
Phytogalactolipid dLGG Inhibits Mouse Melanoma Brain Metastasis through Regulating Oxylipin Activity and Re-Programming Macrophage Polarity in the Tumor Microenvironment

Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Isolation of dLGG
2.3. Cell Viability Assay
2.4. Cell Clonogenic Assay
2.5. Tube Formation Assay
2.6. Cell Cycle and Cell Apoptosis Assay
2.7. Western Blotting
2.8. Differentiation of THP-1 into Macrophages
2.9. Animals
2.10. Establishment of the Melanoma Brain Metastatic Mouse Model and Brain-Seeking Melanoma Cell Lines
2.11. In Vivo Animal Study
2.12. Tight Junction Permeability Analysis
2.13. Hematoxylin and Eosin Staining and Immunohistochemistry
2.14. Ex Vivo 3D Tumor-Brain Organoid Co-Culture Model
2.15. Ultra-Performance Liquid Chromatography–Electrospray Ionization Tandem Mass Spectrometry Analysis of Serum Oxylipin Metabolome
2.16. Statistical Analysis
3. Results
3.1. Establishment of the Melanoma Brain Metastatic Mouse Model and Brain-Seeking Melanoma Cell
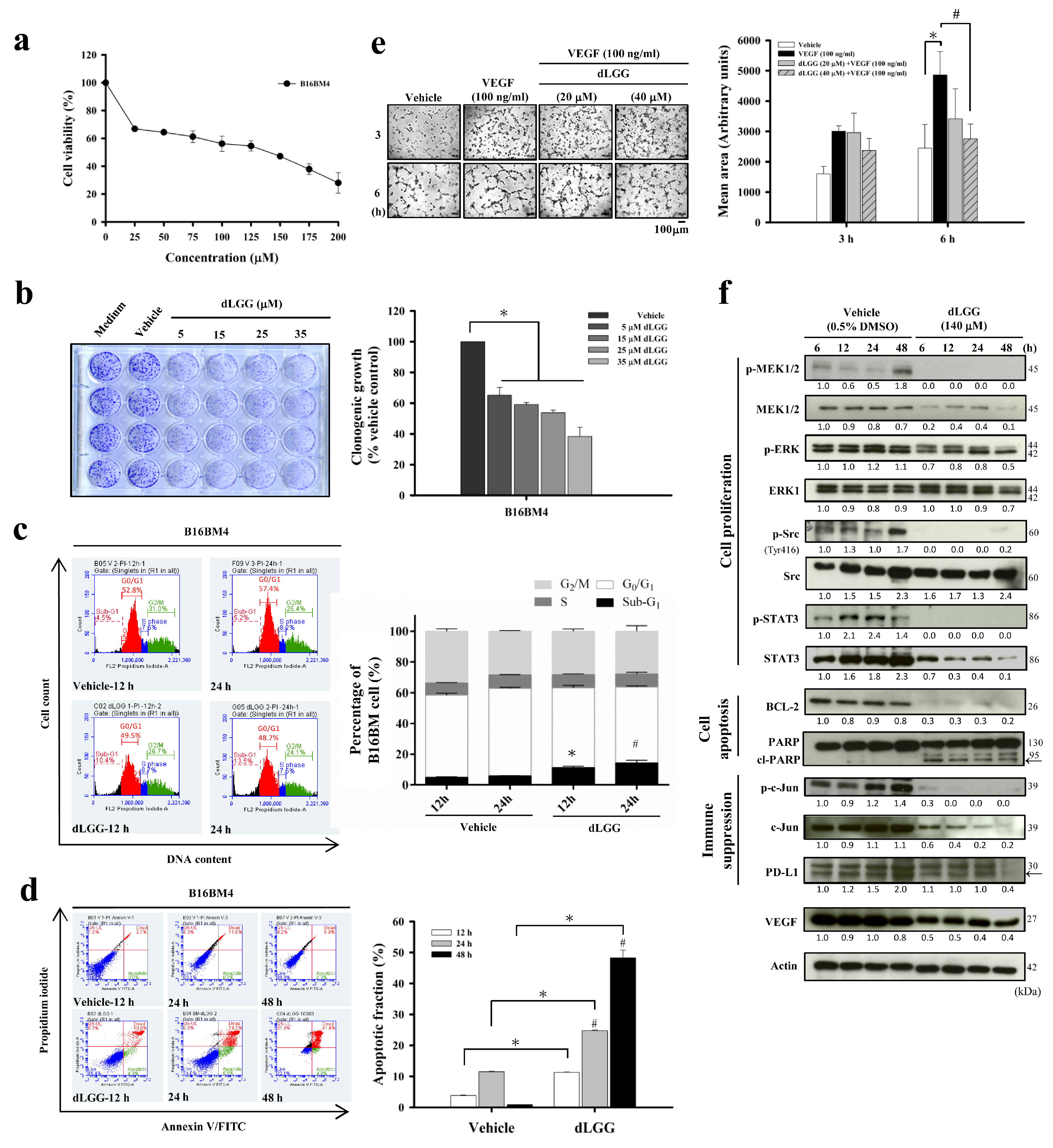
3.2. dLGG Inhibits B16BM4 Brain-Seeking Cell Activities, HUVEC Tube Formation, and Selected Marker Protein Expression
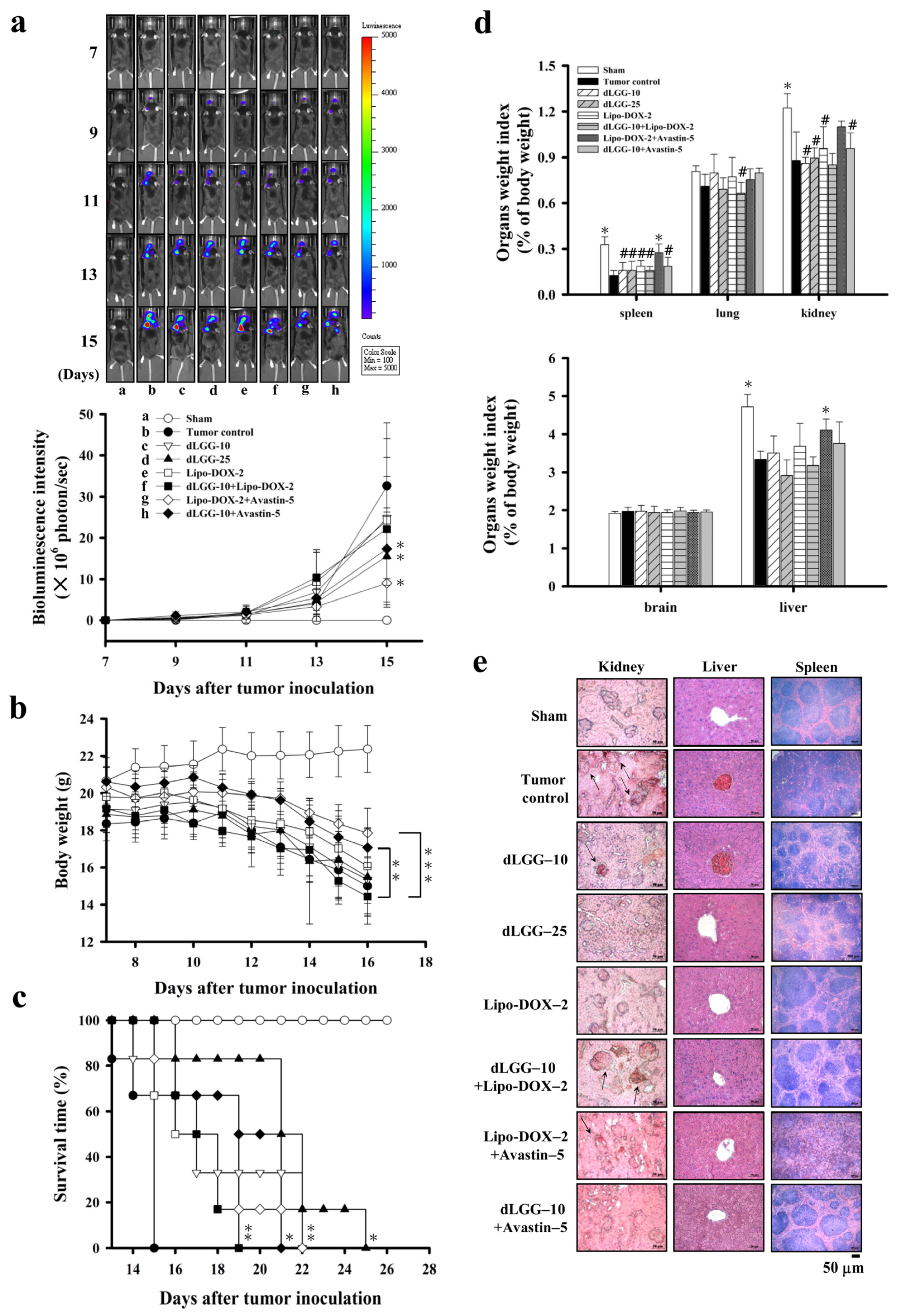
3.3. dLGG Suppresses Melanoma Brain Metastases in Mice
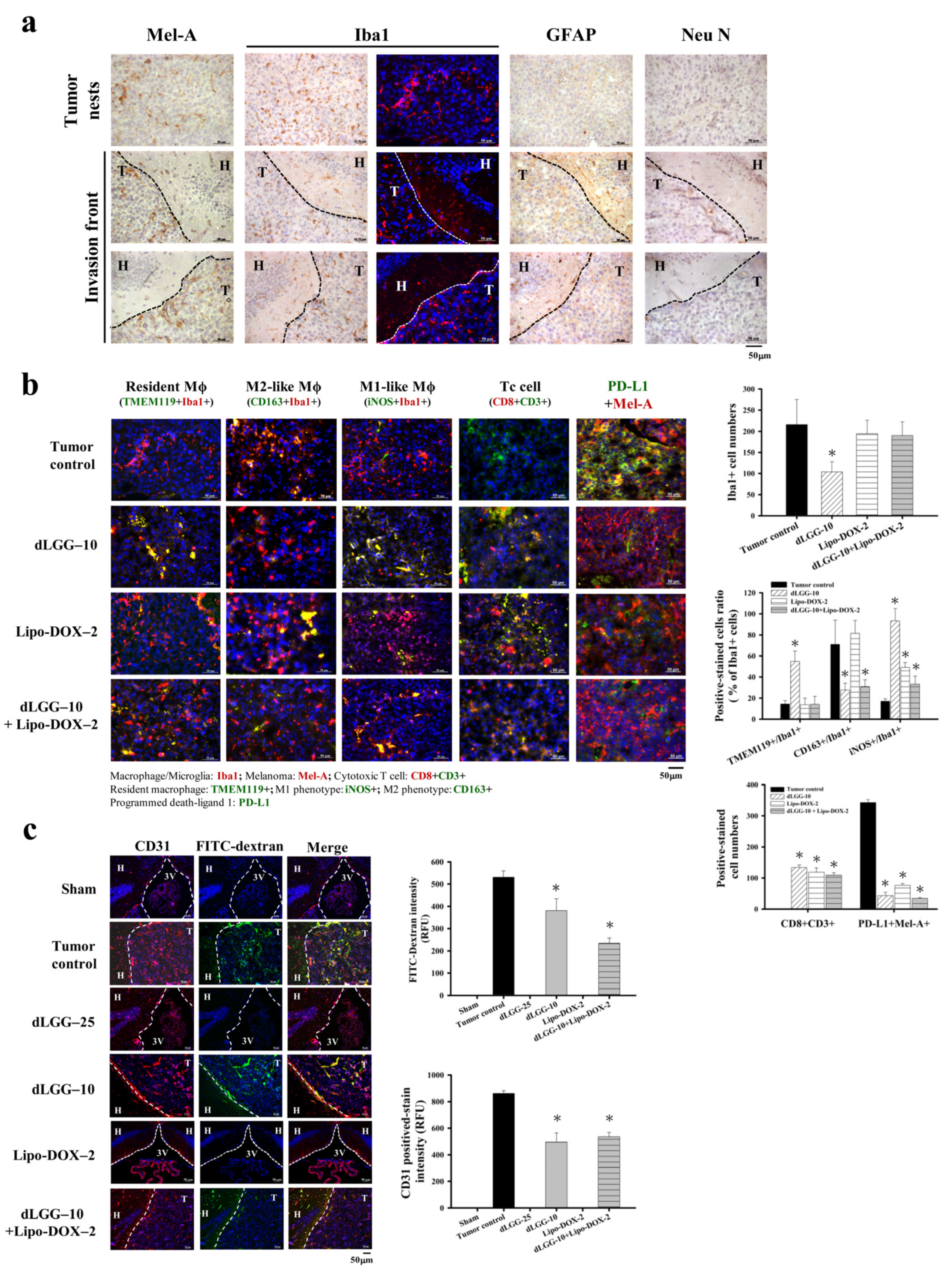
3.4. dLGG Reprograms Tumor-Associated Macrophage Profiles in the Melanoma Tumor Microenvironment in Mouse Brain
3.5. dLGG Inhibits Angiogenesis and Prevents Melanoma Cell-Induced Vascular Tight Junction Permeability
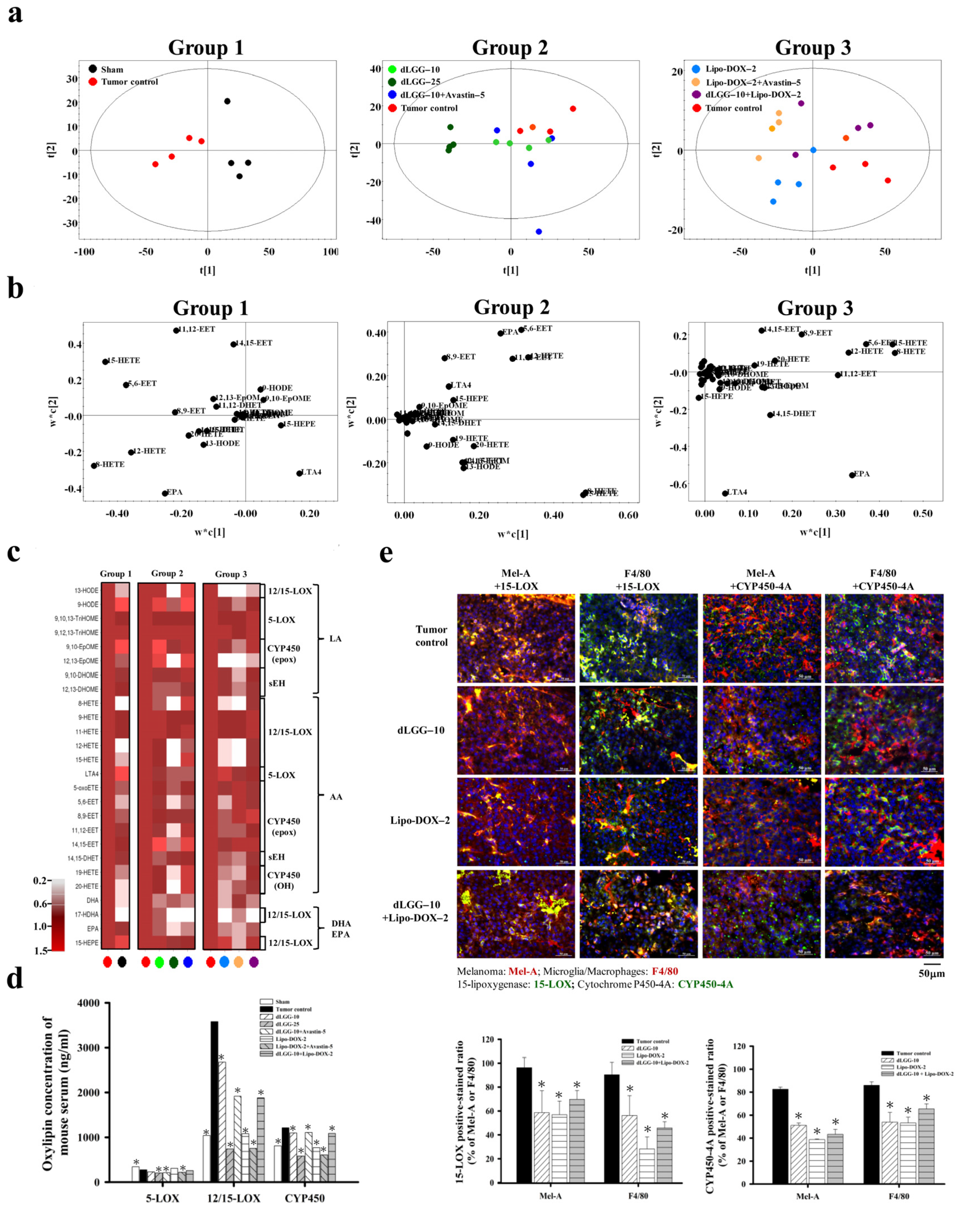
3.6. Serum Oxylipin Metabolome in Tested Mice
3.7. dLGG Inhibits 15-LOX and CYP450-4A Expressions in Brain TME
