
Prevalence of Germline Variants in a Large Cohort of Japanese Patients with Pheochromocytoma and/or Paraganglioma

 ,
,  , ,
, ,  ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
2.1. Clinical Characteristics of the Study Population
2.2. Classification of Profiled Variants
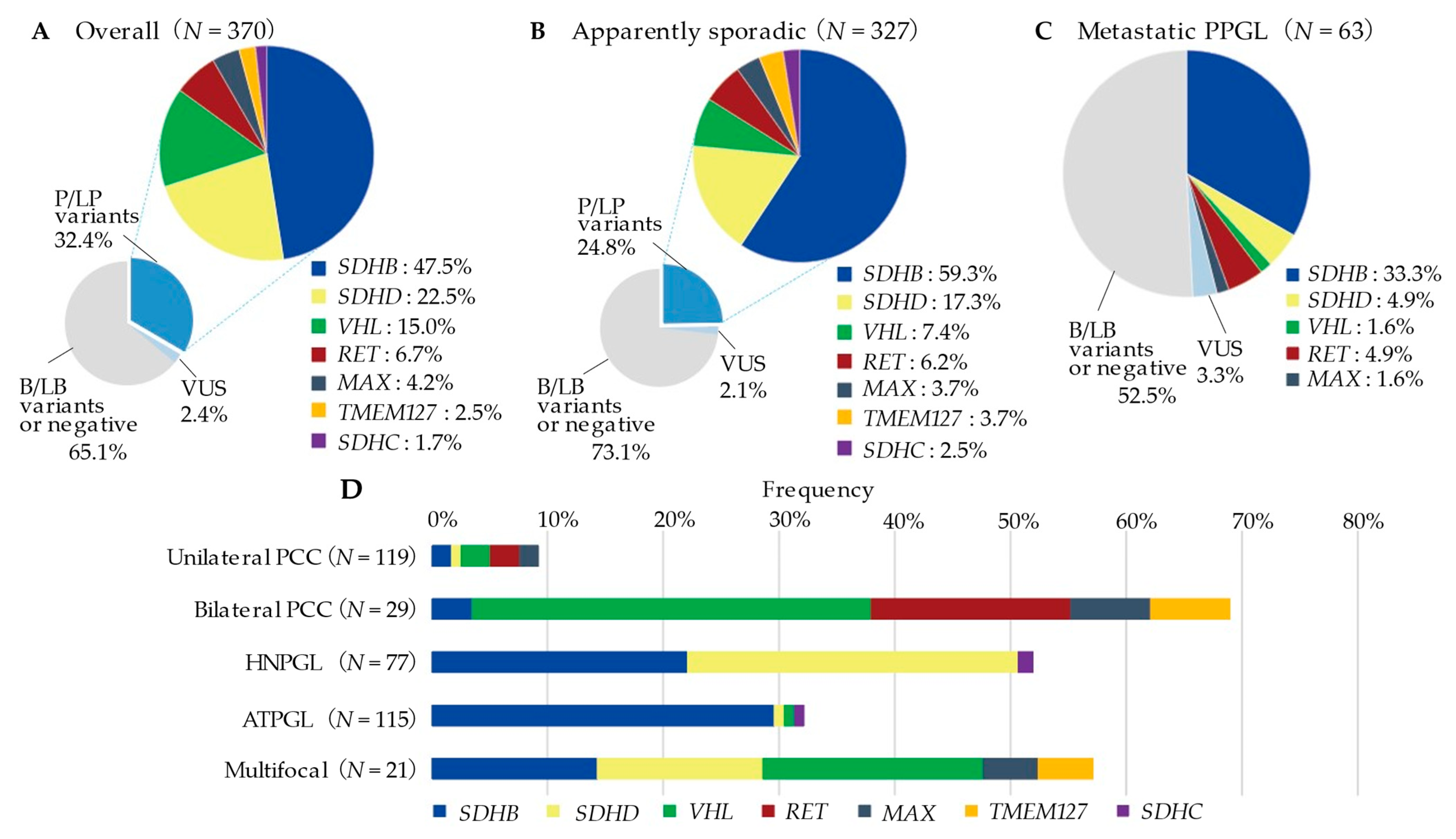
2.3. Frequency of Germline Variants
2.4. Clinical Characteristics of Probands with P/LP Variants
2.5. Genetic and Clinical Characteristics by Specific Susceptibility Genes
2.5.1. SDHB Variants
2.5.2. SDHD Variants
2.5.3. VHL Variants
2.5.4. Minor Variants Genes
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Genetic Analysis
4.3. Variant Classification
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Favier, J.; Amar, L.; Gimenez-Roqueplo, A.P. Paraganglioma and phaeochromocytoma: From genetics to personalized medicine. Nat. Rev. Endocrinol. 2015, 11, 101–111. [Google Scholar] [CrossRef]
- Muth, A.; Crona, J.; Gimm, O.; Elmgren, A.; Filipsson, K.; Stenmark Askmalm, M.; Sandstedt, J.; Tengvar, M.; Tham, E. Genetic testing and surveillance guidelines in hereditary pheochromocytoma and paraganglioma. J. Intern. Med. 2019, 285, 187–204. [Google Scholar] [CrossRef] [PubMed]
- Eisenhofer, G.; Prejbisz, A.; Peitzsch, M.; Pamporaki, C.; Masjkur, J.; Rogowski-Lehmann, N.; Langton, K.; Tsourdi, E.; Pęczkowska, M.; Fliedner, S.; et al. Biochemical Diagnosis of Chromaffin Cell Tumors in Patients at High and Low Risk of Disease: Plasma versus Urinary Free or Deconjugated O-Methylated Catecholamine Metabolites. Clin. Chem. 2018, 64, 1646–1656. [Google Scholar] [CrossRef]
- Lloyd, R.V.; Osamura, R.Y.; Kloppel, G.; Rosai, J. WHO Classification of Tumours: Pathology and Genetics of Tumours of Endocrine Organs, 4th ed.; International Agency for Research on Cancer (IARC): Lyon, France, 2017; Volume 10. [Google Scholar]
- Amar, L.; Bertherat, J.; Baudin, E.; Ajzenberg, C.; Bressac-de Paillerets, B.; Chabre, O.; Chamontin, B.; Delemer, B.; Giraud, S.; Murat, A.; et al. Genetic testing in pheochromocytoma or functional paraganglioma. J. Clin. Oncol. 2005, 23, 8812–8818. [Google Scholar] [CrossRef]
- Mannelli, M.; Castellano, M.; Schiavi, F.; Filetti, S.; Giacchè, M.; Mori, L.; Pignataro, V.; Bernini, G.; Giachè, V.; Bacca, A.; et al. Clinically guided genetic screening in a large cohort of italian patients with pheochromocytomas and/or functional or nonfunctional paragangliomas. J. Clin. Endocrinol. Metab. 2009, 94, 1541–1547. [Google Scholar] [CrossRef]
- Jafri, M.; Whitworth, J.; Rattenberry, E.; Vialard, L.; Kilby, G.; Kumar, A.V.; Izatt, L.; Lalloo, F.; Brennan, P.; Cook, J.; et al. Evaluation of SDHB, SDHD and VHL gene susceptibility testing in the assessment of individuals with non-syndromic phaeochromocytoma, paraganglioma and head and neck paraganglioma. Clin. Endocrinol. 2013, 78, 898–906. [Google Scholar] [CrossRef] [PubMed]
