
Citrate-Coated Superparamagnetic Iron Oxide Nanoparticles Enable a Stable Non-Spilling Loading of T Cells and Their Magnetic Accumulation

 , ,
, ,
Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of SPIONs and Physicochemical Characterization
2.3. Isolation of T Cells from Human Whole Blood
2.4. Determination of T Cell Purity
2.5. Determination of Cell Viability
2.6. Colloidal Stability of SPIONs in Various Media and Cellular Nanoparticle Uptake
2.7. Comparison of Stimulated and Non-Stimulated T Cells
2.8. Determination of Nanoparticle Uptake
2.9. SPION Exchange with Non-Loaded T Cells
2.10. Transmission Electronic Microscopy
2.11. Magnetic Accumulation of SPION-Loaded T Cells under Dynamic Conditions
2.12. T Cell Activation and Proliferation after Polyclonal Stimulation
2.13. Data Analysis and Statistics
3. Results
3.1. Physicochemical Characterization of SPIONs
3.2. Colloidal Stability of SPIONs in Cell Culture Medium
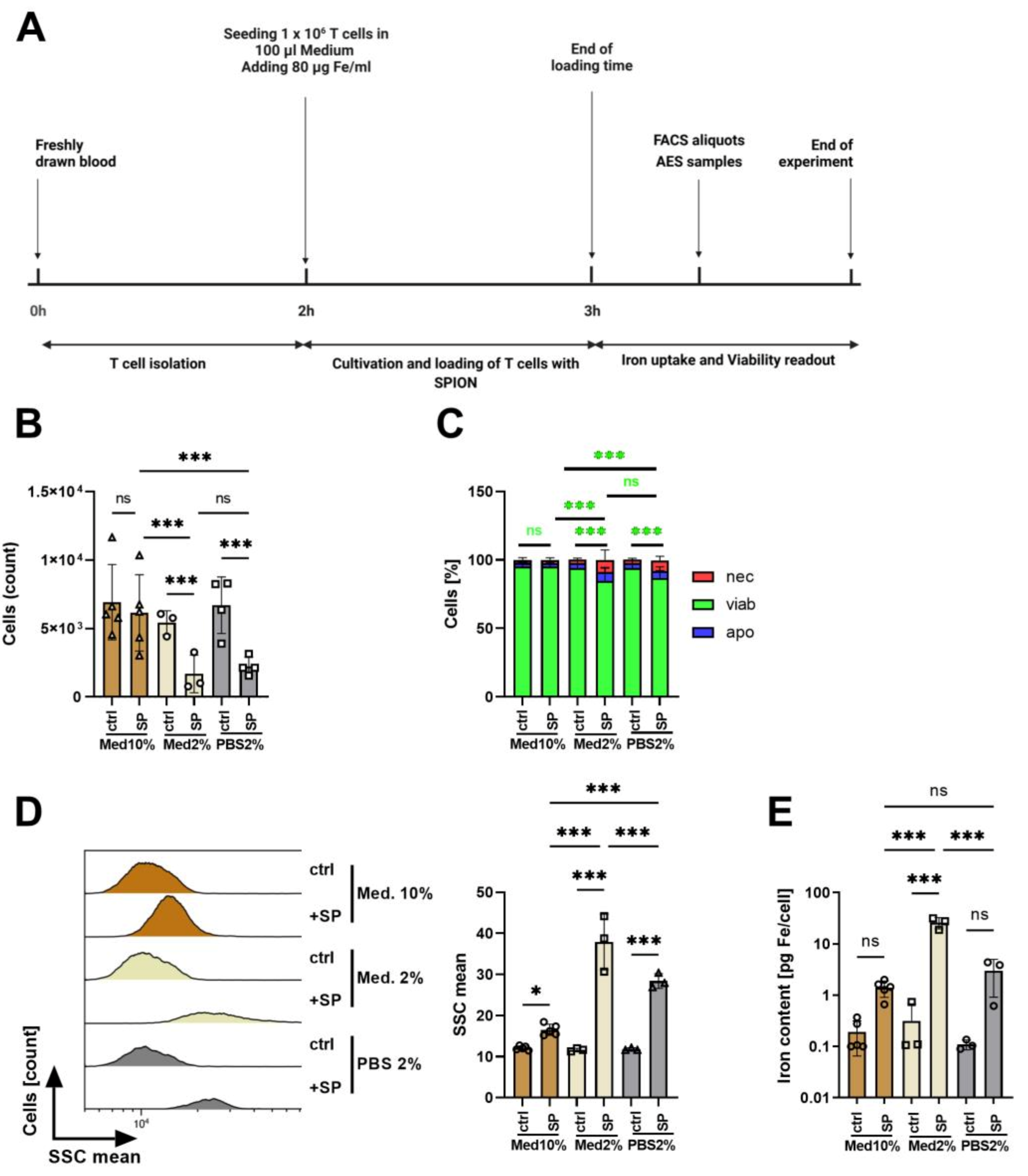
3.3. Influence of Medium Composition on SPION-Loading of T Cells
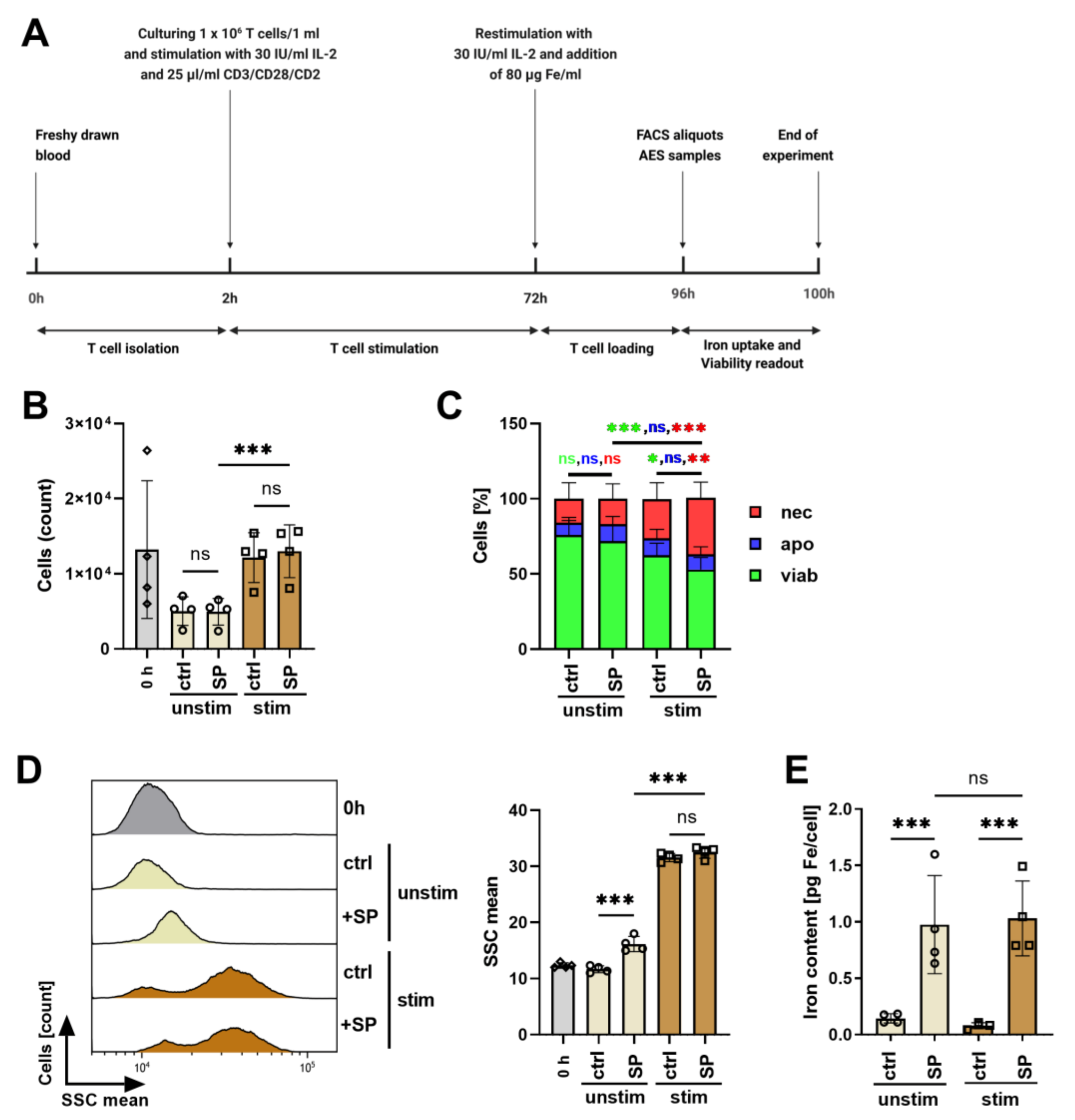
3.4. Influence of Polyclonal Stimulation on SPION-Loading of T Cells
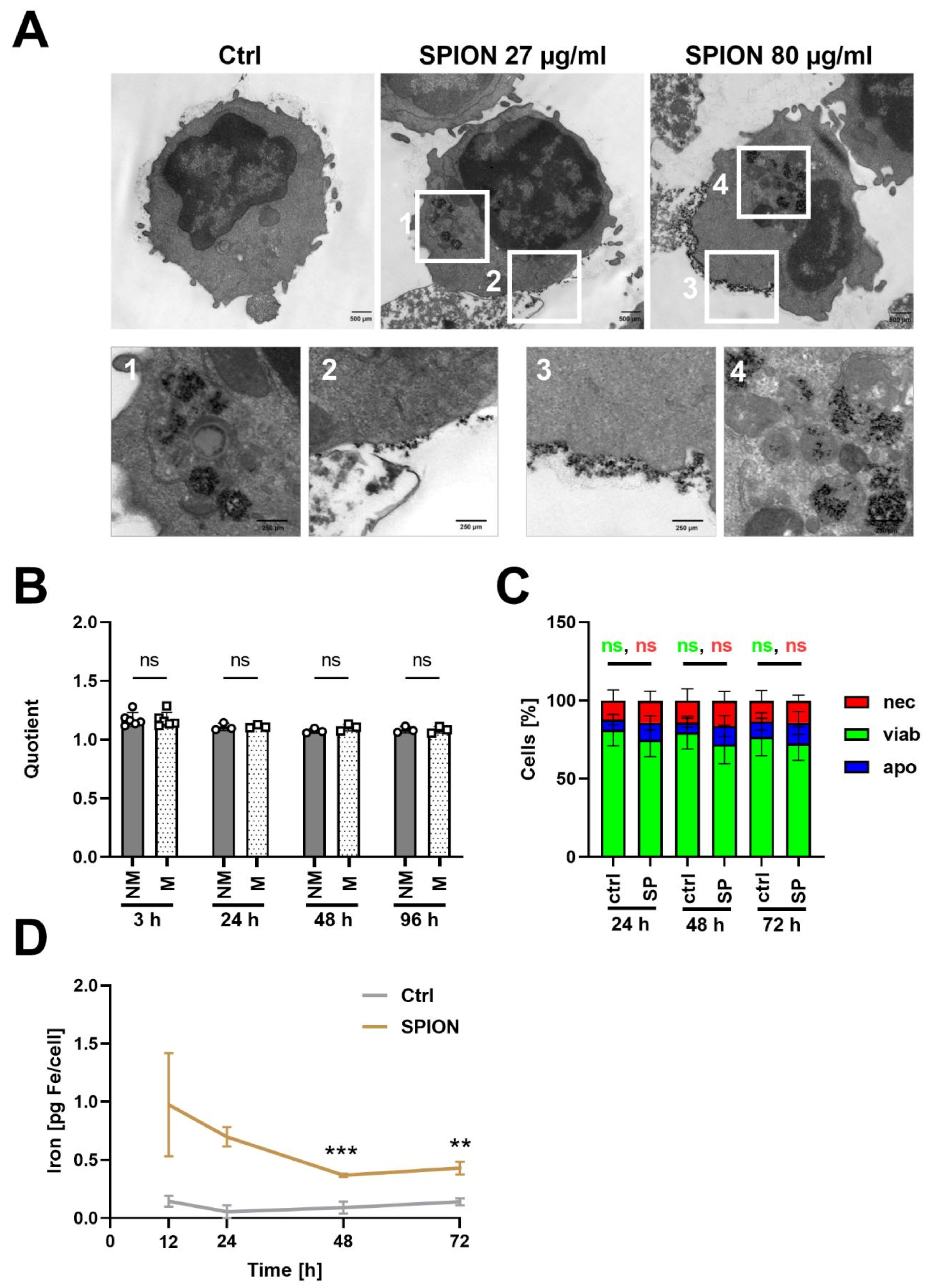
3.5. SPION-Loaded T Cells Do Not Exchange Nanoparticles with Non-Loaded T Cells
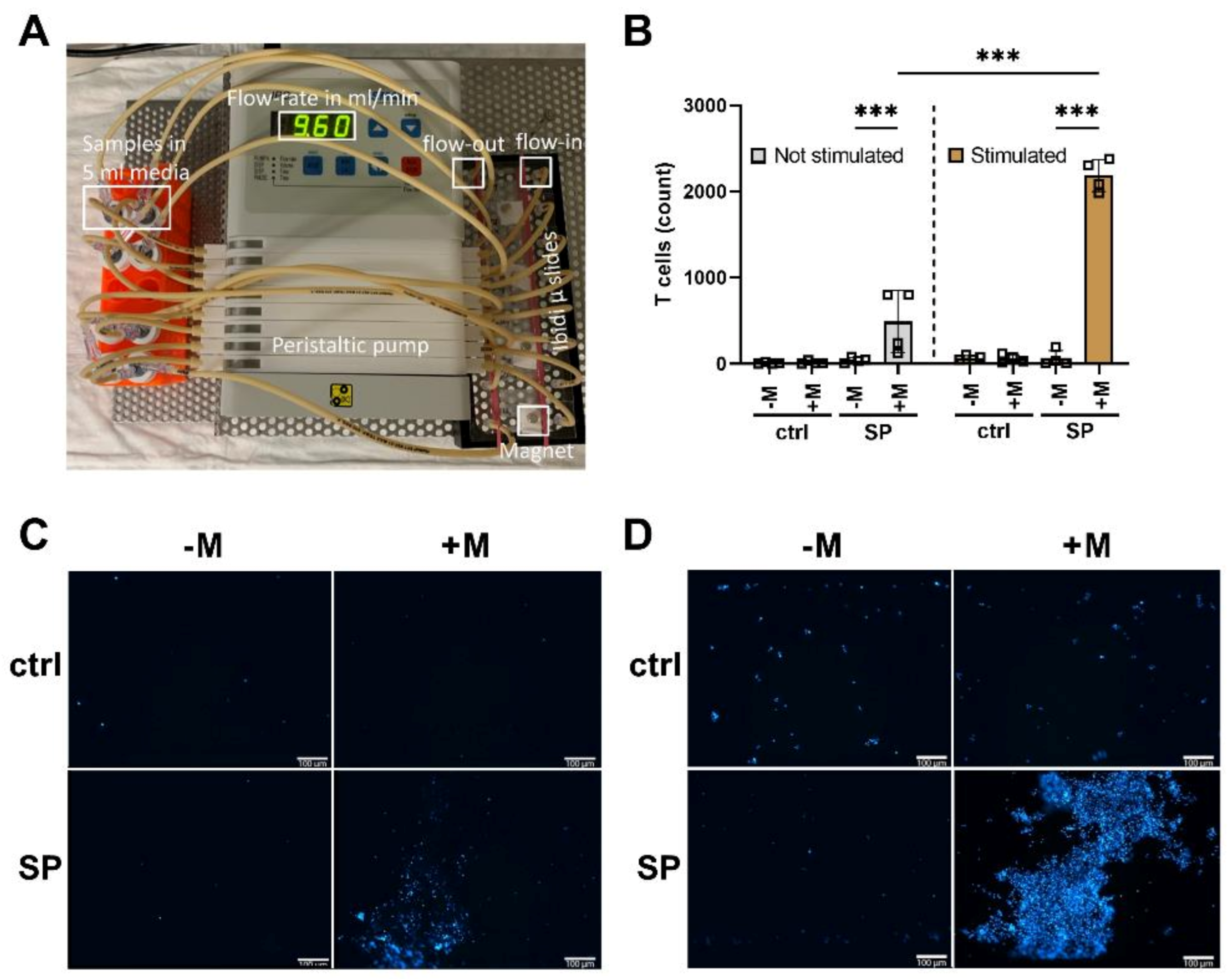
3.6. Loading of T Cells with SPIONs Allows Magnetic Enrichment under Dynamic Conditions
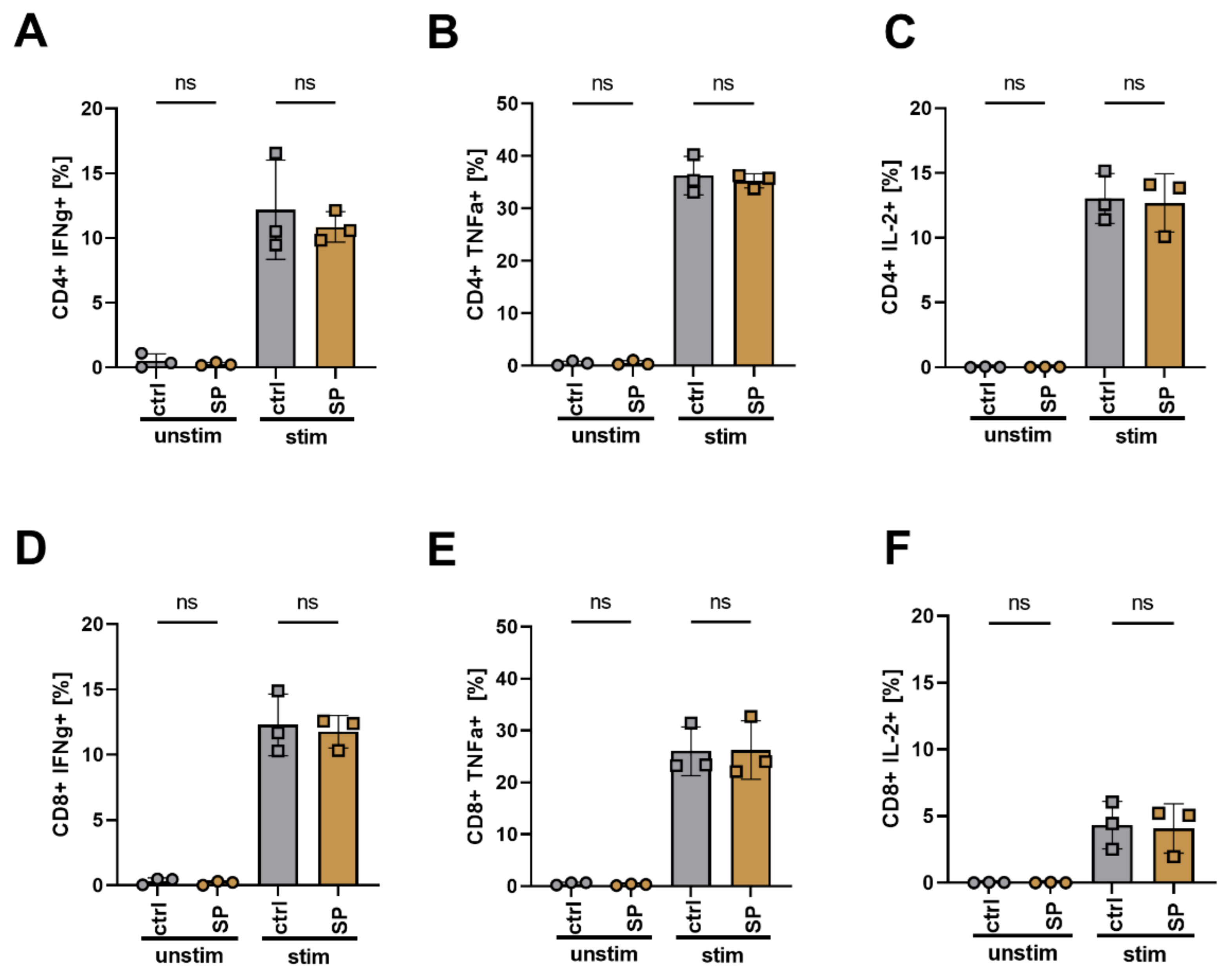
3.7. SPION Loading Does Not Impair Cytokine Release and Differentiation of T Cells after Polyclonal Stimulation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mattiuzzi, C.; Lippi, G. Current Cancer Epidemiology. J. Epidemiol. Glob. Health 2019, 9, 217–222. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pages, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jass, J.R. Lymphocytic infiltration and survival in rectal cancer. J. Clin. Pathol. 1986, 39, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.T.; Jeong, H.; Woo, O.H.; Seo, J.H.; Kim, A.; Lee, E.S.; Shin, S.W.; Kim, Y.H.; Kim, J.S.; Park, K.H. Tumor-infiltrating lymphocytes, tumor characteristics, and recurrence in patients with early breast cancer. Am. J. Clin. Oncol. 2013, 36, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Kmiecik, J.; Poli, A.; Brons, N.H.; Waha, A.; Eide, G.E.; Enger, P.O.; Zimmer, J.; Chekenya, M. Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J. Neuroimmunol. 2013, 264, 71–83. [Google Scholar] [CrossRef] [Green Version]
- Dudley, M.E.; Rosenberg, S.A. Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nat. Rev. Cancer 2003, 3, 666–675. [Google Scholar] [CrossRef] [Green Version]
