
Genomic Mapping of Splicing-Related Genes Identify Amplifications in LSM1, CLNS1A, and ILF2 in Luminal Breast Cancer

 ,
,  , ,
, ,  , and
, and 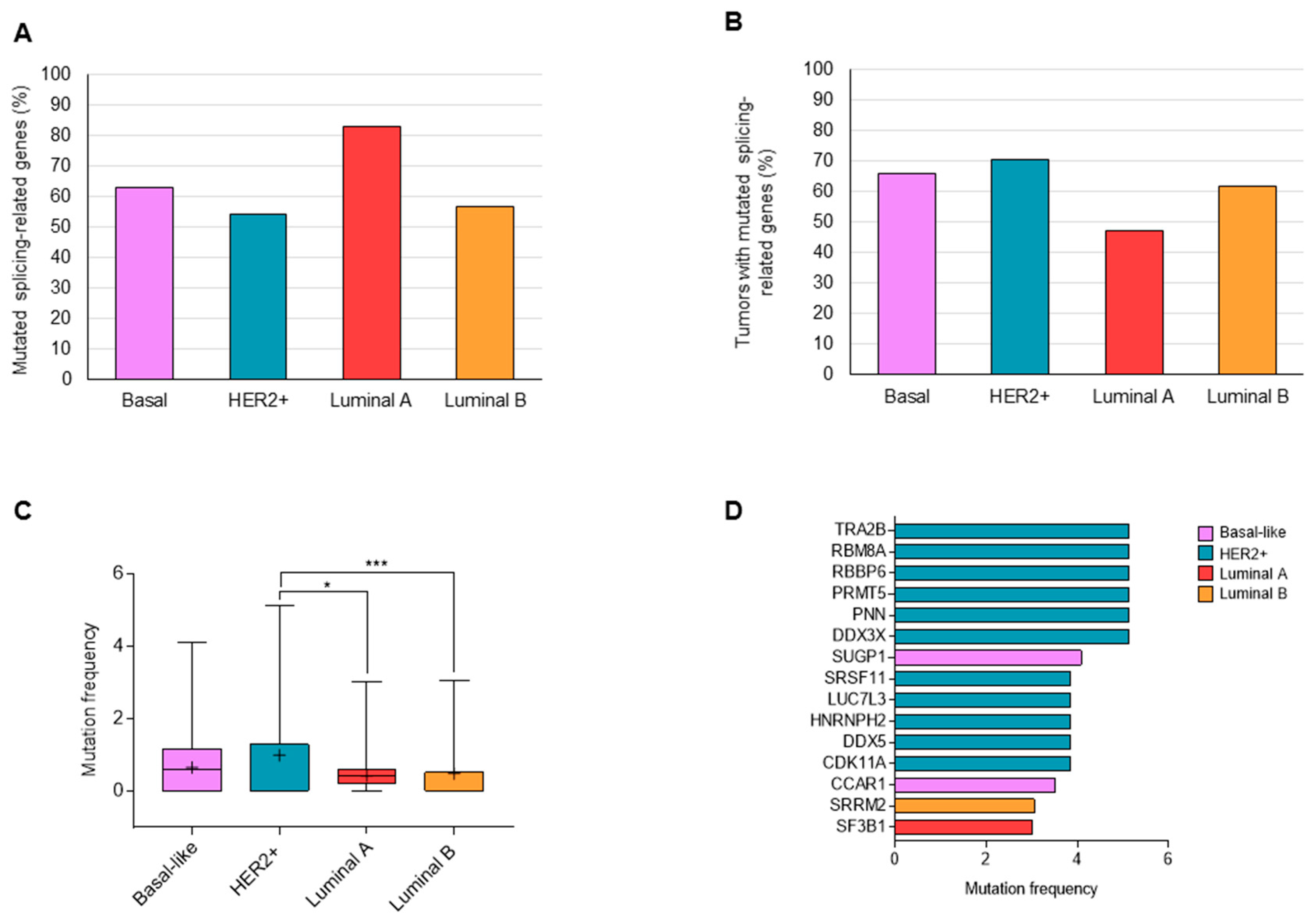{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Collection and Processing
2.2. Outcome Analysis
2.3. Cell Culture and Compounds
2.4. Small Interfering RNA
2.5. Quantitative Reverse-Transcription PCR
2.6. Proliferation MTT Assays
2.7. Growth Studies
2.8. Cell Cycle Assay
2.9. Statistical Analysis
3. Results
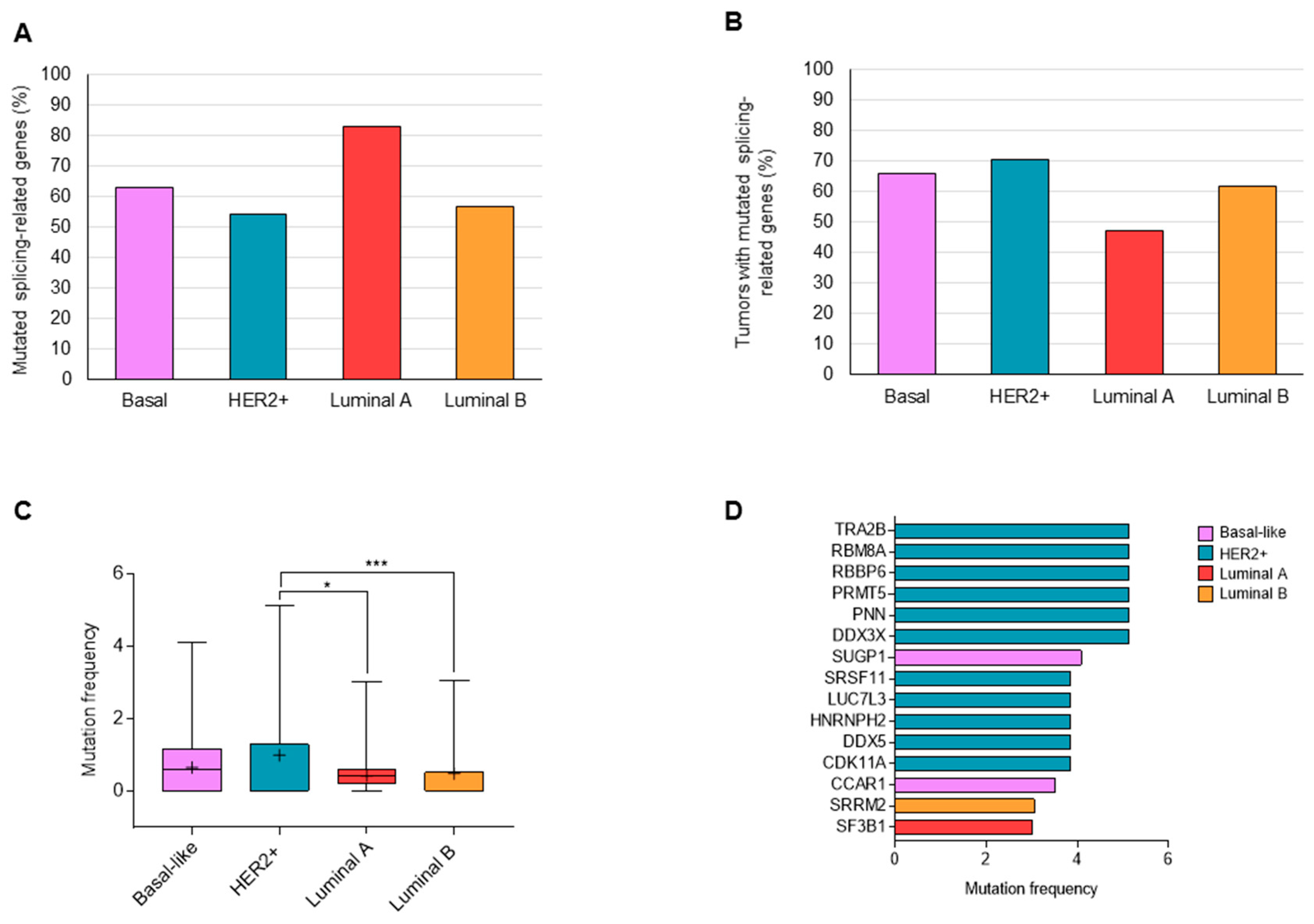
3.1. Mutations in Splicing-Related Genes
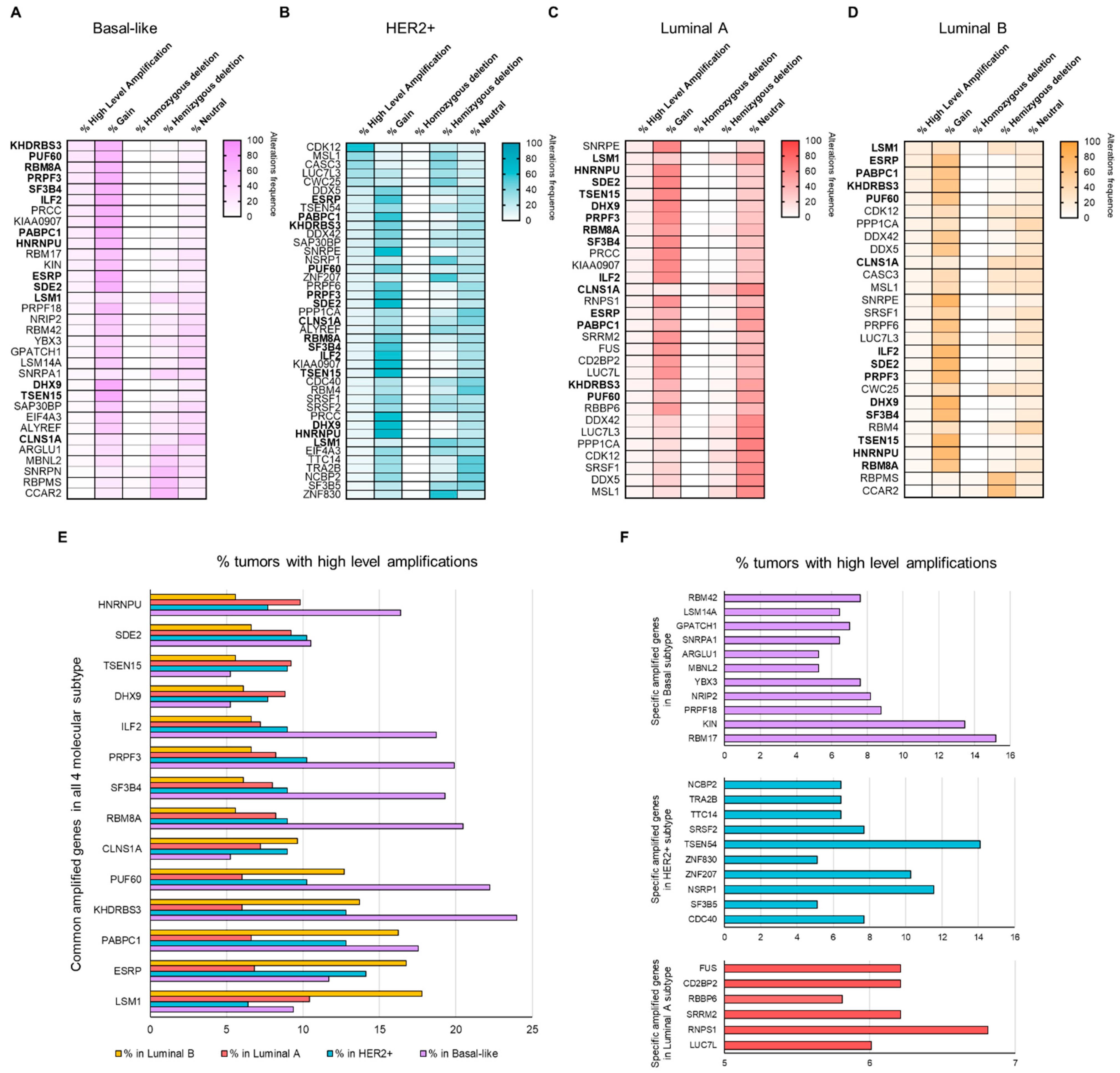
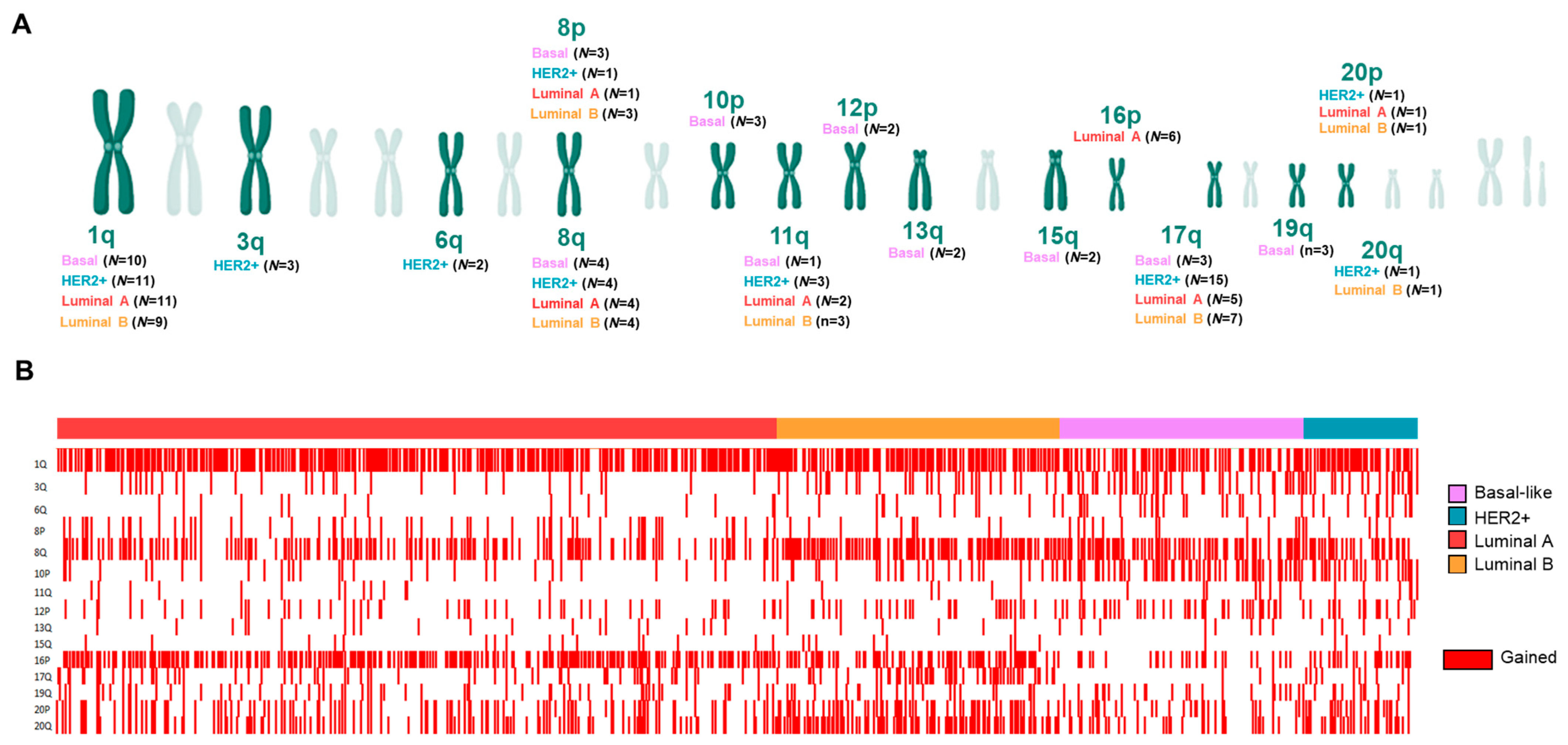
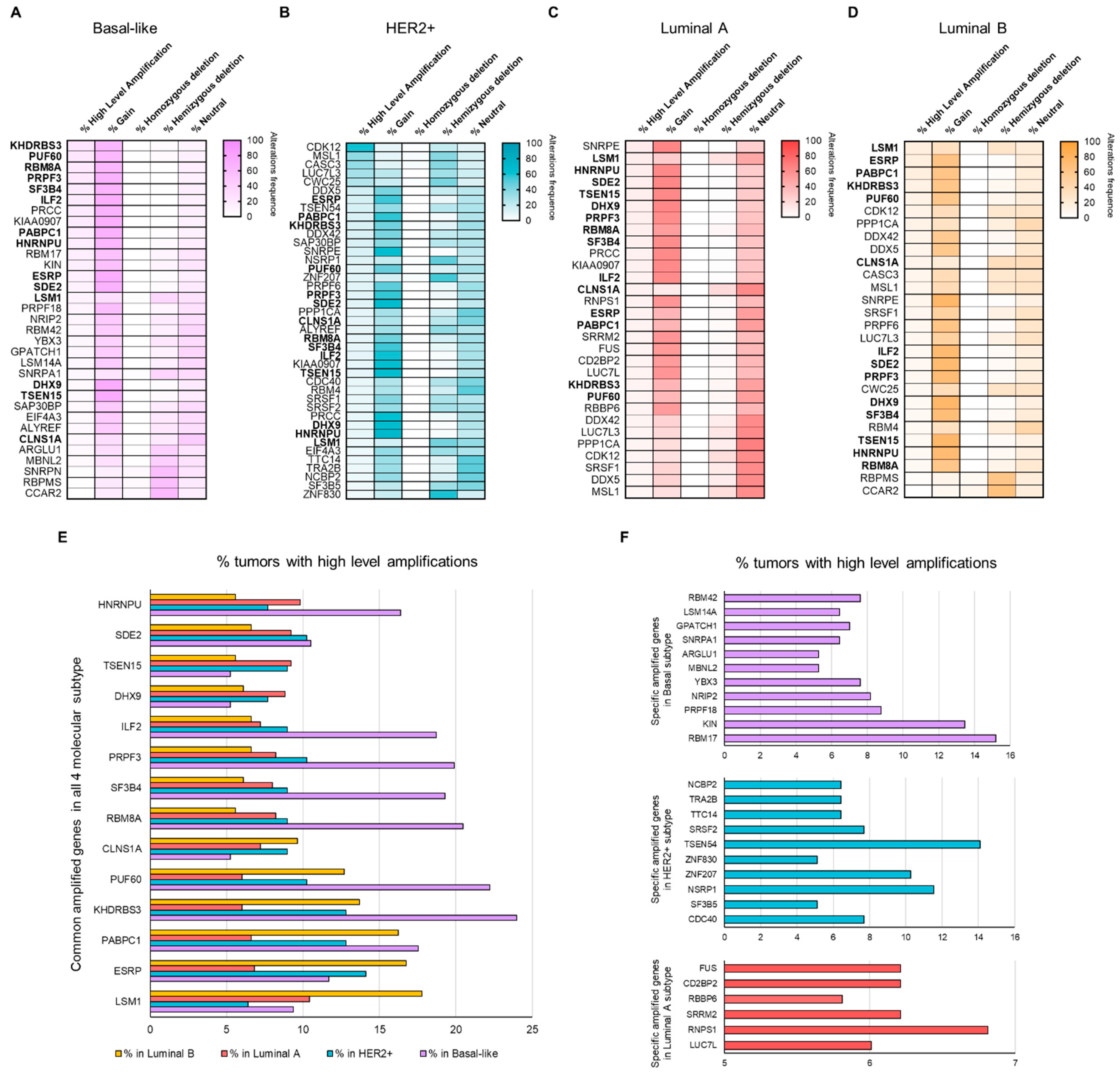
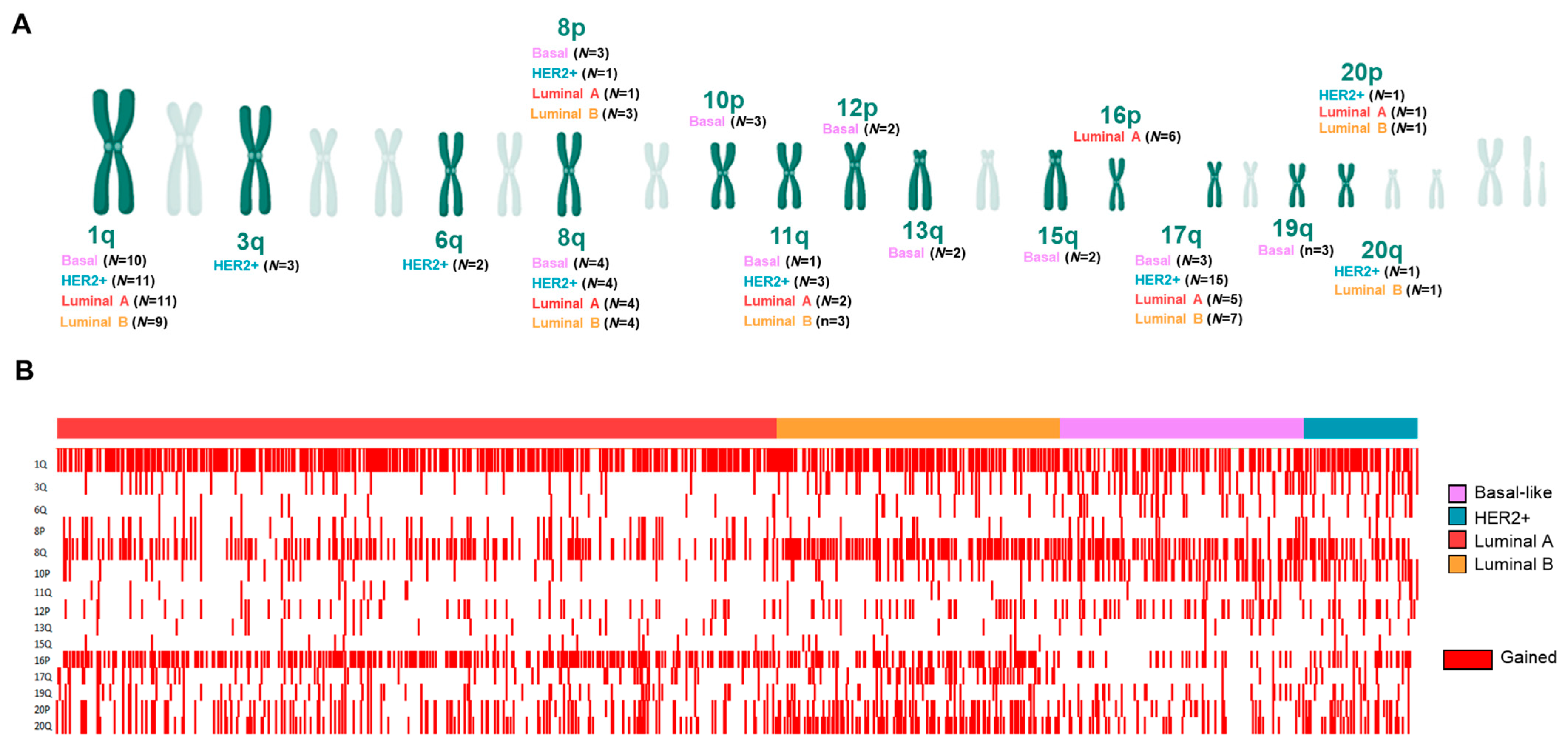
3.2. Copy Number Alterations (CNAs) in Splicing-Related Genes
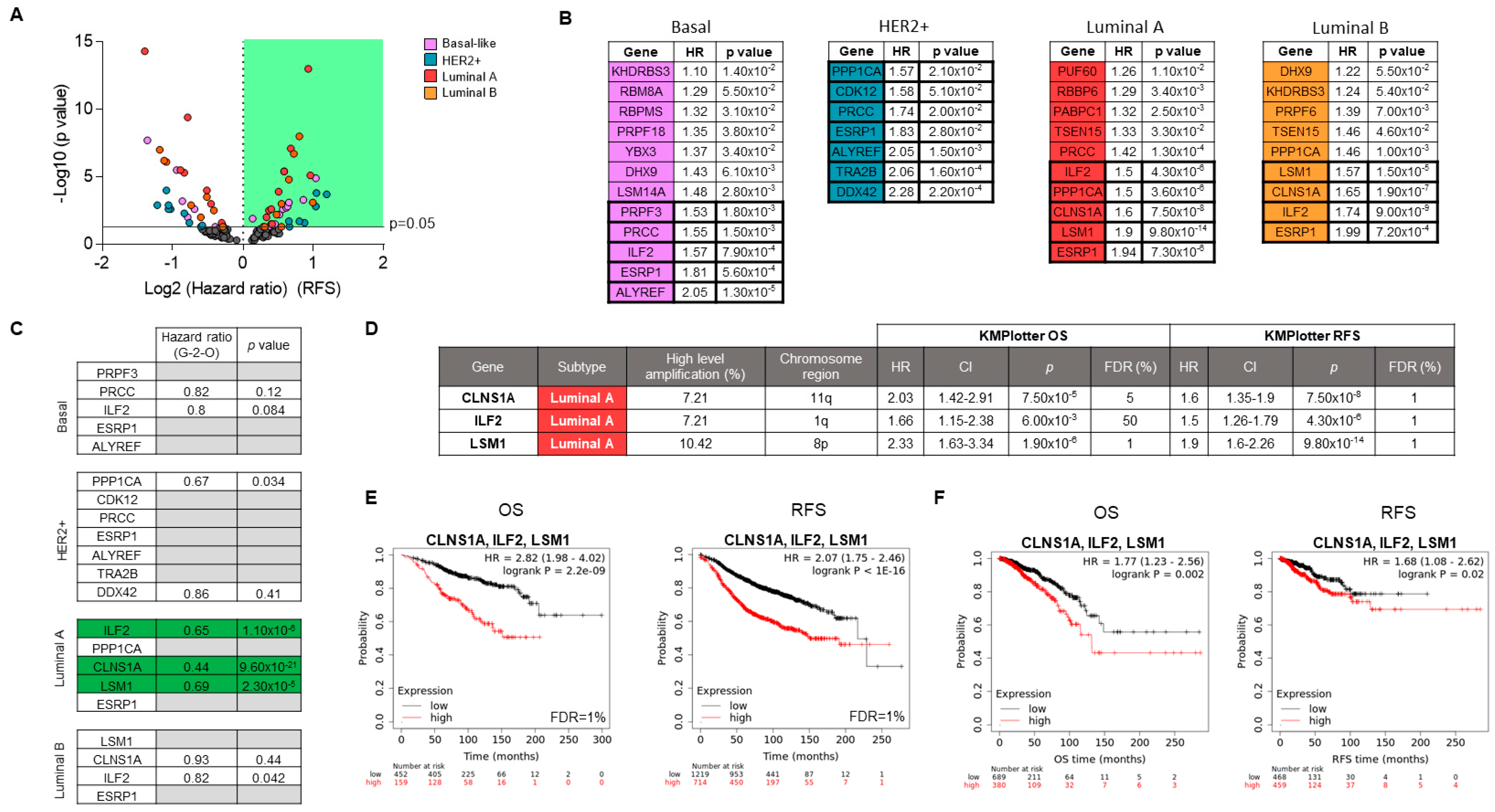
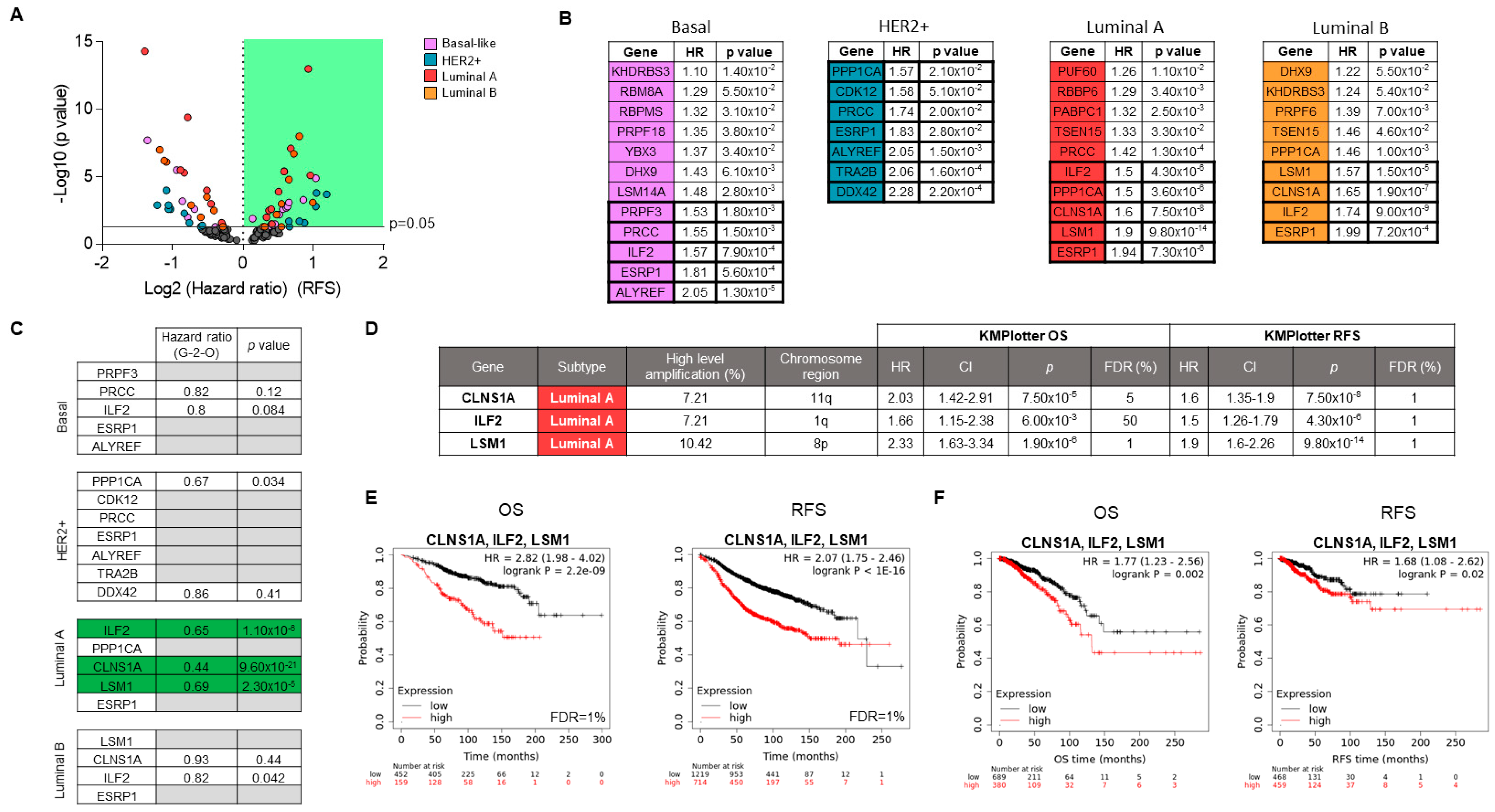
3.3. Associations of Splicing-Related Genes with Clinical Outcome in Patients
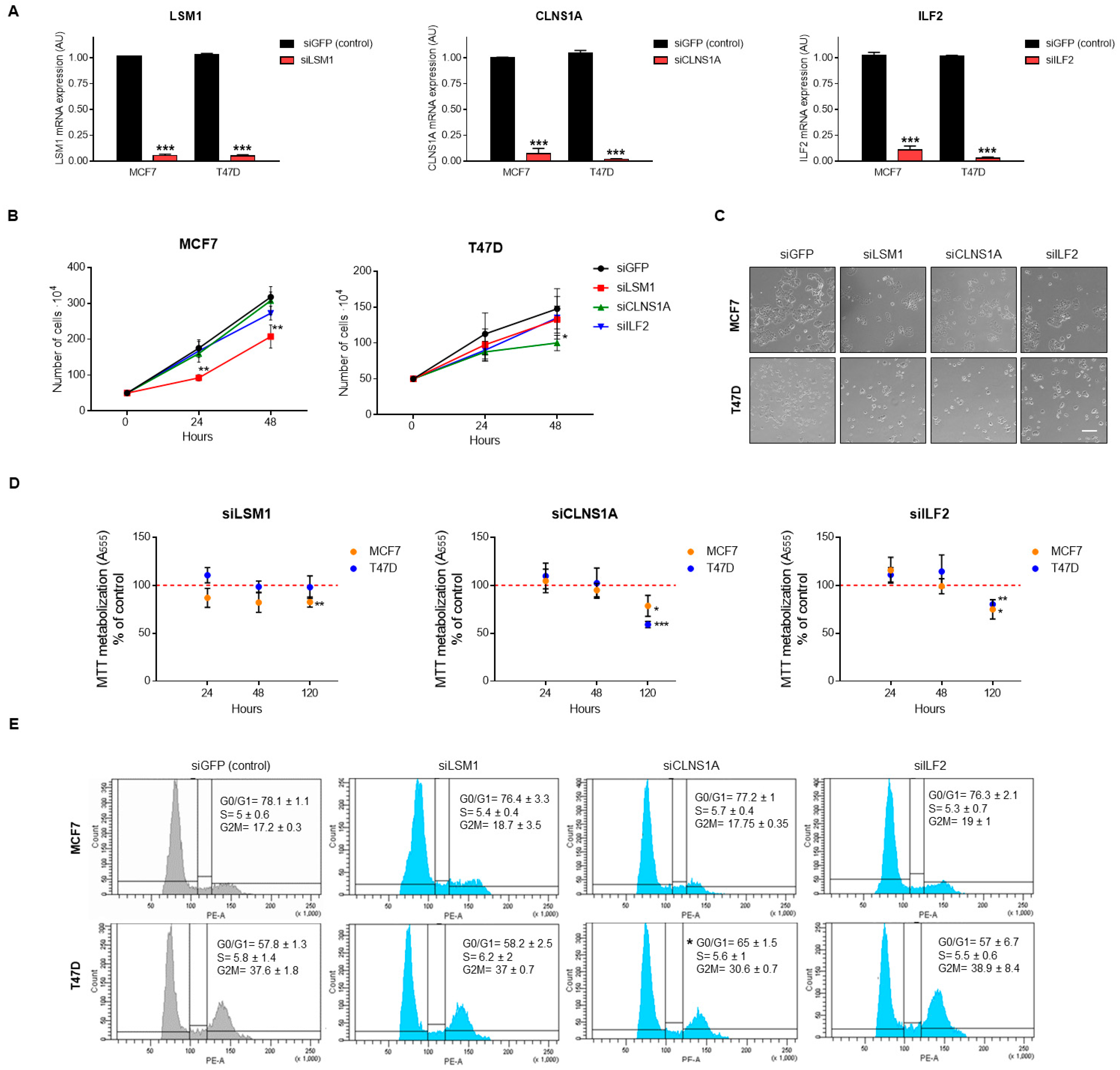
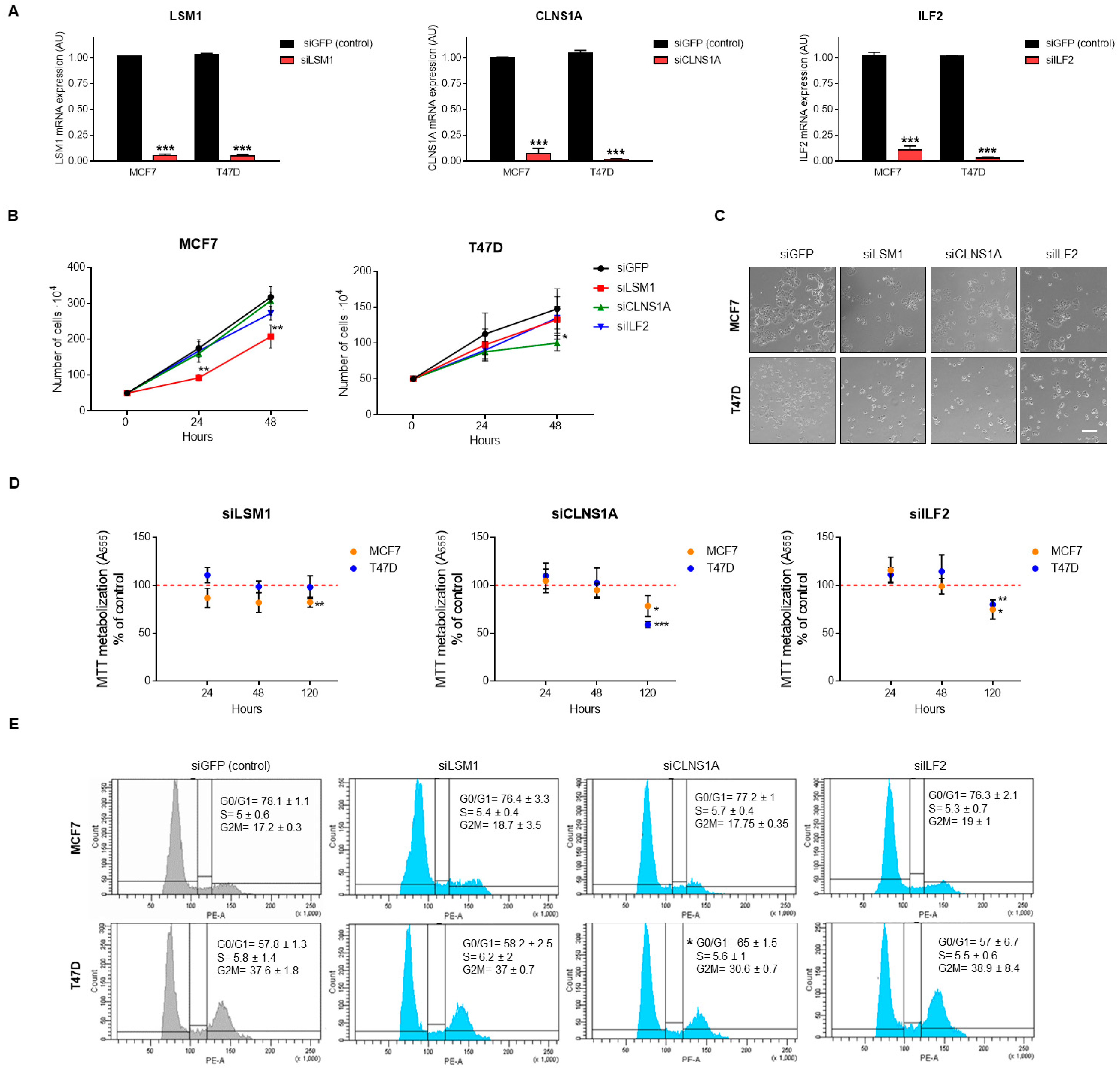
3.4. Genomic Down-Regulation of LSM1, CLNS1A, and ILF2 Reduces Cell Proliferation
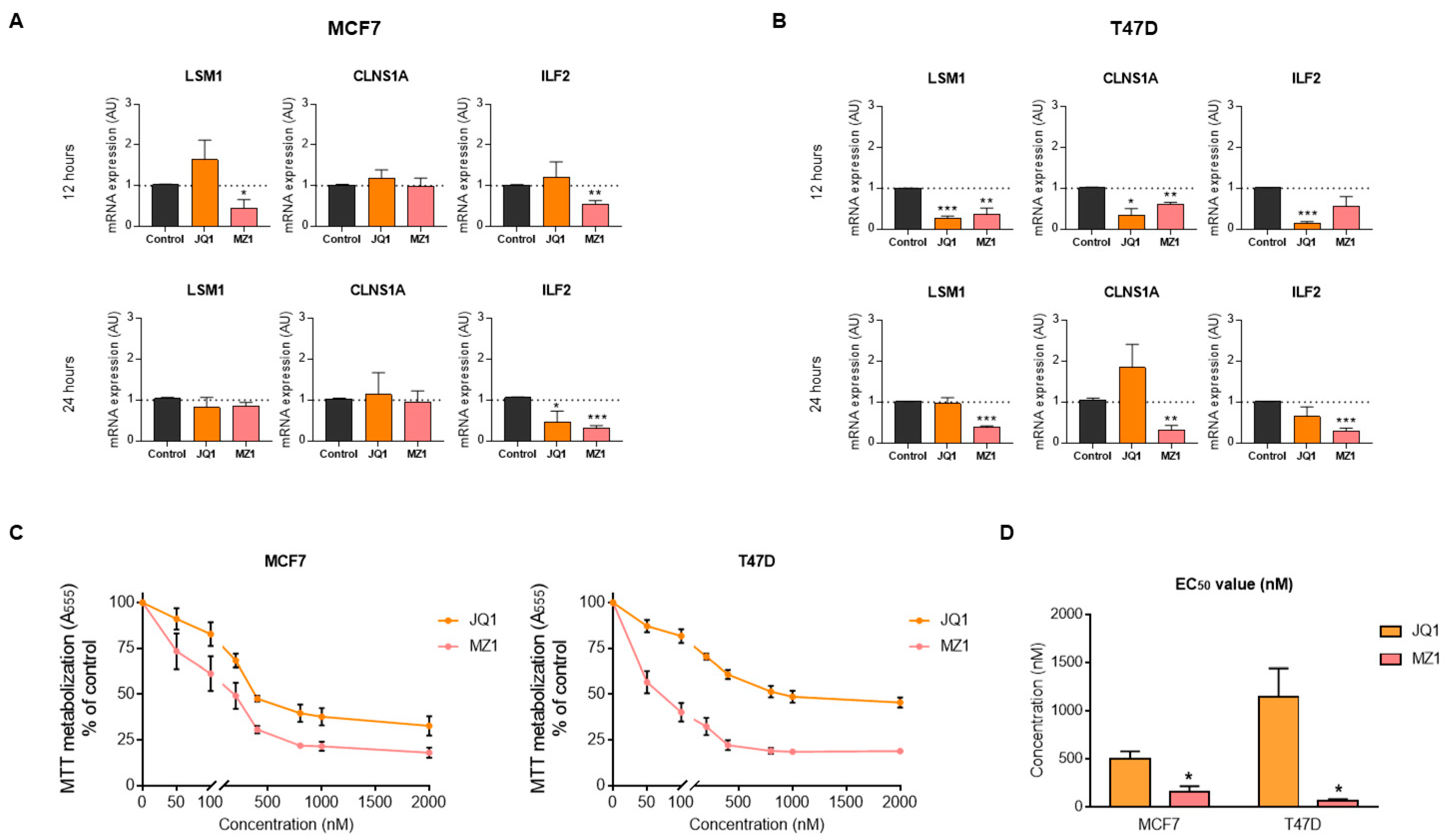
3.5. BET Inhibitors Reduce the Expression of LSM1, CLNS1A, and ILF2
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dvinge, H.; Kim, E.; Abdel-Wahab, O.; Bradley, R.K. RNA splicing factors as oncoproteins and tumour suppressors. Nat. Rev. Cancer 2016, 16, 413–430. [Google Scholar] [CrossRef]
