
Leveraging Genomics, Transcriptomics, and Epigenomics to Understand the Biology and Chemoresistance of Ovarian Cancer



Abstract
:Simple Summary
Abstract
1. Introduction
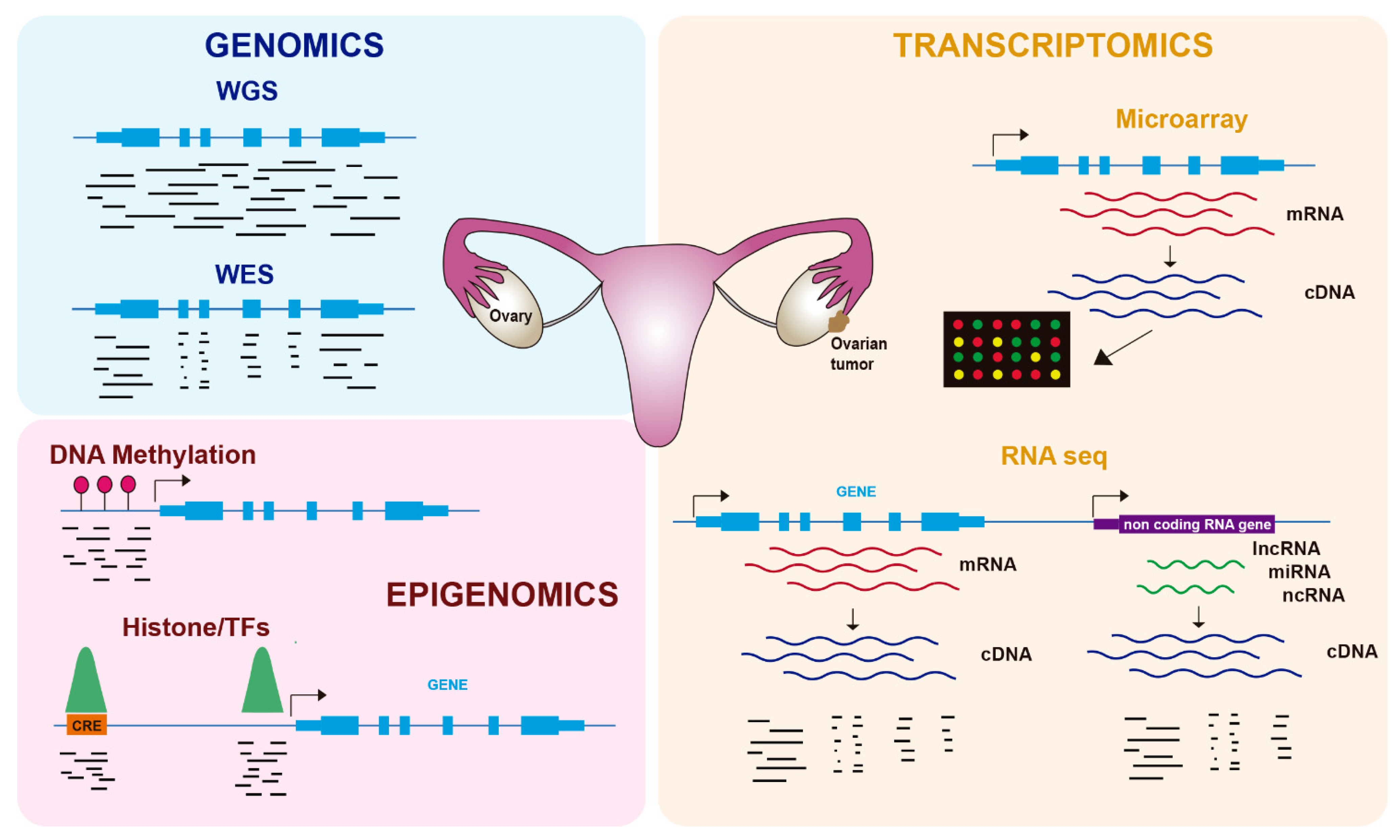
2. Genomics of OC
2.1. OC Genomic Alterations: Mutations, Copy Number Variations and Structural Variants
2.2. OC Tumor Heterogenety and Evolution
2.3. OC Genomics and Chemoresistance

{kind=link}
| Marker | Approach | Alteration | Treatment | Outcome | OC Subtype | Refs. |
|---|---|---|---|---|---|---|
| BRCA1, BRCA2 | WGS | Mutation reversion | Platinum | Resistance | HG-SOC | [13] |
| ADAMTS Family | WES | Mutation | Platinum | Sensitivity | HG-SOC | [34] |
| TMEM205, POLR2A | WES | Mutation | Paclitaxel + Carboplatin | Resistance | HG-SOC | [39] |
| PDL-L1 | WES | Structural variant | Pembrolizumab | Sensitivity | HG-SOC | [43] |
| KRAS | WES, WGS | Mutation | MEK inhibitors | Sensitivity | LG-SOC | [42] |
| MDR1 | RNA-seq | Up-regulated expression | Platinum | Resistance | HG-SOC | [13] |
| SAP25 HLA-DPA1 AKT3 PIK3R5 | RNA-seq | Differential expression | Paclitaxel + Carboplatin | Resistance | HG-SOC | [39] |
| IRF1 | RNA-seq | Up-regulated expression | Platinum | Sensitivity | HG-SOC | [44] |
| DUOXA1 | RNA-seq | Up-regulated expression | Platinum | Resistance | Ovarian cell line | [45] |
| miR-137 | RNA-seq | Down-regulated expression | Cisplatin | Resistance | Ovarian cell line | [46] |
| HIF1A-AS2 | Bru-seq | Up-regulated expression | Olaparid + carboplatin + cisplatin | Sensitivity | Ovarian cell line | [47] |
| BRCA1 | Bisulfite chip | Loss of promoter methylation | Platinum | Resistance | HG-SOC | [13] |
| H3K27me3/H3K4me3 | ChIP-seq, RNA-seq | Down-regulated expression | Platinum | Resistance | HG-SOC | [48] |
| H3K79me | ChIP-seq | Increased deposition | Platinum | Resistance | Ovarian cell line | [49] |
| SOX9 | ChIP-seq RNA-seq | Up-regulated expression by superenhancers | Cisplatin | Resistance | Ovarian cell line | [50] |
| ISL1 | ChIP-seq RNA-seq | Down-regulated expression by superenhancers | Cisplatin | resistance | Ovarian cell line | [51] |
3. Transcriptomics of OC
3.1. Biomarkers for Detection and Outcome of OC
3.2. Unraveling OC Chemoresistance Using RNA-Seq
3.3. A Single-Cell Perspective for the Study of OC Transcriptomics
4. Epigenomics of OC
4.1. Alterations of DNA Methylation in OC
4.2. Histone Modifications, Regulatory Elements and Transcription Factors in OC
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Pignata, S.; Cannella, L.; Leopardo, D.; Pisano, C.; Bruni, G.S.; Facchini, G. Chemotherapy in epithelial ovarian cancer. Cancer Lett. 2011, 303, 73–83. [Google Scholar] [CrossRef]
- Armstrong, D.K.; Bundy, B.; Wenzel, L.; Huang, H.Q.; Baergen, R.; Lele, S.; Copeland, L.J.; Walker, J.L.; Burger, R.A. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N. Engl. J. Med. 2006, 354, 34–43. [Google Scholar] [CrossRef] [Green Version]
- Mallen, A.; Soong, T.R.; Townsend, M.K.; Wenham, R.M.; Crum, C.P.; Tworoger, S.S. Surgical prevention strategies in ovarian cancer. Gynecol. Oncol. 2018, 151, 166–175. [Google Scholar] [CrossRef]
- Kurman, R.J.; Shih, I.M. The Dualistic Model of Ovarian Carcinogenesis: Revisited, Revised, and Expanded. Am. J. Pathol. 2016, 186, 733–747. [Google Scholar] [CrossRef] [Green Version]
- Bamshad, M.J.; Ng, S.B.; Bigham, A.W.; Tabor, H.K.; Emond, M.J.; Nickerson, D.A.; Shendure, J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 2011, 12, 745–755. [Google Scholar] [CrossRef]
- Piraino, S.W.; Furney, S.J. Beyond the exome: The role of non-coding somatic mutations in cancer. Ann. Oncol. 2016, 27, 240–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cirulli, E.T.; Goldstein, D.B. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat. Rev. Genet. 2010, 11, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Nagahashi, M.; Shimada, Y.; Ichikawa, H.; Kameyama, H.; Takabe, K.; Okuda, S.; Wakai, T. Next generation sequencing-based gene panel tests for the management of solid tumors. Cancer Sci. 2019, 110, 6–15. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.A.; Etemadmoghadam, D.; Temple, J.; Lynch, A.G.; Riad, M.; Sharma, R.; Stewart, C.; Fereday, S.; Caldas, C.; Defazio, A.; et al. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J. Pathol. 2010, 221, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Patch, A.M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef]
