
Recent Advances in Enhancing the Therapeutic Index of PARP Inhibitors in Breast Cancer



Abstract
:Simple Summary
Abstract
1. Introduction
2. HR Deficiency (HRD) and BRCAness Phenotype
2.1. Causes of HRD
2.1.1. Germline Mutations of Either BRCA1 or BRCA2
2.1.2. Germline Mutations of Additional DNA Damage Response (DDR)-Associated Genes
2.1.3. Somatic Mutations of BRCA1 or BRCA2
2.1.4. Epigenetic Silencing of BRCA1, BRCA2 or RAD51C
2.2. HRD Diagnosis
2.2.1. Hereditary Breast and Ovarian Cancer Panel
2.2.2. Genomic Signatures
2.2.3. Functional Analyses of HRD
3. Clinical Trials with PARPi
3.1. PARPi in Monotherapy
3.1.1. Patients with Germline BRCA Mutations
3.1.2. Patients without Germline BRCA Mutation but BRCAness Tumours
3.2. PARPi in Combination with Chemotherapy
3.3. PARPi in Combination with Other Agents
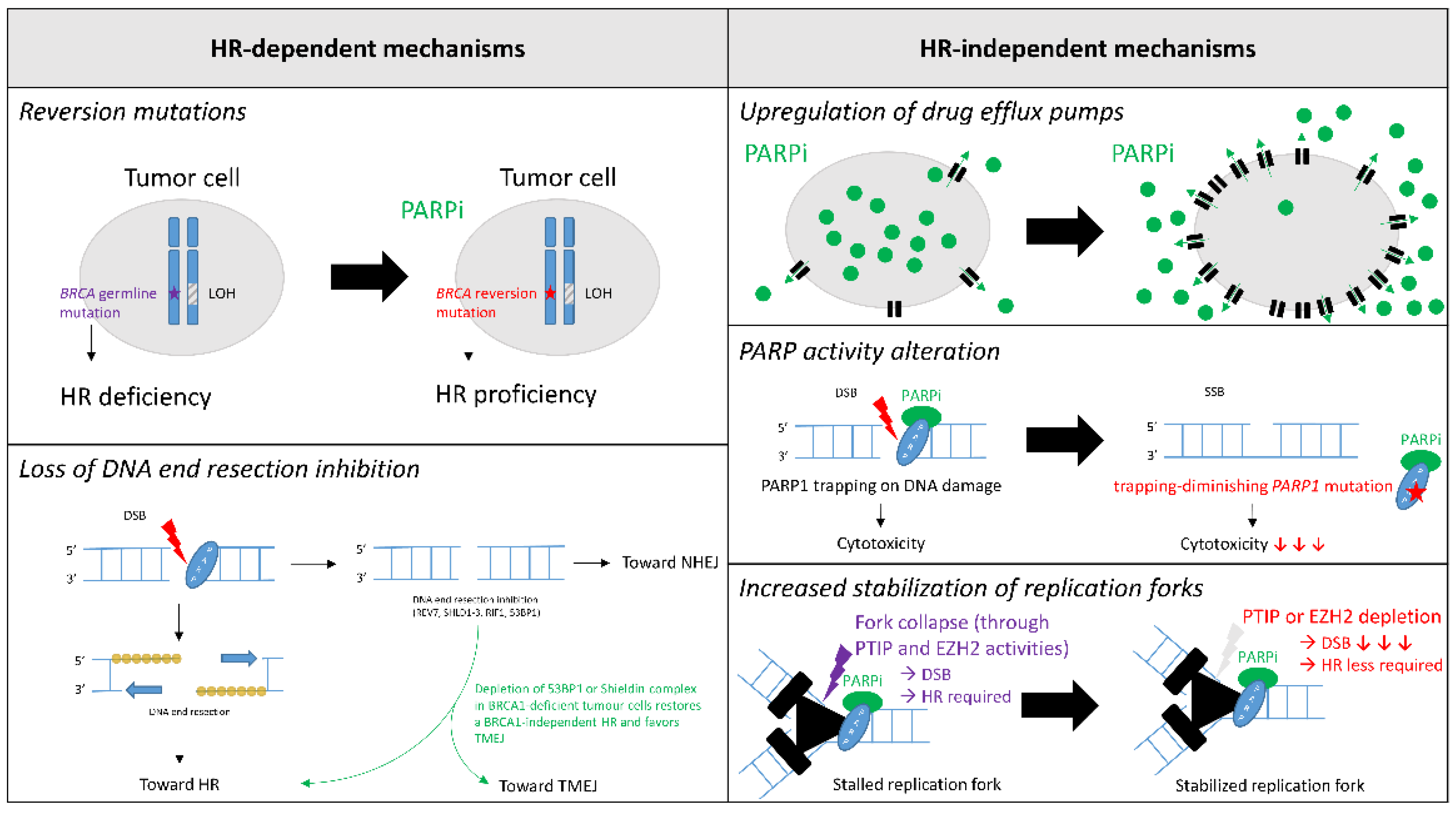
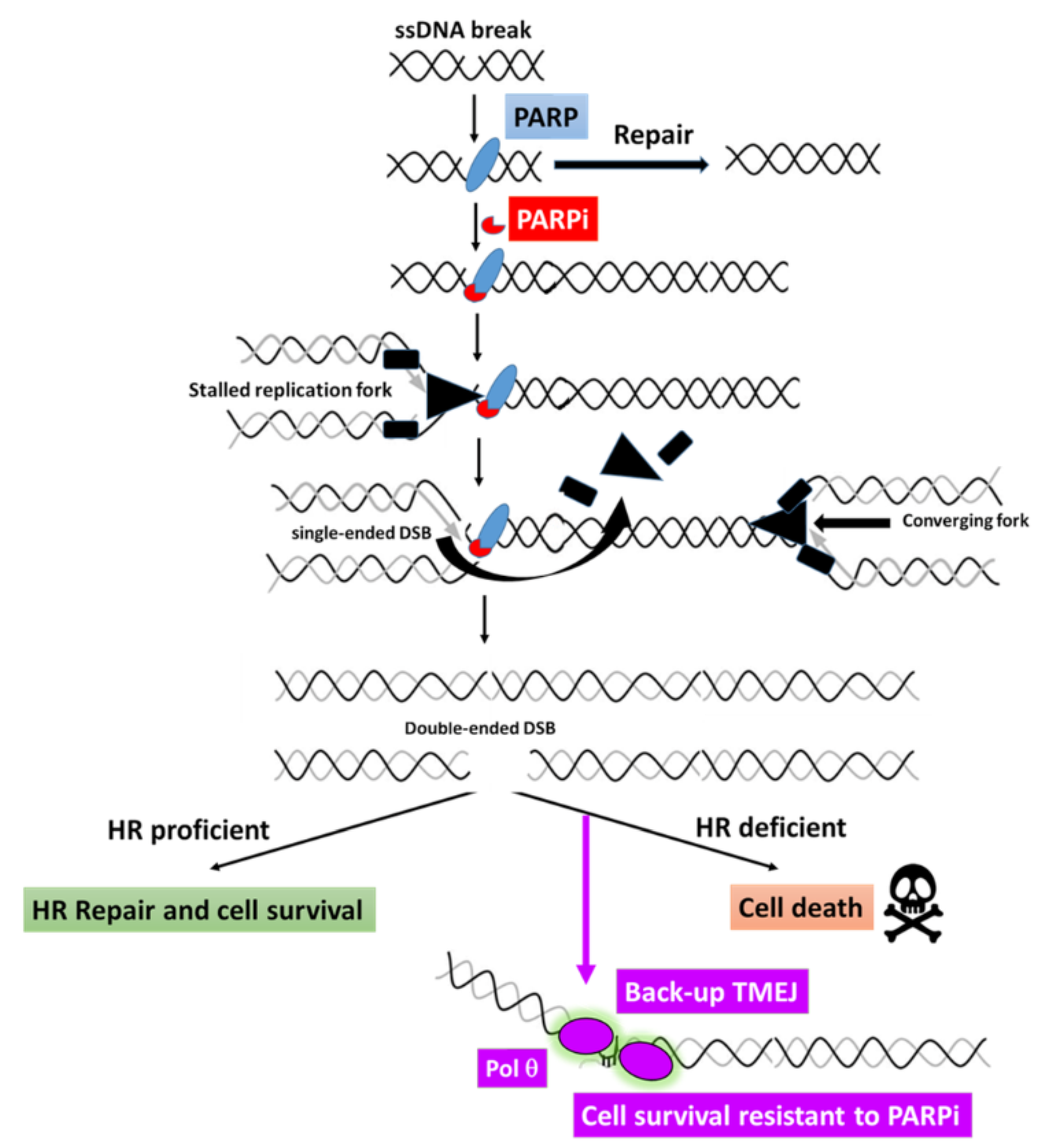
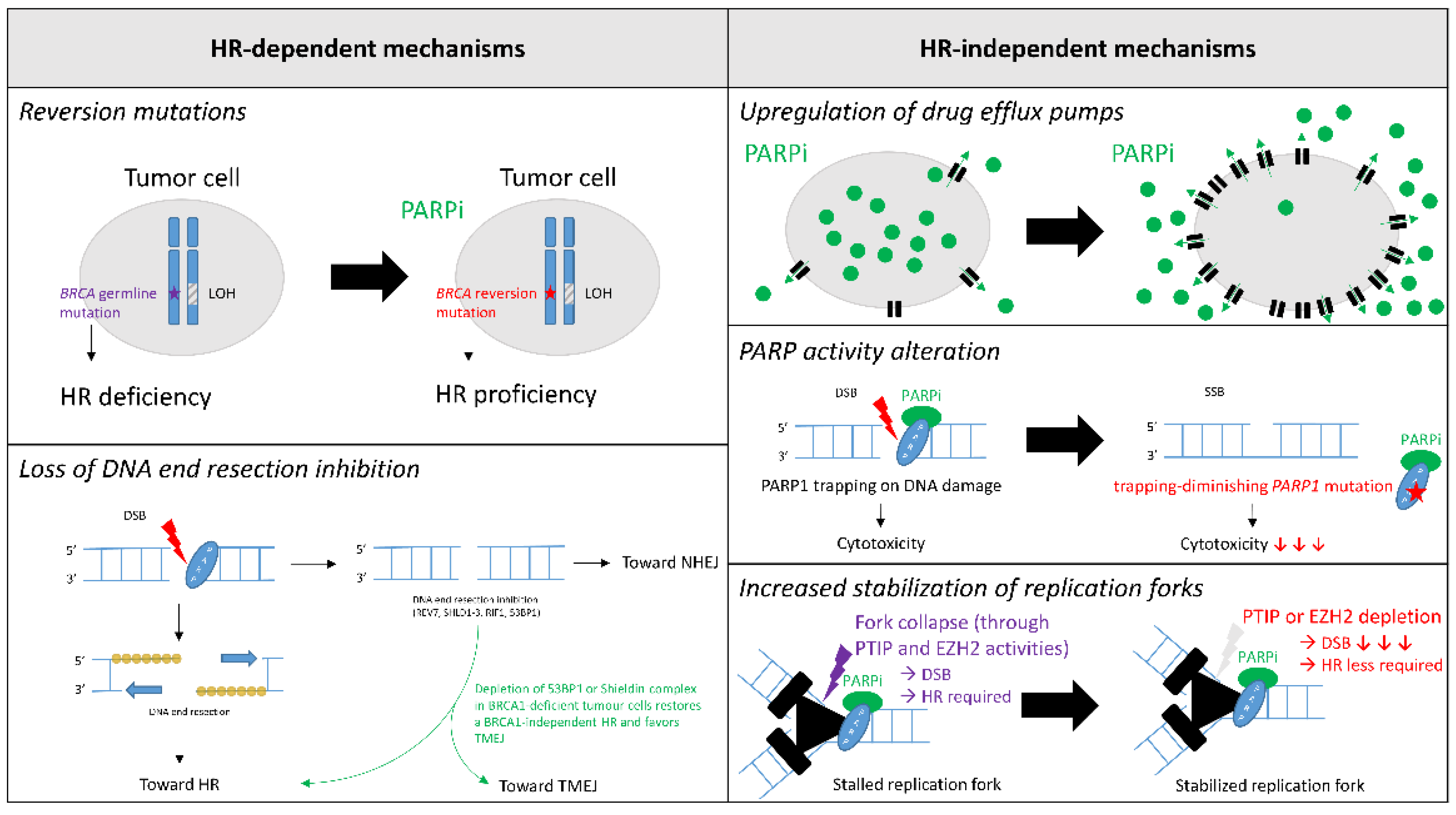
4. Mechanisms of PARPi Resistance
4.1. HR-Dependent Mechanisms
4.1.1. Reversion Mutations
4.1.2. Loss of DNA End Resection Inhibition
4.2. HR-Independent Mechanisms
4.2.1. Upregulation of Drug Efflux Pumps
4.2.2. PARP Activity Alteration
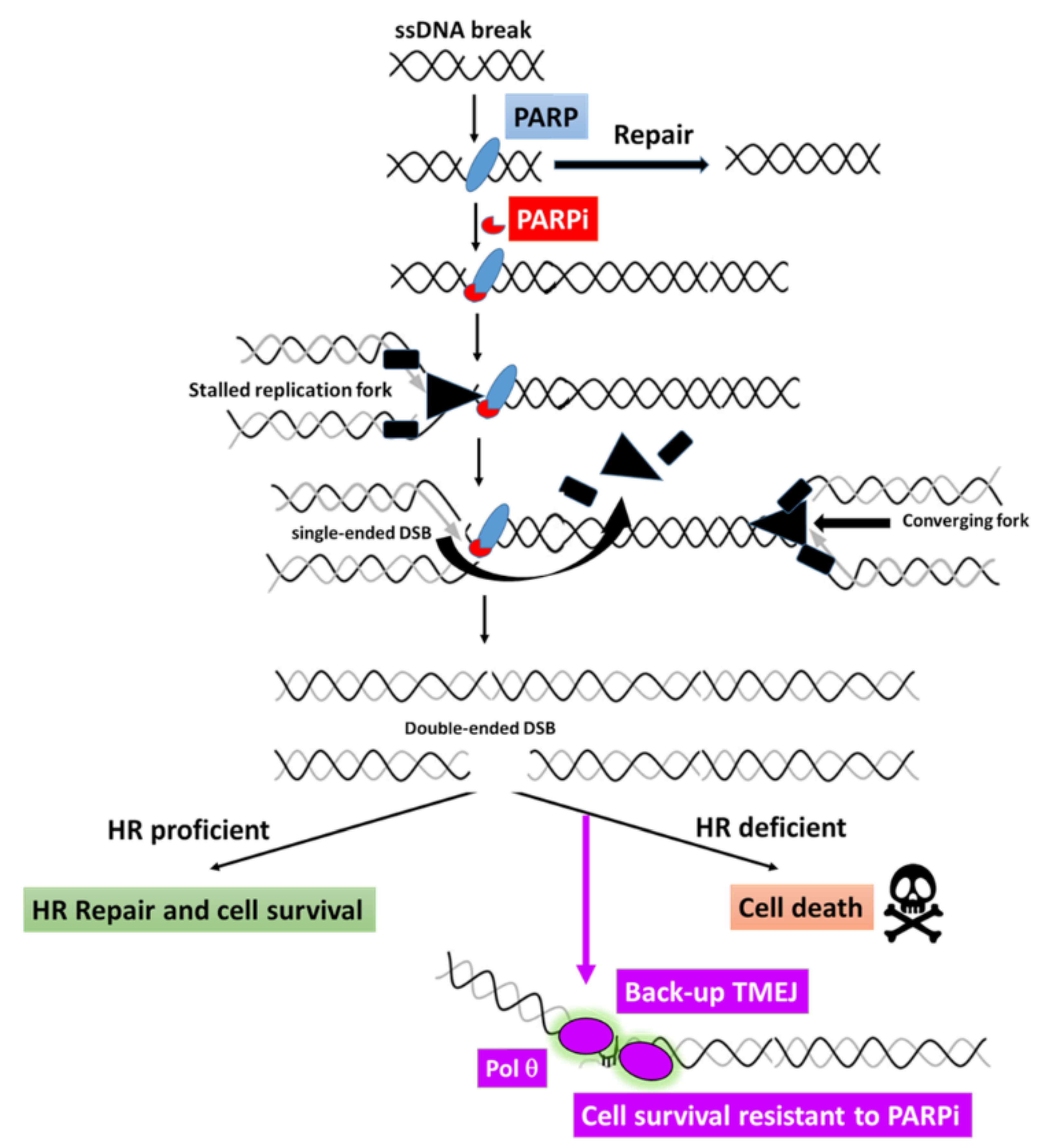
4.2.3. Increased Stabilization of Replication Forks
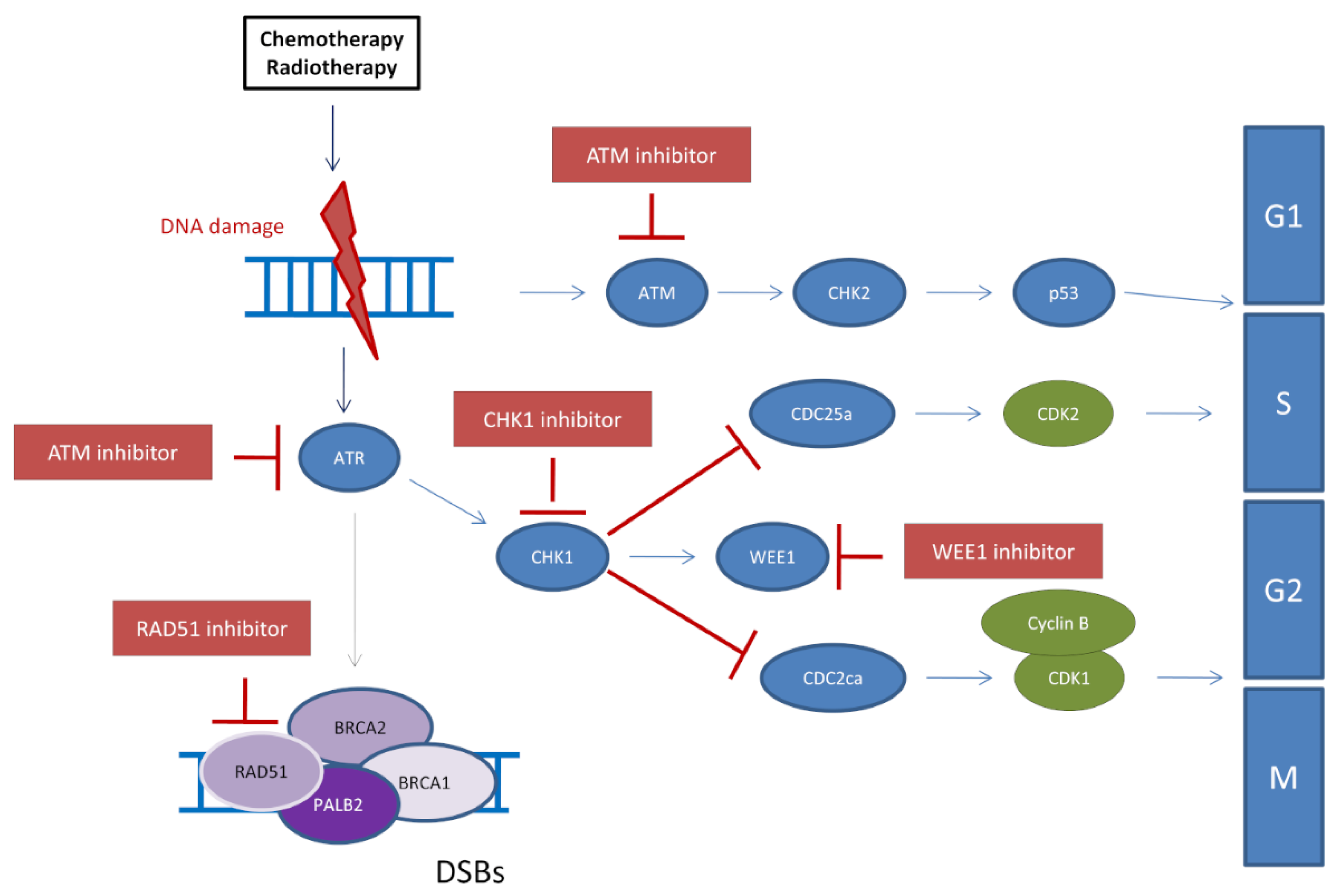
5. Novel Strategies for Future Combination Therapies to Overcome Resistance to PARP Inhibitors in Breast Cancers
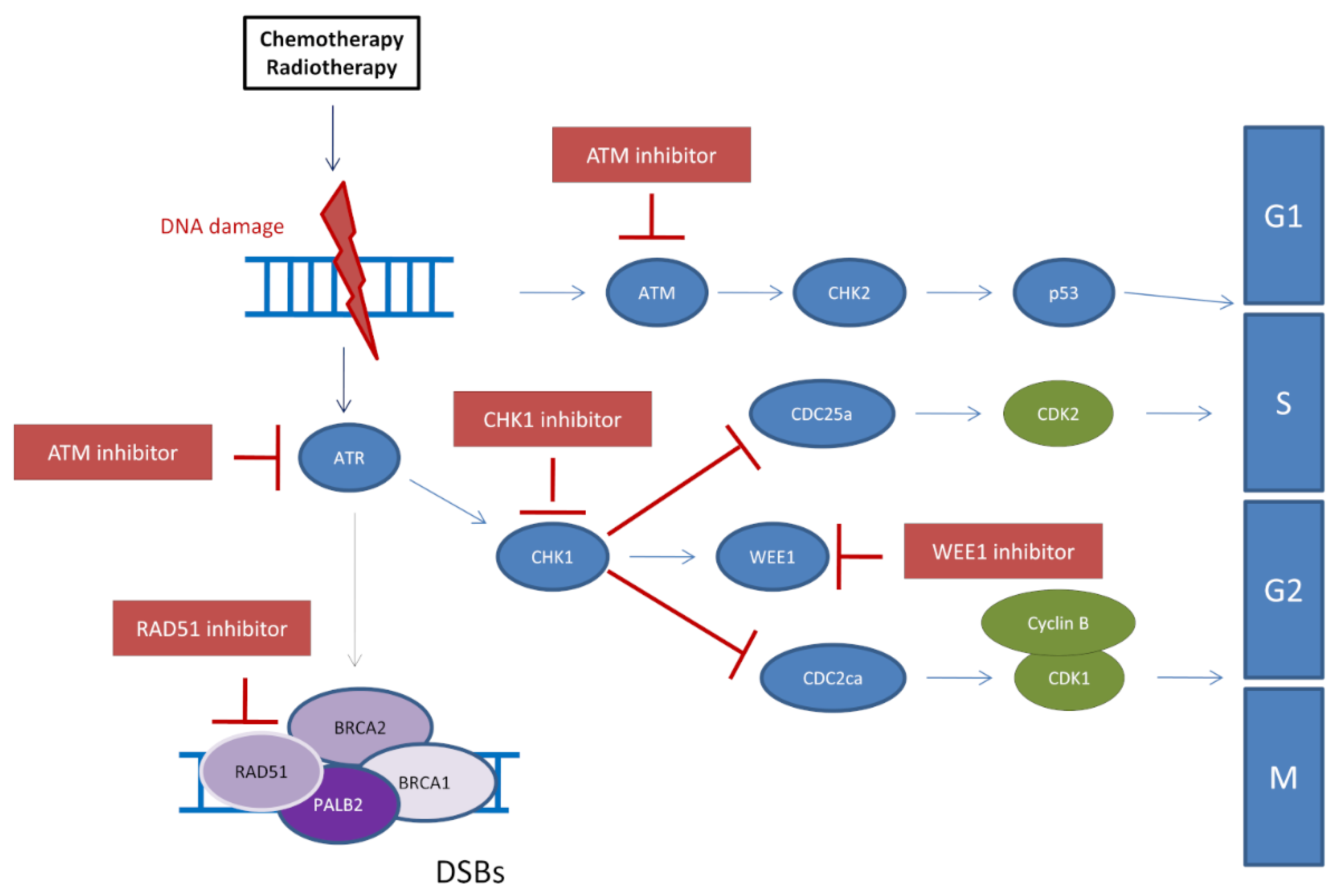
5.1. Inhibition of the ATR/CHK1/WEE1 Pathway to Restore PARPi Sensitivity
5.2. Potential Use of POLQ Inhibitors for PARPi-Resistant Breast Cancers
5.3. Inhibition of Chromatin Remodelers Combined with PARPi
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Li, J.; Wen, W.X.; Eklund, M.; Kvist, A.; Eriksson, M.; Christensen, H.N.; Torstensson, A.; Bajalica-Lagercrantz, S.; Dunning, A.M.; Decker, B.; et al. Prevalence of BRCA1 and BRCA2 Pathogenic Variants in a Large, Unselected Breast Cancer Cohort: BRCA Testing for All Breast Cancer Patients? Int. J. Cancer 2019, 144, 1195–1204. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Polley, E.C.; Yadav, S.; Lilyquist, J.; Shimelis, H.; Na, J.; Hart, S.N.; Goldgar, D.E.; Shah, S.; Pesaran, T.; et al. The Contribution of Germline Predisposition Gene Mutations to Clinical Subtypes of Invasive Breast Cancer From a Clinical Genetic Testing Cohort. J. Natl. Cancer Inst. 2020, 112, 1231–1241. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Hart, S.N.; Gnanaolivu, R.; Huang, H.; Lee, K.Y.; Na, J.; Gao, C.; Lilyquist, J.; Yadav, S.; Boddicker, N.J.; et al. A Population-Based Study of Genes Previously Implicated in Breast Cancer. N. Engl. J. Med. 2021, 384, 440–451. [Google Scholar] [CrossRef]
- Ece Solmaz, A.; Yeniay, L.; Gökmen, E.; Zekioğlu, O.; Haydaroğlu, A.; Bilgen, I.; Özkınay, F.; Onay, H. Clinical Contribution of Next-Generation Sequencing Multigene Panel Testing for BRCA Negative High-Risk Patients With Breast Cancer. Clin. Breast Cancer 2021. [Google Scholar] [CrossRef]
- Lang, G.-T.; Shi, J.-X.; Huang, L.; Cao, A.-Y.; Zhang, C.-H.; Song, C.-G.; Zhuang, Z.-G.; Hu, X.; Huang, W.; Shao, Z.-M. Multiple Cancer Susceptible Genes Sequencing in BRCA-Negative Breast Cancer with High Hereditary Risk. Ann. Transl. Med. 2020, 8, 1417. [Google Scholar] [CrossRef] [PubMed]
- Moscatello, C.; Di Nicola, M.; Veschi, S.; Di Gregorio, P.; Cianchetti, E.; Stuppia, L.; Battista, P.; Cama, A.; Curia, M.C.; Aceto, G.M. Relationship between MUTYH, OGG1 and BRCA1 Mutations and MRNA Expression in Breast and Ovarian Cancer Predisposition. Mol. Clin. Oncol. 2021, 14, 15. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network Comprehensive Molecular Portraits of Human Breast Tumours. Nature 2012, 490, 61–70. [CrossRef] [Green Version]
