
Pancreatic Cancer and Immunotherapy: A Clinical Overview

 , , ,
, , ,  , ,
, ,  ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
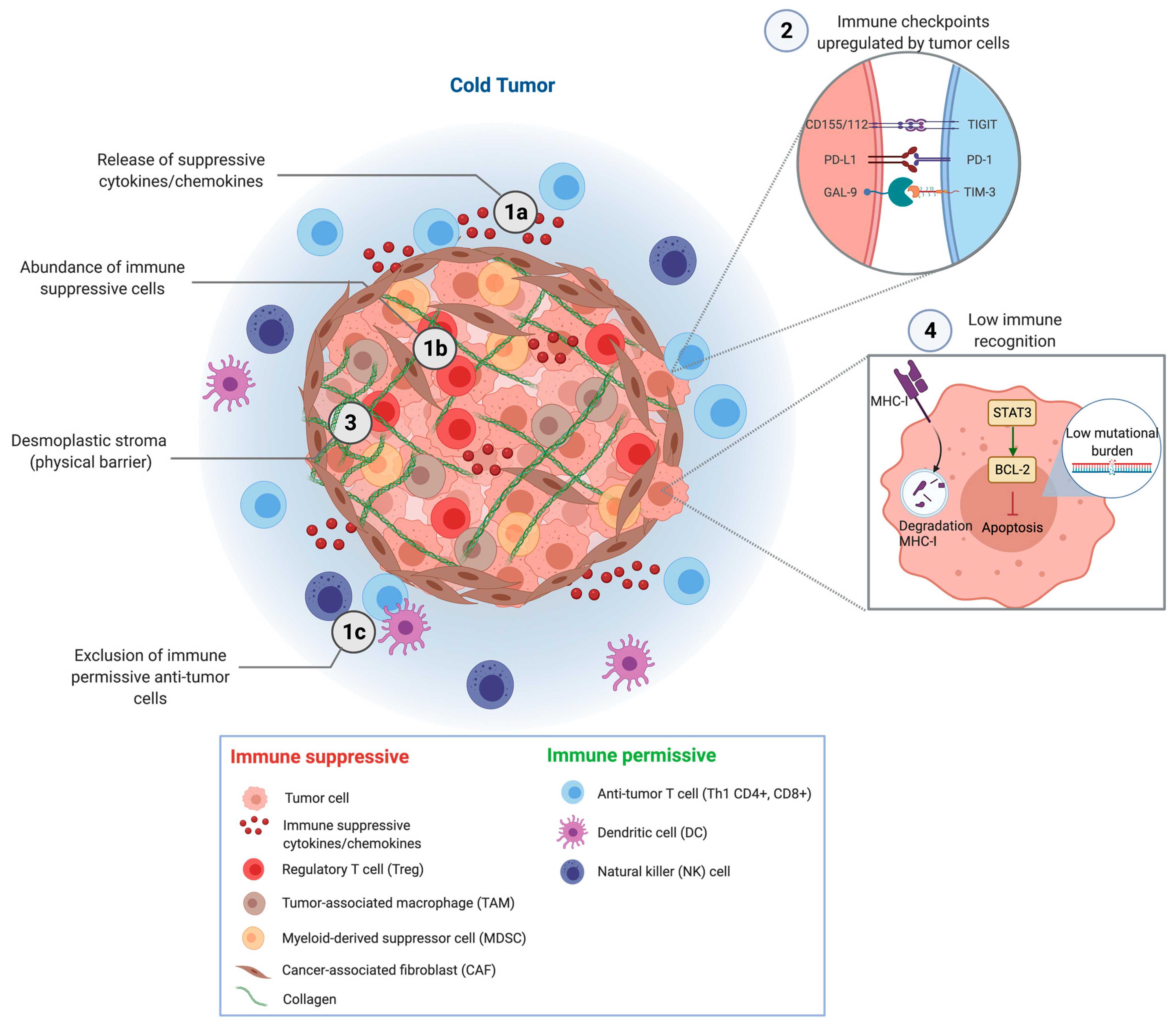
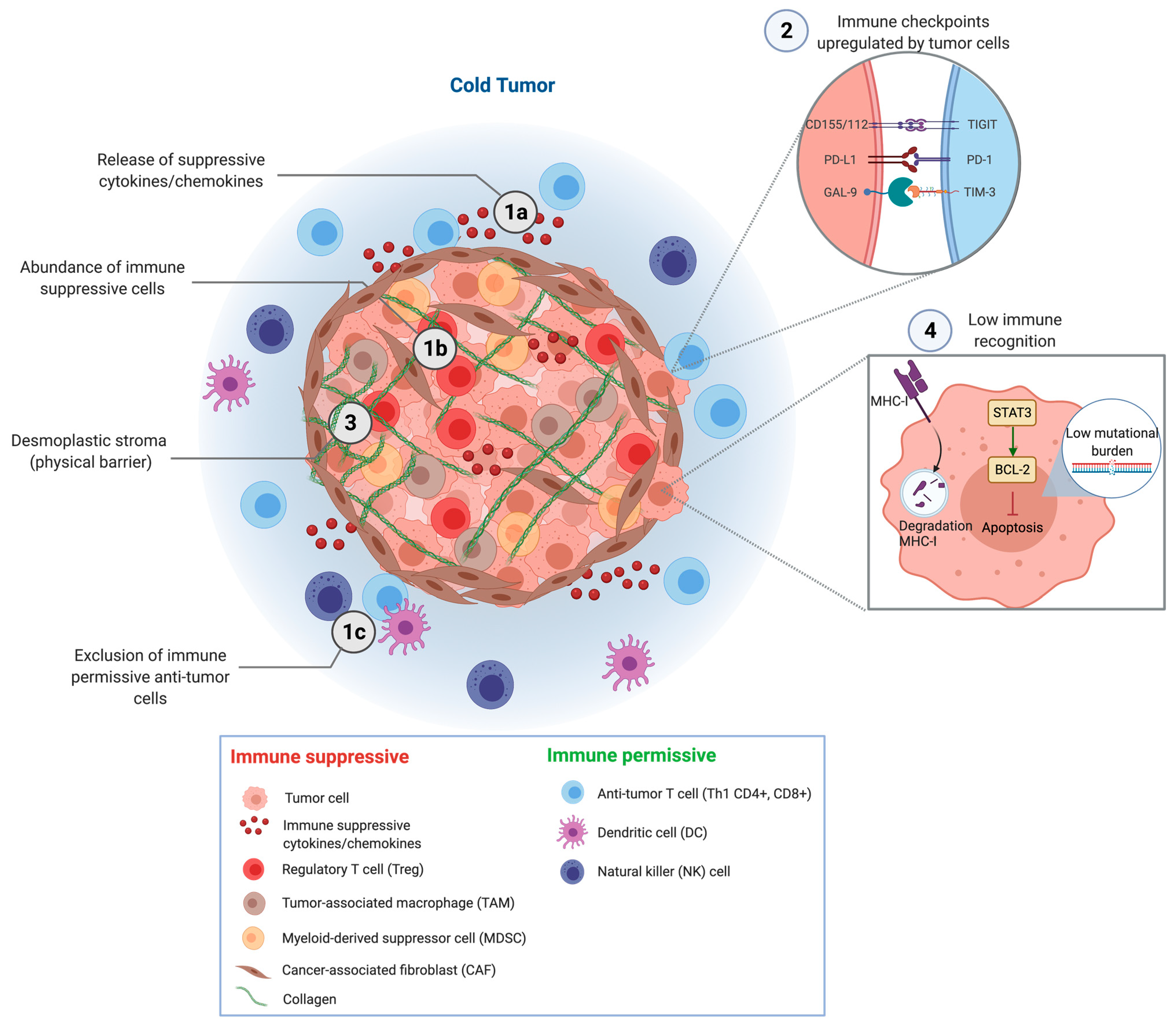
2. Mechanisms of Immune Evasion
3. Regulation of the T Cell Response
4. Immunotherapy
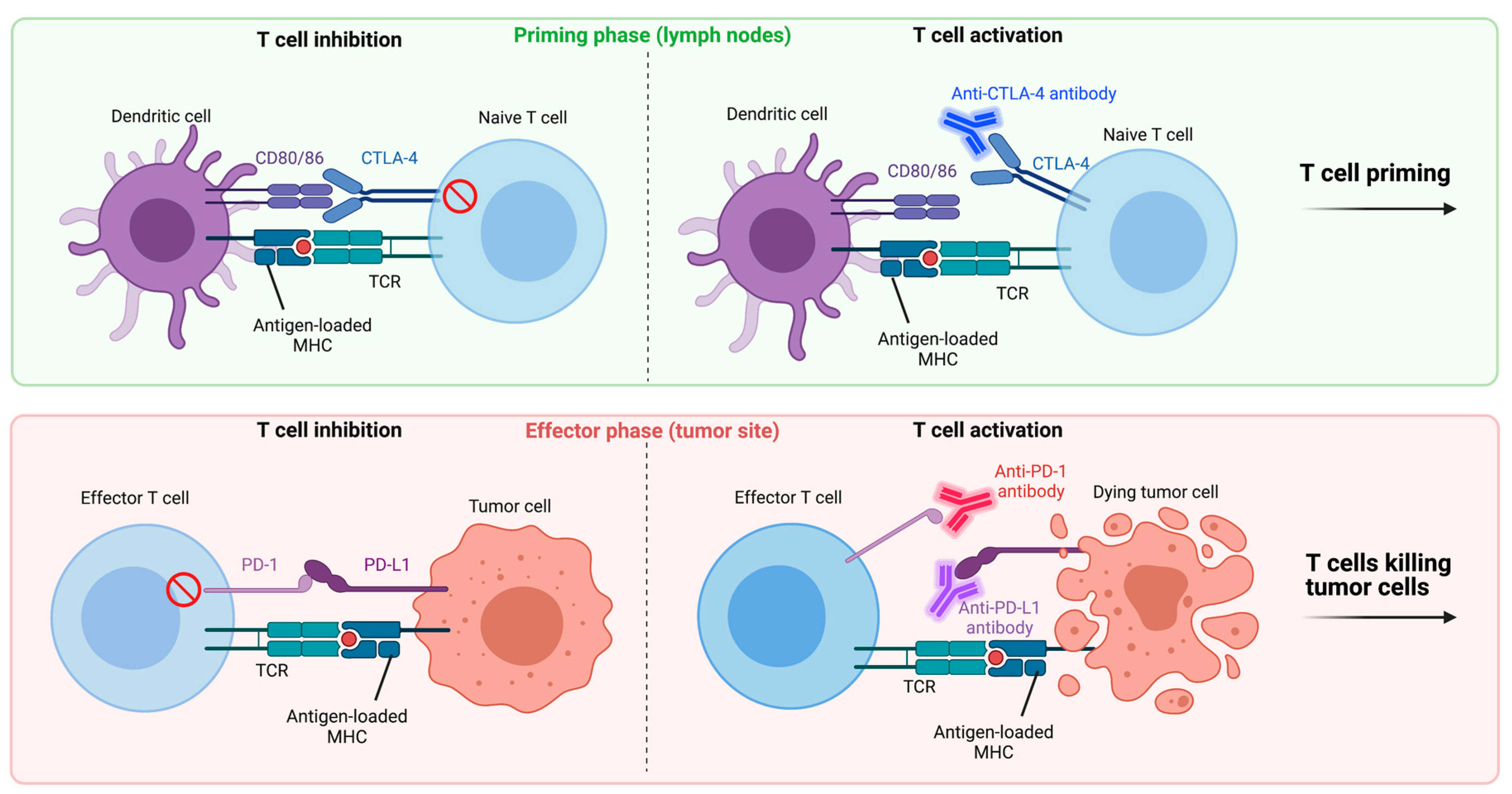
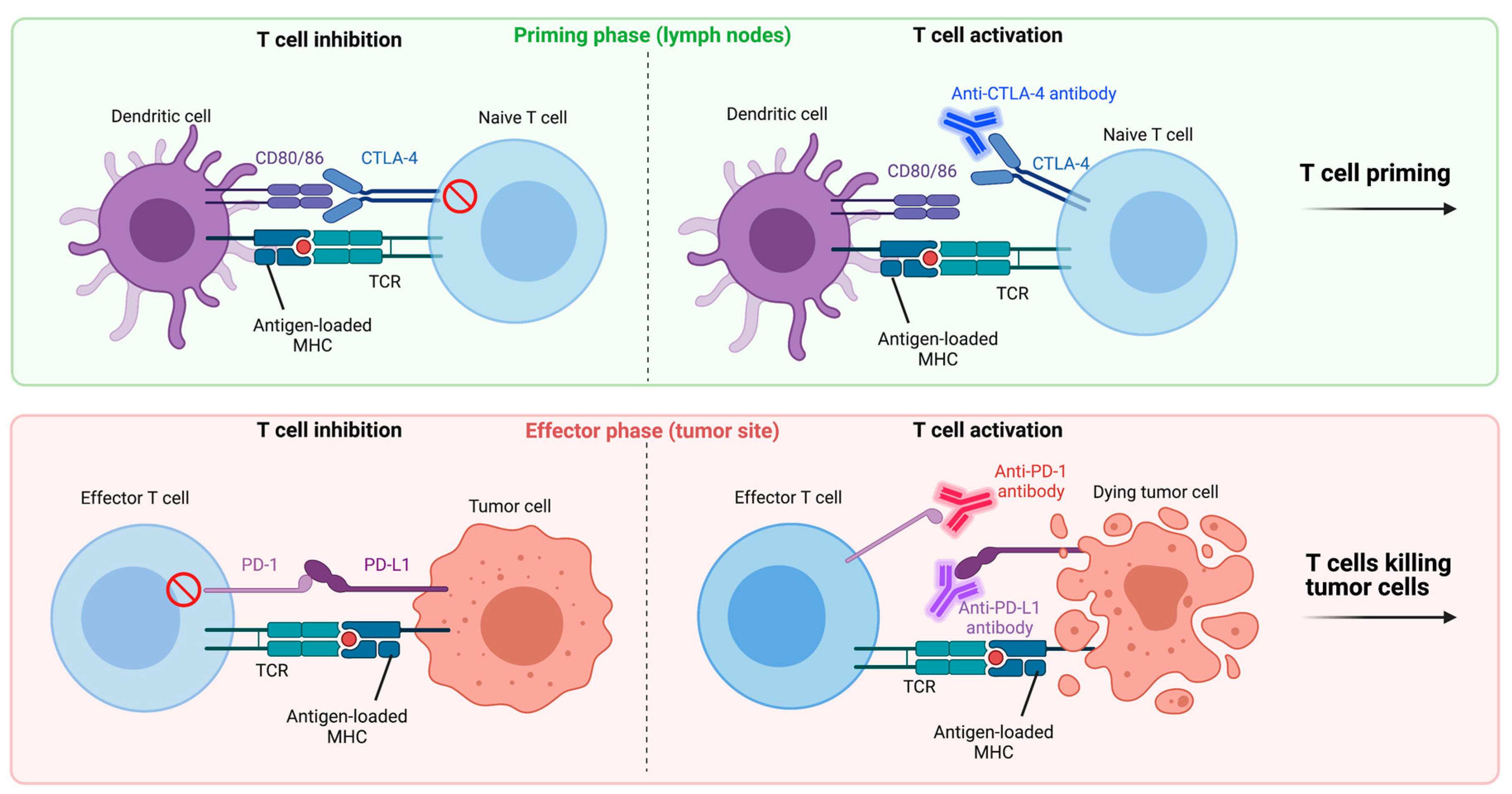
4.1. Immune Checkpoint Inhibitors
4.1.1. Anti-PD-1/Anti-PD-L1
4.1.2. Anti-CTLA-4
4.1.3. Anti-TIM-3
4.1.4. Anti-TIGIT
4.1.5. Anti-LAG-3
4.2. Agonistic Immune Stimulators
4.2.1. MHC Class II Agonist
4.2.2. OX40 Agonist
4.2.3. CD137 (4-1BB) Agonist
4.2.4. ICOS Agonist
4.2.5. CD40 Agonist
4.2.6. CD27 Agonist/Anti-CD70
4.3. Cytokines
4.4. Adjuvants
4.4.1. Toll-Like Receptor (TLR) Agonists
4.4.2. Stimulator of Interferon Genes (STING) Agonists
4.4.3. NOD-Like Receptor (NLR) Agonists
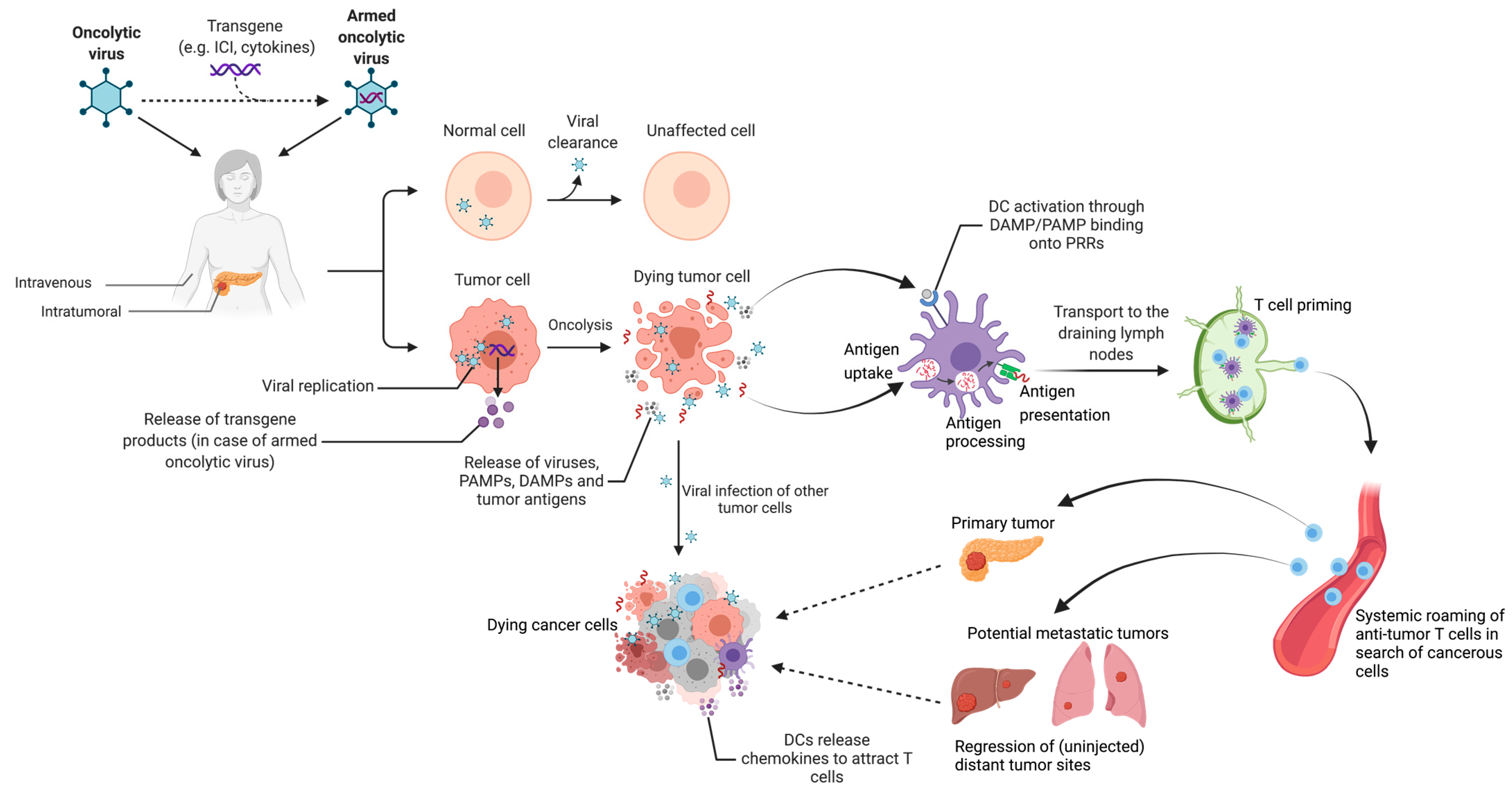
4.5. Oncolytic Viruses
4.6. Adoptive Cell Therapies
4.7. Tumor Vaccines
4.7.1. GVAX
4.7.2. WT-1
4.7.3. KIF20A
4.7.4. VEGFR
4.7.5. Survivin
4.7.6. MUC-1
4.7.7. Mesothelin
4.7.8. hTERT
4.7.9. Neoantigen: Mutant KRAS
4.7.10. Neoantigen: Other
4.7.11. Tumor-Based/Multiple Antigen Vaccines
4.8. Immunotherapy and Local Ablation
4.8.1. Immunotherapy + IRE
4.8.2. Immunotherapy + SABR
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- McGuigan, A.; Kelly, P.; Turkington, R.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 2018, 24, 4846–4861. [Google Scholar] [CrossRef]
- Bükki, J. Pancreatic Adenocarcinoma. N. Engl. J. Med. 2014, 371, 2139–2141. [Google Scholar] [CrossRef] [Green Version]
- Bengtsson, A.; Andersson, R.; Ansari, D. The actual 5-year survivors of pancreatic ductal adenocarcinoma based on real-world data. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; De La Fouchardière, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [Green Version]
- Riquelme, E.; Maitra, A.; McAllister, F. Immunotherapy for Pancreatic Cancer: More than Just a Gut Feeling. Cancer Discov. 2018, 8, 386–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pihlak, R.; Weaver, J.M.J.; Valle, J.W.; McNamara, M.G. Advances in Molecular Profiling and Categorisation of Pancreatic Adenocarcinoma and the Implications for Therapy. Cancers 2018, 10, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, M.; Brehm, C.U.; Gress, T.M.; Buchholz, M.; Alhamwe, B.A.; Von Strandmann, E.P.; Slater, E.P.; Bartsch, J.W.; Bauer, C.; Lauth, M. The Immune Microenvironment in Pancreatic Cancer. Int. J. Mol. Sci. 2020, 21, 7307. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xiao-Zhong, G.; Qi, X.-S. Clinical Outcomes of Specific Immunotherapy in Advanced Pancreatic Cancer: A Systematic Review and Meta-Analysis. J. Immunol. Res. 2017, 2017, 1–16. [Google Scholar] [CrossRef]
- Galluzzi, L.; Humeau, J.; Buqué, A.; Zitvogel, L.; Kroemer, G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2020, 17, 725–741. [Google Scholar] [CrossRef] [PubMed]
- Geboers, B.; Ruarus, A.H.; Nieuwenhuizen, S.; Puijk, R.S.; Scheffer, H.J.; De Gruijl, T.D.; Meijerink, M.R. Needle-guided ablation of locally advanced pancreatic cancer: Cytoreduction or immunomodulation by in vivo vaccination? Chin. Clin. Oncol. 2019, 8, 61. [Google Scholar] [CrossRef]
- Karamitopoulou, E. Tumour microenvironment of pancreatic cancer: Immune landscape is dictated by molecular and histopathological features. Br. J. Cancer 2019, 121, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Zhou, X.; Liu, Y.; Das, A.; Vincent, B.G.; Li, J.; Liu, R.; Huang, L. Tumor neoantigen heterogeneity impacts bystander immune inhibition of pancreatic cancer growth. Transl. Oncol. 2020, 13, 100856. [Google Scholar] [CrossRef]
- Martinez-Bosch, N.; Vinaixa, J.; Navarro, P. Immune Evasion in Pancreatic Cancer: From Mechanisms to Therapy. Cancers 2018, 10, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, R.; Ren, H. Targeting chemokines/chemokine receptors: A promising strategy for enhancing the immunotherapy of pancreatic ductal adenocarcinoma. Signal Transduct. Target. Ther. 2020, 5, 1–2. [Google Scholar] [CrossRef]
- Li, M.; Bharadwaj, U.; Zhang, R.; Zhang, S.; Mu, H.; Fisher, W.E.; Brunicardi, F.C.; Chen, C.; Yao, Q. Mesothelin is a malignant factor and therapeutic vaccine target for pancreatic cancer. Mol. Cancer Ther. 2008, 7, 286–296. [Google Scholar] [CrossRef] [Green Version]
- Hiraoka, N.; Onozato, K.; Kosuge, T.; Hirohashi, S. Prevalence of FOXP3+ Regulatory T Cells Increases During the Progression of Pancreatic Ductal Adenocarcinoma and Its Premalignant Lesions. Clin. Cancer Res. 2006, 12, 5423–5434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabitass, R.F.; Annels, N.E.; Stocken, D.D.; Pandha, H.A.; Middleton, G. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol. Immunother. 2011, 60, 1419–1430. [Google Scholar] [CrossRef] [Green Version]
- Sideras, K.; Braat, H.; Kwekkeboom, J.; van Eijck, C.; Peppelenbosch, M.; Sleijfer, S.; Bruno, M. Role of the immune system in pancreatic cancer progression and immune modulating treatment strategies. Cancer Treat. Rev. 2013, 40, 513–522. [Google Scholar] [CrossRef]
- Lin, J.H.; Huffman, A.P.; Wattenberg, M.M.; Walter, D.; Carpenter, E.L.; Feldser, D.M.; Beatty, G.L.; Furth, E.E.; Vonderheide, R.H. Type 1 conventional dendritic cells are systemically dysregulated early in pancreatic carcinogenesis. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Yamamoto, T.; Yanagimoto, H.; Satoi, S.; Toyokawa, H.; Yamao, J.; Kim, S.; Terakawa, N.; Takahashi, K.; Kwon, A.-H. Circulating Myeloid Dendritic Cells as Prognostic Factors in Patients with Pancreatic Cancer Who Have Undergone Surgical Resection. J. Surg. Res. 2012, 173, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Hirooka, S.; Yanagimoto, H.; Satoi, S.; Yamamoto, T.; Toyokawa, H.; Yamaki, S.; Yui, R.; Inoue, K.; Michiura, T.; Kwon, A.-H. The role of circulating dendritic cells in patients with unresectable pancreatic cancer. Anticancer. Res. 2011, 31, 3827–3834. [Google Scholar]
- Tjomsland, V.; Sandström, P.; Spångeus, A.; Messmer, D.; Emilsson, J.; Falkmer, U.; Falkmer, S.; Magnusson, K.-E.; Borch, K.; Larsson, M. Pancreatic adenocarcinoma exerts systemic effects on the peripheral blood myeloid and plasmacytoid dendritic cells: An indicator of disease severity? BMC Cancer 2010, 10, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, K.; Venida, A.; Yano, J.; Biancur, D.E.; Kakiuchi, M.; Gupta, S.; Sohn, A.S.W.; Mukhopadhyay, S.; Lin, E.Y.; Parker, S.; et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nat. Cell Biol. 2020, 581, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Bowers, J.; Bailey, S.R.; Rubinstein, M.P.; Paulos, C.M.; Camp, E.R. Genomics meets immunity in pancreatic cancer: Current research and future directions for pancreatic adenocarcinoma immunotherapy. Oncol. Rev. 2019, 13, 430. [Google Scholar] [CrossRef]
- Provenzano, P.; Cuevas, C.; Chang, A.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic Targeting of the Stroma Ablates Physical Barriers to Treatment of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [Green Version]
- Haqq, J.; Howells, L.M.; Garcea, G.; Metcalfe, M.S.; Steward, W.P.; Dennison, A. Pancreatic stellate cells and pancreas cancer: Current perspectives and future strategies. Eur. J. Cancer 2014, 50, 2570–2582. [Google Scholar] [CrossRef]
- Whatcott, C.J.; Diep, C.H.; Jiang, P.; Watanabe, A.; LoBello, J.; Sima, C.; Hostetter, G.; Shepard, H.M.; Von Hoff, D.D.; Han, H. Desmoplasia in Primary Tumors and Metastatic Lesions of Pancreatic Cancer. Clin. Cancer Res. 2015, 21, 3561–3568. [Google Scholar] [CrossRef] [Green Version]
- Schizas, D.; Charalampakis, N.; Kole, C.; Economopoulou, P.; Koustas, E.; Gkotsis, E.; Ziogas, D.; Psyrri, A.; Karamouzis, M.V. Immunotherapy for pancreatic cancer: A 2020 update. Cancer Treat. Rev. 2020, 86, 102016. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Xu, C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 2020, 30, 660–669. [Google Scholar] [CrossRef]
- Jung, K.H.; LoRusso, P.; Burris, H.; Gordon, M.; Bang, Y.-J.; Hellmann, M.D.; Cervantes, A.; de Olza, M.O.; Marabelle, A.; Hodi, F.S.; et al. Phase I Study of the Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitor Navoximod (GDC-0919) Administered with PD-L1 Inhibitor (Atezolizumab) in Advanced Solid Tumors. Clin. Cancer Res. 2019, 25, 3220–3228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamphorst, A.O.; Wieland, A.; Nasti, T.; Yang, S.; Zhang, R.; Barber, D.L.; Konieczny, B.T.; Daugherty, C.Z.; Koenig, L.; Yu, K.; et al. Rescue of exhausted CD8 T cells by PD-1–targeted therapies is CD28-dependent. Science 2017, 355, 1423–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, E.; Cheung, J.; Zhu, J.; Su, X.; Taylor, M.J.; Wallweber, H.A.; Sasmal, D.K.; Huang, J.; Kim, J.M.; Mellman, I.; et al. T cell costimulatory receptor CD28 is a primary target for PD-1–mediated inhibition. Science 2017, 355, 1428–1433. [Google Scholar] [CrossRef]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar]