- Xu, Y.; Zhao, W.; Olson, S.D.; Prabhkara, K.S.; Zhou, X. Alternative splicing links histone modifications to stem cell fate decision. Genome Biol. 2018, 19, 133. [Google Scholar] [CrossRef]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Magen, A.; Ast, G. Different levels of alternative splicing among eukaryotes. Nucleic Acids Res. 2007, 35, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Niklas, K.J.; Bondos, S.E.; Dunker, A.K.; Newman, S.A. Rethinking gene regulatory networks in light of alternative splicing, intrinsically disordered protein domains, and post-translational modifications. Front. Cell Dev. Biol. 2015, 3, 8. [Google Scholar] [CrossRef] [PubMed]
- Vohhodina, J.; Barros, E.M.; Savage, A.L.; Liberante, F.G.; Manti, L.; Bankhead, P.; Cosgrove, N.; Madden, A.F.; Harkin, D.P.; Savage, K.I. The RNA processing factors THRAP3 and BCLAF1 promote the DNA damage response through selective mRNA splicing and nuclear export. Nucleic Acids Res. 2017, 45, 12816–12833. [Google Scholar] [CrossRef] [PubMed]
- Mauger, O.; Scheiffele, P. Beyond proteome diversity: Alternative splicing as a regulator of neuronal transcript dynamics. Curr. Opin. Neurobiol. 2017, 45, 162–168. [Google Scholar] [CrossRef]
- Wang, E.; Aifantis, I. RNA Splicing and Cancer. Trends Cancer 2020, 6, 631–644. [Google Scholar] [CrossRef]
- Wan, R.; Bai, R.; Shi, Y. Molecular choreography of pre-mRNA splicing by the spliceosome. Curr. Opin. Struct. Biol. 2019, 59, 124–133. [Google Scholar] [CrossRef]
- Urbanski, L.; Leclair, N.; Anczuków, O. Alternative-splicing defects in cancer: Splicing regulators and their downstream targets, guiding the way to novel cancer therapeutics. WIREs RNA 2018, 9, e1476. [Google Scholar] [CrossRef]
- El Marabti, E.; Younis, I. The Cancer Spliceome: Reprograming of Alternative Splicing in Cancer. Front. Mol. Biosci. 2018, 5, 80. [Google Scholar] [CrossRef]