- Winter, C.; Nilsson, M.P.; Olsson, E.; George, A.M.; Chen, Y.; Kvist, A.; Törngren, T.; Vallon-Christersson, J.; Hegardt, C.; Häkkinen, J.; et al. Targeted Sequencing of BRCA1 and BRCA2 across a Large Unselected Breast Cancer Cohort Suggests That One-Third of Mutations Are Somatic. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2016, 27, 1532–1538. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of Somatic Mutations in 560 Breast Cancer Whole-Genome Sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Meric-Bernstam, F.; Brusco, L.; Daniels, M.; Wathoo, C.; Bailey, A.M.; Strong, L.; Shaw, K.; Lu, K.; Qi, Y.; Zhao, H.; et al. Incidental Germline Variants in 1000 Advanced Cancers on a Prospective Somatic Genomic Profiling Protocol. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2016, 27, 795–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwong, A.; Cheuk, I.W.; Shin, V.Y.; Ho, C.Y.; Au, C.-H.; Ho, D.N.; Wong, E.Y.; Yu, S.W.; Chen, J.; Chan, K.K.; et al. Somatic Mutation Profiling in BRCA-Negative Breast and Ovarian Cancer Patients by Multigene Panel Sequencing. Am. J. Cancer Res. 2020, 10, 2919–2932. [Google Scholar]
- André, F.; Bachelot, T.; Commo, F.; Campone, M.; Arnedos, M.; Dieras, V.; Lacroix-Triki, M.; Lacroix, L.; Cohen, P.; Gentien, D.; et al. Comparative Genomic Hybridisation Array and DNA Sequencing to Direct Treatment of Metastatic Breast Cancer: A Multicentre, Prospective Trial (SAFIR01/UNICANCER). Lancet Oncol. 2014, 15, 267–274. [Google Scholar] [CrossRef]
- Evans, D.G.R.; van Veen, E.M.; Byers, H.J.; Wallace, A.J.; Ellingford, J.M.; Beaman, G.; Santoyo-Lopez, J.; Aitman, T.J.; Eccles, D.M.; Lalloo, F.I.; et al. A Dominantly Inherited 5′ UTR Variant Causing Methylation-Associated Silencing of BRCA1 as a Cause of Breast and Ovarian Cancer. Am. J. Hum. Genet. 2018, 103, 213–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansmann, T.; Pliushch, G.; Leubner, M.; Kroll, P.; Endt, D.; Gehrig, A.; Preisler-Adams, S.; Wieacker, P.; Haaf, T. Constitutive Promoter Methylation of BRCA1 and RAD51C in Patients with Familial Ovarian Cancer and Early-Onset Sporadic Breast Cancer. Hum. Mol. Genet. 2012, 21, 4669–4679. [Google Scholar] [CrossRef] [PubMed]
- Tabano, S.; Azzollini, J.; Pesenti, C.; Lovati, S.; Costanza, J.; Fontana, L.; Peissel, B.; Miozzo, M.; Manoukian, S. Analysis of BRCA1 and RAD51C Promoter Methylation in Italian Families at High-Risk of Breast and Ovarian Cancer. Cancers 2020, 12, 910. [Google Scholar] [CrossRef] [Green Version]
- Poduval, D.B.; Ognedal, E.; Sichmanova, Z.; Valen, E.; Iversen, G.T.; Minsaas, L.; Lønning, P.E.; Knappskog, S. Assessment of Tumor Suppressor Promoter Methylation in Healthy Individuals. Clin. Epigenetics 2020, 12, 131. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, N.; Tokunaga, E.; Kitao, H.; Hitchins, M.; Inoue, Y.; Tanaka, K.; Hisamatsu, Y.; Taketani, K.; Akiyoshi, S.; Okada, S.; et al. Epigenetic Inactivation of BRCA1 Through Promoter Hypermethylation and Its Clinical Importance in Triple-Negative Breast Cancer. Clin. Breast Cancer 2015, 15, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Glodzik, D.; Bosch, A.; Hartman, J.; Aine, M.; Vallon-Christersson, J.; Reuterswärd, C.; Karlsson, A.; Mitra, S.; Niméus, E.; Holm, K.; et al. Comprehensive Molecular Comparison of BRCA1 Hypermethylated and BRCA1 Mutated Triple Negative Breast Cancers. Nat. Commun. 2020, 11, 3747. [Google Scholar] [CrossRef]
- Prajzendanc, K.; Domagała, P.; Hybiak, J.; Ryś, J.; Huzarski, T.; Szwiec, M.; Tomiczek-Szwiec, J.; Redelbach, W.; Sejda, A.; Gronwald, J.; et al. BRCA1 Promoter Methylation in Peripheral Blood Is Associated with the Risk of Triple-Negative Breast Cancer. Int. J. Cancer 2020, 146, 1293–1298. [Google Scholar] [CrossRef]
- Wong, E.M.; Southey, M.C.; Fox, S.B.; Brown, M.A.; Dowty, J.G.; Jenkins, M.A.; Giles, G.G.; Hopper, J.L.; Dobrovic, A. Constitutional Methylation of the BRCA1 Promoter Is Specifically Associated with BRCA1 Mutation-Associated Pathology in Early-Onset Breast Cancer. Cancer Prev. Res. 2011, 4, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Iwamoto, T.; Yamamoto, N.; Taguchi, T.; Tamaki, Y.; Noguchi, S. BRCA1 Promoter Methylation in Peripheral Blood Cells Is Associated with Increased Risk of Breast Cancer with BRCA1 Promoter Methylation. Breast Cancer Res. Treat. 2011, 129, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Vos, S.; van Diest, P.J.; Moelans, C.B. A Systematic Review on the Frequency of BRCA Promoter Methylation in Breast and Ovarian Carcinomas of BRCA Germline Mutation Carriers: Mutually Exclusive, or Not? Crit. Rev. Oncol. Hematol. 