- Bernhard, H.; Neudorfer, J.; Gebhard, K.; Conrad, H.; Hermann, C.; Nahrig, J.; Fend, F.; Weber, W.; Busch, D.H.; Peschel, C. Adoptive transfer of autologous, HER2-specific, cytotoxic T lymphocytes for the treatment of HER2-overexpressing breast cancer. Cancer Immunol. Immunother. 2008, 57, 271–280. [Google Scholar] [CrossRef]
- Pockaj, B.A.; Sherry, R.M.; Wei, J.P.; Yannelli, J.R.; Carter, C.S.; Leitman, S.F.; Carasquillo, J.A.; Steinberg, S.M.; Rosenberg, S.A.; Yang, J.C. Localization of 111indium-labeled tumor infiltrating lymphocytes to tumor in patients receiving adoptive immunotherapy. Augmentation with cyclophosphamide and correlation with response. Cancer 1994, 73, 1731–1737. [Google Scholar] [CrossRef]
- Sanz-Ortega, L.; Rojas, J.M.; Barber, D.F. Improving Tumor Retention of Effector Cells in Adoptive Cell Transfer Therapies by Magnetic Targeting. Pharmaceutics 2020, 12, 812. [Google Scholar] [CrossRef]
- Bargou, R.; Leo, E.; Zugmaier, G.; Klinger, M.; Goebeler, M.; Knop, S.; Noppeney, R.; Viardot, A.; Hess, G.; Schuler, M.; et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science 2008, 321, 974–977. [Google Scholar] [CrossRef]
- Schlegel, P.; Lang, P.; Zugmaier, G.; Ebinger, M.; Kreyenberg, H.; Witte, K.E.; Feucht, J.; Pfeiffer, M.; Teltschik, H.M.; Kyzirakos, C.; et al. Pediatric posttransplant relapsed/refractory B-precursor acute lymphoblastic leukemia shows durable remission by therapy with the T-cell engaging bispecific antibody blinatumomab. Haematologica 2014, 99, 1212–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topp, M.S.; Kufer, P.; Gokbuget, N.; Goebeler, M.; Klinger, M.; Neumann, S.; Horst, H.A.; Raff, T.; Viardot, A.; Schmid, M.; et al. Targeted therapy with the T-cell-engaging antibody blinatumomab of chemotherapy-refractory minimal residual disease in B-lineage acute lymphoblastic leukemia patients results in high response rate and prolonged leukemia-free survival. J. Clin. Oncol. 2011, 29, 2493–2498. [Google Scholar] [CrossRef] [PubMed]
- Klinger, M.; Brandl, C.; Zugmaier, G.; Hijazi, Y.; Bargou, R.C.; Topp, M.S.; Gökbuget, N.; Neumann, S.; Goebeler, M.; Viardot, A.; et al. Immunopharmacologic response of patients with B-lineage acute lymphoblastic leukemia to continuous infusion of T cell-engaging CD19/CD3-bispecific BiTE antibody blinatumomab. Blood 2012, 119, 6226–6233. [Google Scholar] [CrossRef] [PubMed]
- Tietze, R.; Lyer, S.; Durr, S.; Struffert, T.; Engelhorn, T.; Schwarz, M.; Eckert, E.; Goen, T.; Vasylyev, S.; Peukert, W.; et al. Efficient drug-delivery using magnetic nanoparticles—biodistribution and therapeutic effects in tumour bearing rabbits. Nanomedicine 2013, 9, 961–971. [Google Scholar] [CrossRef]