- Fishbein, L.; Leshchiner, I.; Walter, V.; Danilova, L.; Robertson, A.G.; Johnson, A.R.; Lichtenberg, T.M.; Murray, B.A.; Ghayee, H.K.; Else, T.; et al. Comprehensive Molecular Characterization of Pheochromocytoma and Paraganglioma. Cancer Cell 2017, 31, 181–193. [Google Scholar] [CrossRef]
- Buffet, A.; Burnichon, N.; Favier, J.; Gimenez-Roqueplo, A.P. An overview of 20 years of genetic studies in pheochromocytoma and paraganglioma. Best Pract. Res. Clin. Endocrinol. Metab. 2020, 34, 101416. [Google Scholar] [CrossRef] [PubMed]
- Koopman, K.; Gaal, J.; de Krijger, R.R. Pheochromocytomas and Paragangliomas: New Developments with Regard to Classification, Genetics, and Cell of Origin. Cancers 2019, 11, 1070. [Google Scholar] [CrossRef]
- Neumann, H.P.; Bausch, B.; McWhinney, S.R.; Bender, B.U.; Gimm, O.; Franke, G.; Schipper, J.; Klisch, J.; Altehoefer, C.; Zerres, K.; et al. Germ-line mutations in nonsyndromic pheochromocytoma. N. Engl. J. Med. 2002, 346, 1459–1466. [Google Scholar] [CrossRef]
- Brito, J.P.; Asi, N.; Bancos, I.; Gionfriddo, M.R.; Zeballos-Palacios, C.L.; Leppin, A.L.; Undavalli, C.; Wang, Z.; Domecq, J.P.; Prustsky, G.; et al. Testing for germline mutations in sporadic pheochromocytoma/paraganglioma: A systematic review. Clin. Endocrinol. 2015, 82, 338–345. [Google Scholar] [CrossRef]
- Lee, C.H.; Cheung, C.Y.; Chow, W.S.; Woo, Y.C.; Yeung, C.Y.; Lang, B.H.; Fong, C.H.; Kwok, K.H.; Chen, S.P.; Mak, C.M.; et al. Genetics of Apparently Sporadic Pheochromocytoma and Paraganglioma in a Chinese Population. Horm. Metab. Res. 2015, 47, 833–838. [Google Scholar] [CrossRef][Green Version]
- Lenders, J.W.; Duh, Q.Y.; Eisenhofer, G.; Gimenez-Roqueplo, A.P.; Grebe, S.K.; Murad, M.H.; Naruse, M.; Pacak, K.; Young, W.F., Jr. Pheochromocytoma and paraganglioma: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2014, 99, 1915–1942. [Google Scholar] [CrossRef] [PubMed]
- Plouin, P.F.; Amar, L.; Dekkers, O.M.; Fassnacht, M.; Gimenez-Roqueplo, A.P.; Lenders, J.W.; Lussey-Lepoutre, C.; Steichen, O. European Society of Endocrinology Clinical Practice Guideline for long-term follow-up of patients operated on for a phaeochromocytoma or a paraganglioma. Eur. J. Endocrinol. 2016, 174, G1–G10. [Google Scholar] [CrossRef]
- Neumann, H.P.H.; Young, W.F., Jr.; Eng, C. Pheochromocytoma and Paraganglioma. N. Engl. J. Med. 2019, 381, 552–565. [Google Scholar] [CrossRef]
- Andrews, K.A.; Ascher, D.B.; Pires, D.E.V.; Barnes, D.R.; Vialard, L.; Casey, R.T.; Bradshaw, N.; Adlard, J.; Aylwin, S.; Brennan, P.; et al. Tumour risks and genotype-phenotype correlations associated with germline variants in succinate dehydrogenase subunit genes SDHB, SDHC and SDHD. J. Med. Genet. 2018, 55, 384–394. [Google Scholar] [CrossRef]