- Etemadmoghadam, D.; deFazio, A.; Beroukhim, R.; Mermel, C.; George, J.; Getz, G.; Tothill, R.; Okamoto, A.; Raeder, M.B.; Harnett, P.; et al. Integrated genome-wide DNA copy number and expression analysis identifies distinct mechanisms of primary chemoresistance in ovarian carcinomas. Clin. Cancer Res. 2009, 15, 1417–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norquist, B.; Wurz, K.A.; Pennil, C.C.; Garcia, R.; Gross, J.; Sakai, W.; Karlan, B.Y.; Taniguchi, T.; Swisher, E.M. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J. Clin. Oncol. 2011, 29, 3008–3015. [Google Scholar] [CrossRef] [Green Version]
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008, 451, 1116–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, S.; Briasoulis, E.; Linardou, H.; Bafaloukos, D.; Papadimitriou, C. Taxane resistance in breast cancer: Mechanisms, predictive biomarkers and circumvention strategies. Cancer Treat. Rev. 2012, 38, 890–903. [Google Scholar] [CrossRef]
- Lu, H.-M.; Li, S.; Black, M.H.; Lee, S.; Hoiness, R.; Wu, S.; Mu, W.; Huether, R.; Chen, J.; Sridhar, S.; et al. Association of Breast and Ovarian Cancers with Predisposition Genes Identified by Large-Scale Sequencing. JAMA Oncol. 2019, 5, 51–57. [Google Scholar] [CrossRef]
- Macintyre, G.; Goranova, T.E.; De Silva, D.; Ennis, D.; Piskorz, A.M.; Eldridge, M.; Sie, D.; Lewsley, L.-A.; Hanif, A.; Wilson, C.; et al. Copy number signatures and mutational processes in ovarian carcinoma. Nat. Genet. 2018, 50, 1262–1270. [Google Scholar] [CrossRef] [PubMed]
- dos Reis, F.J.C.; Song, H.; Goode, E.L.; Cunningham, J.M.; Fridley, B.L.; Larson, M.C.; Alsop, K.; Dicks, E.; Harrington, P.; Ramus, S.J.; et al. Germline mutation in BRCA1 or BRCA2 and ten-year survival for women diagnosed with epithelial ovarian cancer. Clin. Cancer Res. 2015, 21, 652–657. [Google Scholar] [CrossRef] [Green Version]
- Norquist, B.M.; Brady, M.F.; Harrell, M.I.; Walsh, T.; Lee, M.K.; Gulsuner, S.; Bernards, S.S.; Casadei, S.; Burger, R.A.; Tewari, K.S.; et al. Mutations in Homologous Recombination Genes and Outcomes in Ovarian Carcinoma Patients in GOG 218: An NRG Oncology/Gynecologic Oncology Group Study. Clin. Cancer Res. 2018, 24, 777–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-Y.; Yoon, J.-K.; Kim, B.; Kim, S.; Kim, M.-A.; Lim, H.; Bang, D.; Song, Y.-S. Tumor evolution and intratumor heterogeneity of an epithelial ovarian cancer investigated using next-generation sequencing. BMC Cancer 2015, 15, 85. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, R.F.; Ng, C.K.; Cooke, S.L.; Newman, S.; Temple, J.; Piskorz, A.M.; Gale, D.; Sayal, K.; Murtaza, M.; Baldwin, P.J.; et al. Spatial and temporal heterogeneity in high-grade serous ovarian cancer: A phylogenetic analysis. PLoS Med. 2015, 12, e1001789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooke, S.L.; Ng, C.K.; Melnyk, N.; Garcia, M.J.; Hardcastle, T.; Temple, J.; Langdon, S.; Huntsman, D.; Brenton, J.D. Genomic analysis of genetic heterogeneity and evolution in high-grade serous ovarian carcinoma. Oncogene 2010, 29, 4905–4913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McPherson, A.; Roth, A.; Laks, E.; Masud, T.; Bashashati, A.; Zhang, A.W.; Ha, G.; Biele, J.; Yap, D.; Wan, A.; et al. Divergent modes of clonal spread and intraperitoneal mixing in high-grade serous ovarian cancer. Nat. Genet. 2016, 48, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.W.; McPherson, A.; Milne, K.; Kroeger, D.R.; Hamilton, P.T.; Miranda, A.; Funnell, T.; Little, N.; de Souza, C.P.E.; Laan, S.; et al. Interfaces of Malignant and Immunologic Clonal Dynamics in Ovarian Cancer. Cell 2018, 173, 1755–1769.e22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsushita, H.; Hasegawa, K.; Oda, K.; Yamamoto, S.; Asada, K.; Karasaki, T.; Yabuno, A.; Nishijima, A.; Nejo, T.; Kobayashi, Y.; et al. Neoantigen load and HLA-class I expression identify a subgroup of tumors with a T-cell-inflamed phenotype and favorable prognosis in homologous recombination-proficient high-grade serous ovarian carcinoma. J. Immunother. Cancer 2020, 8, e000375. [Google Scholar] [CrossRef] [PubMed]
