
Stem Cell Theory of Cancer: Origin of Tumor Heterogeneity and Plasticity



{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
“Nature creates unity even in the parts of a whole”, Eugène Delacroix.
1. Baskets and Umbrellas
2. Clonal Origin
2.1. Testicular Cancer
2.2. Breast Cancer
2.3. Lung Cancer
3. Genetics vs. Epigenetics
4. Genomic Subtypes
5. Stem-Ness Subtypes
6. Renal Cell Carcinoma (RCC) Subtypes
7. Evolutionary Subtypes
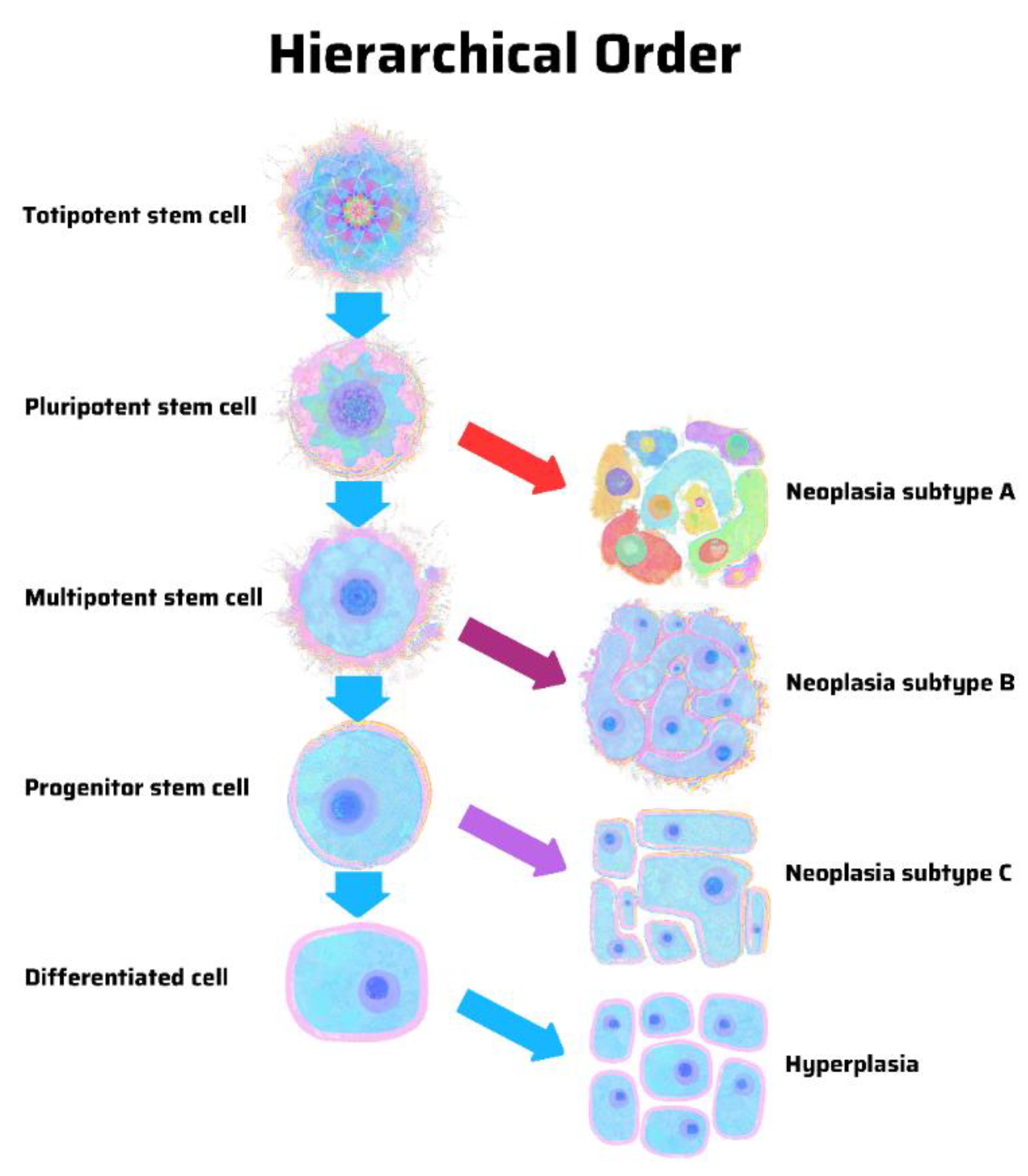
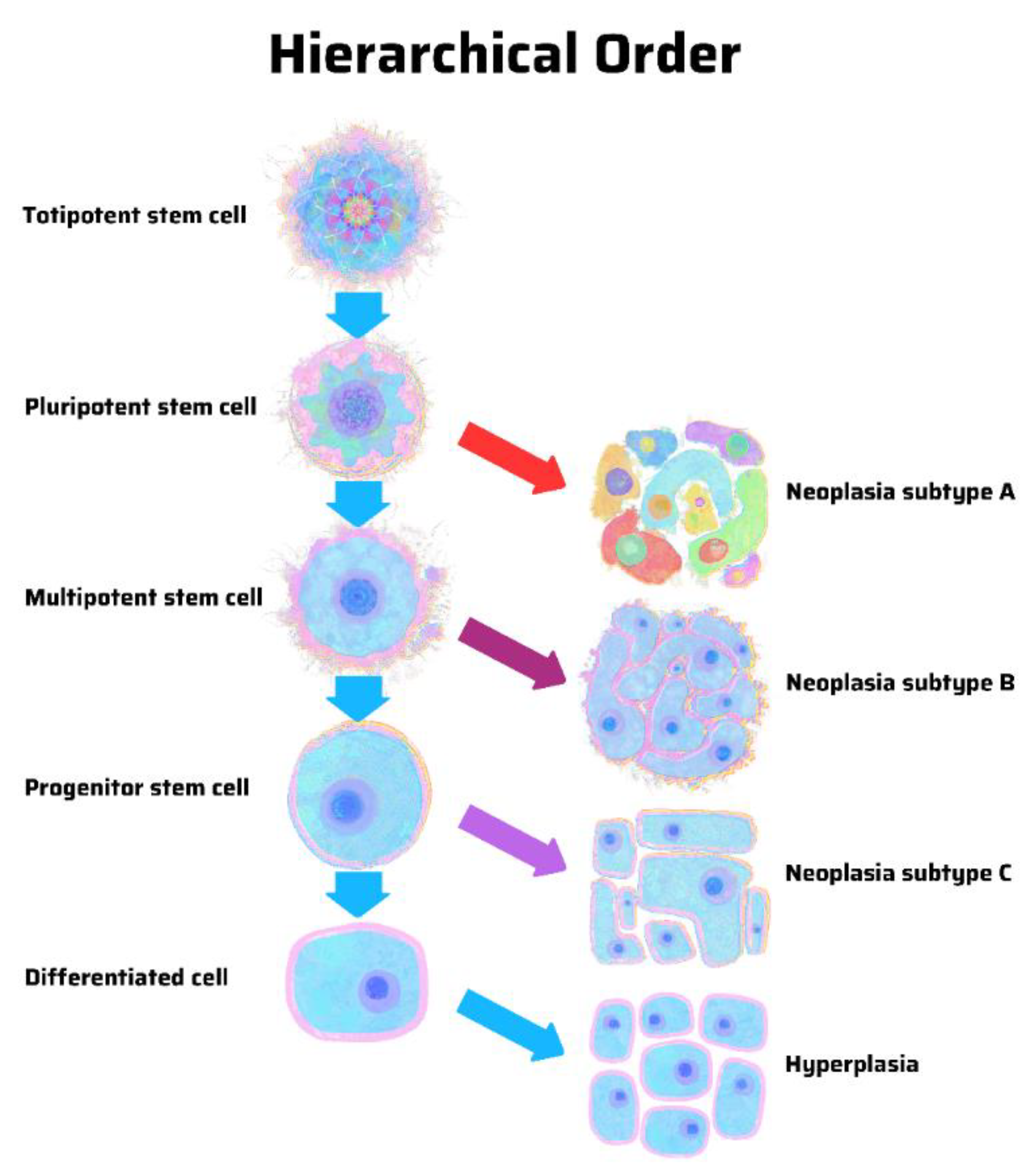
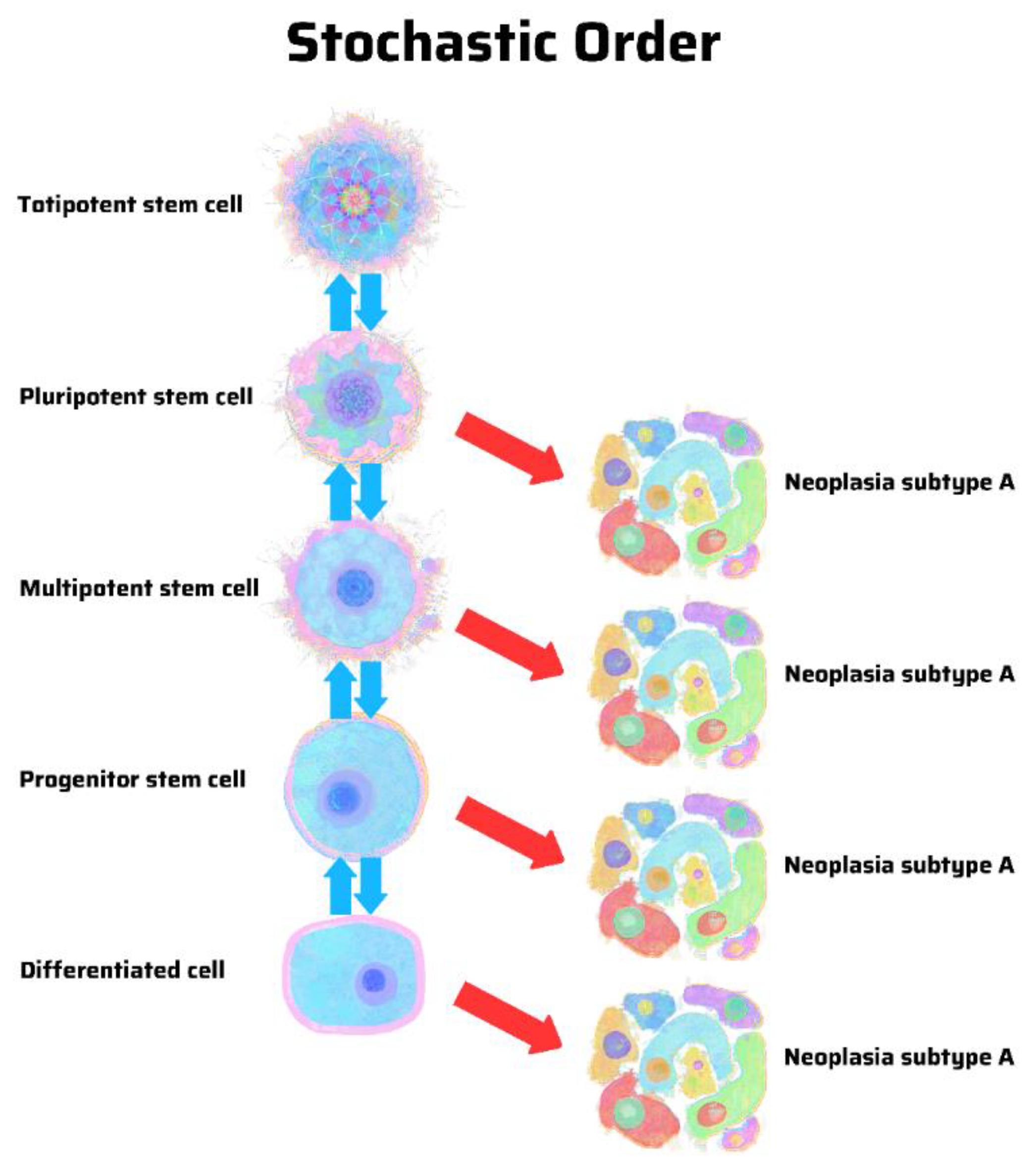
8. Hierarchy vs. Stochasticity
9. Differentiation vs. Dedifferentiation
10. Maturation Arrest
11. Precision Medicine and Targeted Therapy
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Qin, S.; Jiang, J.; Lu, Y.; Nice, E.C.; Huang, C.; Zhang, J.; He, W. Emerging role of tumor cell plasticity in modifying therapeutic response. Signal Transduct. Target. Ther. 2020, 5, 228. [Google Scholar] [CrossRef]
- Vessoni, A.T.; Filippi-Chiela, E.C.; Lenz, G.; Batista, L.F.Z. Tumor propagating cells: Drivers of tumor plasticity, heterogeneity, and recurrence. Oncogene 2020, 39, 2055–2068. [Google Scholar] [CrossRef] [PubMed]
- Boumahdi, S.; de Sauvage, F.J. The great escape: Tumor cell plasticity in resistance to targeted therapy. Nat. Rev. Drug Discov. 2020, 19, 39–56. [Google Scholar] [CrossRef]
- Meacham, C.E.; Morrison, S.J. Tumor heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337. [Google Scholar] [CrossRef] [Green Version]
- Brooks, M.D.; Burness, M.L.; Wicha, M.S. Therapeutic implications of cellular heterogeneity and plasticity in breast cancer. Cell Stem Cell 2015, 17, 260–271. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.K.; Arya, R.K.; Maheshwari, S.; Singh, A.; Meena, S.; Pandey, P.; Dormand, O.; Datta, D. Tumor heterogeneity and cancer stem cell paradigm: Updates in concepts, controversies, and clinical relevance. Int. J. Cancer 2015, 136, 1991–2000. [Google Scholar] [CrossRef]