- Bonnal, S.C.; López-Oreja, I.; Valcárcel, J. Roles and mechanisms of alternative splicing in cancer—Implications for care. Nat. Rev. Clin. Oncol. 2020, 17, 457–474. [Google Scholar] [CrossRef]
- Goodall, G.J.; Wickramasinghe, V.O. RNA in cancer. Nat. Rev. Cancer 2021, 21, 22–36. [Google Scholar] [CrossRef]
- Fish, L.; Khoroshkin, M.; Navickas, A.; Garcia, K.; Culbertson, B.; Hänisch, B.; Zhang, S.; Nguyen, H.C.B.; Soto, L.M.; Dermit, M.; et al. A prometastatic splicing program regulated by SNRPA1 interactions with structured RNA elements. Science 2021, 372, eabc7531. [Google Scholar] [CrossRef]
- Seiler, M.; Peng, S.; Agrawal, A.A.; Palacino, J.; Teng, T.; Zhu, P.; Smith, P.G.; Cancer Genome Atlas Research Network; Buonamici, S.; Yu, L. Somatic Mutational Landscape of Splicing Factor Genes and Their Functional Consequences across 33 Cancer Types. Cell Rep. 2018, 23, 282–296. [Google Scholar] [CrossRef]
- Xiping, Z.; Qingshan, W.; Shuai, Z.; Hongjian, Y.; Xiaowen, D. A summary of relationships between alternative splicing and breast cancer. Oncotarget 2017, 8, 51986–51993. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.; Chin, S.F.; Rueda, O.M.; Vollan, H.K.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.J.; et al. The somatic mutation profiles of 2433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef]
- Brian, W.D.; Helmenstine, E.; Walsh, N.; Gondek, L.P.; Kelkar, D.S.; Read, A.; Natrajan, R.; Christenson, E.S.; Roman, B.; Das, S.; et al. Hotspot SF3B1 mutations induce metabolic reprogramming and vulnerability to serine deprivation. J. Clin. Investig. 2019, 129, 4708–4723. [Google Scholar] [CrossRef]