- Buffet, A.; Venisse, A.; Nau, V.; Roncellin, I.; Boccio, V.; Le Pottier, N.; Boussion, M.; Travers, C.; Simian, C.; Burnichon, N.; et al. A decade (2001–2010) of genetic testing for pheochromocytoma and paraganglioma. Horm. Metab. Res. 2012, 44, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Cascón, A.; Pita, G.; Burnichon, N.; Landa, I.; López-Jiménez, E.; Montero-Conde, C.; Leskelä, S.; Leandro-García, L.J.; Letón, R.; Rodríguez-Antona, C.; et al. Genetics of pheochromocytoma and paraganglioma in Spanish patients. J. Clin. Endocrinol. Metab. 2009, 94, 1701–1705. [Google Scholar] [CrossRef]
- Jiang, J.; Zhang, J.; Pang, Y.; Bechmann, N.; Li, M.; Monteagudo, M.; Calsina, B.; Gimenez-Roqueplo, A.P.; Nölting, S.; Beuschlein, F.; et al. Sino-European Differences in the Genetic Landscape and Clinical Presentation of Pheochromocytoma and Paraganglioma. J. Clin. Endocrinol. Metab. 2020, 105, 3295–3307. [Google Scholar] [CrossRef] [PubMed]
- Albattal, S.; Alswailem, M.; Moria, Y.; Al-Hindi, H.; Dasouki, M.; Abouelhoda, M.; Alkhail, H.A.; Alsuhaibani, E.; Alzahrani, A.S. Mutational profile and genotype/phenotype correlation of non-familial pheochromocytoma and paraganglioma. Oncotarget 2019, 10, 5919–5931. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ma, X.; Li, M.; Tong, A.; Wang, F.; Cui, Y.; Zhang, X.; Zhang, Y.; Chen, S.; Li, Y. Genetic and Clinical Profiles of Pheochromocytoma and Paraganglioma: A Single Center Study. Front. Endocrinol. 2020, 11, 574662. [Google Scholar] [CrossRef]
- Pandit, R.; Khadilkar, K.; Sarathi, V.; Kasaliwal, R.; Goroshi, M.; Khare, S.; Nair, S.; Raghavan, V.; Dalvi, A.; Hira, P.; et al. Germline mutations and genotype-phenotype correlation in Asian Indian patients with pheochromocytoma and paraganglioma. Eur. J. Endocrinol. 2016, 175, X3. [Google Scholar] [CrossRef]
- Kimura, N.; Takekoshi, K.; Horii, A.; Morimoto, R.; Imai, T.; Oki, Y.; Saito, T.; Midorikawa, S.; Arao, T.; Sugisawa, C.; et al. Clinicopathological study of SDHB mutation-related pheochromocytoma and sympathetic paraganglioma. Endocr.-Relat. Cancer 2014, 21, L13–L16. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.; Inaishi, T.; Miyajima, N.; Adachi, Y.; Takano, Y.; Nakanishi, K.; Takeuchi, D.; Noda, S.; Aita, Y.; Takekoshi, K.; et al. Synchronous bilateral pheochromocytomas and paraganglioma with novel germline mutation in MAX: A case report. Surg. Case Rep. 2017, 3, 131. [Google Scholar] [CrossRef] [PubMed]
- Takeichi, N.; Midorikawa, S.; Watanabe, A.; Naing, B.T.; Tamura, H.; Wakakuri-Kano, T.; Ishizaki, A.; Sugihara, H.; Nissato, S.; Saito, Y.; et al. Identical germline mutations in the TMEM127 gene in two unrelated Japanese patients with bilateral pheochromocytoma. Clin. Endocrinol. 2012, 77, 707–714. [Google Scholar] [CrossRef]
- Takekoshi, K.; Isobe, K.; Suzuki, H.; Nissato, S.; Kawakami, Y.; Kawai, K.; Yamada, N. R46Q mutation in the succinate dehydrogenase B gene (SDHB) in a Japanese family with both abdominal and thoracic paraganglioma following metastasis. Endocr. J. 2008, 55, 299–303. [Google Scholar] [CrossRef]