- Rich, J.N. Cancer stem cells: Understanding tumor hierarchy and heterogeneity. Medicine 2016, 95, S2–S7. [Google Scholar] [CrossRef]
- Prasetyanti, P.R.; Medema, J.P. Intratumor heterogeneity from a cancer stem cell perspective. Mol. Cancer 2017, 16, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fanelli, G.N.; Naccarato, A.G.; Scatena, C. Recent advances in cancer plasticity: Cellular mechanism, surveillance strategies, and therapeutic optimization. Front. Oncol. 2020, 10, 569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hass, R.; von der Ohe, J.; Ungefroren, H. Impact of the tumor microenvironment on tumor heterogeneity and consequences for cancer cell plasticity and stemness. Cancers 2020, 12, 3716. [Google Scholar] [CrossRef]
- Tu, S.M. Origin of Cancers. Clinical Perspectives and Implications of a Stem-Cell Theory of Cancer; Cancer Treatment and Research; Rosen, S.T., Ed.; Springer: New York, NY, USA, 2010; Volume 154. [Google Scholar]
- Tu, S.M. Story of Hydra: Portrait of Cancer as a Stem-Cell Disease; Nova: New York, NY, USA, 2019. [Google Scholar]
- Anderson, K.; Lutz, C.; van Delft, F.W.; Bateman, C.M.; Guo, Y.; Colman, S.M.; Kempski, H.; Moorman, A.V.; Titley, I.; Swansbury, J.; et al. Genetic variegation of clonal architecture and propagating cells in leukemia. Nature 2011, 469, 356–361. [Google Scholar] [CrossRef]
- Biswas, A.; De, S. drivers of dynamic intratumor heterogeneity and phenotypic plasticity. Am. J. Physiol. Cell Physiol. 2021, 320, C750–C760. [Google Scholar] [CrossRef]
- Tu, S.M.; Bilen, M.A.; Hess, K.R.; Broaddus, R.R.; Kopetz, S.; Wei, C.; Pagliaro, L.C.; Karam, J.A.; Ward, J.F.; Wood, C.G.; et al. Intratumoral heterogeneity: Role of differentiation in a potentially lethal phenotype of testicular cancer. Cancer 2016, 122, 1836–1843. [Google Scholar] [CrossRef] [PubMed]