3.8. dLGG-Primed Conditioning Medium of B16BM4 Cells Modulates Macrophage Polarity
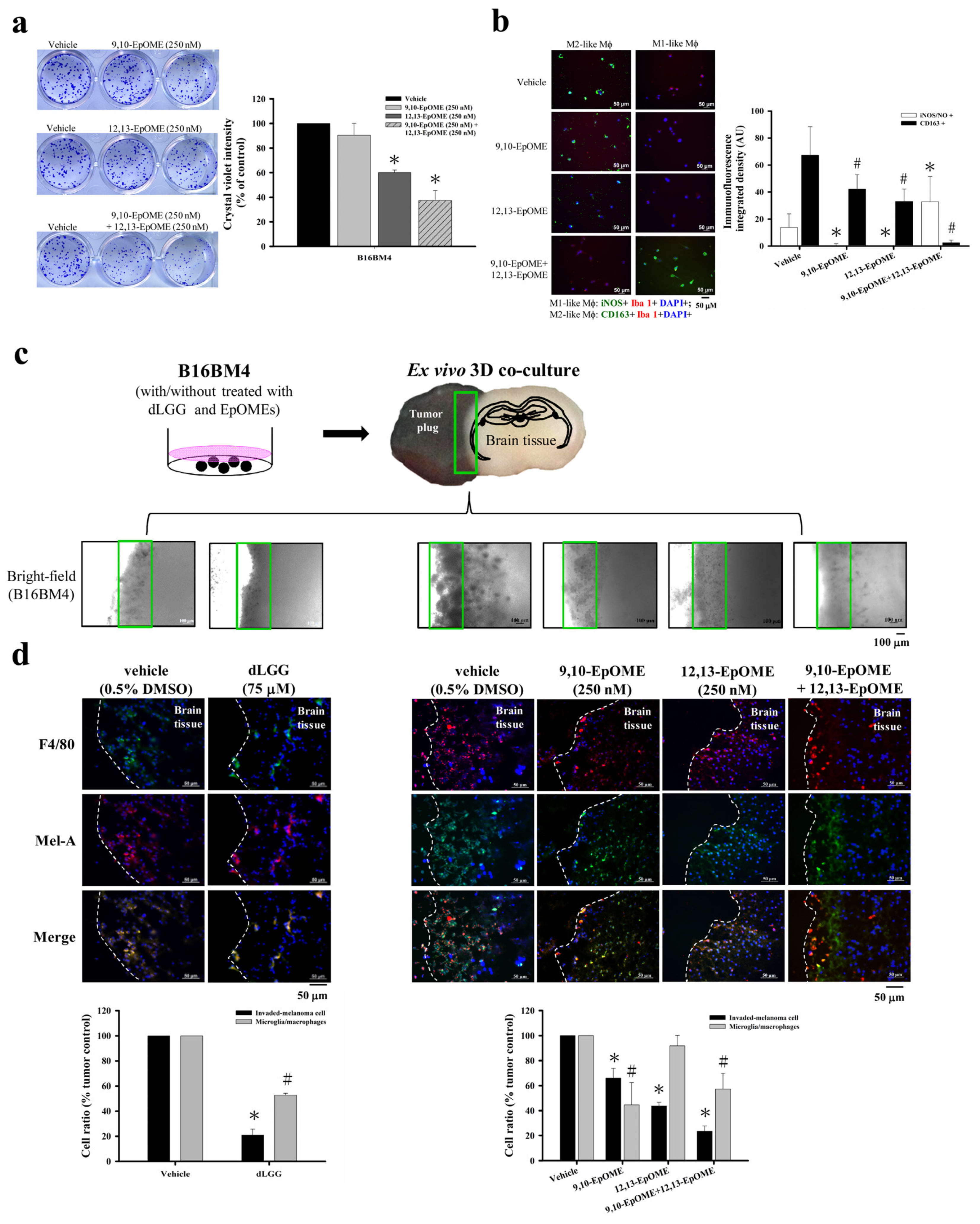
3.9. dLGG and Oxylipins 9,10-EpOME and 12,13-EpOME Inhibit Melanoma Cell Activity and Invade the Brain Parenchyma
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tawbi, H.A.; Forsyth, P.A.; Algazi, A.; Hamid, O.; Hodi, F.S.; Moschos, S.J.; Khushalani, N.I.; Lewis, K.; Lao, C.D.; Postow, M.A.; et al. Combined nivolumab and ipilimumab in melanoma metastatic to the brain. N. Engl. J. Med. 2018, 379, 722–730. [Google Scholar] [CrossRef]
- Yang, Y.; Yan, Z.; Wei, D.; Zhong, J.; Liu, L.; Zhang, L.; Wang, F.; Wei, X.; Xie, C.; Lu, W.; et al. Tumor-penetrating peptide functionalization enhances the anti-glioblastoma effect of doxorubicin liposomes. Nanotechnology 2013, 24, 405101. [Google Scholar] [CrossRef]
- Smylie, M.G.; Wong, R.; Mihalcioiu, C.; Lee, C.; Pouliot, J.-F. A phase II, open label, monotherapy study of liposomal doxorubicin in patients with metastatic malignant melanoma. Investig. New Drugs. 2007, 25, 155–159. [Google Scholar] [CrossRef]
- Ferrucci, P.F.; Minchella, I.; Mosconi, M.; Gandini, S.; Verrecchia, F.; Cocorocchio, E.; Passoni, C.; Pari, C.; Testori, A.; Coco, P.; et al. Dacarbazine in combination with bevacizumab for the treatment of unresectable/metastatic melanoma: A phase II study. Melanoma Res. 2015, 25, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Tiainen, L.; Tanner, M.; Lahdenperä, O.; Vihinen, P.; Jukkola, A.; Karihtala, P.; Paunu, N.; Huttunen, T.; Kellokumpu-Lehtinen, P.L. Bevacizumab combined with docetaxel or paclitaxel as first-line treatment of HER2-negative metastatic breast cancer. Anticancer Res. 2016, 36, 6431–6438. [Google Scholar] [CrossRef]