- Lin, D.Y.-W.; Tanaka, Y.; Iwasaki, M.; Gittis, A.G.; Su, H.-P.; Mikami, B.; Okazaki, T.; Honjo, T.; Minato, N.; Garboczi, D.N. The PD-1/PD-L1 complex resembles the antigen-binding Fv domains of antibodies and T cell receptors. Proc. Natl. Acad. Sci. USA 2008, 105, 3011–3016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarchoan, M.; Albacker, L.A.; Hopkins, A.C.; Montesion, M.; Murugesan, K.; Vithayathil, T.T.; Zaidi, N.; Azad, N.S.; Laheru, D.A.; Frampton, G.M.; et al. PD-L1 expression and tumor mutational burden are independent biomarkers in most cancers. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Nomi, T.; Sho, M.; Akahori, T.; Hamada, K.; Kubo, A.; Kanehiro, H.; Nakamura, S.; Enomoto, K.; Yagita, H.; Azuma, M.; et al. Clinical Significance and Therapeutic Potential of the Programmed Death-1 Ligand/Programmed Death-1 Pathway in Human Pancreatic Cancer. Clin. Cancer Res. 2007, 13, 2151–2157. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Ma, Q.; Chen, X.; Guo, K.; Li, J.; Zhang, M. Clinical Significance of B7-H1 and B7-1 Expressions in Pancreatic Carcinoma. World J. Surg. 2010, 34, 1059–1065. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.-L.; Liu, L.; Qi, Z.-H.; Xu, H.-X.; Wang, W.-Q.; Wu, C.-T.; Zhang, S.-R.; Xu, J.-Z.; Ni, Q.-X.; Yu, X.-J. The clinicopathological and prognostic significance of PD-L1 expression in pancreatic cancer: A meta-analysis. Hepatobiliary Pancreat. Dis. Int. 2018, 17, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Shindo, Y.; Hazama, S.; Suzuki, N.; Iguchi, H.; Uesugi, K.; Tanaka, H.; Aruga, A.; Hatori, T.; Ishizaki, H.; Umeda, Y.; et al. Predictive biomarkers for the efficacy of peptide vaccine treatment: Based on the results of a phase II study on advanced pancreatic cancer. J. Exp. Clin. Cancer Res. 2017, 36, 36. [Google Scholar] [CrossRef] [Green Version]
- Hodi, F.S.; Sileni, V.C.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1480–1492. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.-J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and Activity of Anti–PD-L1 Antibody in Patients with Advanced Cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.-P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.; et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair–Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Oh, D.-Y.; Dhani, N.; Renouf, D.J.; Lee, M.A.; Sun, W.; Fisher, G.; Hezel, A.; Chang, S.-C.; Vlahovic, G.; et al. Durvalumab With or Without Tremelimumab for Patients With Metastatic Pancreatic Ductal Adenocarcinoma. JAMA Oncol. 2019, 5, 1431–1438. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.J.; Waypa, J.; Blaydorn, L.; Coats, J.; McGahey, K.; Sangal, A.; Niu, J.; A Lynch, C.; Farley, J.H.; Khemka, V. A phase Ib study of pembrolizumab plus chemotherapy in patients with advanced cancer (PembroPlus). Br. J. Cancer 2017, 117, 33–40. [Google Scholar] [CrossRef] [Green Version]
- Weiss, G.J.; Blaydorn, L.; Beck, J.; Bornemann-Kolatzki, K.; Urnovitz, H.; Schütz, E.; Khemka, V. Phase Ib/II study of gemcitabine, nab-paclitaxel, and pembrolizumab in metastatic pancreatic adenocarcinoma. Investig. New Drugs 2017, 36, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Bockorny, B.; Semenisty, V.; Macarulla, T.; Borazanci, E.; Wolpin, B.M.; Stemmer, S.M.; Golan, T.; Geva, R.; Borad, M.J.; Pedersen, K.S.; et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: The COMBAT trial. Nat. Med. 2020, 26, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Wilkinson, G.A.; Eng, K.; Fields, P.; Raber, P.; Moseley, J.L.; Cheetham, K.; Coffey, M.; Nuovo, G.; Kalinski, P.; et al. Pembrolizumab in Combination with the Oncolytic Virus Pelareorep and Chemotherapy in Patients with Advanced Pancreatic Adenocarcinoma: A Phase Ib Study. Clin. Cancer Res. 2019, 26, 71–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doi, T.; Muro, K.; Ishii, H.; Kato, T.; Tsushima, T.; Takenoyama, M.; Oizumi, S.; Gemmoto, K.; Suna, H.; Enokitani, K.; et al. A Phase I Study of the Anti-CC Chemokine Receptor 4 Antibody, Mogamulizumab, in Combination with Nivolumab in Patients with Advanced or Metastatic Solid Tumors. Clin. Cancer Res. 2019, 25, 6614–6622. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.; Rasco, D.; Veeder, M.; Luke, J.J.; Chandler, J.; Balmanoukian, A.; George, T.; Munster, P.; Berlin, J.D.; Gutierrez, M.; et al. A Phase 1b/2 Study of the Bruton Tyrosine Kinase Inhibitor Ibrutinib and the PD-L1 Inhibitor Durvalumab in Patients with Pretreated Solid Tumors. Oncology 2019, 97, 102–111. [Google Scholar] [CrossRef]
- Zitvogel, L.; Galluzzi, L.; Smyth, M.J.; Kroemer, G. Mechanism of Action of Conventional and Targeted Anticancer Therapies: Reinstating Immunosurveillance. Immunity 2013, 39, 74–88. [Google Scholar] [CrossRef] [Green Version]
- Cubas, R.; Moskalenko, M.; Cheung, J.; Yang, M.; McNamara, E.; Xiong, H.; Hoves, S.; Ries, C.H.; Kim, J.; Gould, S. Chemotherapy Combines Effectively with Anti–PD-L1 Treatment and Can Augment Antitumor Responses. J. Immunol. 2018, 201, 2273–2286. [Google Scholar] [CrossRef] [Green Version]
- Michelakos, T.; Cai, L.; Villani, V.; Sabbatino, F.; Kontos, F.; Castillo, C.F.-D.; Yamada, T.; Neyaz, A.; Taylor, M.S.; Deshpande, V.; et al. Tumor Microenvironment Immune Response in Pancreatic Ductal Adenocarcinoma Patients Treated With Neoadjuvant Therapy. J. Natl. Cancer Inst. 2020, 113, 182–191. [Google Scholar] [CrossRef]
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I.; et al. Phase 2 Trial of Single Agent Ipilimumab (Anti-CTLA-4) for Locally Advanced or Metastatic Pancreatic Adenocarcinoma. J. Immunother. 2010, 33, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Lutz, E.; Uram, J.N.; Sugar, E.A.; Onners, B.; Solt, S.; Zheng, L.; Diaz, L.; Donehower, R.C.; Jaffee, E.; et al. Evaluation of Ipilimumab in Combination With Allogeneic Pancreatic Tumor Cells Transfected With a GM-CSF Gene in Previously Treated Pancreatic Cancer. J. Immunother. 2013, 36, 382–389. [Google Scholar] [CrossRef] [Green Version]
- Aglietta, M.; Barone, C.; Sawyer, M.B.; Moore, M.J.; Miller, W.H.; Bagalà, C.; Colombi, F.; Cagnazzo, C.; Gioeni, L.; Wang, E.; et al. A phase I dose escalation trial of tremelimumab (CP-675,206) in combination with gemcitabine in chemotherapy-naive patients with metastatic pancreatic cancer. Ann. Oncol. 2014, 25, 1750–1755. [Google Scholar] [CrossRef]
- Mohindra, N.A.; Kircher, S.M.; Nimeiri, H.S.; Benson, A.B.; Rademaker, A.; Alonso, E.; Blatner, N.; Khazaie, K.; Mulcahy, M.F. Results of the phase Ib study of ipilimumab and gemcitabine for advanced pancreas cancer. J. Clin. Oncol. 2015, 33, e15281. [Google Scholar] [CrossRef]
- Kalyan, A.; Kircher, S.M.; Mohindra, N.A.; Nimeiri, H.S.; Maurer, V.; Rademaker, A.; Benson, A.B.; Mulcahy, M.F. Ipilimumab and gemcitabine for advanced pancreas cancer: A phase Ib study. J. Clin. Oncol. 2016, 34, e15747. [Google Scholar] [CrossRef]
- Kamath, S.D.; Kalyan, A.; Kircher, S.; Nimeiri, H.; Fought, A.J.; Benson, A.; Mulcahy, M. Ipilimumab and Gemcitabine for Advanced Pancreatic Cancer: A Phase Ib Study. Oncology 2019, 25, e808–e815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sansom, D. CD28, CTLA-4 and their ligands: Who does what and to whom? Immunology 2000, 101, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Farren, M.; Mace, T.A.; Geyer, S.; Mikhail, S.; Wu, C.; Ciombor, K.K.; Tahiri, S.; Ahn, D.; Noonan, A.; A Villalonacalero, M.; et al. Systemic Immune Activity Predicts Overall Survival in Treatment-Naïve Patients with Metastatic Pancreatic Cancer. Clin. Cancer Res. 2015, 22, 2565–2574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribas, A. Tumor Immunotherapy Directed at PD-1. N. Engl. J. Med. 2012, 366, 2517–2519. [Google Scholar] [CrossRef] [Green Version]
- Vargas, F.A.; Furness, A.J.; Litchfield, K.; Joshi, K.; Rosenthal, R.; Ghorani, E.; Solomon, I.; Lesko, M.H.; Ruef, N.; Roddie, C.; et al. Fc Effector Function Contributes to the Activity of Human Anti-CTLA-4 Antibodies. Cancer Cell 2018, 33, 649–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherpereel, A.; Mazieres, J.; Greillier, L.; Lantuejoul, S.; Dô, P.; Bylicki, O.; Monnet, I.; Corre, R.; Audigier-Valette, C.; Locatelli-Sanchez, M.; et al. Nivolumab or nivolumab plus ipilimumab in patients with relapsed malignant pleural mesothelioma (IFCT-1501 MAPS2): A multicentre, open-label, randomised, non-comparative, phase 2 trial. Lancet Oncol. 2019, 20, 239–253. [Google Scholar] [CrossRef]
- Planchard, D.; Reinmuth, N.; Orlov, S.; Fischer, J.; Sugawara, S.; Mandziuk, S.; Marquez-Medina, D.; Novello, S.; Takeda, Y.; Soo, R.; et al. ARCTIC: Durvalumab with or without tremelimumab as third-line or later treatment of metastatic non-small-cell lung cancer. Ann. Oncol. 2020, 31, 609–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acharya, N.; Sabatos-Peyton, C.; Anderson, A.C. Tim-3 finds its place in the cancer immunotherapy landscape. J. Immunother. Cancer 2020, 8, e000911. [Google Scholar] [CrossRef]
- Du, W.; Yang, M.; Turner, A.; Xu, C.; Ferris, R.L.; Huang, J.; Kane, L.P.; Lu, B. TIM-3 as a Target for Cancer Immunotherapy and Mechanisms of Action. Int. J. Mol. Sci. 2017, 18, 645. [Google Scholar] [CrossRef]
- Peng, P.-J.; Li, Y.; Sun, S. On the significance of Tim-3 expression in pancreatic cancer. Saudi J. Biol. Sci. 2017, 24, 1754–1757. [Google Scholar] [CrossRef]
- Wolf, Y.; Anderson, A.C.; Kuchroo, V.K. TIM3 comes of age as an inhibitory receptor. Nat. Rev. Immunol. 2019, 20, 173–185. [Google Scholar] [CrossRef]
- Cebrián, M.J.G.; Bauden, M.; Andersson, R.; Holdenrieder, S.; Ansari, D. Paradoxical Role of HMGB1 in Pancreatic Cancer: Tumor Suppressor or Tumor Promoter? Anticancer. Res. 2016, 36, 4381–4390. [Google Scholar] [CrossRef] [Green Version]
- Gebauer, F.; Wicklein, D.; Horst, J.; Sundermann, P.; Maar, H.; Streichert, T.; Tachezy, M.; Izbicki, J.R.; Bockhorn, M.; Schumacher, U. Carcinoembryonic Antigen-Related Cell Adhesion Molecules (CEACAM) 1, 5 and 6 as Biomarkers in Pancreatic Cancer. PLoS ONE 2014, 9, e113023. [Google Scholar] [CrossRef] [Green Version]
- Seifert, A.M.; Reiche, C.; Heiduk, M.; Tannert, A.; Meinecke, A.-C.; Baier, S.; von Renesse, J.; Kahlert, C.; Distler, M.; Welsch, T.; et al. Detection of pancreatic ductal adenocarcinoma with galectin-9 serum levels. Oncogene 2020, 39, 3102–3113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauken, K.E.; Wherry, E.J. TIGIT and CD226: Tipping the Balance between Costimulatory and Coinhibitory Molecules to Augment the Cancer Immunotherapy Toolkit. Cancer Cell 2014, 26, 785–787. [Google Scholar] [CrossRef] [Green Version]
