
Genetic Alterations in Childhood Acute Lymphoblastic Leukemia: Interactions with Clinical Features and Treatment Response

Abstract
Simple Summary
Abstract
1. Introduction
2. ALL Genetic Subtypes in 2020 and Beyond
2.1. Favorable-Risk Genetics (FRG) Group
2.2. Intermediate-Risk Genetics (IRG) Group
2.3. High-Risk Genetics (HRG) Group
3. Association of Subtypes with Race and Ethnicity
4. Association of Genetic Subtypes with NCI Criteria of Age and WBC Count
4.1. Infant ALL
4.2. Adolescent and Young Adult ALL (AYA > 10 Years Old)
5. Association of Genetic Subtypes with MRD and Outcomes
5.1. ETV6-RUNX1 and Hyperdiploidy
5.2. PBX1 Fusions Including TCF3-PBX1
5.3. ZNF384-Rearranged (ZNF384-r)
5.4. PAX5
5.5. ETV6-RUNX1-Like
5.6. DUX4
5.7. Philadelphia (Ph, BCR-ABL1)-Positive
5.8. BCR-ABL-Like (Ph-like) with or without CRLF2 Rearrangements
5.9. MEF2D
5.10. KMT2A-Rearranged/MLL
5.11. Low-Hypodiploid and Near-Haploid
5.12. HLF
5.13. iAMP21
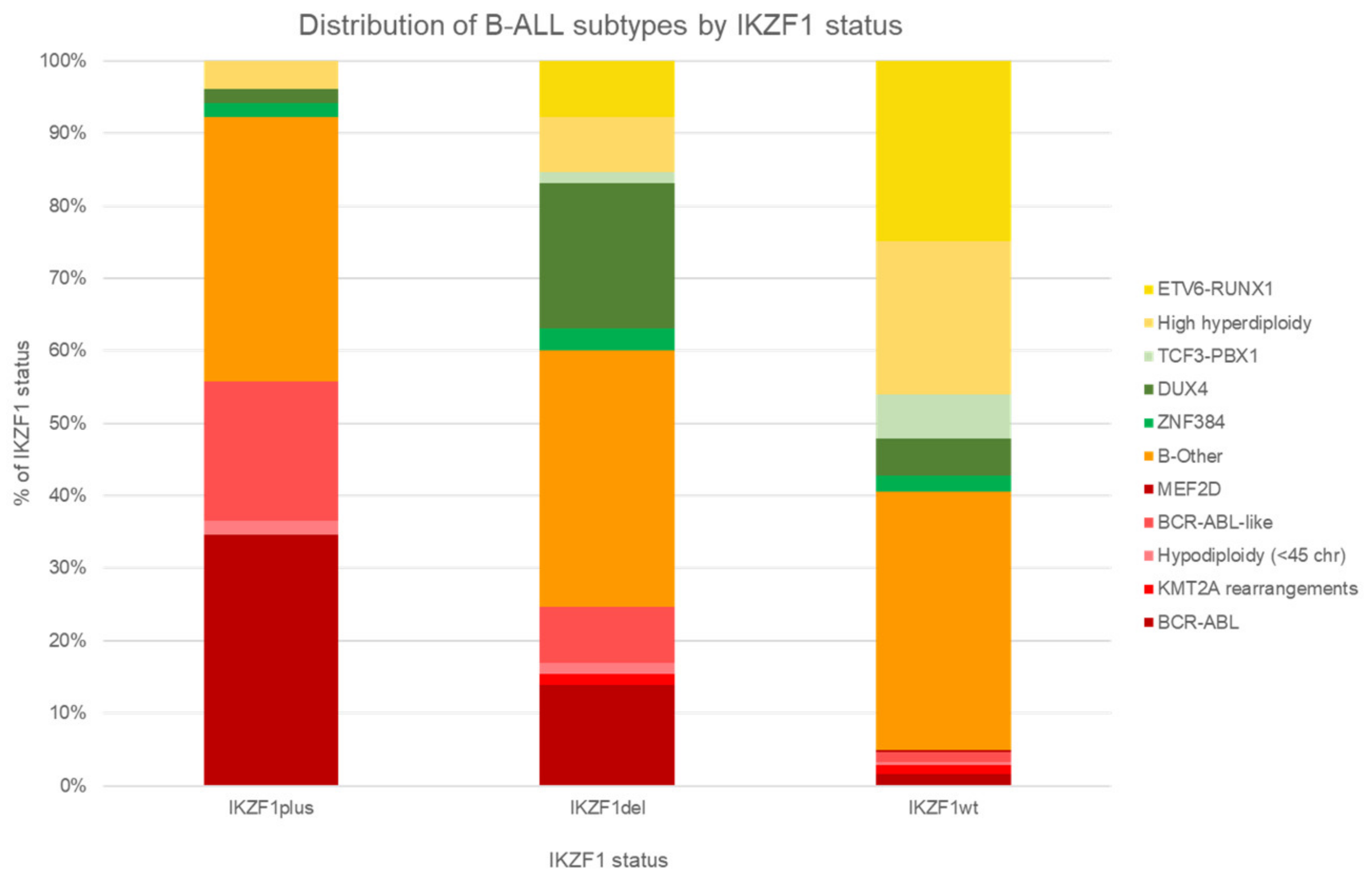
6. IKZF1 Deletion and Interactions with Genetic Subtypes
7. T-ALL
7.1. T-ALL Interaction with Age and WBC
7.2. T-ALL Interaction with MRD and Outcomes
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pui, C.-H.; Nichols, K.E.; Yang, J.J. Somatic and germline genomics in paediatric acute lymphoblastic leukaemia. Nat. Rev. Clin. Oncol. 2019, 16, 227–240. [Google Scholar] [CrossRef]
- Hunger, S.P.; Mullighan, C.G. Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar] [CrossRef]
- Gu, Z.; Churchman, M.L.; Roberts, K.G.; Moore, I.; Zhou, X.; Nakitandwe, J.; Hagiwara, K.; Pelletier, S.; Gingras, S.; Berns, H.; et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat. Genet. 2019, 51, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Iacobucci, I.; Mullighan, C.G. Genetic Basis of Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2017, 35, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Inaba, H.; Mullighan, C.G. Pediatric acute lymphoblastic leukemia. Haematologica 2020, 105, 2524–2539. [Google Scholar] [CrossRef] [PubMed]
- Maloney, K.W.; Devidas, M.; Wang, C.; Mattano, L.A.; Friedmann, A.M.; Buckley, P.; Borowitz, M.J.; Carroll, A.J.; Gastier-Foster, J.M.; Heerema, N.A.; et al. Outcome in Children with Standard-Risk B-Cell Acute Lymphoblastic Leukemia: Results of Children’s Oncology Group Trial AALL0331. J. Clin. Oncol. 2020, 38, 602–612. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, D.; Moorman, A.V.; Wade, R.; Hancock, J.; Tan, R.M.; Bartram, J.; Moppett, J.; Schwab, C.; Patrick, K.; Harrison, C.; et al. Use of Minimal Residual Disease Assessment to Redefine Induction Failure in Pediatric Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2017, 35, 660–667. [Google Scholar] [CrossRef]
- Yeoh, A.E.J.; Ariffin, H.; Chai, E.L.L.; Kwok, C.S.N.; Chan, Y.H.; Ponnudurai, K.; Campana, D.; Tan, P.L.; Chan, M.Y.; Kham, S.K.Y.; et al. Minimal residual disease–guided treatment deintensification for children with acute lymphoblastic leukemia: Results from the Malaysia-Singapore acute lymphoblastic leukemia 2003 Study. J. Clin. Oncol. 2012, 30, 2384–2392. [Google Scholar] [CrossRef] [PubMed]
- Yeoh, A.E.J.; Lu, Y.; Ni Chin, W.H.; Chiew, E.K.H.; Lim, E.H.; Li, Z.; Kham, S.K.Y.; Chan, Y.H.; Abdullah, W.A.; Lin, H.P.; et al. Intensifying Treatment of Childhood B-Lymphoblastic Leukemia with IKZF1 Deletion Reduces Relapse and Improves Overall Survival: Results of Malaysia-Singapore ALL 2010 Study. J. Clin. Oncol. 2018, 36, 2726–2735. [Google Scholar] [CrossRef]
- Teachey, D.T.; Pui, C.-H. Comparative features and outcomes between paediatric T-cell and B-cell acute lymphoblastic leukaemia. Lancet Oncol. 2019, 20, e142–e154. [Google Scholar] [CrossRef]
- Ariffin, H.; Chen, S.P.; Kwok, C.S.; Quah, T.C.; Lin, H.P.; Yeoh, A.E.; Malaysia-Singapore Leukemia Study Group. Ethnic differences in the frequency of subtypes of childhood acute lymphoblastic leukemia: Results of the Malaysia-Singapore Leukemia Study Group. J. Pediatr. Hematol. Oncol. 2007, 29, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.-C.; Shih, L.-Y.; Yang, C.-P.; Hung, I.-J.; Liu, H.-C.; Jaing, T.-H.; Yeh, T.-C.; Liang, S.-T.; Chang, C.-L.; Lee, E.-H.; et al. The Frequencies of ETV6-RUNX1 Fusion and Hyperdiploidy (>50 chromosomes) in Children with Acute Lymphoblastic Leukemia Are Lower in Far East than the West. Blood 2009, 114, 3064. [Google Scholar] [CrossRef]
- Kager, L.; Lion, T.; Attarbaschi, A.; Koenig, M.; Strehl, S.; Haas, O.A.; Dworzak, M.N.; Schrappe, M.; Gadner, H.; Mann, G. Incidence and outcome of TCF3-PBX1-positive acute lymphoblastic leukemia in Austrian children. Haematologica 2007, 92, 1561–1564. [Google Scholar] [CrossRef]
- Pui, C.-H.; Sandlund, J.T.; Pei, D.; Rivera, G.K.; Howard, S.C.; Ribeiro, R.C.; Rubnitz, J.E.; Razzouk, B.; Hudson, M.M.; Cheng, C.; et al. Results of therapy for acute lymphoblastic leukemia in black and white children. JAMA 2003, 290, 2001–2007. [Google Scholar] [CrossRef]
- Kadan-Lottick, N.S. Survival Variability by Race and Ethnicity in Childhood Acute Lymphoblastic Leukemia. JAMA 2003, 290, 2008–2014. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Sather, H.N.; Heerema, N.A.; Trigg, M.E.; Gaynon, P.S.; Robison, L.L. Racial and ethnic differences in survival of children with acute lymphoblastic leukemia. Blood 2002, 100, 1957–1964. [Google Scholar] [CrossRef] [PubMed]
- Harvey, R.; Mullighan, C.G.; Chen, I.-M.; Wharton, W.; Mikhail, F.M.; Carroll, A.J.; Kang, H.; Liu, W.; Dobbin, K.K.; Smith, M.A.; et al. Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood 2010, 115, 5312–5321. [Google Scholar] [CrossRef] [PubMed]
- Medina-Sanson, A.; Núñez-Enríquez, J.C.; Hurtado-Cordova, E.; Pérez-Saldivar, M.L.; Martínez-García, A.; Jiménez-Hernández, E.; Fernández-López, J.C.; Martín-Trejo, J.A.; Pérez-Lorenzana, H.; Flores-Lujano, J.; et al. Genotype-Environment Interaction Analysis of NQO1, CYP2E1, and NAT2 Polymorphisms and the Risk of Childhood Acute Lymphoblastic Leukemia: A Report from the Mexican Interinstitutional Group for the Identification of the Causes of Childhood Leukemia. Front. Oncol. 2020, 10, 571869. [Google Scholar] [CrossRef]
- Lee, S.H.R.; Qian, M.; Yang, W.; Diedrich, J.D.; Raetz, E.; Yang, W.; Dong, Q.; Devidas, M.; Pei, D.; Yeoh, A.; et al. Genome-Wide Association Study of Susceptibility Loci for TCF3-PBX1 Acute Lymphoblastic Leukemia in Children. J. Natl. Cancer Inst. 2021, 113, 933–937. [Google Scholar] [CrossRef]
- Perez-Andreu, V.; Roberts, K.G.; Harvey, R.C.; Yang, W.; Cheng, C.; Pei, D.; Xu, H.; Gastier-Foster, J.; Shuyu, E.; Lim, J.Y.; et al. Inherited GATA3 variants are associated with Ph-like childhood acute lymphoblastic leukemia and risk of relapse. Nat. Genet. 2013, 45, 1494–1498. [Google Scholar] [CrossRef]