- Karnezis, A.N.; Cho, K.R.; Gilks, C.B.; Pearce, C.L.; Huntsman, D.G. The disparate origins of ovarian cancers: Pathogenesis and prevention strategies. Nat. Rev. Cancer 2017, 17, 65–74. [Google Scholar] [CrossRef]
- Munoz-Galvan, S.; Carnero, A. Targeting Cancer Stem Cells to Overcome Therapy Resistance in Ovarian Cancer. Cells 2020, 9, 1402. [Google Scholar] [CrossRef]
- Kuhn, E.; Kurman, R.J.; Vang, R.; Sehdev, A.S.; Han, G.; Soslow, R.; Wang, T.L.; Shih, I.M. TP53 mutations in serous tubal intraepithelial carcinoma and concurrent pelvic high-grade serous carcinoma--evidence supporting the clonal relationship of the two lesions. J. Pathol. 2012, 226, 421–426. [Google Scholar] [CrossRef] [Green Version]
- Labidi-Galy, S.I.; Papp, E.; Hallberg, D.; Niknafs, N.; Adleff, V.; Noe, M.; Bhattacharya, R.; Novak, M.; Jones, S.; Phallen, J.; et al. High grade serous ovarian carcinomas originate in the fallopian tube. Nat. Commun. 2017, 8, 1093. [Google Scholar] [CrossRef] [PubMed]
- Hellner, K.; Miranda, F.; Chedom, D.F.; Herrero-Gonzalez, S.; Hayden, D.M.; Tearle, R.; Artibani, M.; KaramiNejadRanjbar, M.; Williams, R.; Gaitskell, K.; et al. Premalignant SOX2 overexpression in the fallopian tubes of ovarian cancer patients: Discovery and validation studies. EBioMedicine 2016, 10, 137–149. [Google Scholar] [CrossRef] [Green Version]
- Wu, R.C.; Wang, P.; Lin, S.F.; Zhang, M.; Song, Q.; Chu, T.; Wang, B.G.; Kurman, R.J.; Vang, R.; Kinzler, K.; et al. Genomic landscape and evolutionary trajectories of ovarian cancer precursor lesions. J. Pathol. 2019, 248, 41–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Yasukawa, M.; Chen, K.; Hu, L.; Broaddus, R.R.; Ding, L.; Mardis, E.R.; Spellman, P.; Levine, D.A.; Mills, G.B.; et al. Association of Somatic Mutations of ADAMTS Genes with Chemotherapy Sensitivity and Survival in High-Grade Serous Ovarian Carcinoma. JAMA Oncol. 2015, 1, 486–494. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhang, X.; Gao, Y.; Shang, C.; Yu, B.; Wang, T.; Su, J.; Huang, C.; Wu, Y.; Guo, H.; et al. Development of a Genomic Signatures-Based Predictor of Initial Platinum-Resistance in Advanced High-Grade Serous Ovarian Cancer Patients. Front. Oncol. 2020, 10, 625866. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Liu, G.; Xue, F.; Edwards, R.; Sood, A.K.; Zhang, W.; Yang, D. Copy number deletion of RAD50 as predictive marker of BRCAness and PARP inhibitor response in BRCA wild type ovarian cancer. Gynecol. Oncol. 2016, 141, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Papp, E.; Hallberg, D.; Konecny, G.E.; Bruhm, D.C.; Adleff, V.; Noe, M.; Kagiampakis, I.; Palsgrove, D.; Conklin, D.; Kinose, Y.; et al. Integrated Genomic, Epigenomic, and Expression Analyses of Ovarian Cancer Cell Lines. Cell Rep. 2018, 25, 2617–2633. [Google Scholar] [CrossRef] [Green Version]
- Gulhan, D.C.; Lee, J.J.; Melloni, G.E.M.; Cortes-Ciriano, I.; Park, P.J. Detecting the mutational signature of homologous recombination deficiency in clinical samples. Nat. Genet. 2019, 51, 912–919. [Google Scholar] [CrossRef]
- Li, L.Y.; Kim, H.J.; Park, S.A.; Lee, S.H.; Kim, L.K.; Lee, J.Y.; Kim, S.; Kim, Y.T.; Kim, S.W.; Nam, E.J. Genetic Profiles Associated with Chemoresistance in Patient-Derived Xenograft Models of Ovarian Cancer. Cancer Res. Treat. 2019, 51, 1117–1127. [Google Scholar] [CrossRef]