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; Yu, X.; Huseni, M.; Yang, Y.; Park, S.; Javinal, V.; Chiu, H.; Irving, B.; et al. The Immunoreceptor TIGIT Regulates Antitumor and Antiviral CD8 + T Cell Effector Function. Cancer Cell 2014, 26, 923–937. [Google Scholar] [CrossRef] [Green Version]
- Nishiwada, S.; Sho, M.; Yasuda, S.; Shimada, K.; Yamato, I.; Akahori, T.; Kinoshita, S.; Nagai, M.; Konishi, N.; Nakajima, Y. Clinical significance of CD155 expression in human pancreatic cancer. Anticancer. Res. 2015, 35, 2287–2297. [Google Scholar]
- Jin, H.-S.; Ko, M.; Choi, D.-S.; Kim, J.H.; Lee, D.-H.; Kang, S.-H.; Kim, I.; Lee, H.J.; Choi, E.K.; Kim, K.-P.; et al. CD226hiCD8+ T Cells Are a Prerequisite for Anti-TIGIT Immunotherapy. Cancer Immunol. Res. 2020, 8, 912–925. [Google Scholar] [CrossRef] [Green Version]
- Woo, S.-R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune Inhibitory Molecules LAG-3 and PD-1 Synergistically Regulate T-cell Function to Promote Tumoral Immune Escape. Cancer Res. 2011, 72, 917–927. [Google Scholar] [CrossRef] [Green Version]
- Cebada, J.; Flores, A.; Bandala, C.; Lizaliturri-Flores, I.; Villa-Ruano, N.; Perez-Santos, M. Bispecific anti-PD-1/LAG-3 antibodies for treatment of advanced or metastatic solid tumors: A patent evaluation of US2018326054. Expert Opin. Ther. Patents 2020, 30, 487–494. [Google Scholar] [CrossRef]
- Wang-Gillam, A.; Plambeck-Suess, S.; Goedegebuure, P.; Simon, P.O.; Mitchem, J.; Hornick, J.R.; Sorscher, S.; Picus, J.; Suresh, R.; Lockhart, A.C.; et al. A phase I study of IMP321 and gemcitabine as the front-line therapy in patients with advanced pancreatic adenocarcinoma. Investig. New Drugs 2012, 31, 707–713. [Google Scholar] [CrossRef] [Green Version]
- Beatty, G.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 Agonists Alter Tumor Stroma and Show Efficacy against Pancreatic Carcinoma in Mice and Humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef] [Green Version]
- Beatty, G.L.; Torigian, D.A.; Chiorean, E.G.; Saboury, B.; Brothers, A.; Alavi, A.; Troxel, A.; Sun, W.; Teitelbaum, U.R.; Vonderheide, R.H.; et al. A Phase I Study of an Agonist CD40 Monoclonal Antibody (CP-870,893) in Combination with Gemcitabine in Patients with Advanced Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2013, 19, 6286–6295. [Google Scholar] [CrossRef] [Green Version]
- O’Hara, M.H.; O’Reilly, E.M.; Varadhachary, G.; A Wolff, R.; A Wainberg, Z.; Ko, A.H.; Fisher, G.; Rahma, O.; Lyman, J.P.; Cabanski, C.R.; et al. CD40 agonistic monoclonal antibody APX005M (sotigalimab) and chemotherapy, with or without nivolumab, for the treatment of metastatic pancreatic adenocarcinoma: An open-label, multicentre, phase 1b study. Lancet Oncol. 2021, 22, 118–131. [Google Scholar] [CrossRef]
- Dalgleish, A.G.; Stebbing, J.; Adamson, D.J.; Arif, S.S.; Bidoli, P.; Chang, D.; Cheeseman, S.; Diaz-Beveridge, R.; Fernandez-Martos, C.; Glynne-Jones, R.; et al. Randomised, open-label, phase II study of gemcitabine with and without IMM-101 for advanced pancreatic cancer. Br. J. Cancer 2016, 115, 789–796. [Google Scholar] [CrossRef]
- Lu, X. OX40 and OX40L interaction in cancer. Curr. Med. Chem. 2020, 28, 1–13. [Google Scholar] [CrossRef]
- Ma, Y.; Li, J.; Wang, H.; Chiu, Y.; Kingsley, C.V.; Fry, D.; Delaney, S.N.; Wei, S.C.; Zhang, J.; Maitra, A.; et al. Combination of PD-1 Inhibitor and OX40 Agonist Induces Tumor Rejection and Immune Memory in Mouse Models of Pancreatic Cancer. Gastroenterology 2020, 159, 306–319. [Google Scholar] [CrossRef]
- Curti, B.D.; Kovacsovics-Bankowski, M.; Morris, N.; Walker, E.; Chisholm, L.; Floyd, K.; Walker, J.; Gonzalez, I.; Meeuwsen, T.; Fox, B.A.; et al. OX40 Is a Potent Immune-Stimulating Target in Late-Stage Cancer Patients. Cancer Res. 2013, 73, 7189–7198. [Google Scholar] [CrossRef] [Green Version]
- Etxeberria, I.; Glez-Vaz, J.; Teijeira, A.; Melero, I. New emerging targets in cancer immunotherapy: CD137/4-1BB costimulatory axis. ESMO Open 2019, 4, e000733. [Google Scholar] [CrossRef] [PubMed]
- Muth, S.T.; Saung, M.T.; Blair, A.B.; Henderson, M.G.; Thomas, D.L.; Zheng, L. CD137 agonist-based combination immunotherapy enhances activated, effector memory T cells and prolongs survival in pancreatic adenocarcinoma. Cancer Lett. 2020, 499, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Posey, J.A.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Sakellariou-Thompson, D.; Forget, M.-A.; Creasy, C.; Bernard, V.; Zhao, L.; Kim, Y.U.; Hurd, M.W.; Uraoka, N.; Parra, E.R.; Kang, Y.; et al. 4-1BB Agonist Focuses CD8+ Tumor-Infiltrating T-Cell Growth into a Distinct Repertoire Capable of Tumor Recognition in Pancreatic Cancer. Clin. Cancer Res. 2017, 23, 7263–7275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, A.R.; Tanyi, J.L.; O’Hara, M.H.; Gladney, W.L.; Lacey, S.F.; Torigian, D.A.; Soulen, M.C.; Tian, L.; McGarvey, M.; Nelson, A.M.; et al. Phase I Study of Lentiviral-Transduced Chimeric Antigen Receptor-Modified T Cells Recognizing Mesothelin in Advanced Solid Cancers. Mol. Ther. 2019, 27, 1919–1929. [Google Scholar] [CrossRef]
- Solinas, C.; Gu-Trantien, C.; Willard-Gallo, K. The rationale behind targeting the ICOS-ICOS ligand costimulatory pathway in cancer immunotherapy. ESMO Open 2020, 5, e000544. [Google Scholar] [CrossRef] [Green Version]
- Amatore, F.; Gorvel, L.; Olive, D. Inducible Co-Stimulator (ICOS) as a potential therapeutic target for anti-cancer therapy. Expert Opin. Ther. Targets 2018, 22, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Jong, J.M.V.D.-D.; Santegoets, S.J.; Van De Ven, P.M.; Versluis, J.; Verheul, H.; De Gruijl, T.D.; Gerritsen, W.R.; Eertwegh, A.J.M.V.D. Improved efficacy of mitoxantrone in patients with castration-resistant prostate cancer after vaccination with GM-CSF-transduced allogeneic prostate cancer cells. OncoImmunology 2015, 5, e1105431. [Google Scholar] [CrossRef] [Green Version]
- Carrell, R.K.; Stanton, R.A.; Ethier, S.P.; LaRue, A.C.; Soloff, A.C. ICOSL-augmented adenoviral-based vaccination induces a bipolar Th17/Th1 T cell response against unglycosylated MUC1 antigen. Vaccine 2018, 36, 6262–6269. [Google Scholar] [CrossRef]
- Vonderheide, R.H. CD40 Agonist Antibodies in Cancer Immunotherapy. Annu. Rev. Med. 2020, 71, 47–58. [Google Scholar] [CrossRef] [Green Version]
- Lau, S.P.; Van Montfoort, N.; Kinderman, P.; Lukkes, M.; Klaase, L.; Van Nimwegen, M.; Van Gulijk, M.; Dumas, J.; Mustafa, D.A.M.; A Lievense, S.L.; et al. Dendritic cell vaccination and CD40-agonist combination therapy licenses T cell-dependent antitumor immunity in a pancreatic carcinoma murine model. J. Immunother. Cancer 2020, 8, e000772. [Google Scholar] [CrossRef] [PubMed]
- Van Audenaerde, J.R.; Marcq, E.; Von Scheidt, B.; Davey, A.S.; Oliver, A.J.; De Waele, J.; Quatannens, D.; Van Loenhout, J.; Pauwels, P.; Roeyen, G.; et al. Novel combination immunotherapy for pancreatic cancer: Potent anti-tumor effects with CD40 agonist and interleukin-15 treatment. Clin. Transl. Immunol. 2020, 9, e1165. [Google Scholar] [CrossRef] [PubMed]
- Starzer, A.M.; Berghoff, A.S. New emerging targets in cancer immunotherapy: CD27 (TNFRSF7). ESMO Open 2019, 4, e000629. [Google Scholar] [CrossRef] [Green Version]
- Burris, H.A.; Infante, J.R.; Ansell, S.M.; Nemunaitis, J.J.; Weiss, G.R.; Villalobos, V.M.; Sikic, B.I.; Taylor, M.H.; Northfelt, D.W.; Carson, I.W.E.; et al. Safety and Activity of Varlilumab, a Novel and First-in-Class Agonist Anti-CD27 Antibody, in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2017, 35, 2028–2036. [Google Scholar] [CrossRef] [Green Version]
- Ryan, M.C.; Kostner, H.; A Gordon, K.; Duniho, S.; Sutherland, M.K.; Yu, C.; Kim, K.M.; Nesterova, A.; Anderson, M.; A McEarchern, J.; et al. Targeting pancreatic and ovarian carcinomas using the auristatin-based anti-CD70 antibody–drug conjugate SGN-75. Br. J. Cancer 2010, 103, 676–684. [Google Scholar] [CrossRef]
- Claus, C.; Riether, C.; Schürch, C.; Matter, M.; Hilmenyuk, T.; Ochsenbein, A. CD27 Signaling Increases the Frequency of Regulatory T Cells and Promotes Tumor Growth. Cancer Res. 2012, 72, 3664–3676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aftimos, P.; Rolfo, C.C.; Rottey, S.S.; Offner, F.; Bron, D.; Maerevoet, M.; Soria, J.-C.; Moshir, M.M.; Dreier, T.T.; Van Rompaey, L.L.; et al. Phase I Dose-Escalation Study of the Anti-CD70 Antibody ARGX-110 in Advanced Malignancies. Clin. Cancer Res. 2017, 23, 6411–6420. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, J.; Deschoolmeester, V.; Rolfo, C.; Zwaenepoel, K.; Bossche, J.V.D.; Deben, C.; Silence, K.; De Haard, H.; Hermans, C.; Rottey, S.; et al. Preclinical data on the combination of cisplatin and anti-CD70 therapy in non-small cell lung cancer as an excellent match in the era of combination therapy. Oncotarget 2017, 8, 74058–74067. [Google Scholar] [CrossRef] [Green Version]
- Eric, L.; Yeo, C.J.; Lillemoe, K.D.; Biedrzycki, B.; Kobrin, B.; Herman, J.; Sugar, E.; Piantadosi, S.; Cameron, J.L.; Solt, S.; et al. A Lethally Irradiated Allogeneic Granulocyte-Macrophage Colony Stimulating Factor-Secreting Tumor Vaccine for Pancreatic Adenocarcinoma. Ann. Surg. 2011, 253, 328–335. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Wang-Gillam, A.; Picozzi, V.; Greten, T.F.; Crocenzi, T.; Springett, G.; Morse, M.; Zeh, H.; Cohen, D.; Fine, R.L.; et al. Safety and Survival With GVAX Pancreas Prime and Listeria Monocytogenes–Expressing Mesothelin (CRS-207) Boost Vaccines for Metastatic Pancreatic Cancer. J. Clin. Oncol. 2015, 33, 1325–1333. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Picozzi, V.J.; Ko, A.H.; Wainberg, Z.A.; Kindler, H.; Wang-Gillam, A.; Oberstein, P.E.; Morse, M.A.; Zeh, H.J.; Weekes, C.D.; et al. Results from a Phase IIb, Randomized, Multicenter Study of GVAX Pancreas and CRS-207 Compared with Chemotherapy in Adults with Previously Treated Metastatic Pancreatic Adenocarcinoma (ECLIPSE Study). Clin. Cancer Res. 2019, 25, 5493–5502. [Google Scholar] [CrossRef]