- Smith, M.; Arthur, D.; Camitta, B.; Carroll, A.J.; Crist, W.; Gaynon, P.; Gelber, R.; Heerema, N.; Korn, E.L.; Link, M.; et al. Uniform approach to risk classification and treatment assignment for children with acute lymphoblastic leukemia. J. Clin. Oncol. 1996, 14, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Pieters, R.; Schrappe, M.; De Lorenzo, P.; Hann, I.; De Rossi, G.; Felice, M.; Hovi, L.; LeBlanc, T.; Szczepanski, T.; Ferster, A.; et al. A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): An observational study and a multicentre randomised trial. Lancet 2007, 370, 240–250. [Google Scholar] [CrossRef]
- Pieters, R.; De Lorenzo, P.; Ancliffe, P.; Aversa, L.A.; Brethon, B.; Biondi, A.; Campbell, M.; Escherich, G.; Ferster, A.; Gardner, R.A.; et al. Outcome of Infants Younger Than 1 Year with Acute Lymphoblastic Leukemia Treated with the Interfant-06 Protocol: Results from an International Phase III Randomized Study. J. Clin. Oncol. 2019, 37, 2246–2256. [Google Scholar] [CrossRef] [PubMed]
- Andersson, A.K.; Ma, J.; Wang, J.; Chen, X.; Gedman, A.L.; Dang, J.; Nakitandwe, J.; Holmfeldt, L.; Parker, M.; Easton, J.; et al. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat. Genet. 2015, 47, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Winters, A.C.; Bernt, K.M. MLL-Rearranged Leukemias—An Update on Science and Clinical Approaches. Front. Pediatr. 2017, 5, 4. [Google Scholar] [CrossRef]
- Britten, O.; Ragusa, D.; Tosi, S.; Mostafa Kamel, Y. MLL-Rearranged Acute Leukemia with t(4;11)(q21;q23)-Current Treatment Options. Is There a Role for CAR-T Cell Therapy? Cells 2019, 8, 1341. [Google Scholar] [CrossRef] [PubMed]
- Reaman, G.H.; Sposto, R.; Sensel, M.G.; Lange, B.J.; Feusner, J.H.; Heerema, N.A.; Leonard, M.; Holmes, E.J.; Sather, H.N.; Pendergrass, T.W.; et al. Treatment outcome and prognostic factors for infants with acute lymphoblastic leukemia treated on two consecutive trials of the Children’s Cancer Group. J. Clin. Oncol. 1999, 17, 445–455. [Google Scholar] [CrossRef]
- Van der Linden, M.H.; Valsecchi, M.G.; De Lorenzo, P.; Möricke, A.; Janka, G.; Leblanc, T.M.; Felice, M.; Biondi, A.; Campbell, M.; Hann, I.; et al. Outcome of congenital acute lymphoblastic leukemia treated on the Interfant-99 protocol. Blood 2009, 114, 3764–3768. [Google Scholar] [CrossRef]
- Van Dongen, J.J.; Seriu, T.; Panzer-Grümayer, E.R.; Biondi, A.; Pongers-Willemse, M.J.; Corral, L.; Stolz, F.; Schrappe, M.; Masera, G.; Kamps, W.A.; et al. Prognostic value of minimal residual disease in acute lymphoblastic leukaemia in childhood. Lancet 1998, 352, 1731–1738. [Google Scholar] [CrossRef]
- Della Starza, I.; Chiaretti, S.; De Propris, M.S.; Elia, L.; Cavalli, M.; DE Novi, L.A.; Soscia, R.; Messina, M.; Vitale, A.; Guarini, A.; et al. Minimal Residual Disease in Acute Lymphoblastic Leukemia: Technical and Clinical Advances. Front. Oncol. 2019, 9, 726. [Google Scholar] [CrossRef]
- Eckert, C.; Flohr, T.; Koehler, R.; Hagedorn, N.; Moericke, A.; Stanulla, M.; Kirschner-Schwabe, R.; Cario, G.; Stackelberg, A.; Bartram, C.R.; et al. Very early/early relapses of acute lymphoblastic leukemia show unexpected changes of clonal markers and high hetero-geneity in response to initial and relapse treatment. Leukemia 2011, 25, 1305–1313. [Google Scholar] [CrossRef]
- Kotrová, M.; Muzikova, K.; Mejstrikova, E.; Novakova, M.; Bakardjieva-Mihaylova, V.; Fišer, K.; Stuchly, J.; Giraud, M.; Salson, M.; Pott, C.; et al. The predictive strength of next-generation sequencing MRD detection for relapse compared with current methods in childhood ALL. Blood 2015, 126, 1045–1047. [Google Scholar] [CrossRef]
- O’Connor, D.; Enshaei, A.; Bartram, J.; Hancock, J.; Harrison, C.J.; Hough, R.; Samarasinghe, S.; Schwab, C.; Vora, A.; Wade, R.; et al. Genotype-Specific Minimal Residual Disease Interpretation Improves Stratification in Pediatric Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2018, 36, 34–43. [Google Scholar] [CrossRef]
- Pui, C.H.; Pei, D.; Raimondi, S.C.; Coustan-Smith, E.; Jeha, S.; Cheng, C.; Bowman, W.P.; Sandlund, J.T.; Ribeiro, R.C.; Rubnitz, J.E.; et al. Clinical impact of minimal residual disease in children with different subtypes of acute lymphoblastic leukemia treated with Response-Adapted therapy. Leukemia 2017, 31, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Borowitz, M.J.; Devidas, M.; Hunger, S.P.; Bowman, W.P.; Carroll, A.J.; Carroll, W.L.; Linda, S.; Martin, P.L.; Pullen, D.J.; Viswanatha, D.; et al. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: A Children’s Oncology Group study. Blood 2008, 111, 5477–5485. [Google Scholar] [CrossRef] [PubMed]
- Borowitz, M.J.; Wood, B.L.; Devidas, M.; Loh, M.L.; Raetz, E.A.; Salzer, W.L.; Nachman, J.B.; Carroll, A.J.; Heerema, N.A.; Gastier-Foster, J.M.; et al. Prognostic significance of minimal residual disease in high risk B-ALL: A report from Children’s Oncology Group study AALL0232. Blood 2015, 126, 964–971. [Google Scholar] [CrossRef]
- Pieters, R.; de Groot-Kruseman, H.; Van der Velden, V.; Fiocco, M.; Berg, H.V.D.; de Bont, E.; Egeler, R.M.; Hoogerbrugge, P.; Kaspers, G.; Van der Schoot, E.; et al. Successful Therapy Reduction and Intensification for Childhood Acute Lymphoblastic Leukemia Based on Minimal Residual Disease Monitoring: Study ALL10 From the Dutch Childhood Oncology Group. J. Clin. Oncol. 2016, 34, 2591–2601. [Google Scholar] [CrossRef] [PubMed]
- Bhojwani, D.; Pei, D.; Sandlund, J.T.; Jeha, S.; Ribeiro, R.C.; Rubnitz, J.E.; Raimondi, S.C.; Shurtleff, S.; Onciu, M.; Cheng, C.; et al. ETV6-RUNX1-positive childhood acute lymphoblastic leukemia: Improved outcome with contemporary therapy. Leukemia 2012, 26, 265–270. [Google Scholar] [CrossRef]
- Burmeister, T.; Gökbuget, N.; Schwartz, S.; Fischer, L.; Hubert, D.; Sindram, A.; Hoelzer, D.; Thiel, E. Clinical features and prognostic implications of TCF3-PBX1 and ETV6-RUNX1 in adult acute lymphoblastic leu-kemia. Haematologica 2010, 95, 241–246. [Google Scholar] [CrossRef]
- Asai, D.; Imamura, T.; Yamashita, Y.; Suenobu, S.; Moriya-Saito, A.; Hasegawa, D.; Deguchi, T.; Hashii, Y.; Endo, M.; Hatakeyama, N.; et al. Outcome of TCF3-PBX1 positive pediatric acute lymphoblastic leukemia patients in J apan: A collaborative study of Japan Association of Childhood Leukemia Study (JACLS) and Children’s Cancer and Leukemia Study Group (CCLSG). Cancer Med. 2014, 3, 623–631. [Google Scholar] [CrossRef]
- Kager, L.; Lion, T.; Attarbaschi, A.; Koenig, M.; Strehl, S.; Haas, O.A.; Dworzak, M.N.; Schrappe, M.; Gadner, H.; Mann, G. Treatment Response and Outcome in Childhood t(1;19)/TCF3-PBX1 Positive Acute Lymphoblastic Leukemia: A Report from the Austrian BFM Group. Blood 2005, 106, 1458. [Google Scholar] [CrossRef]
- Crist, W.M.; Carroll, A.J.; Shuster, J.J.; Behm, F.G.; Whitehead, M.; Vietti, T.J.; Look, A.T.; Mahoney, D.; Ragab, A.; Pullen, D.J. Poor prognosis of children with pre-B acute lymphoblastic leukemia is associated with the t(1;19)(q23;p13): A Pediatric Oncology Group study. Blood 1990, 76, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Cheng, F.W.; Chiang, A.K.; Luk, C.-W.; Li, R.C.; Ling, A.S.; Cheuk, D.K.; Chang, K.-O.; Ku, D.; Lee, V.; et al. Excellent outcome of acute lymphoblastic leukaemia withTCF3-PBX1rearrangement in Hong Kong. Pediatr. Blood Cancer 2018, 65, e27346. [Google Scholar] [CrossRef] [PubMed]