- Liang, P.C.; Chen, Y.C.; Chiang, C.F.; Mo, L.R.; Wei, S.Y.; Hsieh, W.Y.; Lin, W.L. Doxorubicin-modified magnetic nanoparticles as a drug delivery system for magnetic resonance imaging-monitoring magnet-enhancing tumor chemotherapy. Int. J. Nanomed. 2016, 11, 2021–2037. [Google Scholar]
- Pan, C.; Liu, Y.; Zhou, M.; Wang, W.; Shi, M.; Xing, M.; Liao, W. Theranostic pH-sensitive nanoparticles for highly efficient targeted delivery of doxorubicin for breast tumor treatment. Int. J. Nanomed. 2018, 13, 1119–1137. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Qian, Y.; Dai, Y.; Qiao, S.; Huang, C.; Lu, L.; Luo, Q.; Chen, J.; Zhang, Z. Magnetic Enrichment of Dendritic Cell Vaccine in Lymph Node with Fluorescent-Magnetic Nanoparticles Enhanced Cancer Immunotherapy. Theranostics 2016, 6, 2000–2014. [Google Scholar] [CrossRef]
- Polyak, B.; Fishbein, I.; Chorny, M.; Alferiev, I.; Williams, D.; Yellen, B.; Friedman, G.; Levy, R.J. High field gradient targeting of magnetic nanoparticle-loaded endothelial cells to the surfaces of steel stents. Proc. Natl. Acad. Sci. USA 2008, 105, 698–703. [Google Scholar] [CrossRef] [Green Version]
- Tukmachev, D.; Lunov, O.; Zablotskii, V.; Dejneka, A.; Babic, M.; Sykova, E.; Kubinova, S. An effective strategy of magnetic stem cell delivery for spinal cord injury therapy. Nanoscale 2015, 7, 3954–3958. [Google Scholar] [CrossRef]
- Baeten, K.; Adriaensens, P.; Hendriks, J.; Theunissen, E.; Gelan, J.; Hellings, N.; Stinissen, P. Tracking of myelin-reactive T cells in experimental autoimmune encephalomyelitis (EAE) animals using small particles of iron oxide and MRI. NMR Biomed. 2010, 23, 601–609. [Google Scholar] [CrossRef]
- Liu, L.; Ye, Q.; Wu, Y.; Hsieh, W.Y.; Chen, C.L.; Shen, H.H.; Wang, S.J.; Zhang, H.; Hitchens, T.K.; Ho, C. Tracking T-cells in vivo with a new nano-sized MRI contrast agent. Nanomedicine 2012, 8, 1345–1354. [Google Scholar] [CrossRef] [Green Version]
- Beer, A.J.; Holzapfel, K.; Neudorfer, J.; Piontek, G.; Settles, M.; Kronig, H.; Peschel, C.; Schlegel, J.; Rummeny, E.J.; Bernhard, H. Visualization of antigen-specific human cytotoxic T lymphocytes labeled with superparamagnetic iron-oxide particles. Eur. Radiol. 2008, 18, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Muthana, M.; Kennerley, A.J.; Hughes, R.; Fagnano, E.; Richardson, J.; Paul, M.; Murdoch, C.; Wright, F.; Payne, C.; Lythgoe, M.F.; et al. Directing cell therapy to anatomic target sites in vivo with magnetic resonance targeting. Nat. Commun. 2015, 6, 8009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riegler, J.; Wells, J.A.; Kyrtatos, P.G.; Price, A.N.; Pankhurst, Q.A.; Lythgoe, M.F. Targeted magnetic delivery and tracking of cells using a magnetic resonance imaging system. Biomaterials 2010, 31, 5366–5371. [Google Scholar] [CrossRef] [PubMed]