- Bilen, M.A.; Hess, K.R.; Campbell, M.T.; Wang, J.; Broaddus, R.R.; Karam, J.A.; Ward, J.F.; Wood, C.G.; Choi, S.L.; Rao, P.; et al. Intratumoral heterogeneity and chemoresistance in nonseminomatous germ cell tumor of the testis. Oncotarget 2016, 7, 86280. [Google Scholar] [CrossRef] [Green Version]
- Umbreit, E.C.; Siddiqui, B.A.; Hwang, M.J.; Joon, A.Y.; Maity, T.; Westerman, M.E.; Merriman, K.W.; Alhasson, H.; Uthup, J.; Guo, T.; et al. Origin of subsequent malignant neoplasms in patients with history of testicular germ cell tumor. Cancers 2020, 12, 3755. [Google Scholar] [CrossRef] [PubMed]
- McCart Reed, A.E.; Kutasovic, J.; Nones, K.; Saunus, J.M.; Da Silva, L.; Newell, F.; Kazakoff, S.; Melville, L.; Jayanthan, J.; Vargas, A.C.; et al. Mixed ductal-lobular carcinomas: Evidence for progression from ductal to lobular morphology. J. Pathol. 2018, 244, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, F.R.; Matthews, M.J.; Aisner, S.; Campobasso, O.; Elema, J.D.; Gazdar, A.F.; Mackay, B.; Nasiell, M.; Shimosato, Y.; Steele, R.H.; et al. Histopathologic classification of small cell lung cancer. Changing concepts and terminology. Cancer 1988, 62, 973–977. [Google Scholar] [CrossRef]
- Zhao, X.; McCutcheon, J.N.; Kallakury, B.; Chahine, J.J.; Pratt, D.; Raffeld, M.; Chen, Y.; Wang, C.; Giaccone, G. Combined small cell carcinoma of the lung: Is it a single entity? J. Thorac. Oncol. 2017, 13, 237–245. [Google Scholar] [CrossRef] [Green Version]
- Reiter, J.G.; Makohon-Moore, A.P.; Gerold, J.M.; Heyde, A.; Attiyeh, M.C.; Kohutek, Z.A.; Tokheim, C.J.; Brown, A.; DeBlasio, R.M.; Niyazov, J.; et al. Minimal functional driver gene heterogeneity among untreated metastases. Science 2018, 361, 1033–1037. [Google Scholar] [CrossRef] [Green Version]
- Proietti, S.; Cucina, A.; Pensotti, A.; Fuso, A.; Marchese, C.; Nicolini, A.; Bizzarri, M. Tumor reversion and embryo morphogenetic factors. Semin. Cancer Biol. 2020, in press. [Google Scholar] [CrossRef]