- Borowitz, M.J.; Pullen, D.J.; Shuster, J.J.; Viswanatha, D.; Montgomery, K.; Willman, C.L.; Camitta, B. Minimal residual disease detection in childhood precursor-B-cell acute lymphoblastic leukemia: Relation to other risk factors. A Children’s Oncology Group study. Leukemia 2003, 17, 1566–1572. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.-J.; Wang, Y.; Jia, Y.-P.; Zuo, Y.-X.; Wu, J.; Lu, A.-D.; Zhang, L.-P. The role of minimal residual disease in specific subtypes of pediatric acute lymphoblastic leukemia. Int. J. Hematol. 2021, 113, 547–555. [Google Scholar] [CrossRef]
- Pui, C.-H.; Campana, D.; Pei, D.; Bowman, W.P.; Sandlund, J.T.; Kaste, S.C.; Ribeiro, R.C.; Rubnitz, J.E.; Raimondi, S.C.; Onciu, M.; et al. Treating childhood acute lymphoblastic leukemia without cranial irradiation. N. Engl. J. Med. 2009, 360, 2730–2741. [Google Scholar] [CrossRef]
- Jeha, S.; Pei, D.; Choi, J.; Cheng, C.; Sandlund, J.T.; Coustan-Smith, E.; Campana, D.; Inaba, H.; Rubnitz, J.E.; Ribeiro, R.C.; et al. Improved CNS Control of Childhood Acute Lymphoblastic Leukemia Without Cranial Irradiation: St Jude Total Therapy Study 16. J. Clin. Oncol. 2019, 37, 3377–3391. [Google Scholar] [CrossRef] [PubMed]
- Hirabayashi, S.; Butler, E.; Ohki, K.; Kiyokawa, N.; Bergmann, A.K.; Boer, J.M.; Cavé, H.; Cazzaniga, G.; Yeoh, A.E.J.; Imamura, T.; et al. Acute Lymphoblastic Leukemia with Zinc-Finger Protein 384 (ZNF384)-Related Rearrangements: A Retrospective Analysis from the Ponte Di Legno Childhood ALL Working Group. Blood 2019, 134 (Suppl. 1), 652. [Google Scholar] [CrossRef]
- McClure, B.J.; Heatley, S.L.; Kok, C.H.; Sadras, T.; An, J.; Hughes, T.P.; Lock, R.B.; Yeung, D.; Sutton, R.; White, D.L. Pre-B acute lymphoblastic leukaemia recurrent fusion, EP300-ZNF384, is associated with a distinct gene expression. Br. J. Cancer 2018, 118, 1000–1004. [Google Scholar] [CrossRef]
- Shinsuke, H.; Kentaro, O.; Kazuhiko, N.; Hitoshi, I.; Yukihide, M.; Kohji, O.; Akinori, Y.; Kazuki, T.; Yuya, S.; Ai, Y.; et al. ZNF384-related fusion genes define a subgroup of childhood B-cell precursor acute lymphoblastic leukemia with a characteristic immunotype. Haematologica 2017, 102, 118–129. [Google Scholar] [CrossRef]
- Hirabayashi, S.; Butler, E.R.; Ohki, K.; Kiyokawa, N.; Bergmann, A.K.; Möricke, A.; Boer, J.M.; Cavé, H.; Cazzaniga, G.; Yeoh, A.E.J.; et al. Clinical characteristics and outcomes of B-ALL with ZNF384 rearrangements: A retrospective analysis by the Ponte di Legno Childhood ALL Working Group. Leukemia 2021. [Google Scholar] [CrossRef]
- Nishimura, A.; Hasegawa, D.; Hirabayashi, S.; Kanabuchi, S.; Yamamoto, K.; Aiga, S.; Nishitani, M.; Hosoya, Y.; Noguchi, Y.; Ohki, K.; et al. Very late relapse cases of TCF3-ZNF384-positive acute lymphoblastic leukemia. Pediatric Blood Cancer 2019, 66, e27891. [Google Scholar] [CrossRef] [PubMed]
- Stasevich, I.; Inglott, S.; Austin, N.; Chatters, S.; Chalker, J.; Addy, D.; Dryden, C.; Ancliff, P.; Ford, A.; Williams, O.; et al. PAX5 alterations in genetically unclassified childhood Precursor B-cell acute lymphoblastic leukaemia. Br. J. Haematol. 2015, 171, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Bastian, L.; Schroeder, M.P.; Eckert, C.; Schlee, C.; Tanchez, J.O.; Kämpf, S.; Wagner, D.L.; Schulze, V.; Isaakidis, K.; Lázaro-Navarro, J.; et al. PAX5 biallelic genomic alterations define a novel subgroup of B-cell precursor acute lymphoblastic leukemia. Leukemia 2019, 33, 1895–1909. [Google Scholar] [CrossRef]
- Lilljebjörn, H.; Henningsson, R.; Hyrenius-Wittsten, A.; Olsson, L.; Orsmark-Pietras, C.; Von Palffy, S.; Askmyr, M.; Rissler, M.; Schrappe, M.; Cario, G.; et al. Identification of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute lympho-blastic leukaemia. Nat. Commun. 2016, 7, 11790. [Google Scholar] [CrossRef]
- Zaliova, M.; Kotrova, M.; Bresolin, S.; Stuchly, J.; Stary, J.; Hrusak, O.; Te Kronnie, G.; Trka, J.; Zuna, J.; Vaskova, M. ETV6/RUNX1-like acute lymphoblastic leukemia: A novel B-cell precursor leukemia subtype associated with the CD27/CD44 immunophenotype. Genes Chromosomes Cancer 2017, 56, 608–616. [Google Scholar] [CrossRef]
- Jeha, S.; Choi, J.; Roberts, K.G.; Pei, D.; Coustan-Smith, E.; Inaba, H.; Rubnitz, J.E.; Ribeiro, R.C.; Gruber, T.A.; Raimondi, S.C.; et al. Clinical significance of novel subtypes of acute lymphoblastic leukemia in the context of minimal residual disease–directed therapy. Blood Cancer Discov. 2021, 2, 326–337. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; McCastlain, K.; Yoshihara, H.; Xu, B.; Chang, Y.; Churchman, M.L.; Wu, G.; Li, Y.; Wei, L.; Iacobucci, I.; et al. Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Nat. Genet. 2016, 48, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- Schinnerl, D.; Mejstrikova, E.; Schumich, A.; Zaliova, M.; Fortschegger, K.; Nebral, K.; Attarbaschi, A.; Fiser, K.; Kauer, M.O.; Popitsch, N.; et al. CD371 cell surface expression: A unique feature of DUX4-rearranged acute lymphoblastic leukemia. Haematologica 2019, 104, e352–e355. [Google Scholar] [CrossRef] [PubMed]
- Rehn, J.A.; O’Connor, M.J.; White, D.L.; Yeung, D.T. DUX Hunting—Clinical Features and Diagnostic Challenges Associated with DUX4-Rearranged Leukaemia. Cancers 2020, 12, 2815. [Google Scholar] [CrossRef]
- Marincevic-Zuniga, Y.; Dahlberg, J.; Nilsson, S.; Raine, A.; Nystedt, S.; Lindqvist, C.M.; Berglund, E.C.; Abrahamsson, J.; Cavelier, L.; Forestier, E.; et al. Transcriptome sequencing in pediatric acute lymphoblastic leukemia identifies fusion genes associated with distinct DNA methylation profiles. J. Hematol. Oncol. 2017, 10, 148. [Google Scholar] [CrossRef]
- Clappier, E.; Auclerc, M.-F.; Rapion, J.; Bakkus, M.; Caye, A.; Khemiri, A.; Giroux, C.; Hernandez, L.; Kabongo, E.; Savola, S.; et al. An intragenic ERG deletion is a marker of an oncogenic subtype of B-cell precursor acute lymphoblastic leukemia with a favorable outcome despite frequent IKZF1 deletions. Leukemia 2014, 28, 70–77. [Google Scholar] [CrossRef]
- Zaliova, M.; Zimmermannova, O.; Dörge, P.; Eckert, C.; Möricke, A.; Zimmermann, M.; Stuchly, J.; Teigler-Schlegel, A.; Meissner, B.; Koehler, R.; et al. ERG deletion is associated with CD2 and attenuates the negative impact of IKZF1 deletion in childhood acute lym-phoblastic leukemia. Leukemia 2014, 28, 182–185. [Google Scholar] [CrossRef]
- Vendramini, E.; Giordan, M.; Giarin, E.; Michielotto, B.; Fazio, G.; Cazzaniga, G.; Biondi, A.; Silvestri, D.; Valsecchi, M.G.; Muckenthaler, M.; et al. High expression of miR-125b-2 and SNORD116 noncoding RNA clusters characterize ERG-related B cell precursor acute lymphoblastic leukemia. Oncotarget 2017, 8, 42398–42413. [Google Scholar] [CrossRef]
- Zaliova, M.; Stuchly, J.; Winkowska, L.; Musilova, A.; Fiser, K.; Slamova, M.; Starkova, J.; Vaskova, M.; Hrusak, O.; Sramkova, L.; et al. Genomic landscape of pediatric B-other acute lymphoblastic leukemia in a consecutive European cohort. Haematologica 2019, 104, 1396–1406. [Google Scholar] [CrossRef] [PubMed]