2018, 127, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.M.; Southey, M.C.; Terry, M.B. Integrating DNA Methylation Measures to Improve Clinical Risk Assessment: Are We There yet? The Case of BRCA1 Methylation Marks to Improve Clinical Risk Assessment of Breast Cancer. Br. J. Cancer 2020, 122, 1133–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAlarnen, L.; Stearns, K.; Uyar, D. Challenges of Genomic Testing for Hereditary Breast and Ovarian Cancers. Appl. Clin. Genet. 2021, 14, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Watkins, J.A.; Irshad, S.; Grigoriadis, A.; Tutt, A.N.J. Genomic Scars as Biomarkers of Homologous Recombination Deficiency and Drug Response in Breast and Ovarian Cancers. Breast Cancer Res. BCR 2014, 16, 211. [Google Scholar] [CrossRef] [Green Version]
- Birkbak, N.J.; Wang, Z.C.; Kim, J.-Y.; Eklund, A.C.; Li, Q.; Tian, R.; Bowman-Colin, C.; Li, Y.; Greene-Colozzi, A.; Iglehart, J.D.; et al. Telomeric Allelic Imbalance Indicates Defective DNA Repair and Sensitivity to DNA-Damaging Agents. Cancer Discov. 2012, 2, 366–375. [Google Scholar] [CrossRef] [Green Version]
- Popova, T.; Manié, E.; Rieunier, G.; Caux-Moncoutier, V.; Tirapo, C.; Dubois, T.; Delattre, O.; Sigal-Zafrani, B.; Bollet, M.; Longy, M.; et al. Ploidy and Large-Scale Genomic Instability Consistently Identify Basal-like Breast Carcinomas with BRCA1/2 Inactivation. Cancer Res. 2012, 72, 5454–5462. [Google Scholar] [CrossRef] [Green Version]
- Telli, M.L.; Jensen, K.C.; Vinayak, S.; Kurian, A.W.; Lipson, J.A.; Flaherty, P.J.; Timms, K.; Abkevich, V.; Schackmann, E.A.; Wapnir, I.L.; et al. Phase II Study of Gemcitabine, Carboplatin, and Iniparib As Neoadjuvant Therapy for Triple-Negative and BRCA1/2 Mutation-Associated Breast Cancer With Assessment of a Tumor-Based Measure of Genomic Instability: PrECOG 0105. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 1895–1901. [Google Scholar] [CrossRef] [Green Version]
- Davies, H.; Glodzik, D.; Morganella, S.; Yates, L.R.; Staaf, J.; Zou, X.; Ramakrishna, M.; Martin, S.; Boyault, S.; Sieuwerts, A.M.; et al. HRDetect Is a Predictor of BRCA1 and BRCA2 Deficiency Based on Mutational Signatures. Nat. Med. 2017, 23, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Hollstein, M.; Alexandrov, L.B.; Wild, C.P.; Ardin, M.; Zavadil, J. Base Changes in Tumour DNA Have the Power to Reveal the Causes and Evolution of Cancer. Oncogene 2017, 36, 158–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of Mutational Processes in Human Cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; Van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational Processes Molding the Genomes of 21 Breast Cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.; Martens, J.W.M.; Van Hoeck, A.; Cuppen, E. Pan-Cancer Landscape of Homologous Recombination Deficiency. Nat. Commun. 2020, 11, 5584. [Google Scholar] [CrossRef]
- Chan, S.H.; Yu, A.M.; McVey, M. Dual Roles for DNA Polymerase Theta in Alternative End-Joining Repair of Double-Strand Breaks in Drosophila. PLoS Genet. 2010, 6, e1001005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patterson-Fortin, J.; D’Andrea, A.D. Exploiting the Microhomology-Mediated End-Joining Pathway in Cancer Therapy. Cancer Res. 2020, 80, 4593–4600. [Google Scholar] [CrossRef]
- Gulhan, D.C.; Lee, J.J.-K.; Melloni, G.E.M.; Cortés-Ciriano, I.; Park, P.J. Detecting the Mutational Signature of Homologous Recombination Deficiency in Clinical Samples. Nat. Genet. 2019, 51, 912–919. [Google Scholar] [CrossRef]
- Willers, H.; Taghian, A.G.; Luo, C.-M.; Treszezamsky, A.; Sgroi, D.C.; Powell, S.N. Utility of DNA Repair Protein Foci for the Detection of Putative BRCA1 Pathway Defects in Breast Cancer Biopsies. Mol. Cancer Res. MCR 2009, 7, 1304–1309. [Google Scholar] [CrossRef] [Green Version]