- Saito, T.; Saito, Y.; Matsumura, K.; Tsubota, Y.; Maniwa, T.; Kaneda, H.; Minami, K.; Sakaida, N.; Uemura, Y.; Kawa, G.; et al. Novel mutation (L157X) in the succinate dehydrogenase B gene (SDHB) in a Japanese family with abdominal paraganglioma following lung metastasis. Endocr. J. 2009, 56, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn. JMD 2017, 19, 4–23. [Google Scholar] [CrossRef]
- Hirose, R.; Tsurutani, Y.; Sugisawa, C.; Inoue, K.; Suematsu, S.; Nagata, M.; Hasegawa, N.; Kakuta, Y.; Yonamine, M.; Takekoshi, K.; et al. Hereditary pheochromocytoma/paraganglioma syndrome with a novel mutation in the succinate dehydrogenase subunit B gene in a Japanese family: Two case reports. J. Med. Case Rep. 2021, 15, 282. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Kim, K.J.; Hong, N.; Shin, S.; Choi, J.R.; Kang, S.W.; Lee, S.T.; Rhee, Y. Genetic Analysis and Clinical Characteristics of Hereditary Pheochromocytoma and Paraganglioma Syndrome in Korean Population. Endocrinol. Metab. (Seoul Korea) 2020, 35, 858–872. [Google Scholar] [CrossRef]
- Flores, S.K.; Cheng, Z.; Jasper, A.M.; Natori, K.; Okamoto, T.; Tanabe, A.; Gotoh, K.; Shibata, H.; Sakurai, A.; Nakai, T.; et al. A synonymous VHL variant in exon 2 confers susceptibility to familial pheochromocytoma and von Hippel-Lindau disease. J. Clin. Endocrinol. Metab. 2019, 104, 3826–3834. [Google Scholar] [CrossRef]
- Benn, D.E.; Robinson, B.G.; Clifton-Bligh, R.J. 15 YEARS OF PARAGANGLIOMA: Clinical manifestations of paraganglioma syndromes types 1–5. Endocr.-Relat. Cancer 2015, 22, T91–T103. [Google Scholar] [CrossRef]
- Baysal, B.E. Genomic imprinting and environment in hereditary paraganglioma. Am. J. Med. Genet. Part C Semin. Med. Genet. 2004, 129c, 85–90. [Google Scholar] [CrossRef]
- Buffet, A.; Ben Aim, L.; Leboulleux, S.; Drui, D.; Vezzosi, D.; Libé, R.; Ajzenberg, C.; Bernardeschi, D.; Cariou, B.; Chabolle, F.; et al. Positive Impact of Genetic Test on the Management and Outcome of Patients With Paraganglioma and/or Pheochromocytoma. J. Clin. Endocrinol. Metab. 2019, 104, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Sbardella, E.; Cranston, T.; Isidori, A.M.; Shine, B.; Pal, A.; Jafar-Mohammadi, B.; Sadler, G.; Mihai, R.; Grossman, A.B. Routine genetic screening with a multi-gene panel in patients with pheochromocytomas. Endocrine 2018, 59, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Piccini, V.; Rapizzi, E.; Bacca, A.; Di Trapani, G.; Pulli, R.; Giachè, V.; Zampetti, B.; Lucci-Cordisco, E.; Canu, L.; Corsini, E.; et al. Head and neck paragangliomas: Genetic spectrum and clinical variability in 79 consecutive patients. Endocr.-Relat. Cancer 2012, 19, 149–155. [Google Scholar] [CrossRef]
- Williams, M.D. Paragangliomas of the Head and Neck: An Overview from Diagnosis to Genetics. Head Neck Pathol. 2017, 11, 278–287. [Google Scholar] [CrossRef]
- Benn, D.E.; Gimenez-Roqueplo, A.P.; Reilly, J.R.; Bertherat, J.; Burgess, J.; Byth, K.; Croxson, M.; Dahia, P.L.; Elston, M.; Gimm, O.; et al. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J. Clin. Endocrinol. Metab. 2006, 91, 827–836. [Google Scholar] [CrossRef]