- Mühlberger, M.; Janko, C.; Unterweger, H.; Friedrich, R.P.; Friedrich, B.; Band, J.; Cebulla, N.; Alexiou, C.; Dudziak, D.; Lee, G.; et al. Functionalization of T Lymphocytes with Citrate-Coated Superpara-magnetic Iron Oxide Nanoparticles for Magnetically Controlled Immune Therapy. Int. J. Nanomed. 2019, 14, 8421–8432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mühlberger, M.; Janko, C.; Unterweger, H.; Schreiber, E.; Band, J.; Lehmann, C.; Dudziak, D.; Lee, G.; Alexiou, C.; Tietze, R. Functionalization of T lymphocytes for magnetically controlled immune therapy: Selection of suitable superparamagnetic iron oxide nanoparticles. J. Magn. Magn. Mater. 2019, 473, 61–67. [Google Scholar] [CrossRef]
- Mühlberger, M.; Unterweger, H.; Band, J.; Lehmann, C.; Heger, L.; Dudziak, D.; Alexiou, C.; Lee, G.; Janko, C. Loading of Primary Human T Lymphocytes with Citrate-Coated Superparamagnetic Iron Oxide Nanoparticles Does Not Impair Their Activation after Polyclonal Stimulation. Cells 2020, 9, 342. [Google Scholar] [CrossRef] [Green Version]
- Nie, W.; Wei, W.; Zuo, L.; Lv, C.; Zhang, F.; Lu, G.H.; Li, F.; Wu, G.; Huang, L.L.; Xi, X.; et al. Magnetic Nanoclusters Armed with Responsive PD-1 Antibody Synergistically Improved Adoptive T-Cell Therapy for Solid Tumors. ACS Nano 2019, 13, 1469–1478. [Google Scholar] [CrossRef]
- Pai, A.; Cao, P.; White, E.E.; Hong, B.; Pailevanian, T.; Wang, M.; Badie, B.; Hajimiri, A.; Berlin, J.M. Dynamically Programmable Magnetic Fields for Controlled Movement of Cells Loaded with Iron Oxide Nanoparticles. ACS Appl. Bio Mater. 2020, 3, 4139–4147. [Google Scholar] [CrossRef]
- Sanz-Ortega, L.; Portilla, Y.; Pérez-Yagüe, S.; Barber, D.F. Magnetic targeting of adoptively transferred tumour-specific nanoparticle-loaded CD8(+) T cells does not improve their tumour infiltration in a mouse model of cancer but promotes the retention of these cells in tumour-draining lymph nodes. J. Nanobiotechnol. 2019, 17, 87. [Google Scholar] [CrossRef] [Green Version]
- Sanz-Ortega, L.; Rojas, J.M.; Marcos, A.; Portilla, Y.; Stein, J.V.; Barber, D.F. T cells loaded with magnetic nanoparticles are retained in peripheral lymph nodes by the application of a magnetic field. J. Nanobiotechnol. 2019, 17, 14. [Google Scholar] [CrossRef] [Green Version]