- Novakova, M.; Zaliova, M.; Fiser, K.; Vakrmanova, B.; Slamova, L.; Musilova, A.; Brüggemann, M.; Ritgen, M.; Fronkova, E.; Kalina, T.; et al. DUX4r, ZNF384r and PAX5-P80R mutated B-cell precursor acute lymphoblastic leukemia frequently undergo monocytic switch. Haematologica 2020, 106, 2066–2075. [Google Scholar] [CrossRef]
- Zaliova, M.; Potuckova, E.; Hovorkova, L.; Musilova, A.; Winkowska, L.; Fiser, K.; Stuchly, J.; Mejstrikova, E.; Starkova, J.; Zuna, J.; et al. ERG deletions in childhood acute lymphoblastic leukemia with DUX4 rearrangements are mostly polyclonal, prognostically relevant and their detection rate strongly depends on screening method sensitivity. Haematologica 2019, 104, 1407–1416. [Google Scholar] [CrossRef] [PubMed]
- Schrappe, M.; Hunger, S.P.; Pui, C.-H.; Saha, V.; Gaynon, P.S.; Baruchel, A.; Conter, V.; Otten, J.; Ohara, A.; Versluys, A.B.; et al. Outcomes after induction failure in childhood acute lymphoblastic leukemia. N. Engl. J. Med. 2012, 366, 1371–1381. [Google Scholar] [CrossRef] [PubMed]
- Aricò, M.; Schrappe, M.; Hunger, S.P.; Carroll, W.L.; Conter, V.; Galimberti, S.; Manabe, A.; Saha, V.; Baruchel, A.; Vettenranta, K.; et al. Clinical outcome of children with newly diagnosed philadelphia chromosome–positive acute lymphoblastic leukemia treated between 1995 and 2005. J. Clin. Oncol. 2010, 28, 4755–4761. [Google Scholar] [CrossRef]
- Biondi, A.; Schrappe, M.; De Lorenzo, P.; Castor, A.; Lucchini, G.; Gandemer, V.; Pieters, R.; Stary, J.; Escherich, G.; Campbell, M.; et al. Imatinib after induction for treatment of children and adolescents with Philadelphia-chromosome-positive acute lym-phoblastic leukaemia (EsPhALL): A randomised, open-label, intergroup study. Lancet Oncol. 2012, 13, 936–945. [Google Scholar] [CrossRef]
- Schultz, K.R.; Bowman, W.P.; Aledo, A.; Slayton, W.B.; Sather, H.; Devidas, M.; Wang, C.; Davies, S.M.; Gaynon, P.S.; Trigg, M.; et al. Improved early event-free survival with imatinib in philadelphia chromosome–positive acute lymphoblastic leukemia: A children’s oncology group study. J. Clin. Oncol. 2009, 27, 5175–5181. [Google Scholar] [CrossRef]
- Shen, S.; Chen, X.; Cai, J.; Yu, J.; Gao, J.; Hu, S.; Zhai, X.; Liang, C.; Ju, X.; Jiang, H.; et al. Effect of Dasatinib vs Imatinib in the Treatment of Pediatric Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia: A Randomized Clinical Trial. JAMA Oncol. 2020, 6, 358–366. [Google Scholar] [CrossRef]
- Vora, A.; Goulden, N.; Wade, R.; Mitchell, C.; Hancock, J.; Hough, R.; Rowntree, C.; Richards, S. Treatment reduction for children and young adults with low-risk acute lymphoblastic leukaemia defined by minimal residual disease (UKALL 2003): A randomised controlled trial. Lancet Oncol. 2013, 14, 199–209. [Google Scholar] [CrossRef]
- Vora, A.; Goulden, N.; Mitchell, C.; Hancock, J.; Hough, R.; Rowntree, C.; Moorman, A.; Wade, R. Augmented post-remission therapy for a minimal residual disease-defined high-risk subgroup of children and young people with clinical standard-risk and intermediate-risk acute lymphoblastic leukaemia (UKALL 2003): A randomised controlled trial. Lancet Oncol. 2014, 15, 809–818. [Google Scholar] [CrossRef]
- Conter, V.; Bartram, C.R.; Valsecchi, M.G.; Schrauder, A.; Panzer-Grümayer, R.; Möricke, A.; Aricò, M.; Zimmermann, M.; Mann, G.; De Rossi, G.; et al. Molecular response to treatment redefines all prognostic factors in children and adolescents with B-cell precursor acute lymphoblastic leukemia: Results in 3184 patients of the AIEOP-BFM ALL 2000 study. Blood 2010, 115, 3206–3214. [Google Scholar] [CrossRef] [PubMed]
- Den Boer, M.L.; van Slegtenhorst, M.; De Menezes, R.X.; Cheok, M.H.; Buijs-Gladdines, J.G.; Peters, S.T.; Van Zutven, L.J.; Beverloo, H.B.; Van der Spek, P.J.; Escherich, G.; et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: A genome-wide classification study. Lancet Oncol. 2009, 10, 125–134. [Google Scholar] [CrossRef]
- Roberts, K.G.; Li, Y.; Payne-Turner, D.; Harvey, R.C.; Yang, Y.-L.; Pei, D.; McCastlain, K.; Ding, L.; Lu, C.; Song, G.; et al. Targetable Kinase-Activating Lesions in Ph-like Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2014, 371, 1005–1015. [Google Scholar] [CrossRef]
- Slayton, W.B.; Schultz, K.R.; Kairalla, J.A.; Devidas, M.; Mi, X.; Pulsipher, M.A.; Chang, B.H.; Mullighan, C.; Iacobucci, I.; Silverman, L.B.; et al. Dasatinib Plus Intensive Chemotherapy in Children, Adolescents, and Young Adults with Philadelphia Chromo-some-Positive Acute Lymphoblastic Leukemia: Results of Children’s Oncology Group Trial AALL0622. J. Clin. Oncol. 2018, 36, 2306–2314. [Google Scholar] [CrossRef]
- Schlieben, S.; Borkhardt, A.; Reinisch, I.; Ritterbach, J.; Janssen, J.W.; Ratei, R.; Schrappe, M.; Repp, R.; Zimmermann, M.; Kabisch, H.; et al. Incidence and clinical outcome of children with BCR/ABL-positive acute lymphoblastic leukemia (ALL). A prospective RT-PCR study based on 673 patients enrolled in the German pediatric multicenter therapy trials ALL-BFM-90 and CoALL-05-92. Leukemia 1996, 10, 957–963. [Google Scholar]
- Tasian, S.K.; Loh, M.L.; Hunger, S.P. Philadelphia chromosome–like acute lymphoblastic leukemia. Blood 2017, 130, 2064–2072. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.G.; Pei, D.; Campana, D.; Payne-Turner, D.; Li, Y.; Cheng, C.; Sandlund, J.T.; Jeha, S.; Easton, J.; Becksfort, J.; et al. Outcomes of children with BCR-ABL1–like acute lymphoblastic leukemia treated with risk-directed therapy based on the levels of minimal residual disease. J. Clin. Oncol. 2014, 32, 3012–3020. [Google Scholar] [CrossRef]
- Roberts, K.G. Genetics and prognosis of ALL in children vs adults. Hematol. Am. Soc. Hematol. Educ. Program. 2018, 2018, 137–145. [Google Scholar] [CrossRef]
- Palmi, C.; Vendramini, E.; Silvestri, D.; Longinotti, G.; Frison, D.; Cario, G.; Shochat, C.; Stanulla, M.; Rossi, V.; Di Meglio, A.M.; et al. Poor prognosis for P2RY8-CRLF2 fusion but not for CRLF2 over-expression in children with intermediate risk B-cell precursor acute lymphoblastic leukemia. Leukemia 2012, 26, 2245–2253. [Google Scholar] [CrossRef]
- Reshmi, S.C.; Harvey, R.C.; Roberts, K.G.; Stonerock, E.; Smith, A.; Jenkins, H.; Chen, I.-M.; Valentine, M.; Liu, Y.; Li, Y.; et al. Targetable kinase gene fusions in high-risk B-ALL: A study from the Children’s Oncology Group. Blood 2017, 129, 3352–3361. [Google Scholar] [CrossRef] [PubMed]
- Tasian, S.K.; Loh, M.L. Understanding the Biology of CRLF2-Overexpressing Acute Lymphoblastic Leukemia. Crit. Rev. Oncog. 2011, 16, 13–24. [Google Scholar] [CrossRef]
- Yoda, A.; Yoda, Y.; Chiaretti, S.; Bar-Natan, M.; Mani, K.; Rodig, S.J.; West, N.; Xiao, Y.; Brown, J.R.; Mitsiades, C.; et al. Functional screening identifies CRLF2 in precursor B-cell acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA 2010, 107, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G.; Collins-Underwood, J.R.; Phillips, L.A.; Loudin, M.G.; Liu, W.; Zhang, J.; Ma, J.; Coustan-Smith, E.; Harvey, R.C.; Willman, C.L.; et al. Rearrangement of CRLF2 in B-progenitor– and Down syndrome–associated acute lymphoblastic leukemia. Nat. Genet. 2009, 41, 1243–1246. [Google Scholar] [CrossRef]
