; Strickland, D.K.; Pandey, A.; Coffey, G.; Leeds, J.M.; Levy, G.G.; Curnutte, J.T.; Wagner-Johnston, N.; Flinn, I.W. A Phase I Open-Label, Multi-Dose Escalation Study of the Dual Syk/Jak Inhibitor PRT062070 (Cerdulatinib) in Patients with Relapsed/Refractory B Cell Malignancies. Blood 2014, 124. [Google Scholar] [CrossRef]
- Gerstenberger, B.S.; Ambler, C.; Arnold, E.P.; Banker, M.E.; Brown, M.F.; Clark, J.D.; Dermenci, A.; Dowty, M.E.; Fensome, A.; Fish, S.; et al. Discovery of Tyrosine Kinase 2 (TYK2) Inhibitor (PF-06826647) for the Treatment of Autoimmune Diseases. J. Med. Chem. 2020, 63, 13561–13577. [Google Scholar] [CrossRef] [PubMed]
- Qureshy, Z.; Johnson, D.E.; Grandis, J.R. Targeting the JAK/STAT pathway in solid tumors. JCMT 2020, 6, 27. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, M.; Puig, L.; Torres, T. JAK Inhibitors for Treatment of Psoriasis: Focus on Selective TYK2 Inhibitors. Drugs 2020, 80, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Foley, P.; Gooderham, M.; Gordon, K.; Thaci, D.; Kundu, S.; Kisa, R.; Wei, L.; Banerjee, S. An oral, selective TYK2 inhibitor, deucravacitinib, in patients with moderate-to-severe plaque psoriasis and baseline PASI 15. Australas J. Dermatol. 2021, 62, 55. [Google Scholar]
- Liu, X.; Tan, F.; Liang, C. Preclinical Characterization of Tll018, a Novel, Highly Potent and Selective Jak1/Tyk2 Inhibitor for Treating Autoimmune Diseases. Ann. Rheum. Dis. 2020, 79, 248. [Google Scholar] [CrossRef]
- Wrobleski, S.T.; Moslin, R.; Lin, S.; Zhang, Y.; Spergel, S.; Kempson, J.; Tokarski, J.S.; Strnad, J.; Zupa-Fernandez, A.; Cheng, L.; et al. Highly Selective Inhibition of Tyrosine Kinase 2 (TYK2) for the Treatment of Autoimmune Diseases: Discovery of the Allosteric Inhibitor BMS-986165. J. Med. Chem. 2019, 62, 8973–8995. [Google Scholar] [CrossRef]
- Liosi, M.E.; Krimmer, S.G.; Newton, A.S.; Dawson, T.K.; Puleo, D.E.; Cutrona, K.J.; Suzuki, Y.; Schlessinger, J.; Jorgensen, W.L. Selective Janus Kinase 2 (JAK2) Pseudokinase Ligands with a Diaminotriazole Core. J. Med. Chem. 2020, 63, 5324–5340. [Google Scholar] [CrossRef]
- Okay, M.; Haznedaroglu, I.C. Protein Kinases in Hematological Disorders. Adv. Exp. Med. Biol. 2021, 1275, 383–393. [Google Scholar]
- Papp, K.; Gordon, K.; Thaçi, D.; Morita, A.; Gooderham, M.; Foley, P.; Girgis, I.G.; Kundu, S.; Banerjee, S. Phase 2 Trial of Selective Tyrosine Kinase 2 Inhibition in Psoriasis. N. Engl. J. Med. 2018, 379, 1313–1321. [Google Scholar] [CrossRef]
- Akahane, K.; Li, Z.; Etchin, J.; Berezovskaya, A.; Gjini, E.; Masse, C.E.; Miao, W.; Rocnik, J.; Kapeller, R.; Greenwood, J.R.; et al. Anti-leukaemic activity of the TYK2 selective inhibitor NDI-031301 in T-cell acute lymphoblastic leukaemia. Br. J. Haematol. 2017, 177, 271–282. [Google Scholar] [CrossRef]
- Reader, J.; Williams, N.; Bojdo, J.; Worthington, J.; Mitchell, T. Abstract C086: Immunotherapeutic effects of the TYK2 inhibitor SAR-20351 in syngeneic tumor models. Mol. Cancer Ther. 2019, 18, C086. [Google Scholar] [CrossRef]
- Greenfield, G.; McPherson, S.; Mills, K.; McMullin, M.F. The ruxolitinib effect: Understanding how molecular pathogenesis and epigenetic dysregulation impact therapeutic efficacy in myeloproliferative neoplasms. J. Transl. Med. 2018, 16, 360. [Google Scholar] [CrossRef]
- Tavakoli Shirazi, P.; Eadie, L.N.; Page, E.C.; Heatley, S.L.; Bruning, J.B.; White, D.L. Constitutive JAK/STAT signaling is the primary mechanism of resistance to JAKi in TYK2-rearranged acute lymphoblastic leukemia. Cancer Lett. 2021, 512, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Cervantes, F.; Vannucchi, A.M.; Kiladjian, J.J. Three-year efficacy, safety, and survival findings from COMFORT-II, a phase 3 study comparing ruxolitinib with best available therapy for myelofibrosis. Blood 2016, 128, 3013. [Google Scholar] [CrossRef] [PubMed]