- Brychtova, S.; Bezdekova, M.; Brychta, T.; Tichy, M. The role of vascular endothelial growth factors and their receptors in malignant melanomas. Neoplasma 2008, 55, 273–279. [Google Scholar] [PubMed]
- Piperno-Neumann, S.; Diallo, A.; Etienne-Grimaldi, M.C.; Bidard, F.C.; Rodrigues, M.; Plancher, C.; Mariani, P.; Cassoux, N.; Decaudin, D.; Asselain, B.; et al. Phase II trial of bevacizumab in combination with temozolomide as first-line treatment in patients with metastatic uveal melanoma. Oncologist 2016, 21, 281–282. [Google Scholar] [CrossRef][Green Version]
- Tawbi, H.A.; Boutros, C.; Kok, D.; Robert, C.; McArthur, G. New era in the management of melanoma brain metastases. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 741–750. [Google Scholar] [CrossRef]
- Naidoo, J.; Page, D.B.; Li, B.T.; Connell, L.C.; Schindler, K.; Lacouture, M.A.; Postow, M.A.; Wolchok, J.D. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann. Oncol. 2015, 26, 2375–2391. [Google Scholar] [CrossRef] [PubMed]
- Roesch, S.; Rapp, C.; Dettling, S.; Herold-Mende, C. When immune cells turn bad-tumor-associated microglia/macrophages in glioma. Int. J. Mol. Sci. 2018, 19, 436. [Google Scholar] [CrossRef]
- Falleni, M.; Savi, F.; Tosi, D.; Agape, E.; Cerri, A.; Moneghini, L.; Bulfamante, G.P. M1 and M2 macrophages’ clinicopathological significance in cutaneous melanoma. Melanoma Res. 2017, 27, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Palma, M. Origins of brain tumor macrophages. Cancer Cell. 2016, 30, 832–833. [Google Scholar] [CrossRef] [PubMed]