- Sanz-Ortega, L.; Rojas, J.M.; Portilla, Y.; Pérez-Yagüe, S.; Barber, D.F. Magnetic Nanoparticles Attached to the NK Cell Surface for Tumor Targeting in Adoptive Transfer Therapies Does Not Affect Cellular Effector Functions. Front. Immunol. 2019, 10, 2073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foroozandeh, P.; Aziz, A.A. Insight into Cellular Uptake and Intracellular Trafficking of Nanoparticles. Nanoscale Res. Lett. 2018, 13, 339. [Google Scholar] [CrossRef] [PubMed]
- Waiczies, S.; Niendorf, T.; Lombardi, G. Labeling of cell therapies: How can we get it right? Oncoimmunology 2017, 6, e1345403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sim, T.; Choi, B.; Kwon, S.W.; Kim, K.S.; Choi, H.; Ross, A.; Kim, D.H. Magneto-Activation and Magnetic Resonance Imaging of Natural Killer Cells Labeled with Magnetic Nanocomplexes for the Treatment of Solid Tumors. ACS Nano 2021. [Google Scholar] [CrossRef] [PubMed]
- Zupke, O.; Distler, E.; Jürchott, A.; Paiphansiri, U.; Dass, M.; Thomas, S.; Hartwig, U.F.; Theobald, M.; Landfester, K.; Mailänder, V.; et al. Nanoparticles and antigen-specific T-cell therapeutics: A comprehensive study on uptake and release. Nanomedicine 2015, 10, 1063–1076. [Google Scholar] [CrossRef] [PubMed]
- Elbialy, N.S.; Fathy, M.M.; Khalil, W.M. Doxorubicin loaded magnetic gold nanoparticles for in vivo targeted drug delivery. Int. J. Pharm. 2015, 490, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, R.P.; Janko, C.; Poettler, M.; Tripal, P.; Zaloga, J.; Cicha, I.; Dürr, S.; Nowak, J.; Odenbach, S.; Slabu, I.; et al. Flow cytometry for intracellular SPI-ON quantification: Specificity and sensitivity in comparison with spectroscopic methods. Int. J. Nanomed. 2015, 10, 4185–4201. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.A.; Aberg, C.; Salvati, A.; Dawson, K.A. Role of cell cycle on the cellular uptake and dilution of nanoparticles in a cell population. Nat. Nanotechnol. 2011, 7, 62–68. [Google Scholar] [CrossRef]
- Miltenyi, S.; Muller, W.; Weichel, W.; Radbruch, A. High gradient magnetic cell separation with MACS. Cytometry 1990, 11, 231–238. [Google Scholar] [CrossRef]
- Lynch, I.; Salvati, A.; Dawson, K.A. Proteinnanoparticle interactions: What does the cell see? Nat. Nanotechnol. 2009, 4, 546–547. [Google Scholar] [CrossRef]
- Mahmoudi, M.; Lynch, I.; Ejtehadi, M.R.; Monopoli, M.P.; Bombelli, F.B.; Laurent, S. Protein-nanoparticle interactions: Opportunities and challenges. Chem. Rev. 2011, 111, 5610–5637. [Google Scholar] [CrossRef]
- Nel, A.E.; Mädler, L.; Velegol, D.; Xia, T.; Hoek, E.M.V.; Somasundaran, P.; Klaessig, F.; Castranova, V.; Thompson, M. Understanding biophysicochemical interactions at the nano–bio interface. Nat. Mater. 2009, 8, 543–557. [Google Scholar] [CrossRef] [PubMed]