- Madariaga, A.; Garg, S.; Bruce, J.P.; Thiryayi, S.; Mandilaras, V.; Rath, P.; Oza, A.M.; Dhani, N.C.; Cescon, D.W.; Lee, Y.C.; et al. Biomarkers of outcome to weekly paclitaxel in epithelial ovarian cancer. Gynecol. Oncol. 2020, 159, 539–545. [Google Scholar] [CrossRef]
- Harris, F.R.; Zhang, P.; Yang, L.; Hou, X.; Leventakos, K.; Weroha, S.J.; Vasmatzis, G.; Kovtun, I.V. Targeting HER2 in patient-derived xenograft ovarian cancer models sensitizes tumors to chemotherapy. Mol. Oncol. 2019, 13, 132–152. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, M.L.; Dawson, A.; Hoenisch, J.; Kim, H.; Bamford, S.; Salamanca, C.; DiMattia, G.; Shepherd, T.; Cremona, M.; Hennessy, B.; et al. Markers of MEK inhibitor resistance in low-grade serous ovarian cancer: EGFR is a potential therapeutic target. Cancer Cell Int. 2019, 19, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellone, S.; Buza, N.; Choi, J.; Zammataro, L.; Gay, L.; Elvin, J.; Rimm, D.L.; Liu, Y.; Ratner, E.S.; Schwartz, P.E.; et al. Exceptional Response to Pembrolizumab in a Metastatic, Chemotherapy/Radiation-Resistant Ovarian Cancer Patient Harboring a PD-L1-Genetic Rearrangement. Clin. Cancer Res. 2018, 24, 3282–3291. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.; Mosig, R.; Moshier, E.; Pereira, E.; Rahaman, J.; Prasad-Hayes, M.; Halpert, R.; Billaud, J.N.; Dottino, P.; Martignetti, J.A. Interferon regulatory factor 1 is an independent predictor of platinum resistance and survival in high-grade serous ovarian carcinoma. Gynecol. Oncol. 2014, 134, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Chen, C.W.; Yung, M.M.H.; Sun, W.; Sun, J.; Li, Z.; Li, J.; Li, Z.; Zhou, W.; Liu, S.S.; et al. DUOXA1-mediated ROS production promotes cisplatin resistance by activating ATR-Chk1 pathway in ovarian cancer. Cancer Lett. 2018, 428, 104–116. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Cai, X.; Yung, M.M.; Zhou, W.; Li, J.; Zhang, Y.; Li, Z.; Liu, S.S.; Cheung, A.N.Y.; Ngan, H.Y.S.; et al. miR-137 mediates the functional link between c-Myc and EZH2 that regulates cisplatin resistance in ovarian cancer. Oncogene 2019, 38, 564–580. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Tang, J.; Shrestha, B.; Heath, B.R.; Hong, L.; Lei, Y.L.; Ljungman, M.; Neamati, N. Up-regulation of hypoxia-inducible factor antisense as a novel approach to treat ovarian cancer. Theranostics 2020, 10, 6959–6976. [Google Scholar] [CrossRef]
- Chapman-Rothe, N.; Curry, E.; Zeller, C.; Liber, D.; Stronach, E.; Gabra, H.; Ghaem-Maghami, S.; Brown, R. Chromatin H3K27me3/H3K4me3 histone marks define gene sets in high-grade serous ovarian cancer that distinguish malignant, tumour-sustaining and chemo-resistant ovarian tumour cells. Oncogene 2013, 32, 4586–4592. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Zhang, X.-X.; Li, M.-C.; Cao, C.-H.; Wan, D.-Y.; Xi, B.-X.; Tan, J.-H.; Wang, J.; Yang, Z.-Y.; Feng, X.-X.; et al. C/EBPbeta enhances platinum resistance of ovarian cancer cells by reprogramming H3K79 methylation. Nat. Commun. 2018, 9, 1739. [Google Scholar] [CrossRef]
- Shang, S.; Yang, J.; Jazaeri, A.A.; Duval, A.J.; Tufan, T.; Fischer, N.L.; Benamar, M.; Guessous, F.; Lee, I.; Campbell, R.M.; et al. Chemotherapy-Induced Distal Enhancers Drive Transcriptional Programs to Maintain the Chemoresistant State in Ovarian Cancer. Cancer Res. 2019, 79, 4599–4611. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q.; Yang, F.; Mackintosh, C.; Jayani, R.S.; Oh, S.; Jin, C.; Nair, S.J.; Merkurjev, D.; Ma, W.; Allen, S.; et al. Super-Enhancer Redistribution as a Mechanism of Broad Gene Dysregulation in Repeatedly Drug-Treated Cancer Cells. Cell Rep. 2020, 31, 107532. [Google Scholar] [CrossRef]
- Tothill, R.W.; Tinker, A.V.; George, J.; Brown, R.; Fox, S.B.; Lade, S.; Johnson, D.S.; Trivett, M.K.; Etemadmoghadam, D.; Locandro, B.; et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin. Cancer Res. 2008, 14, 5198–5208. [Google Scholar] [CrossRef] [Green Version]