- Cervantes, F.; Pereira, A. Does ruxolitinib prolong the survival of patients with myelofibrosis? Blood 2017, 129, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Andreoli, A.; Verger, E.; Robin, M.; Raffoux, E.; Zini, J.M.; Rousselot, P.; Socie, G.; Rea, D.; Parquet, N.; Giraudier, S.; et al. Clinical Resistance To Ruxolitinib Is More Frequent In Patients Without MPN-Associated Mutations and Is Rarely Due To Mutations In The JAK2 Kinase Drug-Binding Domain. Blood 2013, 122. [Google Scholar] [CrossRef]
- Koppikar, P.; Bhagwat, N.; Kilpivaara, O.; Manshouri, T.; Adli, M.; Hricik, T.; Liu, F.; Saunders, L.M.; Mullally, A.; Abdel-Wahab, O.; et al. Heterodimeric JAK-STAT activation as a mechanism of persistence to JAK2 inhibitor therapy. Nature 2012, 489, 155–U222. [Google Scholar] [CrossRef]
- Bhagwat, N.; Levine, R.L.; Koppikar, P. Sensitivity and resistance of JAK2 inhibitors to myeloproliferative neoplasms. Int. J. Hematol. 2013, 97, 695–702. [Google Scholar] [CrossRef]
- Hornakova, T.; Springuel, L.; Devreux, J.; Dusa, A.; Constantinescu, S.N.; Knoops, L.; Renauld, J.C. Oncogenic JAK1 and JAK2-activating mutations resistant to ATP-competitive inhibitors. Haematol. Hematol. J. 2011, 96, 845–853. [Google Scholar] [CrossRef][Green Version]
- Winter, P.S.; Sarosiek, K.A.; Lin, K.H.; Meggendorfer, M.; Schnittger, S.; Letai, A.; Wood, K.C. RAS signaling promotes resistance to JAK inhibitors by suppressing BAD-mediated apoptosis. Sci. Signal. 2014, 7, ra122. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.L.; Koppikar, P.; Marubayashi, S.; Bhagwat, N.; Taldone, T.; Park, C.Y.; Chiosis, G. Combination Therapy Using JAK2 and HSP90 Inhibitors Increased Efficacy in Myelofibrosis in Vivo. Blood 2012, 120. [Google Scholar] [CrossRef]
- Chakraborty, S.N.; Leng, X.; Perazzona, B.; Sun, X.; Lin, Y.H.; Arlinghaus, R.B. Combination of JAK2 and HSP90 inhibitors: An effective therapeutic option in drug-resistant chronic myelogenous leukemia. Genes Cancer 2016, 7, 201–208. [Google Scholar] [CrossRef][Green Version]
- Harrison, C.N.; Schaap, N.; Mesa, R.A. Management of myelofibrosis after ruxolitinib failure. Ann. Hematol. 2020, 99, 1177–1191. [Google Scholar] [CrossRef]
- Giordano, G.; Parcesepe, P.; D’Andrea, M.R.; Coppola, L.; Di Raimo, T.; Remo, A.; Manfrin, E.; Fiorini, C.; Scarpa, A.; Amoreo, C.A.; et al. JAK/Stat5-mediated subtype-specific lymphocyte antigen 6 complex, locus G6D (LY6G6D) expression drives mismatch repair proficient colorectal cancer. J. Exp. Clin. Cancer Res. 2019, 38, 28. [Google Scholar] [CrossRef]
- Moscow, J.A.; Fojo, T.; Schilsky, R.L. The evidence framework for precision cancer medicine. Nat. Rev. Clin. Oncol. 2018, 15, 183–192. [Google Scholar] [CrossRef]
- Su, M.; Zhang, Z.; Zhou, L.; Han, C.; Huang, C.H.; Nice, E.C. Proteomics, Personalized Medicine and Cancer. Cancers 2021, 13, 2512. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Study Type | Type of Screen for Discovery | SNP, Variant, Fusion Protein | Disease | Tyk2 Activity | Effect on Disease | Ref. |
|---|---|---|---|---|---|---|
| Proteomic | DAMA staining screen | nd | Breast Cancer | High TYK2 levels | nd | [10] |
| Mass Spectrometry | nd | Squamous Cervical Cancer | High TYK2 levels | nd | [11] | |
| Phospho-tyrosine Mass Spectrometry | nd | Colorectal Cancer | High p-TYK2 levels with HGF stimulation | ↑ Proliferation | [12] | |
| Mass Spectrometry | Splice variant (E971fsX67) | Brain Hematopoietic cancer | Inactivating TYK2 mutations | ↑ Disease risk | [15] | |
| Mass Spectrometry | nd | Breast cancer metastasis to regional lymph nodes | Low TYK2 levels | nd | [16] | |