- Rocker, C.; Potzl, M.; Zhang, F.; Parak, W.J.; Nienhaus, G.U. A quantitative fluorescence study of protein monolayer formation on colloidal nanoparticles. Nat. Nanotechnol. 2009, 4, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Hadjidemetriou, M.; Al-Ahmady, Z.; Mazza, M.; Collins, R.F.; Dawson, K.; Kostarelos, K. In Vivo Biomolecule Corona around Blood-Circulating, Clinically Used and Antibody-Targeted Lipid Bilayer Nanoscale Vesicles. ACS Nano 2015, 9, 8142–8156. [Google Scholar] [CrossRef] [PubMed]
- Lesniak, A.; Fenaroli, F.; Monopoli, M.P.; Aberg, C.; Dawson, K.A.; Salvati, A. Effects of the presence or absence of a protein corona on silica nanoparticle uptake and impact on cells. ACS Nano 2012, 6, 5845–5857. [Google Scholar] [CrossRef] [PubMed]
- Kolosnjaj-Tabi, J.; Wilhelm, C.; Clement, O.; Gazeau, F. Cell labeling with magnetic nanoparticles: Opportunity for magnetic cell imaging and cell manipulation. J. Nanobiotechnol. 2013, 11 (Suppl. 1), S7. [Google Scholar] [CrossRef] [Green Version]
- Charpentier, J.C.; Chen, D.; Lapinski, P.E.; Turner, J.; Grigorova, I.; Swanson, J.A.; King, P.D. Macropinocytosis drives T cell growth by sustaining the activation of mTORC1. Nat. Commun. 2020, 11, 180. [Google Scholar] [CrossRef] [PubMed]
- Dodd, C.H.; Hsu, H.C.; Chu, W.J.; Yang, P.; Zhang, H.G.; Mountz, J.D., Jr.; Zinn, K.; Forder, J.; Josephson, L.; Weissleder, R.; et al. Normal T-cell response and in vivo magnetic resonance imaging of T cells loaded with HIV transactivator-peptide-derived superparamagnetic nanoparticles. J. Immunol. Methods 2001, 256, 89–105. [Google Scholar] [CrossRef]
- Panet, E.; Mashriki, T.; Lahmi, R.; Jacob, A.; Ozer, E.; Vecsler, M.; Lazar, I.; Tzur, A. The interface of nanoparticles with proliferating mammalian cells. Nat. Nanotechnol. 2017, 12, 598–600. [Google Scholar] [CrossRef]
- Rees, P.; Wills, J.W.; Brown, M.R.; Barnes, C.M.; Summers, H.D. The origin of heterogeneous nanoparticle uptake by cells. Nat. Commun. 2019, 10, 2341. [Google Scholar] [CrossRef] [PubMed]
- Frohlich, E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int. J. Nanomed. 2012, 7, 5577–5591. [Google Scholar] [CrossRef] [Green Version]
- Jambhrunkar, S.; Qu, Z.; Popat, A.; Yang, J.; Noonan, O.; Acauan, L.; Ahmad Nor, Y.; Yu, C.; Karmakar, S. Effect of surface functionality of silica nanoparticles on cellular uptake and cytotoxicity. Mol. Pharm. 2014, 11, 3642–3655. [Google Scholar] [CrossRef] [PubMed]