- Voss, M.H.; Kuo, F.; Chen, D.; Marker, M.; Patel, P.; Redzematovic, A.; Riaz, N.; Chan, T.A.; Choueiri, T.K.; Hsieh, J.; et al. Integrated biomarker analysis for 412 renal cell cancer patients treated on the phase 3 COMPARZ trial: Correlating common mutation events in PBRM1 and BAP1 with angiogenesis expression signatures and outcomes on tyrosine kinase inhibitor therapy. J. Clin. Oncol. 2017, 35, 4523. [Google Scholar] [CrossRef]
- Miao, D.; Margolis, C.A.; Gao, W.; Voss, M.H.; Li, W.; Martini, D.J.; Norton, C.; Bosse, D.; Wankowicz, S.M.; Cullen, D.; et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 2018, 359, 801–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, D.A.; Hou, Y.; Bakouny, Z.; Ficial, M.; Sant’Angelo, M.; Forman, J.; Ross-Macdonald, P.; Berger, A.C.; Jegede, O.A.; Elagina, L.; et al. Interplay of somatic alterations and immune infiltration modulates response to PD-1 blockade in advanced clear cell renal cell carcinoma. Nat. Med. 2020, 26, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Tollervey, J.R.; Lunyak, V.V. Epigenetics: Judge, jury, and executioner of stem cell fate. Epigenetics 2012, 7, 823–840. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopalan, A.; Leversha, M.A.; Satagopan, J.M.; Zhou, Q.; Al-Ahmadie, H.A.; Fine, S.W.; Eastham, J.A.; Scardino, P.T.; Scher, H.I.; Tickoo, S.K.; et al. TMPRSS2-ERG gene fusion is not associated with outcome in patients treated by prostatectomy. Cancer Res. 2009, 69, 1400–1406. [Google Scholar] [CrossRef] [Green Version]
- Pettersson, A.; Graff, R.E.; Bauer, S.R.; Pitt, M.J.; Lis, R.T.; Stack, E.C.; Martin, N.E.; Kunz, L.; Penney, K.L.; Ligon, A.H.; et al. The TMPRSS2-ERG rearrangement, ERG expression, and prostate cancer outcome: A cohort study and meta-analysis. Cancer Epidemiol. Biomark. Prev. 2012, 21, 1497–1509. [Google Scholar] [CrossRef] [Green Version]