- Linde, N.; Casanova-Acebes, M.; Sosa, M.S.; Mortha, A.; Rahman, A.; Farias, E.; Harper, K.; Tardio, E.; Reyes Torres, I.; Jones, J.; et al. Macrophages orchestrate breast cancer early dissemination and metastasis. Nat. Commun. 2018, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Tariq, M.; Zhang, J.; Liang, G.; Ding, L.; He, Q.; Yang, B. Macrophage polarization: Anti-canccer strategies to target tumor-associated macrophage in breast cancer. J. Cell Biochem. 2017, 118, 2484–2501. [Google Scholar] [CrossRef] [PubMed]
- Andreou, K.E.; Soto, M.S.; Allen, D.; Economopoulos, V.; De Bernardi, A.; Larkin, J.R.; Sibson, N.R. Anti-inflammatory microglia/macrophages as a potential therapeutic target in brain metastasis. Front. Oncol. 2017, 7, 251. [Google Scholar] [CrossRef] [PubMed]
- Chanmee, T.; Ontong, P.; Konno, K.; Itano, N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers 2014, 6, 1670–1690. [Google Scholar] [CrossRef] [PubMed]
- Zivkovic, A.M.; Yang, J.; Georgi, K.; Hegedus, C.; Nording, M.L.; O’Sullivan, A.; German, J.B.; Hogg, R.J.; Weiss, R.H.; Bay, C.; et al. Serum oxylipin profiles in IgA nephropathy patients reflect kidney functional alterations. Metabolomics 2012, 8, 1102–1113. [Google Scholar] [CrossRef][Green Version]
- Kang, K.H.; Ling, T.Y.; Liou, H.H.; Huang, Y.K.; Hour, M.J.; Liou, H.C.; Fu, W.M. Enhancement role of host 12/15-lipoxygenase in melanoma progression. Eur. J. Cancer 2013, 49, 2747–2759. [Google Scholar] [CrossRef]