- Byron, S.A.; Van Keuren-Jensen, K.R.; Engelthaler, D.M.; Carpten, J.D.; Craig, D.W. Translating RNA sequencing into clinical diagnostics: Opportunities and challenges. Nat. Rev. Genet. 2016, 17, 257–271. [Google Scholar] [CrossRef]
- Qiu, C.; Lu, N.; Wang, X.; Zhang, Q.; Yuan, C.; Yan, S.; Dongol, S.; Li, Y.; Sun, X.; Sun, C.; et al. Gene expression profiles of ovarian low-grade serous carcinoma resemble those of fallopian tube epithelium. Gynecol. Oncol. 2017, 147, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Cheung, H.W.; Cowley, G.S.; Weir, B.A.; Boehm, J.S.; Rusin, S.; Scott, J.A.; East, A.; Ali, L.D.; Lizotte, P.H.; Wong, T.C.; et al. Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage-specific dependencies in ovarian cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 12372–12377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrenson, K.; Fonseca, M.A.S.; Liu, A.Y.; Dezem, F.S.; Lee, J.M.; Lin, X.; Corona, R.I.; Abbasi, F.; Vavra, K.C.; Dinh, H.Q.; et al. A Study of High-Grade Serous Ovarian Cancer Origins Implicates the SOX18 Transcription Factor in Tumor Development. Cell Rep. 2019, 29, 3726–3735.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felder, M.; Kapur, A.; Gonzalez-Bosquet, J.; Horibata, S.; Heintz, J.; Albrecht, R.; Fass, L.; Kaur, J.; Hu, K.; Shojaei, H.; et al. MUC16 (CA125): Tumor biomarker to cancer therapy, a work in progress. Mol. Cancer 2014, 13, 129. [Google Scholar] [CrossRef] [Green Version]
- Mosig, R.A.; Lobl, M.; Senturk, E.; Shah, H.; Cohen, S.; Chudin, E.; Fruscio, R.; Marchini, S.; D’Incalci, M.; Sachidanandam, R.; et al. IGFBP-4 tumor and serum levels are increased across all stages of epithelial ovarian cancer. J. Ovarian Res. 2012, 5, 3. [Google Scholar] [CrossRef] [Green Version]
- Sun, T.; Bi, F.; Liu, Z.; Yang, Q. SLC7A2 serves as a potential biomarker and therapeutic target for ovarian cancer. Aging 2020, 12, 13281–13296. [Google Scholar] [CrossRef] [PubMed]
- Fei, H.; Chen, S.; Xu, C. Bioinformatics analysis of gene expression profile of serous ovarian carcinomas to screen key genes and pathways. J. Ovarian Res. 2020, 13, 82. [Google Scholar] [CrossRef]
- Bagnoli, M.; Canevari, S.; Califano, D.; Losito, S.; Maio, M.D.; Raspagliesi, F.; Carcangiu, M.L.; Toffoli, G.; Cecchin, E.; Sorio, R.; et al. Development and validation of a microRNA-based signature (MiROvaR) to predict early relapse or progression of epithelial ovarian cancer: A cohort study. Lancet Oncol. 2016, 17, 1137–1146. [Google Scholar] [CrossRef]
- Marchini, S.; Cavalieri, D.; Fruscio, R.; Calura, E.; Garavaglia, D.; Nerini, I.F.; Mangioni, C.; Cattoretti, G.; Clivio, L.; Beltrame, L.; et al. Association between miR-200c and the survival of patients with stage I epithelial ovarian cancer: A retrospective study of two independent tumour tissue collections. Lancet Oncol. 2011, 12, 273–285. [Google Scholar] [CrossRef]
- Elias, K.M.; Fendler, W.; Stawiski, K.; Fiascone, S.J.; Vitonis, A.F.; Berkowitz, R.S.; Frendl, G.; Konstantinopoulos, P.; Crum, C.P.; Kedzierska, M.; et al. Diagnostic potential for a serum miRNA neural network for detection of ovarian cancer. Elife 2017, 6, e28932. [Google Scholar] [CrossRef] [PubMed]
- Cardillo, N.; Russo, D.; Newtson, A.; Reyes, H.; Lyons, Y.; Devor, E.; Bender, D.; Goodheart, M.J.; Gonzalez-Bosquet, J. Identification of Novel lncRNAs in Ovarian Cancer and Their Impact on Overall Survival. Int. J. Mol. Sci. 2021, 22, 1079. [Google Scholar] [CrossRef] [PubMed]
- Dobbin, Z.C.; Katre, A.A.; Steg, A.D.; Erickson, B.K.; Shah, M.M.; Alvarez, R.D.; Conner, M.G.; Schneider, D.; Chen, D.; Landen, C.N. Using heterogeneity of the patient-derived xenograft model to identify the chemoresistant population in ovarian cancer. Oncotarget 2014, 5, 8750–8764. [Google Scholar] [CrossRef] [Green Version]