- King, K.S.; Prodanov, T.; Kantorovich, V.; Fojo, T.; Hewitt, J.K.; Zacharin, M.; Wesley, R.; Lodish, M.; Raygada, M.; Gimenez-Roqueplo, A.P.; et al. Metastatic pheochromocytoma/paraganglioma related to primary tumor development in childhood or adolescence: Significant link to SDHB mutations. J. Clin. Oncol. 2011, 29, 4137–4142. [Google Scholar] [CrossRef] [PubMed]
- Turkova, H.; Prodanov, T.; Maly, M.; Martucci, V.; Adams, K.; Widimsky, J., Jr.; Chen, C.C.; Ling, A.; Kebebew, E.; Stratakis, C.A.; et al. Characteristics and outcomes of metastatic sdhb and sporadic pheochromocytoma/paraganglioma: An national institutes of health study. Endocr. Pract. Off. J. Am. Coll. Endocrinol. Am. Assoc. Clin. Endocrinol. 2016, 22, 302–314. [Google Scholar] [CrossRef]
- Accetturo, M.; Bartolomeo, N.; Stella, A. In-silico Analysis of NF1 Missense Variants in ClinVar: Translating Variant Predictions into Variant Interpretation and Classification. Int. J. Mol. Sci. 2020, 21, 721. [Google Scholar] [CrossRef]
- Accetturo, M.; D’Uggento, A.M.; Portincasa, P.; Stella, A. Improvement of MEFV gene variants classification to aid treatment decision making in familial Mediterranean fever. Rheumatology (Oxf. Engl.) 2020, 59, 754–761. [Google Scholar] [CrossRef]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Toledo, R.A.; Burnichon, N.; Cascon, A.; Benn, D.E.; Bayley, J.P.; Welander, J.; Tops, C.M.; Firth, H.; Dwight, T.; Ercolino, T.; et al. Consensus Statement on next-generation-sequencing-based diagnostic testing of hereditary phaeochromocytomas and paragangliomas. Nat. Rev. Endocrinol. 2017, 13, 233–247. [Google Scholar] [CrossRef]
- Miller, D.T.; Lee, K.; Chung, W.K.; Gordon, A.S.; Herman, G.E.; Klein, T.E.; Stewart, D.R.; Amendola, L.M.; Adelman, K.; Bale, S.J.; et al. ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. Off. J. Am. Coll. Med. Genet. 2021. [Google Scholar] [CrossRef]
- Alirezaie, N.; Kernohan, K.D.; Hartley, T.; Majewski, J.; Hocking, T.D. ClinPred: Prediction Tool to Identify Disease-Relevant Nonsynonymous Single-Nucleotide Variants. Am. J. Hum. Genet. 2018, 103, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Desmet, F.O.; Hamroun, D.; Lalande, M.; Collod-Béroud, G.; Claustres, M.; Béroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef]


{kind=link}
| Characterisic | Overall (N = 370) | FS Presentation (N = 43, 11.6%) | AS Presentation (N = 327, 88.4%) | p Value (FS vs. AS) |
|---|---|---|---|---|
| Sex (M:F) | 166:204 | 25:18 | 141:186 | 0.073 |
| Age at diagnosis (years) | 0.001 | |||
| Mean ± S.D. | 43.3 ± 15.8 | 36.0 ± 13.9 | 44.3 ± 15.8 | |
| Range | 6–83 | 10–69 | 6–83 | |
| Tumour size (Mean ± S.D.) (cm) | 5.4 ± 3.1 | 4.3 ± 1.6 | 5.3 ± 3.1 | 0.184 |
| Location of tumour | <0.001 | |||
| Unilateral PCC | 122 (33.0%) | 7 (16.3%) | 115 (35.2%) | |