- Russell, L.; Capasso, M.; Vater, I.; Akasaka, T.; Bernard, O.; Calasanz, M.J.; Chandrasekaran, T.; Chapiro, E.; Gesk, S.; Griffiths, M.; et al. Deregulated expression of cytokine receptor gene, CRLF2, is involved in lymphoid transformation in B-cell precursor acute lymphoblastic leukemia. Blood 2009, 114, 2688–2698. [Google Scholar] [CrossRef] [PubMed]
- Ensor, H.M.; Schwab, C.; Russell, L.; Richards, S.M.; Morrison, H.; Masic, D.; Jones, L.; Kinsey, S.E.; Vora, A.J.; Mitchell, C.D.; et al. Demographic, clinical, and outcome features of children with acute lymphoblastic leukemia and CRLF2 deregulation: Results from the MRC ALL97 clinical trial. Blood 2011, 117, 2129–2136. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-F.; Wang, B.-Y.; Zhang, W.-N.; Huang, J.-Y.; Li, B.-S.; Zhang, M.; Jiang, L.; Li, J.-F.; Wang, M.-J.; Dai, Y.-J.; et al. Genomic Profiling of Adult and Pediatric B-cell Acute Lymphoblastic Leukemia. EBioMedicine 2016, 8, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Churchman, M.; Roberts, K.; Li, Y.; Liu, Y.; Harvey, R.C.; McCastlain, K.; Reshmi, S.C.; Payne-Turner, D.; Iacobucci, I.; et al. Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat. Commun. 2016, 7, 13331. [Google Scholar] [CrossRef] [PubMed]
- Ohki, K.; Kiyokawa, N.; Saito, Y.; Hirabayashi, S.; Nakabayashi, K.; Ichikawa, H.; Momozawa, Y.; Okamura, K.; Yoshimi, A.; Ogata-Kawata, H.; et al. Clinical and molecular characteristics of MEF2D fusion-positive B-cell precursor acute lymphoblastic leukemia in childhood, including a novel translocation resulting in MEF2D-HNRNPH1 gene fusion. Haematologica 2019, 104, 128–137. [Google Scholar] [CrossRef]
- Suzuki, K.; Okuno, Y.; Kawashima, N.; Muramatsu, H.; Okuno, T.; Wang, X.; Kataoka, S.; Sekiya, Y.; Hamada, M.; Murakami, N.; et al. MEF2D-BCL9 Fusion Gene Is Associated with High-Risk Acute B-Cell Precursor Lymphoblastic Leukemia in Ado-lescents. J. Clin. Oncol. 2016, 34, 3451–3459. [Google Scholar] [CrossRef]
- Zhang, C.; Richon, V.; Ni, X.; Talpur, R.; Duvic, M. Selective induction of apoptosis by histone deacetylase inhibitor SAHA in cutaneous T-cell lymphoma cells: Relevance to mechanism of therapeutic action. J. Investig. Dermatol. 2005, 125, 1045–1052. [Google Scholar] [CrossRef]
- Stutterheim, J.; van der Sluis, I.M.; De Lorenzo, P.; Alten, J.; Ancliff, P.; Attarbaschi, A.; Brethon, B.; Biondi, A.; Campbell, M.; Cazzaniga, G.; et al. Clinical Implications of Minimal Residual Disease Detection in Infants with KMT2A-Rearranged Acute Lymphoblastic Leukemia Treated on the Interfant-06 Protocol. Blood 2020, 136, 41–42. [Google Scholar] [CrossRef]
- Hilden, J.M.; Dinndorf, P.A.; Meerbaum, S.O.; Sather, H.; Villaluna, D.; Heerema, N.A.; McGlennen, R.; Smith, F.O.; Woods, W.G.; Salzer, W.L.; et al. Analysis of prognostic factors of acute lymphoblastic leukemia in infants: Report on CCG 1953 from the Children’s Oncology Group. Blood 2006, 108, 441–451. [Google Scholar] [CrossRef]
- Woerden, N.L.R.-V.; Beverloo, H.B.; Veerman, A.J.P.; Camitta, B.M.; Loonen, A.H.; Van Wering, E.R.; Slater, R.M.; Harbott, J.; Boer, M.L.D.; Ludwig, W.D.; et al. In vitro drug-resistance profile in infant acute lymphoblastic leukemia in relation to age, MLL rearrangements and immunophenotype. Leukemia 2004, 18, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Mann, G.; Attarbaschi, A.; Schrappe, M.; De Lorenzo, P.; Peters, C.; Hann, I.; De Rossi, G.; Felice, M.; Lausen, B.; Leblanc, T.; et al. Improved outcome with hematopoietic stem cell transplantation in a poor prognostic subgroup of infants with mixed-lineage-leukemia (MLL)–rearranged acute lymphoblastic leukemia: Results from the Interfant-99 Study. Blood 2010, 116, 2644–2650. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, Z.E.; Dinndorf, P.A.; Camitta, B.; Sather, H.; La, M.K.; Devidas, M.; Hilden, J.M.; Heerema, N.A.; Sanders, J.E.; McGlennen, R.; et al. Analysis of the role of hematopoietic stem-cell transplantation in infants with acute lymphoblastic leukemia in first remission and MLL gene rearrangements: A report from the Children’s Oncology Group. J. Clin. Oncol. 2011, 29, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.-H.; Gaynon, P.S.; Boyett, J.M.; Chessells, J.M.; Baruchel, A.; Kamps, W.; Silverman, L.B.; Biondi, A.; Harms, D.O.; Vilmer, E.; et al. Outcome of treatment in childhood acute lymphoblastic leukaemia with rearrangements of the 11q23 chromosomal region. Lancet 2002, 359, 1909–1915. [Google Scholar] [CrossRef]
- Pui, C.-H.; Chessells, J.M.; Camitta, B.M.; Baruchel, A.; Biondi, A.; Boyett, J.M.; Carroll, A.J.; Eden, O.B.; Evans, W.E.; Gadner, H.; et al. Clinical heterogeneity in childhood acute lymphoblastic leukemia with 11q23 rearrangements. Leukemia 2003, 17, 700–706. [Google Scholar] [CrossRef]
- Basso, G.; Veltroni, M.; Valsecchi, M.G.; Dworzak, M.N.; Ratei, R.; Silvestri, D.; Benetello, A.; Buldini, B.; Maglia, O.; Masera, G.; et al. Risk of relapse of childhood acute lymphoblastic leukemia is predicted by flow cytometric measurement of residual disease on day 15 bone marrow. J. Clin. Oncol. 2009, 27, 5168–5174. [Google Scholar] [CrossRef] [PubMed]
- Schultz, K.R.; Devidas, M.; Bowman, W.P.; Aledo, A.; Slayton, W.B.; Sather, H.; Zheng, H.W.; Davies, S.M.; Gaynon, P.S.; Trigg, M.; et al. Philadelphia chromosome-negative very high-risk acute lymphoblastic leukemia in children and adolescents: Results from Children’s Oncology Group Study AALL0031. Leukemia 2014, 28, 964–967. [Google Scholar] [CrossRef]
- Hann, I.; on behalf of the UK Medical Research Council’s Working Party on Childhood Leukaemia; Vora, A.; Richards, S.; Hill, F.; Gibson, B.; Lilleyman, J.; Kinsey, S.; Mitchell, C.; Eden, O.B. Benefit of intensified treatment for all children with acute lymphoblastic leukaemia: Results from MRC UKALL XI and MRC ALL97 randomised trials. UK Medical Research Council’s Working Party on Childhood Leukemia. Leukemia 2000, 14, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G.; Jeha, S.; Pei, D.; Payne-Turner, D.; Coustan-Smith, E.; Roberts, K.G.; Waanders, E.; Choi, J.K.; Ma, X.; Raimondi, S.C.; et al. Outcome of children with hypodiploid ALL treated with risk-directed therapy based on MRD levels. Blood 2015, 126, 2896–2899. [Google Scholar] [CrossRef]
- Moorman, A.V.; Ensor, H.M.; Richards, S.M.; Chilton, L.; Schwab, C.; Kinsey, S.E.; Vora, A.; Mitchell, C.D.; Harrison, C.J. Prognostic effect of chromosomal abnormalities in childhood B-cell precursor acute lymphoblastic leukaemia: Results from the UK Medical Research Council ALL97/99 randomised trial. Lancet Oncol. 2010, 11, 429–438. [Google Scholar] [CrossRef]
- Safavi, S.; Paulsson, K. Near-haploid and low-hypodiploid acute lymphoblastic leukemia: Two distinct subtypes with consistently poor prognosis. Blood 2017, 129, 420–423. [Google Scholar] [CrossRef]