- Chen, X.W.; Yu, T.J.; Zhang, J.; Li, Y.; Chen, H.L.; Yang, G.F.; Yu, W.; Liu, Y.Z.; Liu, X.X.; Duan, C.F.; et al. CYP4A in tumor-associated macrophages promotes pre-metastatic niche formation and metastasis. Oncogene 2017, 36, 5045–5057. [Google Scholar] [CrossRef]
- Nosaka, T.; Baba, T.; Tanabe, Y.; Sasaki, S.; Nishimura, T.; Imamura, Y.; Yurino, H.; Hashimoto, S.; Arita, M.; Nakamoto, Y.; et al. Alveolar macrophages drive hepatocellular carcinoma lung metastasis by generating leukotriene B4. J. Immunol. 2018, 200, 1839–1852. [Google Scholar] [CrossRef]
- Hou, C.C.; Chen, Y.P.; Wu, J.H.; Huang, C.C.; Wang, S.Y.; Yang, N.S.; Shyur, L.F. A galactolipid possesses novel cancer chemopreventive effects by suppressing inflammatory mediators and mouse B16 melanoma. Cancer Res. 2007, 67, 6907–6915. [Google Scholar] [CrossRef] [PubMed]
- Apaya, M.K.; Lin, C.Y.; Chiou, C.Y.; Yang, C.C.; Ting, C.Y.; Shyur, L.F. Simvastatin and a plant galactolipid protect animals from septic shock by regulating oxylipin mediator dynamics through the MAPK-cPLA2 signaling pathway. Mol. Med. 2016, 21, 88–1001. [Google Scholar] [CrossRef]
- Yang, C.C.; Chang, C.K.; Chang, M.T.; Shyur, L.F. Plant galactolipid dLGG suppresses lung metastasis of melanoma through deregulating TNF-α-mediated pulmonary vascular permeability and circulating oxylipin dynamics in mice. Int. J. Cancer 2018, 143, 3248–3261. [Google Scholar] [CrossRef] [PubMed]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; Bree Van, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef]
- Smith, M.P.; Young, H.; Hurlstone, A.; Wellbrock, C. Differentiation of THP1 cells into macrophages for transwell co-culture assay with melanoma cells. Bio-protocol 2015, 5, e1638. [Google Scholar] [CrossRef]