- Tsujikawa, T.; Crocenzi, T.; Durham, J.N.; Sugar, E.A.; Wu, A.A.; Onners, B.; Nauroth, J.M.; Anders, R.A.; Fertig, E.J.; Laheru, D.A.; et al. Evaluation of Cyclophosphamide/GVAX Pancreas Followed by Listeria-Mesothelin (CRS-207) with or without Nivolumab in Patients with Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 3578–3588. [Google Scholar] [CrossRef] [Green Version]
- Wu, A.A.; Bever, K.M.; Ho, W.J.; Fertig, E.J.; Niu, N.; Zheng, L.; Parkinson, R.M.; Durham, J.N.; Onners, B.L.; Ferguson, A.K.; et al. A Phase II Study of Allogeneic GM-CSF–Transfected Pancreatic Tumor Vaccine (GVAX) with Ipilimumab as Maintenance Treatment for Metastatic Pancreatic Cancer. Clin. Cancer Res. 2020, 26. [Google Scholar] [CrossRef]
- Wedén, S.; Klemp, M.; Gladhaug, I.P.; Møller, M.; Eriksen, J.A.; Gaudernack, G.; Buanes, T. Long-term follow-up of patients with resected pancreatic cancer following vaccination against mutant K-ras. Int. J. Cancer 2010, 128, 1120–1128. [Google Scholar] [CrossRef]
- Palmer, D.H.; Valle, J.W.; Ma, Y.T.; Faluyi, O.; Neoptolemos, J.P.; Gjertsen, T.J.; Iversen, B.; Eriksen, J.A.; Møller, A.-S.; Aksnes, A.-K.; et al. TG01/GM-CSF and adjuvant gemcitabine in patients with resected RAS-mutant adenocarcinoma of the pancreas (CT TG01-01): A single-arm, phase 1/2 trial. Br. J. Cancer 2020, 122, 971–977. [Google Scholar] [CrossRef] [Green Version]
- Middleton, G.; Silcocks, P.; Cox, T.; Valle, J.; Wadsley, J.; Propper, D.; Coxon, F.; Ross, P.; Madhusudan, S.; Roques, T.; et al. Gemcitabine and capecitabine with or without telomerase peptide vaccine GV1001 in patients with locally advanced or metastatic pancreatic cancer (TeloVac): An open-label, randomised, phase 3 trial. Lancet Oncol. 2014, 15, 829–840. [Google Scholar] [CrossRef]
- Caprotti, R.; Brivio, F.; Fumagalli, L.; Nobili, C.; Degrate, L.; Lissoni, P.; Parolini, D.; Messina, G.; Colciago, M.; Scotti, M.; et al. Free-from-progression period and overall short preoperative immunotherapy with IL-2 increases the survival of pancreatic cancer patients treated with macroscopically radical surgery. Anticancer. Res. 2008, 28, 1951–1954. [Google Scholar] [PubMed]
- Lygidakis, N.J.; E Berberabe, A.; Spentzouris, N.; Dedemadi, G.; Kalligas, T.; Loukas, G.; Sotiropoulou, V. A prospective randomized study using adjuvant locoregional chemoimmunotherapy in combination with surgery for pancreatic carcinoma. Hepatogastroenterology 1999, 45, 2376–2381. [Google Scholar]
- Circelli, L.; Tornesello, M.L.; Buonaguro, F.M.; Buonaguro, L. Use of adjuvants for immunotherapy. Hum. Vaccines Immunother. 2017, 13, 1774–1777. [Google Scholar] [CrossRef]
- Khong, H.; Overwijk, W.W. Adjuvants for peptide-based cancer vaccines. J. Immunother. Cancer 2016, 4, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fransen, M.F.; van der Sluis, T.; Ossendorp, F.; Arens, R.; Melief, C.J. Controlled Local Delivery of CTLA-4 Blocking Antibody Induces CD8+ T-Cell–Dependent Tumor Eradication and Decreases Risk of Toxic Side Effects. Clin. Cancer Res. 2013, 19, 5381–5389. [Google Scholar] [CrossRef] [Green Version]
- Francis, D.M.; Manspeaker, M.P.; Schudel, A.; Sestito, L.F.; O’Melia, M.J.; Kissick, H.T.; Pollack, B.P.; Waller, E.K.; Thomas, S.N. Blockade of immune checkpoints in lymph nodes through locoregional delivery augments cancer immunotherapy. Sci. Transl. Med. 2020, 12, eaay3575. [Google Scholar] [CrossRef]
- Nierkens, S.; Brok, M.H.D.; Roelofsen, T.; Wagenaars, J.A.L.; Figdor, C.G.; Ruers, T.J.; Adema, G.J. Route of Administration of the TLR9 Agonist CpG Critically Determines the Efficacy of Cancer Immunotherapy in Mice. PLoS ONE 2009, 4, e8368. [Google Scholar] [CrossRef] [Green Version]
- Krieg, A.M. Toll-like receptor 9 (TLR9) agonists in the treatment of cancer. Oncogene 2008, 27, 161–167. [Google Scholar] [CrossRef] [Green Version]
- Smits, E.; Ponsaerts, P.; Berneman, Z.; Van Tendeloo, V. The Use of TLR7 and TLR8 Ligands for the Enhancement of Cancer Immunotherapy. Oncology 2008, 13, 859–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dajon, M.; Iribarren, K.; Cremer, I. Toll-like receptor stimulation in cancer: A pro- and anti-tumor double-edged sword. Immunobiol. 2017, 222, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Leppänen, J.; Helminen, O.; Huhta, H.; Kauppila, J.H.; Isohookana, J.; Haapasaari, K.-M.; Lehenkari, P.; Saarnio, J.; Karttunen, T.J. High toll-like receptor (TLR) 9 expression is associated with better prognosis in surgically treated pancreatic cancer patients. Virchows Arch. 2017, 470, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Lanki, M.; Seppänen, H.; Mustonen, H.; Hagström, J.; Haglund, C. Toll-like receptor 1 predicts favorable prognosis in pancreatic cancer. PLoS ONE 2019, 14, e0219245. [Google Scholar] [CrossRef] [Green Version]
- A Lanki, M.; E Seppänen, H.; Mustonen, H.K.; Böckelman, C.; Juuti, A.T.; Hagström, J.; Haglund, C.H. Toll-like receptor 2 and Toll-like receptor 4 predict favorable prognosis in local pancreatic cancer. Tumor Biol. 2018, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zambirinis, C.; Levie, E.; Nguy, S.; Avanzi, A.; Barilla, R.; Xu, Y.; Seifert, L.; Daley, D.; Greco, S.H.; Deutsch, M.; et al. TLR9 ligation in pancreatic stellate cells promotes tumorigenesis. J. Exp. Med. 2015, 212, 2077–2094. [Google Scholar] [CrossRef]
- Sun, Y.; Wu, C.; Ma, J.; Yang, Y.; Man, X.; Wu, H.; Li, S. Toll-like receptor 4 promotes angiogenesis in pancreatic cancer via PI3K/AKT signaling. Exp. Cell Res. 2016, 347, 274–282. [Google Scholar] [CrossRef]
- Pandey, S.; Singh, S.; Anang, V.; Bhatt, A.N.; Natarajan, K.; Dwarakanath, B.S. Pattern Recognition Receptors in Cancer Progression and Metastasis. Cancer Growth Metastasis 2015, 8, CGM.S24314. [Google Scholar] [CrossRef]
- Jacobs, C.; Duewell, P.; Heckelsmiller, K.; Wei, J.; Bauernfeind, F.; Ellermeier, J.; Kisser, U.; Bauer, C.A.; Dauer, M.; Eigler, A.; et al. An ISCOM vaccine combined with a TLR9 agonist breaks immune evasion mediated by regulatory T cells in an orthotopic model of pancreatic carcinoma. Int. J. Cancer 2010, 128, 897–907. [Google Scholar] [CrossRef]
- Michaelis, K.A.; Norgard, M.A.; Zhu, X.; Levasseur, P.R.; Sivagnanam, S.; Liudahl, S.M.; Burfeind, K.G.; Olson, B.; Pelz, K.R.; Ramos, D.M.A.; et al. The TLR7/8 agonist R848 remodels tumor and host responses to promote survival in pancreatic cancer. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Zou, B.-B.; Wang, F.; Li, L.; Cheng, F.-W.; Jin, R.; Luo, X.; Zhu, L.-X.; Geng, X.; Zhang, S.-Q. Activation of Toll-like receptor 7 inhibits the proliferation and migration, and induces the apoptosis of pancreatic cancer cells. Mol. Med. Rep. 2015, 12, 6079–6085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pratesi, G.; Petrangolini, G.; Tortoreto, M.; Addis, A.; Belluco, S.; Rossini, A.; Selleri, S.; Rumio, C.; Menard, S.; Balsari, A. Therapeutic Synergism of Gemcitabine and CpG-Oligodeoxynucleotides in an Orthotopic Human Pancreatic Carcinoma Xenograft. Cancer Res. 2005, 65, 6388–6393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schölch, S.; Rauber, C.; Tietz, A.; Rahbari, N.N.; Bork, U.; Schmidt, T.; Kahlert, C.; Haberkorn, U.; Tomai, M.A.; Lipson, K.; et al. Radiotherapy combined with TLR7/8 activation induces strong immune responses against gastrointestinal tumors. Oncotarget 2014, 6, 4663–4676. [Google Scholar] [CrossRef] [Green Version]
- Narayanan, J.S.S.; Ray, P.; Hayashi, T.; Whisenant, T.C.; Vicente, D.; Carson, D.A.; Miller, A.M.; Schoenberger, S.P.; White, R.R. Irreversible Electroporation Combined with Checkpoint Blockade and TLR7 Stimulation Induces Antitumor Immunity in a Murine Pancreatic Cancer Model. Cancer Immunol. Res. 2019, 7, 1714–1726. [Google Scholar] [CrossRef]
- Lorkowski, M.; Atukorale, P.; Bielecki, P.; Tong, K.; Covarrubias, G.; Zhang, Y.; Loutrianakis, G.; Moon, T.; Santulli, A.; Becicka, W.; et al. Immunostimulatory nanoparticle incorporating two immune agonists for the treatment of pancreatic tumors. J. Control. Release 2020, 330, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Geboers, B.; Timmer, F.; Ruarus, A.; Pouw, J.; Schouten, E.; Bakker, J.; Puijk, R.; Nieuwenhuizen, S.; Dijkstra, M.; Tol, M.V.D.; et al. Irreversible Electroporation and Nivolumab Combined with Intratumoral Administration of a Toll-Like Receptor Ligand, as a Means of In Vivo Vaccination for Metastatic Pancreatic Ductal Adenocarcinoma (PANFIRE-III). A Phase-I Study Protocol. Cancers 2021, 13, 3902. [Google Scholar] [CrossRef]
- Jiang, M.; Chen, P.; Wang, L.; Li, W.; Chen, B.; Liu, Y.; Wang, H.; Zhao, S.; Ye, L.; He, Y.; et al. cGAS-STING, an important pathway in cancer immunotherapy. J. Hematol. Oncol. 2020, 13, 1–11. [Google Scholar] [CrossRef]
- Jing, W.; McAllister, D.; Vonderhaar, E.P.; Palen, K.; Riese, M.J.; Gershan, J.; Johnson, B.D.; Dwinell, M.B. STING agonist inflames the pancreatic cancer immune microenvironment and reduces tumor burden in mouse models. J. Immunother. Cancer 2019, 7, 115. [Google Scholar] [CrossRef]
- Kinkead, H.L.; Hopkins, A.; Lutz, E.; Wu, A.A.; Yarchoan, M.; Cruz, K.; Woolman, S.; Vithayathil, T.; Glickman, L.H.; Ndubaku, C.O.; et al. Combining STING-based neoantigen-targeted vaccine with checkpoint modulators enhances antitumor immunity in murine pancreatic cancer. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Foote, J.B.; Kok, M.; Leatherman, J.M.; Armstrong, T.D.; Marcinkowski, B.; Ojalvo, L.S.; Kanne, D.B.; Jaffee, E.; Dubensky, T.W.; Emens, L.A. A STING Agonist Given with OX40 Receptor and PD-L1 Modulators Primes Immunity and Reduces Tumor Growth in Tolerized Mice. Cancer Immunol. Res. 2017, 5, 468–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baird, J.R.; Friedman, D.; Cottam, B.; Dubensky, T.W.; Kanne, D.B.; Bambina, S.; Bahjat, K.S.; Crittenden, M.R.; Gough, M.J. Radiotherapy Combined with Novel STING-Targeting Oligonucleotides Results in Regression of Established Tumors. Cancer Res. 2015, 76, 50–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gogoi, H.; Mansouri, S.; Jin, L. The Age of Cyclic Dinucleotide Vaccine Adjuvants. Vaccines 2020, 8, 453. [Google Scholar] [CrossRef] [PubMed]