- Moorman, A.V.; Chilton, L.; Wilkinson, J.; Ensor, H.M.; Bown, N.; Proctor, S.J. A population-based cytogenetic study of adults with acute lymphoblastic leukemia. Blood 2010, 115, 206–214. [Google Scholar] [CrossRef]
- Pui, C.-H.; Rebora, P.; Schrappe, M.; Attarbaschi, A.; Baruchel, A.; Basso, G.; Cavé, H.; Elitzur, S.; Koh, K.; Liu, H.-C.; et al. Outcome of Children with Hypodiploid Acute Lymphoblastic Leukemia: A Retrospective Multinational Study. J. Clin. Oncol. 2019, 37, 770–779. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C.; Moorman, A.; Broadfield, Z.J.; Cheung, K.L.; Harris, R.L.; Jalali, G.R.; Robinson, H.M.; Barber, K.E.; Richards, S.M.; Mitchell, C.D.; et al. Three distinct subgroups of hypodiploidy in acute lymphoblastic leukaemia. Br. J. Haematol. 2004, 125, 552–559. [Google Scholar] [CrossRef] [PubMed]
- McNeer, J.L.; Devidas, M.; Dai, Y.; Carroll, A.J.; Heerema, N.A.; Gastier-Foster, J.M.; Kahwash, S.; Borowitz, M.J.; Wood, B.L.; Larsen, E.; et al. Hematopoietic Stem-Cell Transplantation Does Not Improve the Poor Outcome of Children With Hypodiploid Acute Lymphoblastic Leukemia: A Report From Children’s Oncology Group. J. Clin. Oncol. 2019, 37, 780–789. [Google Scholar] [CrossRef] [PubMed]
- Nachman, J.B.; Heerema, N.A.; Sather, H.; Camitta, B.; Forestier, E.; Harrison, C.J.; Dastugue, N.; Schrappe, M.; Pui, C.-H.; Basso, G.; et al. Outcome of treatment in children with hypodiploid acute lymphoblastic leukemia. Blood 2007, 110, 1112–1115. [Google Scholar] [CrossRef] [PubMed]
- Schmiegelow, K.; Forestier, E.; Hellebostad, M.; Heyman, M.; Kristinsson, J.; Söderhäll, S.; Taskinen, M. Long-term results of NOPHO ALL-92 and ALL-2000 studies of childhood acute lymphoblastic leukemia. Leukemia 2010, 24, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.; Cao, X.; Devidas, M.; Yang, W.; Cheng, C.; Dai, Y.; Carroll, A.; Heerema, N.A.; Zhang, H.; Moriyama, T.; et al. TP53 Germline Variations Influence the Predisposition and Prognosis of B-Cell Acute Lymphoblastic Leukemia in Children. J. Clin. Oncol. 2018, 36, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Hunger, S.P.; Ohyashiki, K.; Toyama, K.; Cleary, M.L. Hlf, a novel hepatic bZIP protein, shows altered DNA-binding properties following fusion to E2A in t(17;19) acute lymphoblastic leukemia. Genes Dev. 1992, 6, 1608–1620. [Google Scholar] [CrossRef] [PubMed]
- Mouttet, B.; Vinti, L.; Ancliff, P.; Bodmer, N.; Brethon, B.; Cario, G.; Chen-Santel, C.; Elitzur, S.; Hazar, V.; Kunz, J.; et al. Durable remissions in TCF3-HLF positive acute lymphoblastic leukemia with blinatumomab and stem cell transplan-tation. Haematologica 2019, 104, e244–e247. [Google Scholar] [CrossRef] [PubMed]
- Fischer, U.; Forster, M.; Rinaldi, A.; Risch, T.; Sungalee, S.; Warnatz, H.-J.; Bornhauser, B.; Gombert, M.; Kratsch, C.; Stütz, A.M.; et al. Genomics and drug profiling of fatal TCF3-HLF−positive acute lymphoblastic leukemia identifies recurrent mutation patterns and therapeutic options. Nat. Genet. 2015, 47, 1020–1029. [Google Scholar] [CrossRef]
- Harrison, C.J. Blood Spotlight on iAMP21 acute lymphoblastic leukemia (ALL), a high-risk pediatric disease. Blood 2015, 125, 1383–1386. [Google Scholar] [CrossRef]
- Harrison, C.J.; Moorman, A.V.; Schwab, C.; Carroll, A.J.; Raetz, E.A.; Devidas, M.; Strehl, S.; Nebral, K.; Harbott, J.; Teigler-Schlegel, A.; et al. An international study of intrachromosomal amplification of chromosome 21 (iAMP21): Cytogenetic characterization and outcome. Leukemia 2014, 28, 1015–1021. [Google Scholar] [CrossRef]
- Heerema, N.A.; Carroll, A.J.; Devidas, M.; Loh, M.L.; Borowitz, M.J.; Gastier-Foster, J.M.; Larsen, E.C.; Mattano, L.A., Jr.; Maloney, K.W.; Willman, C.L.; et al. Intrachromosomal amplification of chromosome 21 is associated with inferior outcomes in children with acute lymphoblastic leukemia treated in contemporary standard-risk children’s oncology group studies: A report from the children’s oncology group. J. Clin. Oncol. 2013, 31, 3397–3402. [Google Scholar] [CrossRef]
- Attarbaschi, A.; Mann, G.; Panzer-Grümayer, R.; Röttgers, S.; Steiner, M.; König, M.; Csinady, E.; Dworzak, M.N.; Seidel, M.G.; Janousek, D.; et al. Minimal residual disease values discriminate between low and high relapse risk in children with B-cell precursor acute lymphoblastic leukemia and an intrachromosomal amplification of chromosome 21: The Austrian and German acute lymphoblastic leukemia Berlin-Frankfurt-Münster (ALL-BFM) trials. J. Clin. Oncol. 2008, 26, 3046–3050. [Google Scholar] [CrossRef]
- Moorman, A.V.; Robinson, H.; Schwab, C.; Richards, S.M.; Hancock, J.; Mitchell, C.D.; Goulden, N.; Vora, A.; Harrison, C.J. Risk-directed treatment intensification significantly reduces the risk of relapse among children and adolescents with acute lymphoblastic leukemia and intrachromosomal amplification of chromosome 21: A comparison of the MRC ALL97/99 and UKALL2003 trials. J. Clin. Oncol. 2013, 31, 3389–3396. [Google Scholar] [CrossRef]
- Waanders, E.; van der elden, V.H.; van der Schoot, E.; van Leeuwen, F.N.; van Reijmersdal, S.V.; de Haas, V.; Veerman, A.J.; van Kessel, A.G.; Hoogerbrugge, P.M.; Kuiper, R.P.; et al. Integrated use of minimal residual disease classification and IKZF1 alteration status accurately predicts 79% of relapses in pediatric acute lymphoblastic leukemia. Leukemia 2011, 25, 254–258. [Google Scholar] [CrossRef]
- Mullighan, C.G.; Su, X.; Zhang, J.; Radtke, I.; Phillips, L.A.A.; Miller, C.B.; Ma, J.; Liu, W.; Cheng, C.; Schulman, B.A.; et al. Deletion ofIKZF1and prognosis in acute lymphoblastic leukemia. N. Engl. J. Med. 2009, 360, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Asai, D.; Imamura, T.; Suenobu, S.; Saito, A.; Hasegawa, D.; Deguchi, T.; Hashii, Y.; Matsumoto, K.; Kawasaki, H.; Hori, H.; et al. IKZF1deletion is associated with a poor outcome in pediatric B-cell precursor acute lymphoblastic leukemia in Japan. Cancer Med. 2013, 2, 412–419. [Google Scholar] [CrossRef]
- Palmi, C.; Valsecchi, M.G.; Longinotti, G.; Silvestri, D.; Carrino, V.; Conter, V.; Basso, G.; Biondi, A.; Kronnie, G.T.; Cazzaniga, G. What is the relevance of Ikaros gene deletions as a prognostic marker in pediatric Philadelphia-negative B-cell precursor acute lymphoblastic leukemia? Haematologica 2013, 98, 1226–1231. [Google Scholar] [CrossRef] [PubMed]
- Iacobucci, I.; Storlazzi, C.T.; Cilloni, D.; Lonetti, A.; Ottaviani, E.; Soverini, S.; Astolfi, A.; Chiaretti, S.; Vitale, A.; Messa, F.; et al. Identification and molecular characterization of recurrent genomic deletions on 7p12 in the IKZF1 gene in a large cohort of BCR-ABL1–positive acute lymphoblastic leukemia patients: On behalf of Gruppo Italiano Malattie Ematologiche dell’Adulto Acute Leukemia Working Party (GIMEMA AL WP). Blood 2009, 114, 2159–2167. [Google Scholar] [CrossRef]
- Lopes, B.A.; Meyer, C.; Barbosa, T.C.; Stadt, U.Z.; Horstmann, M.; Venn, N.C.; Heatley, S.; White, D.L.; Sutton, R.; Pombo-De-Oliveira, M.S.; et al. COBL is a novel hotspot for IKZF1 deletions in childhood acute lymphoblastic leukemia. Oncotarget 2016, 7, 53064–53073. [Google Scholar] [CrossRef] [PubMed]