- Blazquez, R.; Pukrop, T. 3D coculture model of the brain parenchyma-metastasis interface of brain metastasis. Methods Mol. Biol. 2017, 1612, 213–222. [Google Scholar] [CrossRef]
- Yamasaki, M.; Murakami, I.; Nakano, K.; Doi, M.; Kitaguchi, S.; Kondo, T.; Sakurai, J.; Hattori, N.; Arita, K.I. Carboplatin plus weekly paclitaxel combined with bevacizumab as first-line treatment for non-small cell lung cancer. Anticancer Res. 2017, 37, 923–928. [Google Scholar] [CrossRef][Green Version]
- Bai, L.; Wang, F.; Li, Z.Z.; Ren, C.; Zhang, D.S.; Zhao, Q.; Lu, Y.X.; Wang, D.S.; Ju, H.Q.; Qiu, M.Z.; et al. Chemotherapy plus bevacizumab versus chemotherapy plus cetuximab as first-line treatment for patients with metastatic colorectal cancer: Results of a registry-based cohort analysis. Medicine 2016, 95, e4531. [Google Scholar] [CrossRef] [PubMed]
- Izraely, S.; Ben-Menachem, S.; Sagi-Assif, O.; Telerman, A.; Zubrilov, I.; Ashkenazi, O.; Meshel, T.; Maman, S.; Orozco, J.I.J.; Salomon, M.P.; et al. The metastatic microenvironment: Melanoma-microglia cross-talk promotes the malignant phenotype of melanoma cells. Int. J. Cancer. 2019, 144, 802–817. [Google Scholar] [CrossRef] [PubMed]
- Qiao, S.; Qian, Y.; Xu, G.; Luo, Q.; Zhang, Z. Long-term characterization of activated microglia/macrophages facilitating the development of experimental brain metastasis though intravital microscopic imaging. J. Neuroinflamm. 2019, 16, 4. [Google Scholar] [CrossRef]
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.L.; Edson, K.Z.; Totah, R.A.; Rettie, A.E. Cytochrome P450 ω-Hydroxylases in Inflammation and Cancer. Adv. Pharmacol. 2015, 74, 223–262. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrates | Catalytic Enzyme | Oxylipins | Sham | Tumor | dLGG–10 | dLGG–25 | Lipo-DOX–2 | Avastin–5 + Lipo-DOX–2 | dLGG–10 + Lipo-DOX–2 | dLGG–10 + Avastin–5 |