- Garden, O.A.; Reynolds, P.R.; Yates, J.; Larkman, D.J.; Marelli-Berg, F.M.; Haskard, D.O.; Edwards, A.D.; George, A.J. A rapid method for labelling CD4+ T cells with ultrasmall paramagnetic iron oxide nanoparticles for magnetic resonance imaging that preserves proliferative, regulatory and migratory behaviour in vitro. J. Immunol. Methods 2006, 314, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Lankoff, A.; Arabski, M.; Wegierek-Ciuk, A.; Kruszewski, M.; Lisowska, H.; Banasik-Nowak, A.; Rozga-Wijas, K.; Wojewodzka, M.; Slomkowski, S. Effect of surface modification of silica nanoparticles on toxicity and cellular uptake by human peripheral blood lymphocytes in vitro. Nanotoxicology 2013, 7, 235–250. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, C.L.; Williams, M.; Ryle, L.M.; Smith, T.L.; Tanner, M.; Ho, C. Superparamagnetic iron oxide particles transactivator protein-fluorescein isothiocyanate particle labeling for in vivo magnetic resonance imaging detection of cell migration: Uptake and durability. Transplantation 2003, 76, 1043–1046. [Google Scholar] [CrossRef]
- Kircher, M.F.; Allport, J.R.; Graves, E.E.; Love, V.; Josephson, L.; Lichtman, A.H.; Weissleder, R. In vivo high resolution three-dimensional imaging of antigen-specific cytotoxic T-lymphocyte trafficking to tumors. Cancer Res. 2003, 63, 6838–6846. [Google Scholar]
- Smirnov, P.; Lavergne, E.; Gazeau, F.; Lewin, M.; Boissonnas, A.; Doan, B.-T.; Gillet, B.; Combadière, C.; Combadière, B.; Clément, O. In vivo cellular imaging of lymphocyte trafficking by MRI: A tumor model approach to cell-based anticancer therapy. Magn. Reson. Med. 2006, 56, 498–508. [Google Scholar] [CrossRef]
- Briley-Saebo, K.; Bjornerud, A.; Grant, D.; Ahlstrom, H.; Berg, T.; Kindberg, G.M. Hepatic cellular distribution and degradation of iron oxide nanoparticles following single intravenous injection in rats: Implications for magnetic resonance imaging. Cell Tissue Res. 2004, 316, 315–323. [Google Scholar] [CrossRef]
- Mazuel, F.; Espinosa, A.; Luciani, N.; Reffay, M.; Le Borgne, R.; Motte, L.; Desboeufs, K.; Michel, A.; Pellegrino, T.; Lalatonne, Y.; et al. Massive Intracellular Biodegradation of Iron Oxide Nanoparticles Evidenced Magnetically at Single-Endosome and Tissue Levels. ACS Nano 2016, 10, 7627–7638. [Google Scholar] [CrossRef]
- Feliu, N.; Docter, D.; Heine, M.; Del Pino, P.; Ashraf, S.; Kolosnjaj-Tabi, J.; Macchiarini, P.; Nielsen, P.; Alloyeau, D.; Gazeau, F.; et al. In vivo degeneration and the fate of inorganic nanoparticles. Chem. Soc. Rev. 2016, 45, 2440–2457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olden, B.R.; Cheng, E.; Cheng, Y.; Pun, S.H. Identifying key barriers in cationic polymer gene delivery to human T cells. Biomater. Sci. 2019, 7, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, C.; Riviere, C.; Biais, N. Magnetic control of Dictyostelium aggregation. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2007, 75 Pt 1, 041906. [Google Scholar] [CrossRef]