- Geddes, K.; Magalhães, J.G.; Girardin, S.E. Unleashing the therapeutic potential of NOD-like receptors. Nat. Rev. Drug Discov. 2009, 8, 465–479. [Google Scholar] [CrossRef]
- Hemminki, O.; Dos Santos, J.M.; Hemminki, A. Oncolytic viruses for cancer immunotherapy. J. Hematol. Oncol. 2020, 13, 1–15. [Google Scholar] [CrossRef]
- de Graaf, J.; de Vor, L.; Fouchier, R.; Hoogen, B.V.D. Armed oncolytic viruses: A kick-start for antitumor immunity. Cytokine Growth Factor Rev. 2018, 41, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, W.; Wang, R.; Zhang, N.; Shang, H.; Bi, Y.; Chen, D.; Zhang, C.; Li, L.; Yin, J.; et al. Reshaping the Immune Microenvironment by Oncolytic Herpes Simplex Virus in Murine Pancreatic Ductal Adenocarcinoma. Mol. Ther. 2020, 29, 744–761. [Google Scholar] [CrossRef]
- Watanabe, K.; Luo, Y.; Da, T.; Guedan, S.; Ruella, M.; Scholler, J.; Keith, B.; Young, R.M.; Engels, B.; Sorsa, S.; et al. Pancreatic cancer therapy with combined mesothelin-redirected chimeric antigen receptor T cells and cytokine-armed oncolytic adenoviruses. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Nakao, A.; Kasuya, H.; Sahin, T.T.; Nomura, N.; Kanzaki, A.; Misawa, M.; Shirota, T.; Yamada, S.; Fujii, T.; Sugimoto, H.; et al. A phase I dose-escalation clinical trial of intraoperative direct intratumoral injection of HF10 oncolytic virus in non-resectable patients with advanced pancreatic cancer. Cancer Gene Ther. 2010, 18, 167–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasuya, H.; Kodera, Y.; Nakao, A.; Yamamura, K.; Gewen, T.; Zhiwen, W.; Hotta, Y.; Yamada, S.; Fujii, T.; Fukuda, S.; et al. Phase I Dose-escalation Clinical Trial of HF10 Oncolytic Herpes Virus in 17 Japanese Patients with Advanced Cancer. Hepatogastroenterology 2014, 61, 599–605. [Google Scholar]
- Aguilar, L.K.; Shirley, L.; Chung, V.M.; Marsh, C.; Walker, J.; Coyle, W.; Marx, H.; Bekaii-Saab, T.; Lesinski, G.B.; Swanson, B.; et al. Gene-mediated cytotoxic immunotherapy as adjuvant to surgery or chemoradiation for pancreatic adenocarcinoma. Cancer Immunol. Immunother. 2015, 64, 727–736. [Google Scholar] [CrossRef]
- Noonan, A.; Farren, M.; Geyer, S.M.; Huang, Y.; Tahiri, S.; Ahn, D.; Mikhail, S.; Ciombor, K.K.; Pant, S.; Aparo, S.; et al. Randomized Phase 2 Trial of the Oncolytic Virus Pelareorep (Reolysin) in Upfront Treatment of Metastatic Pancreatic Adenocarcinoma. Mol. Ther. 2016, 24, 1150–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirooka, Y.; Kasuya, H.; Ishikawa, T.; Kawashima, H.; Ohno, E.; Villalobos, I.B.; Naoe, Y.; Ichinose, T.; Koyama, N.; Tanaka, M.; et al. A Phase I clinical trial of EUS-guided intratumoral injection of the oncolytic virus, HF10 for unresectable locally advanced pancreatic cancer. BMC Cancer 2018, 18, 1–9. [Google Scholar] [CrossRef]
- Hajda, J.; Lehmann, M.; Krebs, O.; Kieser, M.; Geletneky, K.; Jäger, D.; Dahm, M.; Huber, B.; Schöning, T.; Sedlaczek, O.; et al. A non-controlled, single arm, open label, phase II study of intravenous and intratumoral administration of ParvOryx in patients with metastatic, inoperable pancreatic cancer: ParvOryx02 protocol. BMC Cancer 2017, 17, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beatty, G.L.; O’Hara, M.; Lacey, S.F.; Torigian, D.A.; Nazimuddin, F.; Chen, F.; Kulikovskaya, I.M.; Soulen, M.C.; McGarvey, M.; Nelson, A.M.; et al. Activity of Mesothelin-Specific Chimeric Antigen Receptor T Cells Against Pancreatic Carcinoma Metastases in a Phase 1 Trial. Gastroenterology 2018, 155, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Aoki, T.; Matsushita, H.; Hoshikawa, M.; Hasegawa, K.; Kokudo, N.; Kakimi, K. Adjuvant combination therapy with gemcitabine and autologous γδ T-cell transfer in patients with curatively resected pancreatic cancer. Cytotherapy 2017, 19, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Kumai, T.; Mizukoshi, E.; Hashiba, T.; Nakagawa, H.; Kitahara, M.; Miyashita, T.; Mochizuki, T.; Goto, S.; Kamigaki, T.; Takimoto, R.; et al. Effect of adoptive T-cell immunotherapy on immunological parameters and prognosis in patients with advanced pancreatic cancer. Cytotherapy 2020, 23, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Valilou, S.F.; Rezaei, N. Chapter 4—Tumor Antigens. In Vaccines for Cancer Immunotherapy; Rezaei, N., Keshavarz-Fathi, M., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 61–74. [Google Scholar] [CrossRef]
- Haen, S.P.; Löffler, M.W.; Rammensee, H.-G.; Brossart, P. Towards new horizons: Characterization, classification and implications of the tumour antigenic repertoire. Nat. Rev. Clin. Oncol. 2020, 17, 1–16. [Google Scholar] [CrossRef]
- Zhang, R.; Billingsley, M.; Mitchell, M.J. Biomaterials for vaccine-based cancer immunotherapy. J. Control. Release 2018, 292, 256–276. [Google Scholar] [CrossRef]
- Kaida, M.; Morita-Hoshi, Y.; Soeda, A.; Wakeda, T.; Yamaki, Y.; Kojima, Y.; Ueno, H.; Kondo, S.; Morizane, C.; Ikeda, M.; et al. Phase 1 Trial of Wilms Tumor 1 (WT1) Peptide Vaccine and Gemcitabine Combination Therapy in Patients With Advanced Pancreatic or Biliary Tract Cancer. J. Immunother. 2011, 34, 92–99. [Google Scholar] [CrossRef] [Green Version]
- Nishida, S.; Koido, S.; Takeda, Y.; Homma, S.; Komita, H.; Takahara, A.; Morita, S.; Ito, T.; Morimoto, S.; Hara, K.; et al. Wilms Tumor Gene (WT1) Peptide–based Cancer Vaccine Combined With Gemcitabine for Patients With Advanced Pancreatic Cancer. J. Immunother. 2014, 37, 105–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koido, S.; Homma, S.; Okamoto, M.; Takakura, K.; Mori, M.; Yoshizaki, S.; Tsukinaga, S.; Odahara, S.; Koyama, S.; Imazu, H.; et al. Treatment with Chemotherapy and Dendritic Cells Pulsed with Multiple Wilms’ Tumor 1 (WT1)–Specific MHC Class I/II–Restricted Epitopes for Pancreatic Cancer. Clin. Cancer Res. 2014, 20, 4228–4239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukinaga, S.; Kajihara, M.; Takakura, K.; Ito, Z.; Kanai, T.; Saito, K.; Takami, S.; Kobayashi, H.; Matsumoto, Y.; Odahara, S.; et al. Prognostic significance of plasma interleukin-6/-8 in pancreatic cancer patients receiving chemoimmunotherapy. World J. Gastroenterol. 2015, 21, 11168–11178. [Google Scholar] [CrossRef] [PubMed]
- Mayanagi, S.; Kitago, M.; Sakurai, T.; Matsuda, T.; Fujita, T.; Higuchi, H.; Taguchi, J.; Takeuchi, H.; Itano, O.; Aiura, K.; et al. Phase I pilot study of Wilms tumor gene 1 peptide-pulsed dendritic cell vaccination combined with gemcitabine in pancreatic cancer. Cancer Sci. 2015, 106, 397–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagisawa, R.; Koizumi, T.; Koya, T.; Sano, K.; Koido, S.; Nagai, K.; Kobayashi, M.; Okamoto, M.; Sugiyama, H.; Shimodaira, S. WT1-pulsed Dendritic Cell Vaccine Combined with Chemotherapy for Resected Pancreatic Cancer in a Phase I Study. Anticancer. Res. 2018, 38, 2217–2225. [Google Scholar] [CrossRef] [Green Version]
- Nishida, S.; Ishikawa, T.; Egawa, S.; Koido, S.; Yanagimoto, H.; Ishii, J.; Kanno, Y.; Kokura, S.; Yasuda, H.; Oba, M.S.; et al. Combination Gemcitabine and WT1 Peptide Vaccination Improves Progression-Free Survival in Advanced Pancreatic Ductal Adenocarcinoma: A Phase II Randomized Study. Cancer Immunol. Res. 2018, 6, 320–331. [Google Scholar] [CrossRef] [Green Version]
- Hanada, S.; Tsuruta, T.; Haraguchi, K.; Okamoto, M.; Sugiyama, H.; Koido, S. Long-term survival of pancreatic cancer patients treated with multimodal therapy combined with WT1-targeted dendritic cell vaccines. Hum. Vaccines Immunother. 2018, 15, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Nagai, K.; Adachi, T.; Harada, H.; Eguchi, S.; Sugiyama, H.; Miyazaki, Y. Dendritic Cell-based Immunotherapy Pulsed With Wilms Tumor 1 Peptide and Mucin 1 as an Adjuvant Therapy for Pancreatic Ductal Adenocarcinoma After Curative Resection: A Phase I/IIa Clinical Trial. Anticancer. Res. 2020, 40, 5765–5776. [Google Scholar] [CrossRef]
- Asahara, S.; Takeda, K.; Yamao, K.; Maguchi, H.; Yamaue, H. Phase I/II clinical trial using HLA-A24-restricted peptide vaccine derived from KIF20A for patients with advanced pancreatic cancer. J. Transl. Med. 2013, 11, 291. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, N.; Hazama, S.; Ueno, T.; Matsui, H.; Shindo, Y.; Iida, M.; Yoshimura, K.; Yoshino, S.; Takeda, K.; Oka, M. A Phase I Clinical Trial of Vaccination With KIF20A-derived Peptide in Combination with Gemcitabine for Patients with Advanced Pancreatic Cancer. J. Immunother. 2014, 37, 36–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazawa, M.; Ohsawa, R.; Tsunoda, T.; Hirono, S.; Kawai, M.; Tani, M.; Nakamura, Y.; Yamaue, H. Phase I clinical trial using peptide vaccine for human vascular endothelial growth factor receptor 2 in combination with gemcitabine for patients with advanced pancreatic cancer. Cancer Sci. 2010, 101, 433–439. [Google Scholar] [CrossRef]