- Van Dessel, L.F.; van Riet, J.; Smits, M.; Zhu, Y.; Hamberg, P.; van der Heijden, M.S.; Bergman, A.M.; van Oort, I.M.; de Wit, R.; Voest, E.E.; et al. The genomic landscape of metastatic castration-resistant prostate cancers reveals multiple distinct genotypes with potential clinical impact. Nat. Commun. 2019, 10, 5251. [Google Scholar] [CrossRef] [Green Version]
- Andre, T.; Shiu, K.K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in microsatellite-instability-high advanced colorectal cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef]
- Mateo, J.; Porta, N.; Bianchini, D.; McGovern, U.; Elliott, T.; Jones, R.; Syndikus, I.; Ralph, C.; Jain, S.; Varughese, M.; et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): A multicenter, open-label, randomized, phase 2 trial. Lancet Oncol. 2020, 21, 162–174. [Google Scholar] [CrossRef]
- Smith, B.A.; Sokolov, A.; Uzunangelov, V.; Baertsch, R.; Newton, Y.; Graim, K.; Mathis, C.; Cheng, D.; Stuart, J.M.; Witte, O.N. A basal stem cell signature identifies aggressive prostate cancer phenotypes. Proc. Natl. Acad. Sci. USA 2015, 112, E6544–E6552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, B.A.; Balanis, N.G.; Nanjundiah, A.; Sheu, K.M.; Tsai, B.L.; Zhang, Q.; Park, J.W.; Thompson, M.; Huang, J.; Witte, O.N.; et al. A human adult stem cell signature marks aggressive variants across epithelial cancers. Cell Rep. 2018, 24, 3353–3366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labrecque, M.P.; Coleman, I.M.; Brown, L.G.; True, L.D.; Kollath, L.; Lakely, B.; Nguyen, H.M.; Yang, Y.C.; da Costa, R.M.G.; Kaipainen, A.; et al. Molecular profiling stratifies diverse phenotypes of treatment refractory metastatic castration-resistant prostate cancer. J. Clin. Investig. 2019, 130, 4492–4505. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, R.; Huang, J.; Alumkal, J.J.; Zhang, L.; Feng, F.Y.; Thomas, G.V.; Weinstein, A.S.; Friedl, V.; Zhang, C.; Witte, O.N.; et al. Clinical and genomic characterization of treatment-emergent small-cell neuroendocrine prostate cancer: A multi-institutional prospective study. J. Clin. Oncol. 2018, 36, 2492–2503. [Google Scholar] [CrossRef] [PubMed]