| Bilateral PCC | 31 (8.4%) | 11 (25.6%) | 20 (6.1%) | |
| Single HNPGL | 73 (19.7%) | 9 (20.9%) | 64 (19.6%) | |
| Bilateral/Multiple HNPGL | 6 (1.6%) | 2 (4.7%) | 4 (1.2%) | |
| Single ATPGL | 106 (28.6%) | 8 (18.6%) | 98 (30.0%) | |
| Multiple ATPGL | 10 (2.7%) | - | 10 (3.1%) | |
| Multifocal (PCC + HNPGL) | 3 (0.8%) | - | 3 (0.9%) | |
| Multifocal (PCC + ATPGL) | 17 (4.6%) | 4 (9.3%) | 13 (4.0%) | |
| Multifocal (HNPGL + ATPGL) | 2 (0.5%) | 2 (4.7%) | - | |
| Metastatic | 63 (17.0%) | 5 (11.6%) | 58 (17.7%) | 0.392 |
| Variant | No. of Probands (FS) | In Silico Prediction | Disease Database | Population Database | ACMG/AMP Class | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Nucleotide Change | Amino Acid Change | REVEL | HSF | HGMD | ClinVar | gnomAD (Global) | gnomAD (East Asian) | jMorp (Japanese) | ||
| SDHB | c.79C>T | p.R27* | 1 | - | - | R | P | 1/147,754 | 1/4946 | NR | LP |
| SDHB | c.137G>A | p.R46Q | 14 (2) | 0.911 | - | R | P/LP | 1/147,812 | 0/4966 | NR | P |
| SDHB | c.183T>G | p.Y61* | 1 | - | - | R | P | NR | NR | NR | P |
| SDHB | c.268C>T | p.R90* | 2 | - | - | R | P | 2/147,886 | 0/4960 | NR | P |
| SDHB | c.470delT | p.L157* | 13 (2) | - | - | R | P | NR | NR | NR | P |
| SDHB | c.502C>T | p.Q168* | 1 | - | - | R | P | NR | NR | NR | P |
| SDHB | c.641A>G | p.Q214R | 3 (1) | 0.973 | - | R | US | NR | NR | NR | LP |
| SDHB | c.642G>C | p.Q214H | 3 (1) | 0.891 | - | R | NR | NR | NR | 1/16,760 | LP |
| SDHB | c.649C>T | p.R217C | 1 | 0.988 | - | R | P/LP | 1/147,922 | 1/4950 | NR | LP |
| SDHB | c.725G>A | p.R242H | 4 | 0.944 | - | R | P | 1/147,882 | 0/4960 | NR | LP |
| SDHB | c.201-2A>C | SSC | 8 | - | −32.0% | NR | LP | 1/147,958 | 1/4968 | 4/16,760 | P |
| SDHB | c.424-2delA # | SSC | 2 (1) | - | −91.4% | NR | NR | NR | NR | NR | P |
| SDHB | c.424-7_427 delinsC | SSC and small Indels | 2 (1) | - | −75.2% | R | NR | NR | NR | NR | P |
| SDHB | Exon 1 Del | - | 1 (1) | - | - | R | P | NR | NR | NR | P |
| SDHB | Exon1 Dup | - | 1 | - | - | R | US | NR | NR | NR | VUS |
| SDHB | Promotor and Exon1-2 Del | - | 1 | - | - | R | NR | NR | NR | NR | LP |
| SDHD | c.1A>G | p.M1V | 1 | 0.734 | - | R | P | NR | NR | NR | P |
| SDHD | c.3G>A | p.M1I | 1 (1) | 0.762 | - | R | P | NR | NR | NR | P |
| SDHD | c.15G>A | p.W5* | 2 (1) | - | - | R | NR | NR | NR | NR | P |
| SDHD | c.57delG | p.L20Cfs*66 | 1 | - | - | R | P | NR | NR | NR | P |
| SDHD | c.112C>T | p.R38* | 1 | - | - | R | P | NR | NR | NR | P |
| SDHD | c.196T>C † | p.W66R | 4 | 0.936 | - | NR | NR | NR | NR | NR | LP |
| SDHD | c.228_239del † | p.L77_L80del | 1 | - | - | NR | NR | NR | NR | NR | VUS |
| SDHD | c.236T>G † | p.L79R | 1 | 0.968 | - | NR | NR | NR | NR | NR | VUS |
| SDHD | c.242C>T | p.P81L | 3 (1) | 0.908 | - | R | P | 1/147,924 | 0/4952 | NR | LP |
| SDHD | c.265_279del † | p.S89_Y93del | 1 | - | - | NR | NR | NR | NR | NR | VUS |
| SDHD | c.285_296del † | p.A96_L99del | 5 (3) | - | - | NR | NR | NR | NR | NR | LP |