- Mutter, R.W.; Riaz, N.; Ng, C.K.; Delsite, R.; Piscuoglio, S.; Edelweiss, M.; Martelotto, L.G.; Sakr, R.A.; King, T.A.; Giri, D.D.; et al. Bi-Allelic Alterations in DNA Repair Genes Underpin Homologous Recombination DNA Repair Defects in Breast Cancer. J. Pathol. 2017, 242, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Castroviejo-Bermejo, M.; Cruz, C.; Llop-Guevara, A.; Gutiérrez-Enríquez, S.; Ducy, M.; Ibrahim, Y.H.; Gris-Oliver, A.; Pellegrino, B.; Bruna, A.; Guzmán, M.; et al. A RAD51 Assay Feasible in Routine Tumor Samples Calls PARP Inhibitor Response beyond BRCA Mutation. EMBO Mol. Med. 2018, 10, e9172. [Google Scholar] [CrossRef]
- Cruz, C.; Castroviejo-Bermejo, M.; Gutiérrez-Enríquez, S.; Llop-Guevara, A.; Ibrahim, Y.H.; Gris-Oliver, A.; Bonache, S.; Morancho, B.; Bruna, A.; Rueda, O.M.; et al. RAD51 Foci as a Functional Biomarker of Homologous Recombination Repair and PARP Inhibitor Resistance in Germline BRCA-Mutated Breast Cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 1203–1210. [Google Scholar] [CrossRef]
- Naipal, K.A.T.; Verkaik, N.S.; Ameziane, N.; van Deurzen, C.H.M.; Ter Brugge, P.; Meijers, M.; Sieuwerts, A.M.; Martens, J.W.; O’Connor, M.J.; Vrieling, H.; et al. Functional Ex Vivo Assay to Select Homologous Recombination-Deficient Breast Tumors for PARP Inhibitor Treatment. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 4816–4826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murai, J.; Pommier, Y. PARP Trapping Beyond Homologous Recombination and Platinum Sensitivity in Cancers. Annu. Rev. Cancer Biol. 2019, 3, 131–150. [Google Scholar] [CrossRef]
- Sizemore, S.T.; Mohammad, R.; Sizemore, G.M.; Yu, H.; Ostrowski, M.C.; Chakravarti, A.; Xia, F. Relationship of Ionizing Radiation-Induced Synthetic Lethality of PARP Inhibition in BRCA1-Proficient Cancer Cells with P53. J. Clin. Oncol. 2016, 34, e14115. [Google Scholar] [CrossRef]
- Murai, J.; Zhang, Y.; Morris, J.; Ji, J.; Takeda, S.; Doroshow, J.H.; Pommier, Y. Rationale for Poly(ADP-Ribose) Polymerase (PARP) Inhibitors in Combination Therapy with Camptothecins or Temozolomide Based on PARP Trapping versus Catalytic Inhibition. J. Pharmacol. Exp. Ther. 2014, 349, 408–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.-H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef]
- Robson, M.E.; Tung, N.; Conte, P.; Im, S.-A.; Senkus, E.; Xu, B.; Masuda, N.; Delaloge, S.; Li, W.; Armstrong, A.; et al. OlympiAD Final Overall Survival and Tolerability Results: Olaparib versus Chemotherapy Treatment of Physician’s Choice in Patients with a Germline BRCA Mutation and HER2-Negative Metastatic Breast Cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 558–566. [Google Scholar] [CrossRef]
- Litton, J.K.; Hurvitz, S.A.; Mina, L.A.; Rugo, H.S.; Lee, K.-H.; Gonçalves, A.; Diab, S.; Woodward, N.; Goodwin, A.; Yerushalmi, R.; et al. Talazoparib versus Chemotherapy in Patients with Germline BRCA1/2-Mutated HER2-Negative Advanced Breast Cancer: Final Overall Survival Results from the EMBRACA Trial. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2020, 31, 1526–1535. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.; Tong, Y.; Jones, D.R.; Walsh, T.; Danso, M.A.; Ma, C.X.; Silverman, P.; King, M.-C.; Badve, S.S.; Perkins, S.M. Cisplatin with or without Rucaparib after Preoperative Chemotherapy in Patients with Triple Negative Breast Cancer: Final Efficacy Results of Hoosier Oncology Group BRE09-146. J. Clin. Oncol. 2015, 33, 1082. [Google Scholar] [CrossRef]
- Telli, M.L.; Litton, J.K.; Beck, J.T.; Jones, J.M.; Andersen, J.; Mina, L.A.; Brig, R.; Danso, M.A.; Yuan, Y.; Symmans, W.F.; et al. Neoadjuvant Talazoparib (TALA) in Patients (Pts) with Germline BRCA1/2 (g BRCA1/2) Mutation-Positive, Early HER2-Negative Breast Cancer (BC): Exploration of Tumor BRCA Mutational Status and Zygosity and Overall Mutational Landscape in a Phase 2 Study. J. Clin. Oncol. 2021, 39, 554. [Google Scholar] [CrossRef]
- Tutt, A.N.J.; Garber, J.E.; Kaufman, B.; Viale, G.; Fumagalli, D.; Rastogi, P.; Gelber, R.D.; de Azambuja, E.; Fielding, A.; Balmaña, J.; et al. Adjuvant Olaparib for Patients with BRCA1- or BRCA2- Mutated Breast Cancer. N. Engl. J. Med. 2021, 384, 2394–2405. [Google Scholar] [CrossRef]