- Alumkal, J.J.; Sun, D.; Lu, E.; Beer, T.M.; Thomas, G.V.; Latour, E.; Aggarwal, R.; Cetnar, J.; Ryan, C.J.; Tabatabaei, S.; et al. Transcriptional profiling identifies an androgen receptor activity-low, stemness program associated with enzalutamide resistance. Proc. Natl. Acad. Sci. USA 2020, 117, 12315–12323. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.M.; Campbell, M.; Shah, A.; Logothetis, C.J. Application of a successful germ cell tumor paradigm to the challenges of common adult solid cancers. J. Cell Sci. Ther. 2021, 12, 301. [Google Scholar]
- Brannon, A.R.; Reddy, A.; Seiler, M.; Arreola, A.; Moore, D.T.; Pruthi, R.S.; Wallen, E.M.; Nielsen, M.E.; Liu, H.; Nathanson, K.L.; et al. Molecular stratification of clear cell carcinoma by consensus clustering reveals distinct subtypes and survival patterns. Genes Cancer 2010, 1, 152–163. [Google Scholar] [CrossRef]
- Dahinden, C.; Ingold, B.; Wild, P.; Boysen, G.; Luu, V.D.; Montani, M.; Kristiansen, G.; Sulser, T.; Buhlmann, P.; Moch, H.; et al. Mining tissue microarray data to uncover combinations of biomarker expression patterns that improve intermediate staging and grading of clear cell renal cell cancer. Clin. Cancer Res. 2010, 16, 88–98. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Ma, Z.; Tibshirani, R.; Higgins, J.P.T.; Ljungberg, B.; Brooks, J.D. Alteration of gene expression signatures of cortical differentiation and wound response in lethal clear cell renal cell carcinomas. PLoS ONE 2009, 4, e6039. [Google Scholar] [CrossRef]
- Turajlic, S.; Xu, H.; Litchfield, K.; Rowan, A.; Horswell, S.; Chambers, T.; O’brien, T.; Lopez, J.I.; Watkins, T.B.K.; Nicol, D.; et al. Deterministic evolutionary trajectories influence primary tumor growth: TRACERx Renal. Cell 2018, 173, 595–610. [Google Scholar] [CrossRef] [Green Version]