- Munoz-Galvan, S.; Felipe-Abrio, B.; Garcia-Carrasco, M.; Dominguez-Pinol, J.; Suarez-Martinez, E.; Verdugo-Sivianes, E.M.; Espinosa-Sanchez, A.; Navas, L.E.; Otero-Albiol, D.; Marin, J.J.; et al. New markers for human ovarian cancer that link platinum resistance to the cancer stem cell phenotype and define new therapeutic combinations and diagnostic tools. J. Exp. Clin. Cancer Res. 2019, 38, 234. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Wang, G.; Zhang, X.; Wu, D.; Yang, L.; Wang, G.; Hao, D. The expression of miRNAs is associated with tumour genome instability and predicts the outcome of ovarian cancer patients treated with platinum agents. Sci. Rep. 2017, 7, 14736. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Zhang, W.; Wang, S.; Liu, K.; Song, F.; Ran, L. A panel of 7 prognosis-related long non-coding RNAs to improve platinum-based chemoresistance prediction in ovarian cancer. Int. J. Oncol. 2018, 53, 866–876. [Google Scholar] [CrossRef]
- Karedath, T.; Ahmed, I.; Al Ameri, W.; Al-Dasim, F.M.; Andrews, S.S.; Samuel, S.; Al-Azwani, I.K.; Mohamoud, Y.A.; Rafii, A.; Malek, J.A. Silencing of ANKRD12 circRNA induces molecular and functional changes associated with invasive phenotypes. BMC Cancer 2019, 19, 565. [Google Scholar] [CrossRef] [Green Version]
- Arakelyan, A.; Melkonyan, A.; Hakobyan, S.; Boyarskih, U.; Simonyan, A.; Nersisyan, L.; Nikoghosyan, M.; Filipenko, M.; Binder, H. Transcriptome Patterns of BRCA1- and BRCA2- Mutated Breast and Ovarian Cancers. Int. J. Mol. Sci. 2021, 22, 1266. [Google Scholar] [CrossRef]
- Shi, Z.; Zhao, Q.; Lv, B.; Qu, X.; Han, X.; Wang, H.; Qiu, J.; Hua, K. Identification of biomarkers complementary to homologous recombination deficiency for improving the clinical outcome of ovarian serous cystadenocarcinoma. Clin. Transl. Med. 2021, 11, e399. [Google Scholar] [CrossRef]
- Gonzalez-Silva, L.; Quevedo, L.; Varela, I. Tumor Functional Heterogeneity Unraveled by scRNA-seq Technologies. Trends Cancer 2020, 6, 13–19. [Google Scholar] [CrossRef] [Green Version]
- Shih, A.J.; Menzin, A.; Whyte, J.; Lovecchio, J.; Liew, A.; Khalili, H.; Bhuiya, T.; Gregersen, P.K.; Lee, A.T. Identification of grade and origin specific cell populations in serous epithelial ovarian cancer by single cell RNA-seq. PLoS ONE 2018, 13, e0206785. [Google Scholar] [CrossRef]
- Dinh, H.Q.; Lin, X.; Abbasi, F.; Nameki, R.; Haro, M.; Olingy, C.E.; Chang, H.; Hernandez, L.; Gayther, S.A.; Wright, K.N.; et al. Single-cell transcriptomics identifies gene expression networks driving differentiation and tumorigenesis in the human fallopian tube. Cell Rep. 2021, 35, 108978. [Google Scholar] [CrossRef]
- Olalekan, S.; Xie, B.; Back, R.; Eckart, H.; Basu, A. Characterizing the tumor microenvironment of metastatic ovarian cancer by single-cell transcriptomics. Cell Rep. 2021, 35, 109165. [Google Scholar] [CrossRef] [PubMed]
- Hornburg, M.; Desbois, M.; Lu, S.; Guan, Y.; Lo, A.A.; Kaufman, S.; Elrod, A.; Lotstein, A.; DesRochers, T.M.; Munoz-Rodriguez, J.L.; et al. Single-cell dissection of cellular components and interactions shaping the tumor immune phenotypes in ovarian cancer. Cancer Cell 2021, 39, 928–944.e6. [Google Scholar] [CrossRef]
- Nath, A.; Cosgrove, P.A.; Mirsafian, H.; Christie, E.L.; Pflieger, L.; Copeland, B.; Majumdar, S.; Cristea, M.C.; Han, E.S.; Lee, S.J.; et al. Evolution of core archetypal phenotypes in progressive high grade serous ovarian cancer. Nat. Commun. 2021, 12, 3039. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Issa, J.-P.; Baylin, S. Targeting the cancer epigenome for therapy. Nat. Rev. Genet. 2016, 17, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Matei, D.; Nephew, K.P. Epigenetic Attire in Ovarian Cancer: The Emperor’s New Clothes. Cancer Res. 2020, 80, 3775–3785. [Google Scholar] [CrossRef] [PubMed]