- Yamaue, H.; Tsunoda, T.; Tani, M.; Miyazawa, M.; Yamao, K.; Mizuno, N.; Okusaka, T.; Ueno, H.; Boku, N.; Fukutomi, A.; et al. Randomized phase II/III clinical trial of elpamotide for patients with advanced pancreatic cancer: PEGASUS—PC Study. Cancer Sci. 2015, 106, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Hazama, S.; Iguchi, H.; Uesugi, K.; Tanaka, H.; Hirakawa, K.; Aruga, A.; Hatori, T.; Ishizaki, H.; Umeda, Y.; et al. Phase II clinical trial of peptide cocktail therapy for patients with advanced pancreatic cancer: VENUS-PC study. Cancer Sci. 2016, 108, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Miyazawa, M.; Katsuda, M.; Maguchi, H.; Katanuma, A.; Ishii, H.; Ozaka, M.; Yamao, K.; Imaoka, H.; Kawai, M.; Hirono, S.; et al. Phase II clinical trial using novel peptide cocktail vaccine as a postoperative adjuvant treatment for surgically resected pancreatic cancer patients. Int. J. Cancer 2016, 140, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Kameshima, H.; Tsuruma, T.; Kutomi, G.; Shima, H.; Iwayama, Y.; Kimura, Y.; Imamura, M.; Torigoe, T.; Takahashi, A.; Hirohashi, Y.; et al. Immunotherapeutic benefit of α-interferon (IFNα) in survivin2B-derived peptide vaccination for advanced pancreatic cancer patients. Cancer Sci. 2012, 104, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Shima, H.; Tsurita, G.; Wada, S.; Hirohashi, Y.; Yasui, H.; Hayashi, H.; Miyakoshi, T.; Watanabe, K.; Murai, A.; Asanuma, H.; et al. Randomized phase II trial of survivin 2B peptide vaccination for patients with HLA -A24-positive pancreatic adenocarcinoma. Cancer Sci. 2019, 110, 2378–2385. [Google Scholar] [CrossRef] [Green Version]
- Rong, Y.; Qin, X.; Jin, D.; Lou, W.; Wu, L.; Wang, D.; Wu, W.; Ni, X.; Mao, Z.; Kuang, T.; et al. A phase I pilot trial of MUC1-peptide-pulsed dendritic cells in the treatment of advanced pancreatic cancer. Clin. Exp Med. 2011, 12, 173–180. [Google Scholar] [CrossRef]
- Le, D.T.; Brockstedt, D.G.; Nir-Paz, R.; Hampl, J.; Mathur, S.; Nemunaitis, J.; Sterman, D.; Hassan, R.; Lutz, E.; Moyer, B.; et al. A Live-Attenuated Listeria Vaccine (ANZ-100) and a Live-Attenuated Listeria Vaccine Expressing Mesothelin (CRS-207) for Advanced Cancers: Phase I Studies of Safety and Immune Induction. Clin. Cancer Res. 2011, 18, 858–868. [Google Scholar] [CrossRef] [Green Version]
- Abou-Alfa, G.K.; Chapman, P.B.; Feilchenfeldt, J.; Brennan, M.; Capanu, M.; Gansukh, B.; Jacobs, G.; Levin, A.; Neville, D.; Kelsen, D.P.; et al. Targeting Mutated K-ras in Pancreatic Adenocarcinoma Using an Adjuvant Vaccine. Am. J. Clin. Oncol. 2011, 34, 321–325. [Google Scholar] [CrossRef]
- Kubuschok, B.; Pfreundschuh, M.; Breit, R.; Hartmann, F.; Sester, M.; Gärtner, B.; König, J.; Murawski, N.; Held, G.; Zwick, C.; et al. Mutated Ras-Transfected, EBV-Transformed Lymphoblastoid Cell Lines as a Model Tumor Vaccine for Boosting T-Cell Responses Against Pancreatic Cancer: A Pilot Trial. Hum. Gene Ther. 2012, 23, 1224–1236. [Google Scholar] [CrossRef]
- Bassani-Sternberg, M.; Digklia, A.; Huber, F.; Wagner, D.; Sempoux, C.; Stevenson, B.J.; Thierry, A.-C.; Michaux, J.; Pak, H.; Racle, J.; et al. A Phase Ib Study of the Combination of Personalized Autologous Dendritic Cell Vaccine, Aspirin, and Standard of Care Adjuvant Chemotherapy Followed by Nivolumab for Resected Pancreatic Adenocarcinoma—A Proof of Antigen Discovery Feasibility in Three Patients. Front. Immunol. 2019, 10, 1832. [Google Scholar] [CrossRef] [Green Version]
- Noguchi, M.; Yanagimoto, H.; Shiomi, H.; Satoi, S.; Mine, T.; Toyokawa, H.; Yamamoto, T.; Tani, T.; Yamada, A.; Kwon, A.-H.; et al. A phase II study of personalized peptide vaccination combined with gemcitabine for non-resectable pancreatic cancer patients. Oncol. Rep. 2010, 24, 795–801. [Google Scholar] [CrossRef]
- Bauer, C.; Dauer, M.; Saraj, S.; Schnurr, M.; Bauernfeind, F.; Sterzik, A.; Junkmann, J.; Jakl, V.; Kiefl, R.; Oduncu, F.; et al. Dendritic cell-based vaccination of patients with advanced pancreatic carcinoma: Results of a pilot study. Cancer Immunol. Immunother. 2011, 60, 1097–1107. [Google Scholar] [CrossRef]
- Kimura, Y.; Tsukada, J.; Tomoda, T.; Takahashi, H.; Imai, K.; Shimamura, K.; Sunamura, M.; Yonemitsu, Y.; Shimodaira, S.; Koido, S.; et al. Clinical and Immunologic Evaluation of Dendritic Cell–Based Immunotherapy in Combination With Gemcitabine and/or S-1 in Patients With Advanced Pancreatic Carcinoma. Pancreas 2012, 41, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Yutani, S.; Komatsu, N.; Yoshitomi, M.; Matsueda, S.; Yonemoto, K.; Mine, T.; Noguchi, M.; Ishihara, Y.; Yamada, A.; Itoh, K.; et al. A phase II study of a personalized peptide vaccination for chemotherapy-resistant advanced pancreatic cancer patients. Oncol. Rep. 2013, 30, 1094–1100. [Google Scholar] [CrossRef]
- Qiu, Y.; Yun, M.M.; Xu, M.B.; Wang, Y.Z.; Yun, S. Pancreatic carcinoma-specific immunotherapy using synthesised alpha-galactosyl epitope-activated immune responders: Findings from a pilot study. Int. J. Clin. Oncol. 2012, 18, 657–665. [Google Scholar] [CrossRef]
- Lin, M.; Yuan, Y.-Y.; Liu, S.-P.; Shi, J.-J.; Long, X.-A.; Niu, L.-Z.; Chen, J.-B.; Li, Q.; Xu, K.-C. Prospective study of the safety and efficacy of a pancreatic cancer stem cell vaccine. J. Cancer Res. Clin. Oncol. 2015, 141, 1827–1833. [Google Scholar] [CrossRef]
- Zhang, L.; Zhu, W.; Li, J.; Yang, X.; Ren, Y.; Niu, J.; Pang, Y. Clinical outcome of immunotherapy with dendritic cell vaccine and cytokine-induced killer cell therapy in hepatobiliary and pancreatic cancer. Mol. Clin. Oncol. 2015, 4, 129–133. [Google Scholar] [CrossRef] [Green Version]
- Mehrotra, S.; Britten, C.D.; Chin, S.; Garrett-Mayer, E.; Cloud, C.A.; Li, M.; Scurti, G.; Salem, M.; Nelson, M.H.; Thomas, M.B.; et al. Vaccination with poly(IC:LC) and peptide-pulsed autologous dendritic cells in patients with pancreatic cancer. J. Hematol. Oncol. 2017, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ushach, I.; Zlotnik, A. Biological role of granulocyte macrophage colony-stimulating factor (GM-CSF) and macrophage colony-stimulating factor (M-CSF) on cells of the myeloid lineage. J. Leukoc. Biol. 2016, 100, 481–489. [Google Scholar] [CrossRef]
- Cheever, M.A.; Allison, J.; Ferris, A.S.; Finn, O.J.; Hastings, B.M.; Hecht, T.T.; Mellman, I.; Prindiville, S.A.; Viner, J.L.; Weiner, L.M.; et al. The Prioritization of Cancer Antigens: A National Cancer Institute Pilot Project for the Acceleration of Translational Research. Clin. Cancer Res. 2009, 15, 5323–5337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oji, Y.; Nakamori, S.; Fujikawa, M.; Nakatsuka, S.-I.; Yokota, A.; Tatsumi, N.; Abeno, S.; Ikeba, A.; Takashima, S.; Tsujie, M.; et al. Overexpression of the Wilms’ tumor gene WT1 in pancreatic ductal adenocarcinoma. Cancer Sci. 2004, 95, 583–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koido, S.; Okamoto, M.; Kobayashi, M.; Shimodaira, S.; Sugiyama, H. Significance of Wilms’ tumor 1 antigen as a cancer vaccine for pancreatic cancer. Discov. Med. 2017, 24, 41–49. [Google Scholar]
- Taniuchi, K.; Nakagawa, H.; Nakamura, T.; Eguchi, H.; Ohigashi, H.; Ishikawa, O.; Katagiri, T.; Nakamura, Y. Down-regulation of RAB6KIFL/KIF20A, a kinesin involved with membrane trafficking of discs large homologue 5, can attenuate growth of pancreatic cancer cell. Cancer Res. 2005, 65, 105–112. [Google Scholar] [PubMed]
- Imai, K.; Hirata, S.; Irie, A.; Senju, S.; Ikuta, Y.; Yokomine, K.; Harao, M.; Inoue, M.; Tomita, Y.; Tsunoda, T.; et al. Identification of HLA-A2-restricted CTL epitopes of a novel tumour-associated antigen, KIF20A, overexpressed in pancreatic cancer. Br. J. Cancer 2010, 104, 300–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapisarda, A.; Melillo, G. Role of the VEGF/VEGFR Axis in Cancer Biology and Therapy. Adv. Cancer Res. 2012, 114, 237–267. [Google Scholar] [CrossRef]
- Seo, Y.; Baba, H.; Fukuda, T.; Takashima, M.; Sugimachi, K. High expression of vascular endothelial growth factor is associated with liver metastasis and a poor prognosis for patients with ductal pancreatic adenocarcinoma. Cancer 2000, 88, 2239–2245. [Google Scholar] [CrossRef]
- Liang, Q.-L.; Wang, B.-R.; Chen, G.-Q.; Li, G.-H.; Xu, Y.-Y. Clinical significance of vascular endothelial growth factor and connexin43 for predicting pancreatic cancer clinicopathologic parameters. Med. Oncol. 2009, 27, 1164–1170. [Google Scholar] [CrossRef]
- Dong, H.; Qian, N.; Wang, Y.; Meng, L.; Chen, D.; Ji, X.; Feng, W. Survivin expression and serum levels in pancreatic cancer. World J. Surg. Oncol. 2015, 13, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, M.; Zhang, W.; Yan, D.; Kenath, R.; Le, L.T.T.; Wang, H.; Delitto, D.; Ostrov, D.; Robertson, K.; Liu, C.; et al. The role of survivin in the progression of pancreatic ductal adenocarcinoma (PDAC) and a novel survivin-targeted therapeutic for PDAC. PLoS ONE 2020, 15, e0226917. [Google Scholar] [CrossRef] [PubMed]
- Suh, H.; Pillai, K.; Morris, D.L. Mucins in pancreatic cancer: Biological role, implications in carcinogenesis and applications in diagnosis and therapy. Am. J. Cancer Res. 2017, 7, 1372–1383. [Google Scholar]
- Lv, J.; Li, P. Mesothelin as a biomarker for targeted therapy. Biomark. Res. 2019, 7, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.-H.; Hung, W.-C.; Wang, P.; Paul, C.; Konstantopoulos, K. Mesothelin Binding to CA125/MUC16 Promotes Pancreatic Cancer Cell Motility and Invasion via MMP-7 Activation. Sci. Rep. 2013, 3, srep01870. [Google Scholar] [CrossRef] [Green Version]
- Jafri, M.A.; Ansari, S.A.; Alqahtani, M.H.; Shay, J.W. Roles of telomeres and telomerase in cancer, and advances in telomerase-targeted therapies. Genome Med. 2016, 8, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Reber, H.A.; Dry, S.M.; Elashoff, D.; Chen, S.L.; Umetani, N.; Kitago, M.; Hines, O.J.; Kazanjian, K.K.; Hiramatsu, S.; et al. Unfavourable prognosis associated with K-ras gene mutation in pancreatic cancer surgical margins. Gut 2006, 55, 1598–1605. [Google Scholar] [CrossRef] [Green Version]
- Uesaka, K.; Boku, N.; Fukutomi, A.; Okamura, Y.; Konishi, M.; Matsumoto, I.; Kaneoka, Y.; Shimizu, Y.; Nakamori, S.; Sakamoto, H.; et al. Adjuvant chemotherapy of S-1 versus gemcitabine for resected pancreatic cancer: A phase 3, open-label, randomised, non-inferiority trial (JASPAC 01). Lancet 2016, 388, 248–257. [Google Scholar] [CrossRef]