- Wang, Y.; Bernhardy, A.J.; Cruz, C.; Krais, J.J.; Nacson, J.; Nicolas, E.; Peri, S.; van der Gulden, H.; van der Heijden, I.; O’Brien, S.W.; et al. The BRCA1-delta11q Alternative Splice Isoform Bypasses Germline Mutations and Promotes Therapeutic Resistance to PARP Inhibition and Cisplatin. Cancer Res. 2016, 76, 2778–2790. [Google Scholar] [CrossRef] [PubMed]
- Koedoot, E.; Wolters, L.; van de Water, B.; Le Dévédec, S.E. Splicing regulatory factors in breast cancer hallmarks and disease progression. Oncotarget 2019, 10, 6021–6037. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, I.; Chakraborty, A.; Saulnier, O.; Benoit-Pilven, C.; Vacher, S.; Labiod, D.; Lam, E.W.F.; Bièche, I.; Delattre, O.; Pouzoulet, F.; et al. ZRANB2 and SYF2-mediated splicing programs converging on ECT2 are involved in breast cancer cell resistance to doxorubicin. Nucleic Acids Res. 2020, 48, 2676–2693. [Google Scholar] [CrossRef]
- Gabriel, M.; Delforge, Y.; Deward, A.; Habraken, Y.; Hennuy, B.; Piette, J.; Klinck, R.; Chabot, B.; Colige, A.; Lambert, C.; et al. Role of the splicing factor SRSF4 in cisplatin-induced modifications of pre-mRNA splicing and apoptosis. BMC Cancer 2015, 15, 227. [Google Scholar] [CrossRef]
- Marchesini, M.; Ogoti, Y.; Fiorini, E.; Aktas Samur, A.; Nezi, L.; D’Anca, M.; Storti, P.; Samur, M.K.; Ganan-Gomez, I.; Fulciniti, M.T.; et al. ILF2 Is a Regulator of RNA Splicing and DNA Damage Response in 1q21-Amplified Multiple Myeloma. Cancer Cell 2017, 32, 88–100. [Google Scholar] [CrossRef]
- Lobel, J.H.; Tibble, R.W.; Gross, J.D. Pat1 activates late steps in mRNA decay by multiple mechanisms. Proc. Natl. Acad. Sci. USA 2019, 116, 23512–23517. [Google Scholar] [CrossRef] [PubMed]