- Park, K. Facing the truth about nanotechnology in drug delivery. ACS Nano 2013, 7, 7442–7447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef] [PubMed]
- Torrice, M. Does Nanomedicine Have a Delivery Problem? ACS Cent. Sci. 2016, 2, 434–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilhelm, C.; Gazeau, F. Universal cell labelling with anionic magnetic nanoparticles. Biomaterials 2008, 29, 3161–3174. [Google Scholar] [CrossRef] [PubMed]
- Blumler, P.; Friedrich, R.P.; Pereira, J.; Baun, O.; Alexiou, C.; Mailander, V. Contactless Nanoparticle-Based Guiding of Cells by Controllable Magnetic Fields. Nanotechnol. Sci. Appl. 2021, 14, 91–100. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Physicochemical Feature | Batch 1 | Batch 2 | Batch 3 |
|---|---|---|---|
| Magnetic susceptibility (10−3) | 4.08 ± 0.00 | 4.12 ± 0.00 | 4.10 ± 0.00 |
| Z-average size (nm) in H2O | 58 ± 0.1 | 52 ± 0.1 | 53 ± 0.2 |
| Polydispersity index (PDI) | 0.143 ± 0.005 | 0.151 ± 0.08 | 0.152 ± 0.07 |
| Zeta potential (mV) at pH 7.3 | −48.5 ± 0.5 | −53.7 ± 0.4 | −51.8 ± 0.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boosz, P.; Pfister, F.; Stein, R.; Friedrich, B.; Fester, L.; Band, J.; Mühlberger, M.; Schreiber, E.; Lyer, S.; Dudziak, D.; et al. Citrate-Coated Superparamagnetic Iron Oxide Nanoparticles Enable a Stable Non-Spilling Loading of T Cells and Their Magnetic Accumulation. Cancers 2021, 13, 4143. https://doi.org/10.3390/cancers13164143
Boosz P, Pfister F, Stein R, Friedrich B, Fester L, Band J, Mühlberger M, Schreiber E, Lyer S, Dudziak D, et al. Citrate-Coated Superparamagnetic Iron Oxide Nanoparticles Enable a Stable Non-Spilling Loading of T Cells and Their Magnetic Accumulation. Cancers. 2021; 13(16):4143. https://doi.org/10.3390/cancers13164143
Chicago/Turabian StyleBoosz, Philipp, Felix Pfister, Rene Stein, Bernhard Friedrich, Lars Fester, Julia Band, Marina Mühlberger, Eveline Schreiber, Stefan Lyer, Diana Dudziak, and et al. 2021. "Citrate-Coated Superparamagnetic Iron Oxide Nanoparticles Enable a Stable Non-Spilling Loading of T Cells and Their Magnetic Accumulation" Cancers 13, no. 16: 4143. https://doi.org/10.3390/cancers13164143
APA StyleBoosz, P., Pfister, F., Stein, R., Friedrich, B., Fester, L., Band, J., Mühlberger, M., Schreiber, E., Lyer, S., Dudziak, D., Alexiou, C., & Janko, C. (2021). Citrate-Coated Superparamagnetic Iron Oxide Nanoparticles Enable a Stable Non-Spilling Loading of T Cells and Their Magnetic Accumulation. Cancers, 13(16), 4143. https://doi.org/10.3390/cancers13164143