- Tung, N.M.; Robson, M.E.; Ventz, S.; Santa-Maria, C.A.; Nanda, R.; Marcom, P.K.; Shah, P.D.; Ballinger, T.J.; Yang, E.S.; Vinayak, S.; et al. TBCRC 048: Phase II Study of Olaparib for Metastatic Breast Cancer and Mutations in Homologous Recombination-Related Genes. J. Clin. Oncol. 2020, 38, 4274–4282. [Google Scholar] [CrossRef]
- Chopra, N.; Tovey, H.; Pearson, A.; Cutts, R.; Toms, C.; Proszek, P.; Hubank, M.; Dowsett, M.; Dodson, A.; Daley, F.; et al. Homologous Recombination DNA Repair Deficiency and PARP Inhibition Activity in Primary Triple Negative Breast Cancer. Nat. Commun. 2020, 11, 2662. [Google Scholar] [CrossRef] [PubMed]
- Diéras, V.; Han, H.S.; Kaufman, B.; Wildiers, H.; Friedlander, M.; Ayoub, J.-P.; Puhalla, S.L.; Bondarenko, I.; Campone, M.; Jakobsen, E.H.; et al. Veliparib with Carboplatin and Paclitaxel in BRCA-Mutated Advanced Breast Cancer (BROCADE3): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2020, 21, 1269–1282. [Google Scholar] [CrossRef]
- Rugo, H.S.; Olopade, O.I.; DeMichele, A.; Yau, C.; van ’t Veer, L.J.; Buxton, M.B.; Hogarth, M.; Hylton, N.M.; Paoloni, M.; Perlmutter, J.; et al. Adaptive Randomization of Veliparib–Carboplatin Treatment in Breast Cancer. N. Engl. J. Med. 2016, 375, 23–34. [Google Scholar] [CrossRef]
- Loibl, S.; O’Shaughnessy, J.; Untch, M.; Sikov, W.M.; Rugo, H.S.; McKee, M.D.; Huober, J.; Golshan, M.; von Minckwitz, G.; Maag, D.; et al. Addition of the PARP Inhibitor Veliparib plus Carboplatin or Carboplatin Alone to Standard Neoadjuvant Chemotherapy in Triple-Negative Breast Cancer (BrighTNess): A Randomised, Phase 3 Trial. Lancet Oncol. 2018, 19, 497–509. [Google Scholar] [CrossRef]
- Fasching, P.A.; Link, T.; Hauke, J.; Seither, F.; Jackisch, C.; Klare, P.; Schmatloch, S.; Hanusch, C.; Huober, J.; Stefek, A.; et al. Neoadjuvant Paclitaxel/Olaparib in Comparison to Paclitaxel/Carboplatinum in Patients with HER2-Negative Breast Cancer and Homologous Recombination Deficiency (GeparOLA Study). Ann. Oncol. 2021, 32, 49–57. [Google Scholar] [CrossRef]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.-K.; Hsu, J.-M.; Hsu, J.L.; Yu, W.-H.; Du, Y.; Lee, H.-H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Kim, H.-J.; Wang, Q.; Kearns, M.; Jiang, T.; Ohlson, C.E.; Li, B.B.; Xie, S.; Liu, J.F.; Stover, E.H.; et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep. 2018, 25, 2972–2980.e5. [Google Scholar] [CrossRef] [Green Version]
- Domchek, S.M.; Postel-Vinay, S.; Im, S.-A.; Park, Y.H.; Delord, J.-P.; Italiano, A.; Alexandre, J.; You, B.; Bastian, S.; Krebs, M.G.; et al. Olaparib and Durvalumab in Patients with Germline BRCA-Mutated Metastatic Breast Cancer (MEDIOLA): An Open-Label, Multicentre, Phase 1/2, Basket Study. Lancet Oncol. 2020, 21, 1155–1164. [Google Scholar] [CrossRef]
- Vinayak, S.; Tolaney, S.M.; Schwartzberg, L.; Mita, M.; McCann, G.; Tan, A.R.; Wahner-Hendrickson, A.E.; Forero, A.; Anders, C.; Wulf, G.M.; et al. Open-Label Clinical Trial of Niraparib Combined With Pembrolizumab for Treatment of Advanced or Metastatic Triple-Negative Breast Cancer. JAMA Oncol. 2019, 5, 1132–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barber, L.J.; Sandhu, S.; Chen, L.; Campbell, J.; Kozarewa, I.; Fenwick, K.; Assiotis, I.; Rodrigues, D.N.; Reis Filho, J.S.; Moreno, V.; et al. Secondary Mutations in BRCA2 Associated with Clinical Resistance to a PARP Inhibitor. J. Pathol. 2013, 229, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Gornstein, E.L.; Sandefur, S.; Chung, J.H.; Gay, L.M.; Holmes, O.; Erlich, R.L.; Soman, S.; Martin, L.K.; Rose, A.V.; Stephens, P.J.; et al. BRCA2 Reversion Mutation Associated With Acquired Resistance to Olaparib in Estrogen Receptor-Positive Breast Cancer Detected by Genomic Profiling of Tissue and Liquid Biopsy. Clin. Breast Cancer 2018, 18, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Comino-Méndez, I.; de Bruijn, I.; Tian, L.; Meisel, J.L.; García-Murillas, I.; Fribbens, C.; Cutts, R.; Martelotto, L.G.; Ng, C.K.Y.; et al. Diverse BRCA1 and BRCA2 Reversion Mutations in Circulating Cell-Free DNA of Therapy-Resistant Breast or Ovarian Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 6708–6720. [Google Scholar] [CrossRef] [Green Version]