- Dörge, P.; Meissner, B.; Zimmermann, M.; Möricke, A.; Schrauder, A.; Bouquin, J.P.; Schewe, D.; Harbott, J.; Teigler-Schlegel, A.; Ratei, R.; et al. IKZF1 deletion is an independent predictor of outcome in pediatric acute lymphoblastic leukemia treated according to the ALL-BFM 2000 protocol. Haematologica 2013, 98, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Clappier, E.; Grardel, N.; Bakkus, M.; Rapion, J.; De Moerloose, B.; Kastner, P.; Caye, A.; Vivent, J.; Costa, V.; Ferster, A.; et al. IKZF1 deletion is an independent prognostic marker in childhood B-cell precursor acute lymphoblastic leukemia, and distinguishes patients benefiting from pulses during maintenance therapy: Results of the EORTC Children’s Leukemia Group study 58951. Leukemia 2015, 29, 2154–2161. [Google Scholar] [CrossRef] [PubMed]
- Buitenkamp, T.D.; Pieters, R.; Gallimore, N.E.; van der Veer, A.; Meijerink, J.P.; Beverloo, H.B.; Zimmermann, M.; de Haas, V.; Richards, S.M.; Vora, A.J.; et al. Outcome in children with Down’s syndrome and acute lymphoblastic leukemia: Role of IKZF1 deletions and CRLF2 aberrations. Leukemia 2012, 26, 2204–2211. [Google Scholar] [CrossRef] [PubMed]
- Stanulla, M.; Dagdan, E.; Zaliova, M.; Möricke, A.; Palmi, C.; Cazzaniga, G.; Eckert, C.; Te Kronnie, G.; Bourquin, J.P.; Bornhauser, B.; et al. IKZF1plus Defines a New Minimal Residual Disease–Dependent Very-Poor Prognostic Profile in Pediatric B-Cell Precursor Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2018, 36, 1240–1249. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-L.; Hung, C.-C.; Chen, J.-S.; Lin, K.-H.; Jou, S.-T.; Hsiao, C.-C.; Sheen, J.-M.; Cheng, C.-N.; Wu, K.-H.; Lin, S.-R.; et al. IKZF1 deletions predict a poor prognosis in children with B-cell progenitor acute lymphoblastic leukemia: A multicenter analysis in Taiwan. Cancer Sci. 2011, 102, 1874–1881. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, R.P.; Waanders, E.; Van Der Velden, V.H.J.; Van Reijmersdal, S.V.; Venkatachalam, R.; Scheijen, B.; Sonneveld, E.; Van Dongen, J.J.M.; Veerman, A.J.P.; Van Leeuwen, F.N.; et al. IKZF1 deletions predict relapse in uniformly treated pediatric precursor B-ALL. Leukemia 2010, 24, 1258–1264. [Google Scholar] [CrossRef] [PubMed]
- Van Der Veer, A.; Zaliova, M.; Mottadelli, F.; De Lorenzo, P.; Te Kronnie, G.; Harrison, C.J.; Cavé, H.; Trka, J.; Saha, V.; Schrappe, M.; et al. IKZF1 status as a prognostic feature in BCR-ABL1-positive childhood ALL. Blood 2014, 123, 1691–1698. [Google Scholar] [CrossRef] [PubMed]
- Van Der Veer, A.; Waanders, E.; Pieters, R.; Willemse, M.; Van Reijmersdal, S.; Russell, L.; Harrison, C.; Evans, W.; Van Der Velden, V.; Hoogerbrugge, P.; et al. Independent prognostic value of BCR-ABL1-like signature and IKZF1 deletion, but not high CRLF2 expression, in children with B-cell precursor ALL. Blood 2013, 122, 2622–2629. [Google Scholar] [CrossRef]
- Irving, J.A.E.; Enshaei, A.; Parker, C.A.; Sutton, R.; Kuiper, R.P.; Erhorn, A.; Minto, L.; Venn, N.C.; Law, T.; Yu, J.; et al. Integration of genetic and clinical risk factors improves prognostication in relapsed childhood B-cell precursor acute lymphoblastic leukemia. Blood 2016, 128, 911–922. [Google Scholar] [CrossRef] [PubMed]
- Krentz, S.; Hof, J.; Mendioroz, A.; Vaggopoulou, R.; Dörge, P.; Lottaz, C.; Engelmann, J.C.; Groeneveld, T.; Körner, G.; Seeger, K.; et al. Prognostic value of genetic alterations in children with first bone marrow relapse of childhood B-cell precursor acute lymphoblastic leukemia. Leukemia 2012, 27, 295–304. [Google Scholar] [CrossRef]
- Churchman, M.L.; Mullighan, C.G. Ikaros: Exploiting and targeting the hematopoietic stem cell niche in B-progenitor acute lym-phoblastic leukemia. Exp. Hematol. 2017, 46, 1–8. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Churchman, M.L.; Low, J.; Qu, C.; Paietta, E.M.; Kasper, L.H.; Chang, Y.; Payne-Turner, D.; Althoff, M.J.; Song, G.; Chen, S.-C.; et al. Efficacy of Retinoids in IKZF1-Mutated BCR-ABL1 Acute Lymphoblastic Leukemia. Cancer Cell 2015, 28, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Churchman, M.L.; Qian, M.; Kronnie, G.T.; Zhang, R.; Yang, W.; Zhang, H.; Lana, T.; Tedrick, P.; Baskin, R.; Verbist, K.; et al. Germline Genetic IKZF1 Variation and Predisposition to Childhood Acute Lymphoblastic Leukemia. Cancer Cell 2018, 33, 937–948.e8. [Google Scholar] [CrossRef]
- Gianni, F.; Belver, L.; Ferrando, A. The Genetics and Mechanisms of T-Cell Acute Lymphoblastic Leukemia. Cold Spring Harb. Perspect. Med. 2020, 10, a035246. [Google Scholar] [CrossRef]
- Ferrando, A.A.; Neuberg, D.S.; Staunton, J.; Loh, M.L.; Huard, C.; Raimondi, S.C.; Behm, F.G.; Pui, C.-H.; Downing, J.R.; Gilliland, D.; et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell 2002, 1, 75–87. [Google Scholar] [CrossRef]
- Coustan-Smith, E.; Mullighan, C.G.; Onciu, M.; Behm, F.G.; Raimondi, S.C.; Pei, D.; Cheng, C.; Su, X.; Rubnitz, J.E.; Basso, G.; et al. Early T-cell precursor leukaemia: A subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol. 2009, 10, 147–156. [Google Scholar] [CrossRef]
- Chonghaile, T.N.; Roderick, J.E.; Glenfield, C.; Ryan, J.; Sallan, S.E.; Silverman, L.B.; Loh, M.L.; Hunger, S.P.; Wood, B.; DeAngelo, D.J.; et al. Maturation stage of T-cell acute lymphoblastic leukemia determines BCL-2 versus BCL-XL dependence and sensitivity to ABT-199. Cancer Discov. 2014, 4, 1074–1087. [Google Scholar] [CrossRef] [PubMed]
- Jaime-Pérez, J.C.; Santos, J.A.H.-D.L.; Gómez-Almaguer, D. Childhood T-cell acute lymphoblastic leukemia in a single Latin American center: Impact of improved treatment scheme and support therapy on survival. Hematol. Transfus. Cell Ther. 2020, 42, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-H.; Wang, H.-S.; Qian, X.-W.; Fan, C.-Q.; Li, J.; Miao, H.; Zhu, X.-H.; Yu, Y.; Meng, J.-H.; Cao, P.; et al. Genetic variants and clinical significance of pediatric acute lymphoblastic leukemia. Ann. Transl. Med. 2019, 7, 296. [Google Scholar] [CrossRef] [PubMed]
- D’Angiò, M.; Valsecchi, M.G.; Testi, A.M.; Conter, V.; Nunes, V.; Parasole, R.; Colombini, A.; Santoro, N.; Varotto, S.; Caniglia, M.; et al. Clinical features and outcome of SIL/TAL1-positive T-cell acute lymphoblastic leukemia in children and adolescents: A 10-year experience of the AIEOP group. Haematologica 2015, 100, e10–e13. [Google Scholar] [CrossRef]
- Patrick, K.; Wade, R.; Goulden, N.; Mitchell, C.; Moorman, A.V.; Rowntree, C.; Jenkinson, S.; Hough, R.; Vora, A. Outcome for children and young people with Early T-cell precursor acute lymphoblastic leukaemia treated on a con-temporary protocol, UKALL 2003. Br. J. Haematol. 2014, 166, 421–424. [Google Scholar] [CrossRef]
- Schrappe, M.; Valsecchi, M.G.; Bartram, C.R.; Schrauder, A.; Panzer-Grümayer, R.; Möricke, A.; Parasole, R.; Zimmermann, M.; Dworzak, M.; Buldini, B.; et al. Late MRD response determines relapse risk overall and in subsets of childhood T-cell ALL: Results of the AIEOP-BFM-ALL 2000 study. Blood 2011, 118, 2077–2084. [Google Scholar] [CrossRef]