| SDHD | c.317G>A | p.G106D | 3 (3) | 0.945 | - | R | NR | NR | NR | NR | LP |
| SDHD | c.337_340del | p.D113Mfs*21 | 2 (1) | - | - | R | P | NR | NR | NR | P |
| SDHD | c.352del | p.D118Mfs*17 | 1 (1) | - | - | R | P | NR | NR | NR | P |
| SDHD | c.412G>A | p.G138R | 1 (1) | 0.978 | - | NR | LP | NR | NR | NR | LP |
| SDHD | c.315-1G>A | SSC | 1 (1) | - | −30.7% | NR | LP | NR | NR | NR | P |
| SDHD | Exon 4 Del | - | 1 | - | - | R | P | NR | NR | NR | P |
| VHL | c.191G>C | p.R64P | 1 | 0.938 | - | R | P/LP | NR | NR | NR | LP |
| VHL | c.235C>G | p.R79G | 1 | 0.792 | - | NR | US | NR | NR | NR | VUS |
| VHL | c.250G>A | p.V84M | 1 | 0.758 | - | R | LP/US | NR | NR | NR | LP |
| VHL | c.250G>C | p.V84L | 1 | 0.626 | - | R | P | 1/147,942 | 0/4948 | NR | LP |
| VHL | c.293A>G | p.Y98C | 1 (1) | 0.938 | - | R | P | NR | NR | NR | LP |
| VHL | c.370A>C † | p.T124P | 1 (1) | 0.88 | - | NR | NR | NR | NR | NR | LP |
| VHL | c.371C>T | p.T124I | 2 (2) | 0.924 | - | R | LP | NR | NR | NR | LP |
| VHL | c.407T>G | p.F136C | 1 | 0.821 | - | R | P/US | NR | NR | NR | LP |
| VHL | c.414A>G | p.P138= | 4 (4) | - | NR | P | NR | NR | NR | P | |
| VHL | c.482G>A | p.R161Q | 1 (1) | 0.797 | - | R | P | NR | NR | NR | LP |
| VHL | c.496G>T | p.V166F | 1 | 0.848 | - | R | P | NR | NR | NR | LP |
| VHL | c.499C>T | p.R167W | 1 (1) | 0.868 | - | R | P | NR | NR | NR | LP |
| VHL | c.500G>A | p.R167Q | 1 (1) | 0.874 | - | R | P/US | NR | NR | NR | LP |
| VHL | c.524A>G | p.Y175C | 1 (1) | 0.892 | - | R | P/LP | NR | NR | NR | LP |
| VHL | c.548C>T | p.S183L | 1 | 0.620 | - | R | US | 2/147,912 | 0/4960 | 3/16,760 | VUS |
| RET | c.1891G>T | p.D631Y | 1 | - | - | R | P | NR | NR | NR | LP |
| RET | c.1892A>T | p.D631V | 1 | 0.837 | - | NR | US | NR | NR | NR | LP |
| RET | c.1900T>C | p.C634R | 2 | 0.972 | - | R | P | 2/147,640 | 0/4906 | NR | LP |
| RET | c.1901G>A | p.C634Y | 4 (3) | 0.917 | - | R | P | NR | NR | NR | LP |
| MAX | c.3G>A $ | p.M1I | 1 | 0.919 | - | NR | NR | NR | NR | NR | P |
| MAX | c.70_73del † | p.K24Gfs*40 | 1 | - | - | NR | NR | NR | NR | NR | LP |
| MAX | c.97C>T | p.R33* | 1 | - | - | R | P | NR | NR | NR | P |
| MAX | c.223C>T | p.R75* | 2 (2) | - | - | R | P | NR | NR | NR | P |
| MAX | c.284T>C | p.L95P | 1 (1) | 0.939 | - | NR | US | NR | NR | NR | VUS |
| TMEM127 | c.116_119del | p.I41Rfs*39 | 2 | - | - | R | P | 3/147,988 | 0/4964 | NR | P |
| TMEM127 | c.119C>T | p.S40F | 1 (1) | 0.715 | - | R | US | NR | NR | NR | VUS |
| TMEM127 | c.232dupG † | p.D78Gfs*30 | 1 | - | - | NR | NR | NR | NR | NR | LP |
| TMEM127 | c.280C>T | p.R94W | 1 | 0.738 | - | R | US | 9/147,980 | 0/4960 | 1/16,760 | VUS |
| SDHC | c.43C>T | p.R15* | 1 | - | - | R | P | 3/147,874 | 0/4964 | NR | P |
| SDHC | c.204dupC † | p.I69Hfs*29 | 1 | - | - | NR | NR | NR | NR | NR | LP |
| Characteristic | Variant-Negative (N = 241) | P/LP-Positive (N = 120) | p Value # | SDHB (N = 57) | SDHD (N = 27) | VHL (N = 18) | RET (N = 8) | MAX (N = 5) | TMEM127 (N = 3) | SDHC (N = 2) |