- Tobalina, L.; Armenia, J.; Irving, E.; O’Connor, M.J.; Forment, J.V. A Meta-Analysis of Reversion Mutations in BRCA Genes Identifies Signatures of DNA End-Joining Repair Mechanisms Driving Therapy Resistance. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2021, 32, 103–112. [Google Scholar] [CrossRef]
- Waks, A.G.; Cohen, O.; Kochupurakkal, B.; Kim, D.; Dunn, C.E.; Buendia Buendia, J.; Wander, S.; Helvie, K.; Lloyd, M.R.; Marini, L.; et al. Reversion and Non-Reversion Mechanisms of Resistance to PARP Inhibitor or Platinum Chemotherapy in BRCA1/2-Mutant Metastatic Breast Cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2020, 31, 590–598. [Google Scholar] [CrossRef]
- Bunting, S.F.; Callén, E.; Wong, N.; Chen, H.-T.; Polato, F.; Gunn, A.; Bothmer, A.; Feldhahn, N.; Fernandez-Capetillo, O.; Cao, L.; et al. 53BP1 Inhibits Homologous Recombination in Brca1-Deficient Cells by Blocking Resection of DNA Breaks. Cell 2010, 141, 243–254. [Google Scholar] [CrossRef] [Green Version]
- Bouwman, P.; Aly, A.; Escandell, J.M.; Pieterse, M.; Bartkova, J.; van der Gulden, H.; Hiddingh, S.; Thanasoula, M.; Kulkarni, A.; Yang, Q.; et al. 53BP1 Loss Rescues BRCA1 Deficiency and Is Associated with Triple-Negative and BRCA-Mutated Breast Cancers. Nat. Struct. Mol. Biol. 2010, 17, 688–695. [Google Scholar] [CrossRef] [Green Version]
- Bruna, A.; Rueda, O.M.; Greenwood, W.; Batra, A.S.; Callari, M.; Batra, R.N.; Pogrebniak, K.; Sandoval, J.; Cassidy, J.W.; Tufegdzic-Vidakovic, A.; et al. A Biobank of Breast Cancer Explants with Preserved Intra-Tumor Heterogeneity to Screen Anticancer Compounds. Cell 2016, 167, 260–274.e22. [Google Scholar] [CrossRef] [Green Version]
- Militello, A.M.; Zielli, T.; Boggiani, D.; Michiara, M.; Naldi, N.; Bortesi, B.; Zanelli, P.; Uliana, V.; Giuliotti, S.; Musolino, A. Mechanism of Action and Clinical Efficacy of CDK4/6 Inhibitors in BRCA-Mutated, Estrogen Receptor-Positive Breast Cancers: Case Report and Literature Review. Front. Oncol. 2019, 9, 759. [Google Scholar] [CrossRef] [Green Version]
- Noordermeer, S.M.; Adam, S.; Setiaputra, D.; Barazas, M.; Pettitt, S.J.; Ling, A.K.; Olivieri, M.; Álvarez-Quilón, A.; Moatti, N.; Zimmermann, M.; et al. The Shieldin Complex Mediates 53BP1-Dependent DNA Repair. Nature 2018, 560, 117–121. [Google Scholar] [CrossRef]
- Zatreanu, D.; Robinson, H.M.R.; Alkhatib, O.; Boursier, M.; Finch, H.; Geo, L.; Grande, D.; Grinkevich, V.; Heald, R.A.; Langdon, S.; et al. Polθ Inhibitors Elicit BRCA-Gene Synthetic Lethality and Target PARP Inhibitor Resistance. Nat. Commun. 2021, 12, 3636. [Google Scholar] [CrossRef]
- Nakada, S.; Yonamine, R.M.; Matsuo, K. RNF8 Regulates Assembly of RAD51 at DNA Double-Strand Breaks in the Absence of BRCA1 and 53BP1. Cancer Res. 2012, 72, 4974–4983. [Google Scholar] [CrossRef] [Green Version]
- Luijsterburg, M.S.; Typas, D.; Caron, M.-C.; Wiegant, W.W.; van den Heuvel, D.; Boonen, R.A.; Couturier, A.M.; Mullenders, L.H.; Masson, J.-Y.; van Attikum, H. A PALB2-Interacting Domain in RNF168 Couples Homologous Recombination to DNA Break-Induced Chromatin Ubiquitylation. eLife 2017, 6, e20922. [Google Scholar] [CrossRef] [Green Version]
- Zong, D.; Adam, S.; Wang, Y.; Sasanuma, H.; Callén, E.; Murga, M.; Day, A.; Kruhlak, M.J.; Wong, N.; Munro, M.; et al. BRCA1 Haploinsufficiency Is Masked by RNF168-Mediated Chromatin Ubiquitylation. Mol. Cell 2019, 73, 1267–1281.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rottenberg, S.; Nygren, A.O.H.; Pajic, M.; van Leeuwen, F.W.B.; van der Heijden, I.; van de Wetering, K.; Liu, X.; de Visser, K.E.; Gilhuijs, K.G.; van Tellingen, O.; et al. Selective Induction of Chemotherapy Resistance of Mammary Tumors in a Conditional Mouse Model for Hereditary Breast Cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 12117–12122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christie, E.L.; Pattnaik, S.; Beach, J.; Copeland, A.; Rashoo, N.; Fereday, S.; Hendley, J.; Alsop, K.; Brady, S.L.; Lamb, G.; et al. Multiple ABCB1 Transcriptional Fusions in Drug Resistant High-Grade Serous Ovarian and Breast Cancer. Nat. Commun. 2019, 10, 1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Dréan, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-Wide and High-Density CRISPR-Cas9 Screens Identify Point Mutations in PARP1 Causing PARP Inhibitor Resistance. Nat. Commun. 2018, 9, 1849. [Google Scholar] [CrossRef]