- Guderian, G.; Peter, C.; Wiesner, J.; Sickmann, A.; Schulze-Osthoff, K.; Fischer, U.; Grimmler, M. RioK1, a new interactor of protein arginine methyltransferase 5 (PRMT5), competes with pICln for binding and modulates PRMT5 complex composition and substrate specificity. J. Biol. Chem. 2011, 286, 1976–1986. [Google Scholar] [CrossRef]
- Hegele, A.; Kamburov, A.; Grossmann, A.; Sourlis, C.; Wowro, S.; Weimann, M.; Will, C.L.; Pena, V.; Lührmann, R.; Stelzlet, U. Dynamic protein-protein interaction wiring of the human spliceosome. Mol. Cell 2012, 45, 567–580. [Google Scholar] [CrossRef]
- Mermel, C.; Schumacher, S.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011, 12, R41. [Google Scholar] [CrossRef] [PubMed]
- Fuentes-Antrás, J.; Alcaraz-Sanabria, A.L.; Morafraile, E.C.; Noblejas-López, M.D.M.; Galán-Moya, E.M.; Baliu-Pique, M.; López-Cade, I.; García-Barberán, V.; Pérez-Segura, P.; Manzano, A.; et al. Mapping of Genomic Vulnerabilities in the Post-Translational Ubiquitination, SUMOylation and Neddylation Machinery in Breast Cancer. Cancers 2021, 13, 833. [Google Scholar] [CrossRef] [PubMed]
- Nagy, A.; Munkacsy, G.; Győrffy, B. Pancancer survival analysis of cancer hallmark genes. Sci. Rep. 2021, 11, 6047. [Google Scholar] [CrossRef] [PubMed]
- Pongor, L.; Kormos, M.; Hatzis, C.; Pusztai, L.; Szabó, A.; Győrffy, B. A genome-wide approach to link genotype to OS by utilizing next generation sequencing and gene chip data of 6697 breast cancer patients. Genome Med. 2015, 7, 104. [Google Scholar] [CrossRef]
- Ciriello, G.; Gatza, M.L.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.K.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015, 163, 506–519. [Google Scholar] [CrossRef] [PubMed]