- Skvortsova, K.; Iovino, N.; Bogdanovic, O. Functions and mechanisms of epigenetic inheritance in animals. Nat. Rev. Mol. Cell Biol. 2018, 19, 774–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Earp, M.A.; Cunningham, J.M. DNA methylation changes in epithelial ovarian cancer histotypes. Genomics 2015, 106, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Balch, C.; Montgomery, J.S.; Jeong, M.; Chung, J.H.; Yan, P.; Huang, T.H.; Kim, S.; Nephew, K.P. Integrated analysis of DNA methylation and gene expression reveals specific signaling pathways associated with platinum resistance in ovarian cancer. BMC Med. Genom. 2009, 2, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Klinkebiel, D.; Barger, C.J.; Pandey, S.; Guda, C.; Miller, A.; Akers, S.N.; Odunsi, K.; Karpf, A.R. Global DNA Hypomethylation in Epithelial Ovarian Cancer: Passive Demethylation and Association with Genomic Instability. Cancers 2020, 12, 764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akers, S.N.; Moysich, K.; Zhang, W.; Lai, G.C.; Miller, A.; Lele, S.; Odunsi, K.; Karpf, A.R. LINE1 and Alu repetitive element DNA methylation in tumors and white blood cells from epithelial ovarian cancer patients. Gynecol. Oncol. 2014, 132, 462–467. [Google Scholar] [CrossRef] [Green Version]
- Pisanic, T.R., 2nd; Asaka, S.; Lin, S.-F.; Yen, T.-T.; Sun, H.; Bahadirli-Talbott, A.; Wang, T.-H.; Burns, K.H.; Wang, T.-L.; Shih, I.-M. Long Interspersed Nuclear Element 1 Retrotransposons Become Deregulated during the Development of Ovarian Cancer Precursor Lesions. Am. J. Pathol. 2019, 189, 513–520. [Google Scholar] [CrossRef] [Green Version]
- Tucker, D.W.; Getchell, C.R.; McCarthy, E.T.; Ohman, A.W.; Sasamoto, N.; Xu, S.; Ko, J.Y.; Gupta, M.; Shafrir, A.; Medina, J.E.; et al. Epigenetic Reprogramming Strategies to Reverse Global Loss of 5-Hydroxymethylcytosine, a Prognostic Factor for Poor Survival in High-grade Serous Ovarian Cancer. Clin. Cancer Res. 2018, 24, 1389–1401. [Google Scholar] [CrossRef] [Green Version]
- Fang, F.; Cardenas, H.; Huang, H.; Jiang, G.; Perkins, S.M.; Zhang, C.; Keer, H.N.; Liu, Y.; Nephew, K.P.; Matei, D. Genomic and Epigenomic Signatures in Ovarian Cancer Associated with Resensitization to Platinum Drugs. Cancer Res. 2018, 78, 631–644. [Google Scholar] [CrossRef] [Green Version]
- Nesic, K.; Kondrashova, O.; Hurley, R.M.; McGehee, C.D.; Vandenberg, C.J.; Ho, G.Y.; Lieschke, E.; Dall, G.; Bound, N.; Shield-Artin, K.; et al. Acquired RAD51C promoter methylation loss causes PARP inhibitor resistance in high grade serous ovarian carcinoma. Cancer Res. 2021. [Google Scholar] [CrossRef]
- Andersson, R.; Sandelin, A. Determinants of enhancer and promoter activities of regulatory elements. Nat. Rev. Genet. 2020, 21, 71–87. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curry, E.; Zeller, C.; Masrour, N.; Patten, D.K.; Gallon, J.; Wilhelm-Benartzi, C.S.; Ghaem-Maghami, S.; Bowtell, D.D.; Brown, R. Genes Predisposed to DNA Hypermethylation during Acquired Resistance to Chemotherapy Are Identified in Ovarian Tumors by Bivalent Chromatin Domains at Initial Diagnosis. Cancer Res. 2018, 78, 1383–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, Z.L.; Yamamoto, T.M.; McMellen, A.; Kim, H.; Hughes, C.J.; Wheeler, L.J.; Post, M.D.; Behbakht, K.; Bitler, B.G. Histone methyltransferases EHMT1 and EHMT2 (GLP/G9A) maintain PARP inhibitor resistance in high-grade serous ovarian carcinoma. Clin. Epigenetics 2019, 11, 165. [Google Scholar] [CrossRef] [PubMed]
- Hooda, J.; Novak, M.; Salomon, M.P.; Matsuba, C.; Ramos, R.I.; MacDuffie, E.; Song, M.; Hirsch, M.S.; Lester, J.; Parkash, V.; et al. Early Loss of Histone H2B Monoubiquitylation Alters Chromatin Accessibility and Activates Key Immune Pathways That Facilitate Progression of Ovarian Cancer. Cancer Res. 2019, 79, 760–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, M.R.; Peng, P.-C.; Coetzee, S.G.; Tyrer, J.; Reyes, A.L.P.; Corona, R.I.; Davis, B.; Chen, S.; Dezem, F.; Seo, J.-H.; et al. Ovarian Cancer Risk Variants Are Enriched in Histotype-Specific Enhancers and Disrupt Transcription Factor Binding Sites. Am. J. Hum. Genet. 2020, 107, 622–635. [Google Scholar] [CrossRef]
- Laury, A.R.; Hornick, J.L.; Perets, R.; Krane, J.F.; Corson, J.; Drapkin, R.; Hirsch, M.S. PAX8 reliably distinguishes ovarian serous tumors from malignant mesothelioma. Am. J. Surg. Pathol. 2010, 34, 627–635. [Google Scholar] [CrossRef] [Green Version]