- Zandarashvili, L.; Langelier, M.-F.; Velagapudi, U.K.; Hancock, M.A.; Steffen, J.D.; Billur, R.; Hannan, Z.M.; Wicks, A.J.; Krastev, D.B.; Pettitt, S.J.; et al. Structural Basis for Allosteric PARP-1 Retention on DNA Breaks. Science 2020, 368, eaax6367. [Google Scholar] [CrossRef] [PubMed]
- Gogola, E.; Duarte, A.A.; de Ruiter, J.R.; Wiegant, W.W.; Schmid, J.A.; de Bruijn, R.; James, D.I.; Guerrero Llobet, S.; Vis, D.J.; Annunziato, S.; et al. Selective Loss of PARG Restores PARylation and Counteracts PARP Inhibitor-Mediated Synthetic Lethality. Cancer Cell 2018, 33, 1078–1093.e12. [Google Scholar] [CrossRef] [Green Version]
- Noordermeer, S.M.; van Attikum, H. PARP Inhibitor Resistance: A Tug-of-War in BRCA-Mutated Cells. Trends Cell Biol. 2019, 29, 820–834. [Google Scholar] [CrossRef] [Green Version]
- Rondinelli, B.; Gogola, E.; Yücel, H.; Duarte, A.A.; van de Ven, M.; van der Sluijs, R.; Konstantinopoulos, P.A.; Jonkers, J.; Ceccaldi, R.; Rottenberg, S.; et al. EZH2 Promotes Degradation of Stalled Replication Forks by Recruiting MUS81 through Histone H3 Trimethylation. Nat. Cell Biol. 2017, 19, 1371–1378. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.-E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication Fork Stability Confers Chemoresistance in BRCA-Deficient Cells. Nature 2016, 535, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J. DNA Repair Dysregulation from Cancer Driver to Therapeutic Target. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef]
- Dobbelstein, M.; Sørensen, C.S. Exploiting Replicative Stress to Treat Cancer. Nat. Rev. Drug Discov. 2015, 14, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Otto, T.; Sicinski, P. Cell Cycle Proteins as Promising Targets in Cancer Therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; George, E.; Ragland, R.; Rafail, S.; Zhang, R.; Krepler, C.; Morgan, M.; Herlyn, M.; Brown, E.; Simpkins, F. Targeting the ATR/CHK1 Axis with PARP Inhibition Results in Tumor Regression in BRCA-Mutant Ovarian Cancer Models. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 3097–3108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yazinski, S.A.; Comaills, V.; Buisson, R.; Genois, M.-M.; Nguyen, H.D.; Ho, C.K.; Todorova Kwan, T.; Morris, R.; Lauffer, S.; Nussenzweig, A.; et al. ATR Inhibition Disrupts Rewired Homologous Recombination and Fork Protection Pathways in PARP Inhibitor-Resistant BRCA-Deficient Cancer Cells. Genes Dev. 2017, 31, 318–332. [Google Scholar] [CrossRef]
- Parmar, K.; Kochupurakkal, B.S.; Lazaro, J.-B.; Wang, Z.C.; Palakurthi, S.; Kirschmeier, P.T.; Yang, C.; Sambel, L.A.; Färkkilä, A.; Reznichenko, E.; et al. The CHK1 Inhibitor Prexasertib Exhibits Monotherapy Activity in High-Grade Serous Ovarian Cancer Models and Sensitizes to PARP Inhibition. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 6127–6140. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-Homologous DNA End Joining and Alternative Pathways to Double-Strand Break Repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Ranjha, L.; Howard, S.M.; Cejka, P. Main Steps in DNA Double-Strand Break Repair: An Introduction to Homologous Recombination and Related Processes. Chromosoma 2018, 127, 187–214. [Google Scholar] [CrossRef] [Green Version]
- Mateos-Gomez, P.A.; Gong, F.; Nair, N.; Miller, K.M.; Lazzerini-Denchi, E.; Sfeir, A. Mammalian Polymerase θ Promotes Alternative NHEJ and Suppresses Recombination. Nature 2015, 518, 254–257. [Google Scholar] [CrossRef] [Green Version]
- Zahn, K.E.; Jensen, R.B.; Wood, R.D.; Doublié, S. Human DNA Polymerase θ Harbors DNA End-Trimming Activity Critical for DNA Repair. Mol. Cell 2021, 81, 1534–1547.e4. [Google Scholar] [CrossRef]
- Maiorano, D.; El Etri, J.; Franchet, C.; Hoffmann, J.-S. Translesion Synthesis or Repair by Specialized DNA Polymerases Limits Excessive Genomic Instability upon Replication Stress. Int. J. Mol. Sci. 2021, 22, 3924. [Google Scholar] [CrossRef]