| Phospho-tyrosine Mass Spectrometry | nd | Metastatic Prostate Cancer | High p-TYK2 levels | nd | [13] | |
| Transcriptomic | RNA-Seq | nd | Stomach Adenocarcinoma | High TYK2 levels | Prognostic Biomarker | [14] |
| RNAseq Screen | NFκB2-TYK2 PABPC4-TYK2 fusion proteins | ACLC | Constitutively Active TYK2 | ↑ Proliferation ↑ Disease risk | [17] | |
| RNAi screen | Point Mutation in FERM, JH2, Kinase Domain | T-ALL | GOF mutations | ↑ Proliferation ↓ Apoptosis ↑ Disease risk | [18] | |
| Whole Transcriptome Sequencing | NPMI-TYK2 fusion protein | T-cell lymphoma | Constitutively active TYK2 | ↑ Disease risk | [19] | |
| Genomic | NGS | rs34536443 (P1104A) | MPNST sarcomas | High TYK2 levels | ↑ Proliferation ↓ Apoptosis ↑ Disease risk | [20] |
| NGS | TYK2 mutation | Sarcomas | nd | ↑ Disease risk | [21] | |
| NGS | rs2304256 (V362F) | Colorectal cancer metastasis | nd | ↑ Disease risk | [22] | |
| Whole Exome Sequencing | G761V P760L (in JH2 domain) | Pediatric ALL | Constitutively active TYK2 | ↑ Disease risk | [23,24] | |
| NGS | Multiple kinase activating mutations MYB-TYK2 | Ph-like ALL | High TYK2 activity | ↑ Disease risk | [25,26] | |
| High Throughput Sequencing | rs2304255 (G363S) | AML | nd | nd | [27] | |
| Amplicon-based NGS | TYK2 variants with lower TYK2 gene expression | B-ALL | Catalytic LOF | ↑ Disease risk | [28] | |
| Genomic | GWAS | rs74956615 | COVID-19 | High TYK2 levels | ↑ Severe disease risk | [29] |
| Immunochip meta-analysis | rs74956615 | RA, T1D, SSc | nd | ↑ Disease risk | [30] | |
| GWAS, Immunochip | rs34536443 (P1104A) | MS, IBD, AS, psoriasis | nd | Protective | [31,32,33] | |
| Immunochip | rs9797854 rs12720356 (I684S) | MS, IBD, AS, psoriasis | nd | Variable depending on disease | [31,32,33] | |
| eQTL analysis | rs2304256 (V362F) rs12720270 | SLE, T1D, psoriasis | Increased TYK2 binding due to exon 8 | Protective | [34] | |
| Immunochip & Exomechip genotyping, Exon Sequencing | rs34536443 (P1104A) rs35018800 (A928V) rs12720356 (I684S) | RA, SLE, IBD | Protective | [35] |
| Drug Name (Generic/Commercial) | Targeted JAKs/TYK2 | Targeted Domains | FDA-Approval/Disease | References |
|---|---|---|---|---|
| Ruxolitinib (Jakafi) | JAK1/2 TYK2 JAK3 | JH1 |
| [133,134,135,136,137] |
| Baricitinib (Olumiant) | JAK1/2 | JH1 |
| [129,138] |
| Tofacitinib (Xeljanz) | JAK1/3 JAK2 TYK2 | JH1 |
| [129] |
| Upadacintinib (Rinvoq) | JAK 1/2 JAK3 TYK2 | JH1 |
| [129,139] |
| Fedratinib (Inrebic) | JAK 2 JAK 1/3 | Substrate binding site |
| [140] |
| Deucravacitinib(BMS-986165) | TYK2 | JH2 |
| [141] |
| Momelotinib | JAK 1/2 | ATP-Competitive inhibitor |
| [142] |
| Filgotinib (Jyseleca) | JAK 1 | JH1 |
| [139] |
| Cerdulatinib | JAK1 JAK2 JAK3 TYK2 | SYK/JAK Kinase Inhibitor |
| [143,144] |
| Tyk2-IN-8, compound 10 (PF-06826647) | JAK 1/2 TYK2 | JH1 |
| [145] |
| AZD1480 | JAK2 JAK3 TYK2 JAK1 | ATP-Competitive inhibitor |
| [146] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borcherding, D.C.; He, K.; Amin, N.V.; Hirbe, A.C. TYK2 in Cancer Metastases: Genomic and Proteomic Discovery. Cancers 2021, 13, 4171. https://doi.org/10.3390/cancers13164171
Borcherding DC, He K, Amin NV, Hirbe AC. TYK2 in Cancer Metastases: Genomic and Proteomic Discovery. Cancers. 2021; 13(16):4171. https://doi.org/10.3390/cancers13164171
Chicago/Turabian StyleBorcherding, Dana C., Kevin He, Neha V. Amin, and Angela C. Hirbe. 2021. "TYK2 in Cancer Metastases: Genomic and Proteomic Discovery" Cancers 13, no. 16: 4171. https://doi.org/10.3390/cancers13164171
APA StyleBorcherding, D. C., He, K., Amin, N. V., & Hirbe, A. C. (2021). TYK2 in Cancer Metastases: Genomic and Proteomic Discovery. Cancers, 13(16), 4171. https://doi.org/10.3390/cancers13164171