- Elias, K.M.; Emori, M.M.; Westerling, T.; Long, H.; Budina-Kolomets, A.; Li, F.; MacDuffie, E.; Davis, M.R.; Holman, A.; Lawney, B.; et al. Epigenetic remodeling regulates transcriptional changes between ovarian cancer and benign precursors. JCI Insight 2016, 1, e87988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz-Galvan, S.; Felipe-Abrio, B.; Verdugo-Sivianes, E.M.; Perez, M.; Jimenez-Garcia, M.P.; Suarez-Martinez, E.; Estevez-Garcia, P.; Carnero, A. Downregulation of MYPT1 increases tumor resistance in ovarian cancer by targeting the Hippo pathway and increasing the stemness. Mol. Cancer 2020, 19, 7. [Google Scholar] [CrossRef] [Green Version]
- Corona, R.I.; Seo, J.H.; Lin, X.; Hazelett, D.J.; Reddy, J.; Fonseca, M.A.S.; Abassi, F.; Lin, Y.G.; Mhawech-Fauceglia, P.Y.; Shah, S.P.; et al. Non-coding somatic mutations converge on the PAX8 pathway in ovarian cancer. Nat. Commun. 2020, 11, 2020. [Google Scholar] [CrossRef]
- Zhang, Z.; Tu, K.; Liu, F.; Liang, M.; Yu, K.; Wang, Y.; Luo, Y.; Yang, B.; Qin, Y.; He, D.; et al. FoxM1 promotes the migration of ovarian cancer cell through KRT5 and KRT7. Gene 2020, 757, 144947. [Google Scholar] [CrossRef]
- Carles, A.; Trigo-Gonzalez, G.; Cao, Q.; Cheng, S.G.; Moksa, M.; Bilenky, M.; Huntsman, D.G.; Morin, G.B.; Hirst, M. The Pathognomonic FOXL2 C134W Mutation Alters DNA-Binding Specificity. Cancer Res. 2020, 80, 3480–3491. [Google Scholar] [CrossRef]
- Tomar, S.; Plotnik, J.P.; Haley, J.; Scantland, J.; Dasari, S.; Sheikh, Z.; Emerson, R.; Lenz, D.; Hollenhorst, P.C.; Mitra, A.K. ETS1 induction by the microenvironment promotes ovarian cancer metastasis through focal adhesion kinase. Cancer Lett. 2018, 414, 190–204. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, C.; Hu, Y.; Li, X.; Jin, S.; Liu, O.; Gou, R.; Zhuang, Y.; Guo, Q.; Nie, X.; et al. ZNF703 promotes tumor progression in ovarian cancer by interacting with HE4 and epigenetically regulating PEA15. J. Exp. Clin. Cancer Res. 2020, 39, 264. [Google Scholar] [CrossRef] [PubMed]

| Histology | Epithelial Subtypes | Type | Clinical Features | Molecular Features | Stages at Detection | 5-Year OS |
|---|---|---|---|---|---|---|
| Epithelial (90%) | High-grade serous (70–80%) | Type II | High grade, aggressive, bilateral, disseminated to omentum and peritoneum | GIN, ubiquitous mutations in TP53 and frequent in BRCA1/2, HR deficiency | Stage III–IV | 43% |
| Low-grade serous (<5%) | Type I | Low grade (except clear cell), indolent, unilateral, restricted to ovary at diagnosis | No GIN, mutations in KRAS, BRAF, ERBB2, PI3K, PTEN, ARID1A, β-catenin | Stage I–II | ||
| Endometroid (10–15%) | Stage I–II | 82% | ||||
| Mucinous (3–4%) | Stage I–II | 71% | ||||
| Clear cell (10%) | Stage I–II | 66% | ||||
| Sex cord stromal cell (2%) | Indolent | Stage I–II | 88% | |||
| Germ cell tumors (3%) | Indolent | Stage I–II | 94% | |||
| Others or unspecified (5%) | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muñoz-Galván, S.; Carnero, A. Leveraging Genomics, Transcriptomics, and Epigenomics to Understand the Biology and Chemoresistance of Ovarian Cancer. Cancers 2021, 13, 4029. https://doi.org/10.3390/cancers13164029
Muñoz-Galván S, Carnero A. Leveraging Genomics, Transcriptomics, and Epigenomics to Understand the Biology and Chemoresistance of Ovarian Cancer. Cancers. 2021; 13(16):4029. https://doi.org/10.3390/cancers13164029
Chicago/Turabian StyleMuñoz-Galván, Sandra, and Amancio Carnero. 2021. "Leveraging Genomics, Transcriptomics, and Epigenomics to Understand the Biology and Chemoresistance of Ovarian Cancer" Cancers 13, no. 16: 4029. https://doi.org/10.3390/cancers13164029
APA StyleMuñoz-Galván, S., & Carnero, A. (2021). Leveraging Genomics, Transcriptomics, and Epigenomics to Understand the Biology and Chemoresistance of Ovarian Cancer. Cancers, 13(16), 4029. https://doi.org/10.3390/cancers13164029