- Setiaputra, D.; Durocher, D. Shieldin—The Protector of DNA Ends. EMBO Rep. 2019. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Liu, J.C.; Amunugama, R.; Hajdu, I.; Primack, B.; Petalcorin, M.I.R.; O’Connor, K.W.; Konstantinopoulos, P.A.; Elledge, S.J.; Boulton, S.J.; et al. Homologous-Recombination-Deficient Tumours Are Dependent on Polθ-Mediated Repair. Nature 2015, 518, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Gelot, C.; Pantelidou, C.; Li, A.; Yücel, H.; Davis, R.E.; Farkkila, A.; Kochupurakkal, B.; Syed, A.; Shapiro, G.I.; et al. A First-in-Class Polymerase Theta Inhibitor Selectively Targets Homologous-Recombination-Deficient Tumors. Nat. Cancer 2021, 2, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Edwards, S.L.; Brough, R.; Lord, C.J.; Natrajan, R.; Vatcheva, R.; Levine, D.A.; Boyd, J.; Reis-Filho, J.S.; Ashworth, A. Resistance to Therapy Caused by Intragenic Deletion in BRCA2. Nature 2008, 451, 1111–1115. [Google Scholar] [CrossRef]
- Feng, W.; Simpson, D.A.; Carvajal-Garcia, J.; Price, B.A.; Kumar, R.J.; Mose, L.E.; Wood, R.D.; Rashid, N.; Purvis, J.E.; Parker, J.S.; et al. Genetic Determinants of Cellular Addiction to DNA Polymerase Theta. Nat. Commun. 2019, 10, 4286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaspers, J.E.; Kersbergen, A.; Boon, U.; Sol, W.; van Deemter, L.; Zander, S.A.; Drost, R.; Wientjens, E.; Ji, J.; Aly, A.; et al. Loss of 53BP1 Causes PARP Inhibitor Resistance in Brca1-Mutated Mouse Mammary Tumors. Cancer Discov. 2013, 3, 68–81. [Google Scholar] [CrossRef] [Green Version]
- Gogola, E.; Rottenberg, S.; Jonkers, J. Resistance to PARP Inhibitors: Lessons from Preclinical Models of BRCA-Associated Cancer. Annu. Rev. Cancer Biol. 2019, 3, 235–254. [Google Scholar] [CrossRef]
- Verma, P.; Zhou, Y.; Cao, Z.; Deraska, P.V.; Deb, M.; Arai, E.; Li, W.; Shao, Y.; Puentes, L.; Li, Y.; et al. ALC1 Links Chromatin Accessibility to PARP Inhibitor Response in Homologous Recombination-Deficient Cells. Nat. Cell Biol. 2021, 23, 160–171. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Clinical Trial | Phase | Patient Population | PARPi | Other Anti-Tumoural Therapy |
|---|---|---|---|---|
| DORA NCT03167619 | II | mTNBC after carboplatine | olaparib in maintenance | durvalumab |
| NCT03801369 | II | mTNBC | olaparib | durvalumab |
| DOLAF NCT04053322 | II | ER+/HER2-negative mBC with gBRCA mutation or HRD+ | olaparib | durvalumab and fulvestrant |
| NCT02849496 | II | HER2-negative and HRD+ mBC | olaparib | atezolizumab |
| NCT02484404 | I-II | mTNBC | olaparib | durvalumab and cediranib (anti-angiogenic) |
| NCT03025035 | II | gBRCA mutation mBC | olaparib | pembrolizumab |
| NCT03598257 | II | inflammatory BC | olaparib | radiotherapy |
| UNITY NCT03945721 | I | early TNBC with residual disease after NACT | niraparib | radiotherapy |
| NCT03542175 | I | early TNBC with residual disease after NACT | rucaparib | radiotherapy |
| NCT01623349 | I | mTNBC | olaparib | BKM120 ou BYL719 |
| NCT04586335 | I | HRD+ and/or PI3K mutation solid cancer | olaparib | CYH33 |
| NCT04039230 | I-II | mTNBC | talazoparib | Sacituzumab-govitecan |
| NCT03901469 | II | mTNBC without gBRCA mutation | talazoparib | ZEN003694 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franchet, C.; Hoffmann, J.-S.; Dalenc, F. Recent Advances in Enhancing the Therapeutic Index of PARP Inhibitors in Breast Cancer. Cancers 2021, 13, 4132. https://doi.org/10.3390/cancers13164132
Franchet C, Hoffmann J-S, Dalenc F. Recent Advances in Enhancing the Therapeutic Index of PARP Inhibitors in Breast Cancer. Cancers. 2021; 13(16):4132. https://doi.org/10.3390/cancers13164132
Chicago/Turabian StyleFranchet, Camille, Jean-Sébastien Hoffmann, and Florence Dalenc. 2021. "Recent Advances in Enhancing the Therapeutic Index of PARP Inhibitors in Breast Cancer" Cancers 13, no. 16: 4132. https://doi.org/10.3390/cancers13164132
APA StyleFranchet, C., Hoffmann, J.-S., & Dalenc, F. (2021). Recent Advances in Enhancing the Therapeutic Index of PARP Inhibitors in Breast Cancer. Cancers, 13(16), 4132. https://doi.org/10.3390/cancers13164132