- Győrffy, B.; Lanczky, A.; Eklund, A.C.; Denkert, C.; Budczies, J.; Li, Q.; Szallasi, Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1809 patients. Breast Cancer Res. Treat. 2010, 123, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Ocaña, A.; Pandiella, A. Proteolysis targeting chimeras (PROTACs) in cancer therapy. J. Exp. Clin. Cancer Res. 2020, 39, 189. [Google Scholar] [CrossRef] [PubMed]
- Ken-Ichi, F.; Takaki, I.; Mizuki, M.; Masashi, K.; Seiji, M. Regulating Divergent Transcriptomes through mRNA Splicing and Its Modulation Using Various Small Compounds. Int. J. Mol. Sci. 2020, 21, 2026. [Google Scholar] [CrossRef]
- Denichenko, P.; Mogilevsky, M.; Cléry, A.; Welte, T.; Biran, J.; Shimshon, O.; Barnabas, G.D.; Danan-Gotthold, M.; Kumar, S.; Yavin, E.; et al. Specific inhibition of splicing factor activity by decoy RNA oligonucleotides. Nat. Commun. 2019, 10, 1590. [Google Scholar] [CrossRef]
- Wang, E.; Lu, S.X.; Pastore, A.; Chen, X.; Imig, J.; Chun-Wei Lee, S.; Hockemeyer, K.; Ghebrechristos, Y.E.; Yoshimi, A.; Inoue, D.; et al. Targeting an RNA-Binding Protein Network in Acute Myeloid Leukemia. Cancer Cell 2019, 35, 369–384.e7. [Google Scholar] [CrossRef] [PubMed]
- Pavlyukov, M.S.; Yu, H.; Bastola, S.; Minata, M.; Shender, V.O.; Lee, Y.; Zhang, S.; Wang, J.; Komarova, S.; Wang, J.; et al. Apoptotic Cell-Derived Extracellular Vesicles Promote Malignancy of Glioblastoma Via Intercellular Transfer of Splicing Factors. Cancer Cell 2018, 34, 119–135. [Google Scholar] [CrossRef]
- Voutsadakis, I.A. 8p11.23 Amplification in Breast Cancer: Molecular Characteristics, Prognosis and Targeted Therapy. J. Clin. Med. 2020, 9, 3079. [Google Scholar] [CrossRef]