|---|---|---|---|---|---|---|---|---|---|---|
| LA | 15-LOX | 13-HODE | 0.5 | 1 | 1.0 | 0.3 | 0.3 | 0.2 | 0.5 | 1.2 |
| 9-LOX | 9-HODE | 1.4 | 1 | 1.2 | 0.7 | 0.8 | 0.6 | 0.9 | 1.5 | |
| 9,10,13-TriHOME | 0.9 | 1 | 0.9 | 0.9 | 0.9 | 0.9 | 1.0 | 0.9 | ||
| 9,12,13-TriHOME | 1.0 | 1 | 1.0 | 1.0 | 1.0 | 0.9 | 1.0 | 1.0 | ||
| CYP450 (epox) | 9,10-EpOME | 1.5 | 1 | 1.3 | 0.8 | 0.8 | 0.6 | 1.0 | 1.0 | |
| 12,13-EpOME | 0.7 | 1 | 1.2 | 0.3 | 0.3 | 0.3 | 0.5 | 1.2 | ||
| sEH | 9,10-DHOME | 0.8 | 1 | 0.8 | 0.8 | 0.8 | 0.5 | 0.9 | 1.0 | |
| 12,13-DHOME | 0.9 | 1 | 0.7 | 0.7 | 0.8 | 0.6 | 0.8 | 0.8 | ||
| AA | 12/15-LOX | 8-HETE | 0.3 | 1 | 0.7 | 0.2 | 0.4 | 0.3 | 0.8 | 1.1 |
| 9-HETE | 0.9 | 1 | 1.0 | 0.9 | 0.9 | 0.9 | 1.0 | 0.9 | ||
| 11-HETE | 0.9 | 1 | 1.0 | 0.9 | 0.9 | 0.9 | 1.0 | 1.0 | ||
| 12-HETE | 0.3 | 1 | 0.9 | 0.2 | 0.4 | 0.3 | 0.8 | 0.7 | ||
| 15-HETE | 0.5 | 1 | 0.9 | 0.2 | 0.4 | 0.3 | 0.8 | 1.1 | ||
| 5-LOX | LTA4 | 1.3 | 1 | 0.8 | 0.7 | 1.1 | 0.8 | 0.9 | 0.7 | |
| 5-oxoETE | 0.7 | 1 | 0.9 | 0.7 | 0.8 | 0.7 | 0.8 | 0.9 | ||
| CYP450 (epoxy) | 5,6-EET | 0.5 | 1 | 0.8 | 0.4 | 0.5 | 0.4 | 0.8 | 0.7 | |
| 8,9-EET | 0.7 | 1 | 0.9 | 0.8 | 0.7 | 0.6 | 1.1 | 0.8 | ||
| 11,12-EET | 0.8 | 1 | 0.8 | 0.4 | 0.7 | 0.5 | 0.9 | 1.0 | ||
| 14,15-EET | 0.9 | 1 | 1.3 | 0.6 | 0.8 | 0.7 | 1.0 | 1.2 | ||
| sEH | 11,12-DHET | 0.8 | 1 | 0.8 | 0.4 | 0.7 | 0.5 | 0.9 | 1.0 | |
| 14,15-DHET | 0.9 | 1 | 1.3 | 0.6 | 0.8 | 0.7 | 1.0 | 1.2 | ||