- Neoptolemos, J.P.; Stocken, D.D.; Bassi, C.; Ghaneh, P.; Cunningham, D.; Goldstein, D.; Padbury, R.; Moore, M.J.; Gallinger, S.; Mariette, C.; et al. Adjuvant Chemotherapy With Fluorouracil Plus Folinic Acid vs. Gemcitabine Following Pancreatic Cancer Resection. JAMA 2010, 304, 1073–1081. [Google Scholar] [CrossRef]
- Oettle, H.; Neuhaus, P.; Hochhaus, A.; Hartmann, J.T.; Gellert, K.; Ridwelski, K.; Niedergethmann, M.; Zülke, C.; Fahlke, J.; Arning, M.B.; et al. Adjuvant Chemotherapy With Gemcitabine and Long-term Outcomes Among Patients With Resected Pancreatic Cancer. JAMA 2013, 310, 1473–1481. [Google Scholar] [CrossRef] [Green Version]
- Van Laethem, J.-L.; Hammel, P.; Mornex, F.; Azria, D.; Van Tienhoven, G.; Vergauwe, P.; Peeters, M.; Polus, M.; Praet, M.; Mauer, M.; et al. Adjuvant Gemcitabine Alone Versus Gemcitabine-Based Chemoradiotherapy After Curative Resection for Pancreatic Cancer: A Randomized EORTC-40013-22012/FFCD-9203/GERCOR Phase II Study. J. Clin. Oncol. 2010, 28, 4450–4456. [Google Scholar] [CrossRef]
- Ott, P.A.; Shuqiang, L.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nat. Cell Biol. 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu-Lieskovan, S.; Chmielowski, B.; Govindan, R.; Naing, A.; Bhardwaj, N.; Margolin, K.; Awad, M.M.; Hellmann, M.D.; Lin, J.J.; et al. A Phase Ib Trial of Personalized Neoantigen Therapy Plus Anti-PD-1 in Patients with Advanced Melanoma, Non-small Cell Lung Cancer, or Bladder Cancer. Cell 2020, 183, 347–362. [Google Scholar] [CrossRef]
- Keskin, D.B.; Anandappa, A.; Sun, J.; Tirosh, I.; Mathewson, N.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nat. Cell Biol. 2018, 565, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Blass, E.; Ott, P.A. Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat. Rev. Clin. Oncol. 2021, 18, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Homma, Y.; Matsuyama, R.; Mori, R.; Miyake, K.; Tanaka, Y.; Den, K.; Nagashima, Y.; Nakazawa, M.; Hiroshima, Y.; et al. Neoadjuvant chemoradiotherapy of pancreatic cancer induces a favorable immunogenic tumor microenvironment associated with increased major histocompatibility complex class I-related chain A/B expression. J. Surg. Oncol. 2017, 116, 416–426. [Google Scholar] [CrossRef]
- Carvalho, R.C.V.H.D.A.; Villar, R.C. Radiotherapy and immune response: The systemic effects of a local treatment. Clinics 2018, 73, e557s. [Google Scholar] [CrossRef]
- Lin, M.; Liang, S.; Wang, X.; Liang, Y.; Zhang, M.; Chen, J.; Niu, L.; Xu, K. Percutaneous irreversible electroporation combined with allogeneic natural killer cell immunotherapy for patients with unresectable (stage III/IV) pancreatic cancer: A promising treatment. J. Cancer Res. Clin. Oncol. 2017, 143, 2607–2618. [Google Scholar] [CrossRef]
- Lin, M.; Zhang, X.; Liang, S.; Luo, H.; Alnaggar, M.; Liu, A.; Yin, Z.; Chen, J.; Niu, L.; Jiang, Y. Irreversible electroporation plus allogenic Vγ9Vδ2 T cells enhances antitumor effect for locally advanced pancreatic cancer patients. Signal Transduct. Target. Ther. 2020, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, C.; Hayat, T.; Hamm, J.; Healey, M.; Zheng, Q.; Li, Y.; Martin, R.C. A phase 1b trial of concurrent immunotherapy and irreversible electroporation in the treatment of locally advanced pancreatic adenocarcinoma. Surgery 2020, 168, 610–616. [Google Scholar] [CrossRef]
- Lin, C.; Verma, V.; Lazenby, A.; Ly, Q.P.; Berim, L.D.; Schwarz, J.K.; Madiyalakan, M.; Nicodemus, C.F.; Hollingsworth, M.A.; Meza, J.L.; et al. Phase I/II Trial of Neoadjuvant Oregovomab-based Chemoimmunotherapy Followed by Stereotactic Body Radiotherapy and Nelfinavir For Locally Advanced Pancreatic Adenocarcinoma. Am. J. Clin. Oncol. 2019, 42, 755–760. [Google Scholar] [CrossRef]
- Xie, C.; Duffy, A.G.; Brar, G.; Fioravanti, S.; Mabry-Hrones, D.; Walker, M.; Bonilla, C.M.; Wood, B.J.; Citrin, D.E.; Gil Ramirez, E.M.; et al. Immune Checkpoint Blockade in Combination with Stereotactic Body Radiotherapy in Patients with Metastatic Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2020, 26, 2318–2326. [Google Scholar] [CrossRef] [Green Version]
- Scheffer, H.J.; Stam, A.G.; Geboers, B.; Vroomen, L.G.; Ruarus, A.; De Bruijn, B.; Tol, M.P.V.D.; Kazemier, G.; Meijerink, M.R.; De Gruijl, T.D. Irreversible electroporation of locally advanced pancreatic cancer transiently alleviates immune suppression and creates a window for antitumor T cell activation. OncoImmunology 2019, 8, 1652532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandit, H.; Hong, Y.K.; Li, Y.; Rostas, J.; Pulliam, Z.; Li, S.P.; Martin, R.C.G. Evaluating the Regulatory Immunomodulation Effect of Irreversible Electroporation (IRE) in Pancreatic Adenocarcinoma. Ann. Surg. Oncol. 2019, 26, 800–806. [Google Scholar] [CrossRef]
- Geboers, B.; Scheffer, H.J.; Graybill, P.M.; Ruarus, A.H.; Nieuwenhuizen, S.; Puijk, R.S.; Tol, P.M.V.D.; Davalos, R.V.; Rubinsky, B.; De Gruijl, T.D.; et al. High-Voltage Electrical Pulses in Oncology: Irreversible Electroporation, Electrochemotherapy, Gene Electrotransfer, Electrofusion, and Electroimmunotherapy. Radiology 2020, 295, 254–272. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Eresen, A.; Shangguan, J.; Ma, Q.; Yaghmai, V.; Zhang, Z. Irreversible electroporation ablation overcomes tumor-associated immunosuppression to improve the efficacy of DC vaccination in a mice model of pancreatic cancer. OncoImmunology 2021, 10, 1875638. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wen, X.; Tian, L.; Li, T.; Xu, C.; Wen, X.; Melancon, M.P.; Gupta, S.; Shen, B.; Peng, W.; et al. Irreversible electroporation reverses resistance to immune checkpoint blockade in pancreatic cancer. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Liu, Y.; He, C.; Hu, W.; Liu, W.; Huang, X.; Wu, J.; Xie, F.; Chen, C.; Wang, J.; et al. Combining NanoKnife with M1 oncolytic virus enhances anticancer activity in pancreatic cancer. Cancer Lett. 2021, 502, 9–24. [Google Scholar] [CrossRef]
- Mills, B.N.; Connolly, K.A.; Ye, J.; Murphy, J.; Uccello, T.; Han, B.J.; Zhao, T.; Drage, M.G.; Murthy, A.; Qiu, H.; et al. Stereotactic Body Radiation and Interleukin-12 Combination Therapy Eradicates Pancreatic Tumors by Repolarizing the Immune Microenvironment. Cell Rep. 2019, 29, 406–421. [Google Scholar] [CrossRef] [Green Version]
- Yasmin-Karim, S.; Bruck, P.T.; Moreau, M.; Kunjachan, S.; Chen, G.Z.; Kumar, R.; Grabow, S.; Dougan, S.K.; Ngwa, W. Radiation and Local Anti-CD40 Generate an Effective in situ Vaccine in Preclinical Models of Pancreatic Cancer. Front. Immunol. 2018, 9, 2030. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen Presenting Cell or Tumor Cell (Ligand) | T Cell (Receptor) |
|---|---|
| MHC class I or II (antigen loaded) | TCR (signal 1), LAG-3 |
| PD-L1 (B7-H1), PD-L2 (B7-DC) | PD-1 |
| Gal-9 | TIM-3 |
| CD155, CD112 | TIGIT |
| CD80 (B7-1), CD86 (B7-2) | CD28 (signal 2), CTLA-4 |
| ICOSL | ICOS |
| CD137L | CD137 (4-1BB) |
| OX40L | OX40 |
| CD70 | CD27 |
| Antigen Presenting Cell or Tumor Cell (Receptor) | T Cell (Ligand) |
| CD40 | CD40L |
| Authors | Year | Phase | Immunotherapy | Combined with | No. of Patients | Patient Population | mPFS (Months) | mOS (Months) |
|---|---|---|---|---|---|---|---|---|
| Anti-PD-1/ PD-L1 | ||||||||
| Brahmer [44] | 2012 | 1 | Anti-PD-L1 | - | 14 | Previously treated LAPC/mPDAC | NR | NR |
| Weiss [47] | 2017 | 1b | Anti-PD-1 | + chemo | 11 | Previously treated mPDAC | NR | 8 (x) |
| Weiss [48] | 2018 | 1b/2 | Anti-PD-1 | + gem/nab-pac | 17 | mPDAC, 11/17 chemotherapy-naïve | 9.1 | 15 (x) |
| Doi [51] | 2019 | 1 | Anti-PD-1 | + anti-CCR4 | 15 | Previously treated mPDAC | 1.8 | 6.5 (x) |
| Hong [52] | 2019 | 1b/2 | Anti-PD-L1 | + TKI | 49 | Previously treated LAPC/mPDAC | 1.7 | 4.2 (t) |
| O’Reilly [46] | 2019 | 2 | Anti-PD-L1 Anti-CTLA-4 | Randomization: (1) − (2) + anti-CTLA-4 | 65 | Previously treated mPDAC | NR | NR |
| Bockorny [49] | 2020 | 2a | Anti-PD-1 | Two cohorts: (1) + anti-CXCR4 (2) + anti-CXCR4 + chemo | 59 | Previously treated mPDAC | NR | (1) 3.3 (t) (2) 7.8 (t) |
| Marabelle [45] | 2020 | 2 | Anti-PD-1 | - | 22 | Previously treated MSI-H LAPC/mPDAC | NR | NR |
| Mahalingam [50] | 2020 | 1b | Anti-PD-1 | + oncolytic virus + chemo | 11 | Previously treated LAPC/mPDAC | 2 | 3.1 (x) |
| Anti-CTLA-4 | ||||||||
| Royal [56] | 2010 | 2 | Anti-CTLA-4 | 27 | Previously treated LAPC/mPDAC | NR | NR | |
| Le [57] | 2013 | 1b | Anti-CTLA-4 | Randomization: (1) − (2) + GVAX | 30 | Previously treated LAPC/mPDAC | (1) 3.6 (t) (2) 5.7 (t) | |
| Aglietta [58] | 2014 | 1 | Anti-CTLA-4 | + gem | 34 | chemotherapy naïve mPDAC | NR | 7.4 (x) |
| Mohindra [59] | 2015 | 1b | Anti-CTLA-4 | + gem | 13 | Previously treated LAPC/mPDAC | NR | NR |
| Kalyan [60] | 2016 | 1b | Anti-CTLA-4 | + gem | 16 | Previously treated LAPC/mPDAC | 2.5 | 8.3 (x) |
| Kamath [61] | 2020 | 1b | Anti-CTLA-4 | + gem | 21 | Previously treated LAPC/mPDAC | 2.8 | 6.9 (x) |
| Authors | Year | Phase | Immunotherapy | Combined with | No. of Patients | Patient Population | mPFS (Months) | mOS (Months) |
|---|---|---|---|---|---|---|---|---|
| MHC-II agonist | ||||||||
| Wang-Gillam [82] | 2013 | 1 | MHC-II agonist | (1) + gem (2) gem alone (control) | 18 | Treatment-naïve LAPC/mPDAC | (1) 2–5.3 (2) 10.2 | (1) 5.6–6.4 (x) (2) 16.7 (x) |
| CD40 agonist | ||||||||
| Beatty [83,84] | 2011, 2013 | 1 | CD40 agonist | + gem | 22 | Chemotherapy-naïve LAPC/mPDAC | 5.2 | 8.4 (t) |
| O’Hara [85] | 2021 | 1b | CD40 agonist | + gem ± anti-PD1 | 24 | Recurrent or synchronous mPDAC | 10.8–12.5 | 12.7–20.1 (t) |
| TLR agonist | ||||||||
| Dalgleish [86] | 2016 | 2 | TLR2/1 agonist (i.t.) | (1) + gem (2) gem alx one (control) | 110 | Chemotherapy-naïve LAPC/mPDAC | NR | mPDAC: (1) 7 (t) (2) 4.4 (t) |
| Authors | Year | Phase | Immunotherapy | Admin. Site | Combined with | No. of Patients | Patient Population | mPFS (Months) | mOS (Months) |
|---|---|---|---|---|---|---|---|---|---|