- Kufel, J.; Bousquet-Antonelli, C.; Beggs, J.D.; Tollervey, D. Nuclear Pre-mRNA Decapping and 5′ Degradation in Yeast Require the Lsm2-8p Complex. Mol. Cell Biol. 2004, 24, 9646–9657. [Google Scholar] [CrossRef] [PubMed]
- Tharun, S.; He, W.; Mayes, A.E.; Lennertz, P.; Beggs, J.D.; Parker, R. Yeast Sm-like proteins function in mRNA decapping and decay. Nature 2000, 404, 515–518. [Google Scholar] [CrossRef]
- Little, E.C.; Camp, E.R.; Wang, C.; Watson, P.M.; Watson, D.K.; Cole, D.J. The CaSm (LSm1) oncogene promotes transformation, chemoresistance and metastasis of pancreatic cancer cells. Oncogenesis 2016, 5, e182. [Google Scholar] [CrossRef] [PubMed]
- Chari, A.; Golas, M.M.; Klingenhäger, M.; Neuenkirchen, N.; Sander, B.; Englbrecht, C.; Sickmann, A.; Stark, H.; Fischer, U. An assembly chaperone collaborates with the SMN complex to generate spliceosomal SnRNPs. Cell 2008, 135, 497–509. [Google Scholar] [CrossRef]
- Beketova, E.; Fang, S.; Owens, J.L.; Liu, S.; Chen, X.; Zhang, Q.; Asberry, A.M.; Deng, X.; Malola, J.; Huang, J.; et al. Protein Arginine Methyltransferase 5 Promotes pICln-Dependent Androgen Receptor Transcription in Castration-Resistant Prostate Cancer. Cancer Res. 2020, 80, 4904–4917. [Google Scholar] [CrossRef] [PubMed]
- Braun, C.J.; Stanciu, M.; Boutz, P.L.; Patterson, J.C.; Calligaris, D.; Higuchi, F.; Neupane, R.; Fenoglio, S.; Cahill, D.P.; Wakimoto, H.; et al. Coordinated Splicing of Regulatory Detained Introns within Oncogenic Transcripts Creates an Exploitable Vulnerability in Malignant Glioma. Cancer Cell 2017, 32, 411–426. [Google Scholar] [CrossRef] [PubMed]