|---|---|---|---|---|---|---|---|---|---|---|
| Sex (M:F) | 102:139 | 59:61 | 0.228 | 23:34 | 18:9 | 10:8 | 4:4 | 1:4 | 3:0 | 0:2 |
| Familial presentation | 2 | 38 | 9 | 13 | 11 | 3 | 2 | 0 | 0 | |
| Syndromic presentation | - | 5 | - | - | 3 | 2 | - | - | ||
| Age at diagnosis (years) | <0.001 | |||||||||
| Mean ± S.D. | 46.0 ± 15.6 | 38.2 ± 15.2 | 37.2 ± 14.5 † | 44.6 ± 12.0 | 27.1 ± 13.8 † | 42.1 ± 15.2 | 39.0 ± 21.1 | 50.3 ± 10.3 | 44.5 ± 1.5 | |
| Range | 6–83 | 10–74 | 12–74 | 23–65 | 10–64 | 25–68 | 13–69 | 39–64 | 43–46 | |
| Tumour size (cm) (Mean ± S.D.) | 5.3 ± 3.1 | 4.9 ± 2.6 | 5.5 ± 2.9 | 3.4 ± 1.2 | 5.2 ± 1.9 | 7.1 ± 2.8 | 4.6 ± 3.9 | 3.0 ± 0.5 | 5.4 ± 1.4 | |
| Location of tumour | <0.001 | |||||||||
| Unilateral PCC | 108 | 11 * | 2 | 1 | 3 | 3 | 2 | - | - | |
| Bilateral PCC | 9 | 20 * | 1 | - | 10 | 5 | 2 | 2 | - | |
| Single HNPGL | 36 | 35 * | 17 | 18 | - | - | - | - | - | |
| Bilateral HNPGL | 1 | 5 | - | 4 | - | - | - | - | 1 | |
| Single ATPGL | 72 | 33 | 30 | 1 | 1 | - | - | - | 1 | |
| Multiple ATPGL | 6 | 4 | 4 | - | - | - | - | - | - | |
| PCC + HNPGL | 2 | 1 | - | 1 | - | - | - | - | - | |
| PCC + ATPGL | 7 | 9 | 3 | - | 4 | - | 1 | 1 | - | |
| HNPGL + ATPGL | - | 2 | - | 2 | - | - | - | - | - | |
| Metastatic | 32 (13.4%) | 29 (24.2%) | 0.029 | 21 (36.8%) ‡ | 3 (11.1%) | 1 (5.6%) | 3 (37.5%) | 1 (20.0%) | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yonamine, M.; Wasano, K.; Aita, Y.; Sugasawa, T.; Takahashi, K.; Kawakami, Y.; Shimano, H.; Nishiyama, H.; Hara, H.; Naruse, M.; et al. Prevalence of Germline Variants in a Large Cohort of Japanese Patients with Pheochromocytoma and/or Paraganglioma. Cancers 2021, 13, 4014. https://doi.org/10.3390/cancers13164014
Yonamine M, Wasano K, Aita Y, Sugasawa T, Takahashi K, Kawakami Y, Shimano H, Nishiyama H, Hara H, Naruse M, et al. Prevalence of Germline Variants in a Large Cohort of Japanese Patients with Pheochromocytoma and/or Paraganglioma. Cancers. 2021; 13(16):4014. https://doi.org/10.3390/cancers13164014
Chicago/Turabian StyleYonamine, Masato, Koichiro Wasano, Yuichi Aita, Takehito Sugasawa, Katsutoshi Takahashi, Yasushi Kawakami, Hitoshi Shimano, Hiroyuki Nishiyama, Hisato Hara, Mitsuhide Naruse, and et al. 2021. "Prevalence of Germline Variants in a Large Cohort of Japanese Patients with Pheochromocytoma and/or Paraganglioma" Cancers 13, no. 16: 4014. https://doi.org/10.3390/cancers13164014
APA StyleYonamine, M., Wasano, K., Aita, Y., Sugasawa, T., Takahashi, K., Kawakami, Y., Shimano, H., Nishiyama, H., Hara, H., Naruse, M., Okamoto, T., Matsuda, T., Kosugi, S., Horiguchi, K., Tanabe, A., Watanabe, A., Kimura, N., Nakamura, E., Sakurai, A., ... Takekoshi, K. (2021). Prevalence of Germline Variants in a Large Cohort of Japanese Patients with Pheochromocytoma and/or Paraganglioma. Cancers, 13(16), 4014. https://doi.org/10.3390/cancers13164014