| Oncolytic viruses | |||||||||
| Nakao [151], Kasuya [152] | 2011, 2014 | 1 | HF10 (HSV) | i.t. | - | 6 | Unresectable (intraoperative) PDAC | NR | 6.2 (x) |
| Aguilar [153] | 2015 | 1 | Adenoviral vector expressing HSV-tk gene | i.t. | + anti-herpetic prodrug | 24 | As adjuvant of: (1) Surgical resection (2) LAPC: CRT | (1) NR (2) 5.8 | (1) NR (2) 12 (t) |
| Noonan [154] | 2016 | 2 | Pelareorep | i.v. | (1) + carb/pac (2) carb/pac alone | 73 | Treatment-naïve mPDAC | (1) 4.9 (2) 5.2 | (1) 7.3 (t) (2) 8 (t) |
| Hirooka [155] | 2018 | 1 | HF10 (HSV) | i.t. | + gem + erlotinib | 10 | Chemotherapy-naïve LAPC | 6.3 | 15.5 (t) |
| Authors | Year | Phase | Immunotherapy | Combined with | No. of Patients | Patient Population | mPFS (Months) | mOS (Months) |
|---|---|---|---|---|---|---|---|---|
| T cell therapy | ||||||||
| Aoki [158] | 2017 | 1 | Vγ9Vδ2 T cells | (1) + gem (2) gem alone | 40 | Resected PDAC | NR | NR |
| Beatty [157] | 2018 | 1 | CAR T cells: mesothelin-directed | - | 6 | Previously treated mPDAC | NR | NR |
| Haas [94] | 2019 | 1 | CAR T cells: Transduced with mesothelin, 4-1BB and CD3ζ | - | 5 | Previously treated mPDAC | NR | NR |
| Kumai [159] | 2021 | NR | αβ T cells | ± chemo | 77 | Previously treated LAPC/mPDAC | NR | 11.3 (t) 18.7 (d) |
| Authors | Year | Phase | Immunotherapy | Type of Vaccine | Admin. Site | Combined with | No. of Patients | Patient Population | mPFS (Months) | mOS (Months) |
|---|---|---|---|---|---|---|---|---|---|---|
| GVAX | ||||||||||
| Lutz [108] | 2011 | 2 | GVAX (+ GM-CSF) | Whole-tumor-cell | i.d. | + resection + CRT | 60 | Resected PDAC | 17.3 | 24.8 (t) |
| Le [109] | 2015 | 2 | GVAX (+ GM-CSF) | Whole-tumor-cell | i.d. | (1) + Cy + CRS-207 (2) + Cy | 90 | Pre-treated mPDAC | NR | (1) 6.1 (t) (2) 3.9 (t) |
| Le [110] | 2019 | 2b | GVAX (+ GM-CSF) | Whole-tumor-cell | i.d. | (1) + Cy + CRS-207 (2) + Cy (3) chemo only | 169 | Pre-treated mPDAC | (1) 2.3 (2) 2.1 (3) 2.1 | (1) 3.7 (t) (2) 5.4 (t) (3) 4.6 (t) |
| Tsujikawa [111] | 2020 | 2 | GVAX (+ GM-CSF) | Whole-tumor-cell | i.d. | (1) + Cy + CRS-207 + anti-PD-1 (2) + Cy + CRS-207 | 93 | Pre-treated mPDAC | (1) 2.2 (2) 2.2 | (1) 5.9 (t) (2) 6.1 (t) |
| Wu [112] | 2020 | 2 | GVAX (+ GM-CSF) | Whole-tumor-cell | i.d. | (1) + anti-CTLA-4 (2) FFX only (continuation) | 82 | Pre-treated mPDAC (4–6 doses FFX) | (1) 2.4 (2) 5.6 | (1) 9.4 (t) (2) 14.7 (t) |
| WT-1 | ||||||||||
| Kaida [163] | 2011 | 1 | WT-1 vaccine | Peptide | i.d. | + gem | 9 | Gem-naïve LAPC/mPDAC | NR | 8.2 (t) |
| Nishida [164] | 2014 | 1 | WT-1 vaccine | Peptide | i.d. | + gem | 32 | Treatment-naïve LAPC/mPDAC and treated recurrent disease | 4.2 | 8.1 (t) |
| Koido [165] | 2014 | 1 | WT-1 vaccine | DC | i.d. | + gem | 10 | mPDAC: Treatment-naïve newly diagnosed or recurrence after resection | NR | NR |
| Tsukinaga [166] | 2015 | NR | WT-1 vaccine | DC | i.d. | + gem | 7 | Treatment-naïve mPDAC | 6.8 | 10.7 (t) |
| Mayanagi [167] | 2015 | 1 | WT-1 vaccine | DC | i.d. | + gem | 10 | Treatment-naïve LAPC/mPDAC | NR | 8 (t) |
| Yanagisawa [168] | 2018 | 1 | WT-1 vaccine | DC | i.d. | + chemo | 8 | Resected, chemo-naïve PDAC | NR | NR |
| Nishida [169] | 2018 | 2 | WT-1 vaccine | Peptide | i.d. | (1) + gem (2) gem alone | 85 | Treatment-naïve LAPC, mPDAC or recurrence after resection | (1) 5.2 (2) 3.3 | (1) 9.6 (t) (2) 8.9 (t) |
| Hanada [170] | 2020 | NR | WT-1 vaccine | DC | i.d. | - | 6 | Pre-treated (CRT) PDAC: Resected, mPDAC, recurrent | 19.9 | 59 (x) |
| Nagai [171] | 2020 | 1/2a | WT-1/MUC-1 vaccine | DC | i.d. | + gem | 10 | Resected PDAC | 17.7 | 46.4 (t) |
| KIF20A | ||||||||||
| Asahara [172] | 2013 | 1/2 | KIF20A vaccine | Peptide | s.c. | (1) -(2) no treatment (BSC) | 110 | Chemo-refractory, LAPC/mPDAC or recurrence after resection | (1) 1.8 (2) NR | (1) 4.7 (x) (2) 2.1–2.7 (x) |
| Suzuki [173] | 2014 | 1 | KIF20A vaccine | Peptide | s.c. | + gem | 9 | Pre-treated LAPC/mPDAC | NR | 5.7 (t) 18 (d) |
| VEGFR | ||||||||||
| Miyazawa [174] | 2010 | 1 | VEGFR2 vaccine | Peptide | s.c. | + gem | 18 | LAPC/mPDAC, 17% treatment-naïve | 3.9 | 7.7 (t) |
| Yamaue [175] | 2015 | 2/3 | VEGFR2 vaccine | Peptide | s.c. | (1) + gem (2) gem only | 153 | Treatment-naïve LAPC/mPDAC | (1) 3.7 (2) 3.8 | (1) 8.4 (t) (2) 8.5 (t) |
| KIF20A + VEGFR | ||||||||||
| Suzuki [176] | 2017 | 2 | KIF20A + VEGFR1/2 vaccine | Peptide | s.c. | + gem | 68 | Chemo-naïve LAPC/mPDAC | 4.7–5.2 | 9–10 (t) |
| Miyazawa [177] | 2017 | 2 | KIF20A + VEGFR1/2 vaccine | Peptide | s.c. | + gem | 30 | Resected PDAC | 15.8 | NR |
| Survivin | ||||||||||
| Kameshima [178] | 2013 | NR | Survivin vaccine | Peptide | s.c. | + IFA, IFN-α | 6 | LAPC/mPDAC/recurrence, unknown prior treatments | NR | NR |
| Shima [179] | 2019 | 2 | Survivin vaccine | Peptide | s.c. | (1) + IFA, IFN-β (2) + IFA(3) placebo only | 83 | Pre-treated LAPC/mPDAC | (1) 2.2 (3) 2.3 | (1) 3.4 (t) (2) 3.2 (t) (3) 3.6 (t) |
| MUC-1 | ||||||||||
| Rong [180] | 2012 | 1 | MUC-1 vaccine | DC | i.d. | - | 6 | Pre-treated LAPC/mPDAC/recurrence | NR | NR |
| Mesothelin | ||||||||||
| Le [181] | 2012 | 1 | Mesothelin-expressing Lm vaccine | Lm | i.v. | - | 9 | Treatment-refractory PDAC | NR | 7 (x) |
| hTERT | ||||||||||
| Middleton [115] | 2014 | 1 | Telomerase vaccine | Peptide | i.d. | (1) chemo only (2) + chemo + GM-CSF (sequential) (3) + chemo + GM-CSF (concurrent) | 1062 | Treatment-naïve LAPC/mPDAC | (1) 6.4 (2) 4.5 (3) 6.6 | (1) 7.9 (t) (2) 6.9 (t) (3) 8.4 (t) |
| Neoantigen:mutant KRAS | ||||||||||
| Wedén [113] | 2011 | 1/2 | KRAS vaccine | Peptide | i.d. | + GM-CSF | 23 | Resected PDAC | NR | 27.5 (t) |
| Abou-Alfa [182] | 2011 | Peptide | i.d. | + GM-CSF | 24 | Resected PDAC | 8.6 | 20.3 (t) | ||
| Kubuschok [183] | 2012 | 1 | KRAS vaccine | LCL | s.c. | - | 7 | mPDAC (recurrent or newly diagnosed) | 3.1 | 4.5 (t) |
| Palmer [114] | 2020 | 1/2 | KRAS vaccine | Peptide | i.d. | + GM-CSF + gem | 32 | Resected PDAC | 13.9–19.5 | 33.1–34.2 (t) |
| Neoantigen:other | ||||||||||
| Bassani-Sternberg [184] | 2019 | 1b | Neoantigens | DC | s.c. | + chemo + anti-PD-1 + aspirin | 3 | Resected PDAC | NR | NR |
| Tumor-based | ||||||||||
| Yanagimoto [185] | 2010 | 2 | Personalized vaccine | Peptide | s.c. | + gem | 21 | Treatment-naïve LAPC/mPDAC | 7 | 9 (t) |
| Bauer [186] | 2011 | 1 | Tumor lysate vaccine | DC—Whole-tumor-cell | i.d. | + gem | 12 | Recurrence after resection | NR | 10.5 (dr) |
| Kimura [187] | 2012 | NS | Personalized and/or tumor lysate vaccine | DC | i.t. | + chemo ± LAK cell therapy | 49 | Chemo-refractory LAPC/mPDAC | NR | 11.8 (t) |
| Yutani [188] | 2013 | 2 | Personalized vaccine | Peptide | s.c. | + chemo | 41 | Chemo-refractory mPDAC, newly diagnosed and recurrence after resection | NR | 7.9 (t) |
| Qiu [189] | 2013 | 1 | Tumor lysate expressing α-Gal | DC | i.d. | + CIK cell therapy | 14 | Pre-treated LAPC/mPDAC | NR | 24.7 (d) |
| Lin [190] | 2015 | NS | Pancreatic cancer stem cell lysate | Whole-tumor-cell | s.c. | - | 90 | Pre-treated stage II PDAC, LAPC, mPDAC | NR | NR |
| Zhang [191] | 2016 | R | Various tumor antigens (unspecified) | DC | i.v. | (1) + CIK (2) no further treatment after baseline therapy | 150 | Pre-treated PDAC | NR | (1) 4.4 (t) (2) 3.8 (t) |
| Mehrotra [192] | 2017 | 1 | hTERT, CEA, survivin vaccine | DC | i.d. | + TLR-3 agonist (i.m.) | 12 | Pre-treated LAPC/mPDAC | 3 | 7.7 (t) |
| Authors | Year | Phase | Immunotherapy | Ablative Therapy | Approach | Treatment | No. of Patients | Patient Population | mPFS (Months) | mOS (Months) |
|---|---|---|---|---|---|---|---|---|---|---|
| IT + IRE | ||||||||||
| Lin [219] | 2017 | NR | NK cell therapy | IRE | Perc. | (1) IRE only (2) IRE + NK cells | 67 | LAPC/mPDAC, 73% pre-treated with CRT | (1) 4.8–7.9 (2) 5.3–9.1 | (1) 9.1–12.2 (t) (2) 10.2–13.6 (t) |
| Lin [220] | 2020 | NR | Vγ9Vδ2 T cell therapy | IRE | Perc. | (1) IRE only (2) IRE + T cells | 62 | LAPC, 79% pre-treated with chemo | (1) 8 (2) 11 | (1) 11 (t) (2) 14.5 (t) |
| O’Neill [221] | 2020 | 1b | Anti-PD-1 | IRE | Open | IRE + anti-PD-1 | 10 | LAPC, pre-treated with chemo | 6.3 | 18 (x) |
| IT + SABR | ||||||||||
| Lin [222] | 2019 | 1/2 | Anti-CA-125 | SABR | - | SABR + chemo + nelfinavir + anti-CA-125 ± resection | 11 | LAPC/mPDAC, treatment-naïve | 8.6 | 13 (t) |
| Xie [223] | 2020 | 1 | Anti-PD-L1 Anti-CTLA-4 | SABR | - | (1) SABR + anti-PD-L1 (2) SABR + anti-PD-L1 + anti-CTLAA-4 | 59 | LAPC/mPDAC, pre-treated chemo | (1) 1.7–2.5 (2) 0.9–2.3 | (1) 3.3–9 (t) (2) 2.1–4.2 (t) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Timmer, F.E.F.; Geboers, B.; Nieuwenhuizen, S.; Dijkstra, M.; Schouten, E.A.C.; Puijk, R.S.; de Vries, J.J.J.; van den Tol, M.P.; Bruynzeel, A.M.E.; Streppel, M.M.; et al. Pancreatic Cancer and Immunotherapy: A Clinical Overview. Cancers 2021, 13, 4138. https://doi.org/10.3390/cancers13164138
Timmer FEF, Geboers B, Nieuwenhuizen S, Dijkstra M, Schouten EAC, Puijk RS, de Vries JJJ, van den Tol MP, Bruynzeel AME, Streppel MM, et al. Pancreatic Cancer and Immunotherapy: A Clinical Overview. Cancers. 2021; 13(16):4138. https://doi.org/10.3390/cancers13164138
Chicago/Turabian StyleTimmer, Florentine E. F., Bart Geboers, Sanne Nieuwenhuizen, Madelon Dijkstra, Evelien A. C. Schouten, Robbert S. Puijk, Jan J. J. de Vries, M. Petrousjka van den Tol, Anna M. E. Bruynzeel, Mirte M. Streppel, and et al. 2021. "Pancreatic Cancer and Immunotherapy: A Clinical Overview" Cancers 13, no. 16: 4138. https://doi.org/10.3390/cancers13164138
APA StyleTimmer, F. E. F., Geboers, B., Nieuwenhuizen, S., Dijkstra, M., Schouten, E. A. C., Puijk, R. S., de Vries, J. J. J., van den Tol, M. P., Bruynzeel, A. M. E., Streppel, M. M., Wilmink, J. W., van der Vliet, H. J., Meijerink, M. R., Scheffer, H. J., & de Gruijl, T. D. (2021). Pancreatic Cancer and Immunotherapy: A Clinical Overview. Cancers, 13(16), 4138. https://doi.org/10.3390/cancers13164138