| CYP450 (OH) | 19-HETE | 0.6 | 1 | 0.9 | 0.4 | 0.6 | 0.5 | 0.9 | 1.1 | |
| 20-HETE | 0.4 | 1 | 0.9 | 0.3 | 0.5 | 0.5 | 0.8 | 1.1 | ||
| DHA | 0.4 | 1 | 0.6 | 0.7 | 0.5 | 0.3 | 0.6 | 0.6 | ||
| 12/15-LOX | 17-HDHA | 0.2 | 1 | 0.7 | 0.2 | 0.2 | 0.1 | 0.3 | 0.2 | |
| EPA | 0.8 | 1 | 0.7 | 0.4 | 0.6 | 0.4 | 0.7 | 0.6 | ||
| 12/15-LOX | 15-HEPE | 1.2 | 1 | 1.0 | 0.8 | 1.1 | 1.0 | 1.0 | 0.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, C.-C.; Chang, M.-T.; Chang, C.-K.; Shyur, L.-F. Phytogalactolipid dLGG Inhibits Mouse Melanoma Brain Metastasis through Regulating Oxylipin Activity and Re-Programming Macrophage Polarity in the Tumor Microenvironment. Cancers 2021, 13, 4120. https://doi.org/10.3390/cancers13164120
Yang C-C, Chang M-T, Chang C-K, Shyur L-F. Phytogalactolipid dLGG Inhibits Mouse Melanoma Brain Metastasis through Regulating Oxylipin Activity and Re-Programming Macrophage Polarity in the Tumor Microenvironment. Cancers. 2021; 13(16):4120. https://doi.org/10.3390/cancers13164120
Chicago/Turabian StyleYang, Chung-Chih, Meng-Ting Chang, Cheng-Kuei Chang, and Lie-Fen Shyur. 2021. "Phytogalactolipid dLGG Inhibits Mouse Melanoma Brain Metastasis through Regulating Oxylipin Activity and Re-Programming Macrophage Polarity in the Tumor Microenvironment" Cancers 13, no. 16: 4120. https://doi.org/10.3390/cancers13164120
APA StyleYang, C.-C., Chang, M.-T., Chang, C.-K., & Shyur, L.-F. (2021). Phytogalactolipid dLGG Inhibits Mouse Melanoma Brain Metastasis through Regulating Oxylipin Activity and Re-Programming Macrophage Polarity in the Tumor Microenvironment. Cancers, 13(16), 4120. https://doi.org/10.3390/cancers13164120