- Teachey, D.T.; O’Connor, D. How I treat newly diagnosed T-cell acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma in children. Blood 2020, 135, 159–166. [Google Scholar] [CrossRef]
- Dunsmore, K.P.; Winter, S.S.; Devidas, M.; Wood, B.L.; Esiashvili, N.; Chen, Z.; Eisenberg, N.; Briegel, N.; Hayashi, R.J.; Gastier-Foster, J.M.; et al. Children’s Oncology Group AALL0434: A Phase III Randomized Clinical Trial Testing Nelarabine in Newly Diagnosed T-Cell Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2020, 38, 3282–3293. [Google Scholar] [CrossRef]
- Goldberg, J.M.; Silverman, L.B.; Levy, D.E.; Dalton, V.K.; Gelber, R.D.; Lehmann, L.; Cohen, H.J.; Sallan, S.E.; Asselin, B.L. Childhood T-cell acute lymphoblastic leukemia: The Dana-Farber Cancer Institute acute lymphoblastic leukemia consortium experience. J. Clin. Oncol. 2003, 21, 3616–3622. [Google Scholar] [CrossRef]
- Winter, S.S.; Dunsmore, K.P.; Devidas, M.; Wood, B.L.; Esiashvili, N.; Chen, Z.; Eisenberg, N.; Briegel, N.; Hayashi, R.J.; Gastier-Foster, J.M.; et al. Improved Survival for Children and Young Adults With T-Lineage Acute Lymphoblastic Leukemia: Results From the Children’s Oncology Group AALL0434 Methotrexate Randomization. J. Clin. Oncol. 2018, 36, 2926–2934. [Google Scholar] [CrossRef] [PubMed]
- Steinherz, P.G.; Gaynon, P.S.; Breneman, J.C.; Cherlow, J.M.; Grossman, N.J.; Kersey, J.H.; Johnstone, H.S.; Sather, H.N.; Trigg, M.E.; Uckun, F.M.; et al. Treatment of patients with acute lymphoblastic leukemia with bulky extramedullary disease and T-cell phenotype or other poor prognostic features. Cancer 1998, 82, 600–612. [Google Scholar] [CrossRef]
- Reismüller, B.; Attarbaschi, A.; Peters, C.; Dworzak, M.N.; Pötschger, U.; Urban, C.; Fink, F.-M.; Meister, B.; Schmitt, K.; Dieckmann, K.; et al. Long-term outcome of initially homogenously treated and relapsed childhood acute lymphoblastic leukaemia in Austria—A population-based report of the Austrian Berlin-Frankfurt-Münster (BFM) Study Group. Br. J. Haematol. 2009, 144, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Raetz, E.A.; Teachey, D.T. T-cell acute lymphoblastic leukemia. Hematology 2016, 2016, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Gocho, Y.; Liu, J.; Hu, J.; Yang, W.; Dharia, N.V.; Zhang, J.; Shi, H.; Du, G.; John, A.; Lin, T.-N.; et al. Network-based systems pharmacology reveals heterogeneity in LCK and BCL2 signaling and therapeutic sensitivity of T-cell acute lymphoblastic leukemia. Nat. Cancer 2021, 2, 284–299. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Risk Group | Subtype | Proportion of B-ALL | Median Age at Presentation, Years (Range) | Median Presenting WBC, ×109/L (Range) | Proportion of MRD-Negative at EOI, % (No of Patients) | Proportion of MRD-Negative at EOC, % (No of Patients) | Proportion with IKZF1del, % (No of Patients) | Interaction with IKZF1del | 5-y CIR, % (Range) | 5-y OS, % (Range) |
|---|---|---|---|---|---|---|---|---|---|---|
| FRG | ETV6-RUNX1 | 20% | 4.0 (1.6–14) | 12 (1–285) | 76% (52/86) | 98% (60/61) | 7% (5/70) | Possible attenuating | 5.2 (1.3 to 13) | 100% |
| Hyperdiploidy | 24% | 3.7 (1.4–12.2) | 9 (1–608) | 54% (46/85) | 92% (58/63) | 6% (4/66) | Possible attenuating | 5.5% (1.7 to 12.6) | 98.8% (91.8 to 99.8) | |
| IRG | TCF3-PBX1 | 5% | 4.8 (1.5–15.6) | 56 (6–224) | 58% (11/19) | 94% (17/18) | 0% (0/18) | Possible attenuating | 5.6% (0.3 to 23.1) | 94.4% (66.6 to 99.2) |
| DUX4 | 14% | 9.8 (2.4–16.7) | 10 (2–142) | 22% (11/50) | 82% (33/40) | 28% (13/46) | Possible attenuating | 8.9% (2.8 to 19.5) | 97.8% (85.3 to 99.7) | |
| ETV6-RUNX1-like | 2% | 2.7 (1.4–12.6) | 69 (1–278) | 57% (4/7) | 83% (5/6) | 62% (5/8) | None | 12.7% (0.5 to 45.3) | 88.9% (43.3 to 98.4) | |
| ZNF384 | 5% | 6.8 (2.1–15.7) | 37 (5–140) | 18% (3/17) | 77% (10/13) | 19% (3/16) | None | 6.3% (0.4 to 25.5) | 93.3% (61.3 to 99.0) | |
| ZNF384-like | 1% | 5.1 (2.5–7.7) | 76 (62–90) | 50% (1/2) | 50% (1/2) | 0% (0/1) | None | - | - | |
| NUTM1 | 1% | 2.4 (0.8–11.3) | 33 (11–53) | 100% (3/3) | 100% (1/1) | 0% (0/3) | None | 0.0% | 100.0% | |
| PAX5alt | 10% | 3.9 (0.7–17.4) | 24 (2–509) | 39% (12/31) | 89% (25/28) | 28% (9/32) | Poorer prognosis, IKZFplus | 18.1% (6.3 to 34.7) | 92.8% (73.7 to 98.2) | |
| PAX5-P80R | 1% | 5.7 (5.0–6.3) | 3 (2–5) | 0% (0/2) | 100% (1/1) | 0% (0/2) | Poorer prognosis, IKZFplus | - | - | |
| B-Others | 7% | 5.1 (0.6–13.0) | 8 (1–124) | 45% (10/22) | 89% (16/18) | 0% (0/21) | None | 20.7% (7.3 to 39.0) | 94.1% (65.0 to 99.1) | |
| IGH-CEBPE | <1% | 3.8 (3.8–3.8) | 32 (32–32) | 0 (0/1) | 100% (1/1) | 0% (0/1) | None | - | - | |
| HRG | Ph (BCR-ABL1) | 2% | 10.6 (2.7–15.2) | 180 (7–708) | 44% (4/9) | 83% (5/6) | 44% (4/9) | Poorer prognosis | 37.5% (7.2 to 69.4) | 75.0% (31.5 to 93.1) |
| Ph-like (BCR-ABL-like) | 2% | 8.0 (2.4–14.1) | 22 (4–518) | 12% (1/8) | 60% (3/5) | 60% (3/5) | Poorer prognosis | 37.5% (6.9 to 69.8) | 75.0% (31.5 to 93.1) | |
| MLL (KMT2A) | 3% | 0.5 (0.2–3.4) | 42 (5–247) | 11% (1/9) | 43% (3/7) | 0% (0/10) | None | 54.3% (16.7 to 81.2) | 64.8% (25.3 to 87.2) | |
| Hypodiploidy | 1% | 15.1 (13.8–16.4) | 9 (6–12) | 0% (0/1) | 0% (0/1) | 0% (0/1) | None | 50.0% (0.0 to 96.0) | 50.0% (0.6 to 91.0) | |
| Near-haploidy | 1% | 6.6 (4.3–8.3) | 26 (4–246) | 100% (3/3) | 100% (3/3) | 0% (0/2) | None | 50.0% (0.0 to 96.0) | 50.0% (0.6 to 91.0) | |
| MEF2D | 1% | 11.0 (4.9–12.4) | 7 (5–11) | 100% (4/4) | 100% (3/3) | 0% (0/4) | None | 0.0% | 100.0% | |
| HLF-r | <1% | 5.2 (5.2–5.2) | 183 (183–183) | 0% (0/1) | N.A | 0% (0/1) | None | - | - | |
| CRLF2 | 3% | 8.3 (3.0–17.3) | 59 (11–145) | 22% (2/9) | 88% (7/8) | 80% (8/10) | Poorer prognosis | 20.0% (2.6 to 49.2) | 59.1% (16.0 to 86.0) |
| EOI MRD <0.01% | Total 15 (Day 19) | Total 16 (Day 15) | COG | UKALL 2003 | MS2003/2010 |
|---|---|---|---|---|---|
| ETV6-RUNX1 | 58% | 54% | 90% | 73% | 76% |
| Hyperdiploidy | 44% | 31% | 80% | 52% | 54% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.H.R.; Li, Z.; Tai, S.T.; Oh, B.L.Z.; Yeoh, A.E.J. Genetic Alterations in Childhood Acute Lymphoblastic Leukemia: Interactions with Clinical Features and Treatment Response. Cancers 2021, 13, 4068. https://doi.org/10.3390/cancers13164068
Lee SHR, Li Z, Tai ST, Oh BLZ, Yeoh AEJ. Genetic Alterations in Childhood Acute Lymphoblastic Leukemia: Interactions with Clinical Features and Treatment Response. Cancers. 2021; 13(16):4068. https://doi.org/10.3390/cancers13164068
Chicago/Turabian StyleLee, Shawn H. R., Zhenhua Li, Si Ting Tai, Bernice L. Z. Oh, and Allen E. J. Yeoh. 2021. "Genetic Alterations in Childhood Acute Lymphoblastic Leukemia: Interactions with Clinical Features and Treatment Response" Cancers 13, no. 16: 4068. https://doi.org/10.3390/cancers13164068
APA StyleLee, S. H. R., Li, Z., Tai, S. T., Oh, B. L. Z., & Yeoh, A. E. J. (2021). Genetic Alterations in Childhood Acute Lymphoblastic Leukemia: Interactions with Clinical Features and Treatment Response. Cancers, 13(16), 4068. https://doi.org/10.3390/cancers13164068