- Read, A.; Natrajan, R. Splicing dysregulation as a driver of breast cancer. Endocr. Relat. Cancer 2018, 25, R467–R478. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noblejas-López, M.d.M.; López-Cade, I.; Fuentes-Antrás, J.; Fernández-Hinojal, G.; Esteban-Sánchez, A.; Manzano, A.; García-Sáenz, J.Á.; Pérez-Segura, P.; la Hoya, M.d.; Pandiella, A.; et al. Genomic Mapping of Splicing-Related Genes Identify Amplifications in LSM1, CLNS1A, and ILF2 in Luminal Breast Cancer. Cancers 2021, 13, 4118. https://doi.org/10.3390/cancers13164118
Noblejas-López MdM, López-Cade I, Fuentes-Antrás J, Fernández-Hinojal G, Esteban-Sánchez A, Manzano A, García-Sáenz JÁ, Pérez-Segura P, la Hoya Md, Pandiella A, et al. Genomic Mapping of Splicing-Related Genes Identify Amplifications in LSM1, CLNS1A, and ILF2 in Luminal Breast Cancer. Cancers. 2021; 13(16):4118. https://doi.org/10.3390/cancers13164118
Chicago/Turabian StyleNoblejas-López, María del Mar, Igor López-Cade, Jesús Fuentes-Antrás, Gonzalo Fernández-Hinojal, Ada Esteban-Sánchez, Aránzazu Manzano, José Ángel García-Sáenz, Pedro Pérez-Segura, Miguel de la Hoya, Atanasio Pandiella, and et al. 2021. "Genomic Mapping of Splicing-Related Genes Identify Amplifications in LSM1, CLNS1A, and ILF2 in Luminal Breast Cancer" Cancers 13, no. 16: 4118. https://doi.org/10.3390/cancers13164118
APA StyleNoblejas-López, M. d. M., López-Cade, I., Fuentes-Antrás, J., Fernández-Hinojal, G., Esteban-Sánchez, A., Manzano, A., García-Sáenz, J. Á., Pérez-Segura, P., la Hoya, M. d., Pandiella, A., Győrffy, B., García-Barberán, V., & Ocaña, A. (2021). Genomic Mapping of Splicing-Related Genes Identify Amplifications in LSM1, CLNS1A, and ILF2 in Luminal Breast Cancer. Cancers, 13(16), 4118. https://doi.org/10.3390/cancers13164118