- Turajlic, S.; Xu, H.; Litchfield, K.; Rowan, A.; Chambers, T.; Lopez, J.I.; Nicol, D.; O’Brien, T.; Larkin, J.; Horswell, S.; et al. Tracking cancer evolution reveals constrained routes to metastases: TRACERx Renal. Cell 2018, 173, 581–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogelstein, B.; Fearon, E.R.; Hamilton, S.R.; Kern, S.E.; Preisinger, A.C.; Leppert, M.; Nakamura, Y.; White, R.; Smits, A.M.; Bos, J.L. Genetic alterations during colorectal-tumor development. N. Engl. J. Med. 1988, 319, 525–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solter, D. From teratocarcinomas to embryonic stem cells and beyond: A history of embryonic stem cell (ESC) research. Nat. Rev. Genet. 2006, 7, 319–327. [Google Scholar] [CrossRef]
- Andrews, P.W.; Martin, M.M.; Bahrami, A.R.; Damjanov, I. Embryonic stem cells and embryonal carcinoma cells: Opposite sides of the same coin. Biochem. Soc. Trans. 2005, 33, 1526–1530. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Knoepfler, P.S. Deconstructing stem cell tumorigenicity: A roadmap to safe regenerative medicine. Stem Cells 2009, 27, 1050–1056. [Google Scholar] [CrossRef] [Green Version]
- Riggs, J.W.; Barrilleaux, B.L.; Varlahhanova, N.; Bush, K.M.; Chan, V.; Knoepfler, P.S. Induced pluripotency and oncogenic transformation are related processes. Stem Cells Dev. 2013, 22, 37–50. [Google Scholar] [CrossRef]
- Abad, M.; Mosteiro, L.; Pantoja, C.; Canamero, M.; Rayon, T.; Ors, I.; Grana, O.; Megias, D.; Dominguez, O.; Martinez, D.; et al. Reprogramming in vivo produces teratomas and iPS cells with totipotency features. Nature 2013, 502, 340–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohnishi, K.; Semi, K.; Yamamoto, T.; Shimizu, M.; Tanaka, A.; Mitsunaga, K.; Okita, K.; Osafune, K.; Arioka, Y.; Maeda, T.; et al. Premature termination of reprogramming in vivo leads to cancer development through altered epigenetic regulation. Cell 2014, 156, 663–677. [Google Scholar] [CrossRef] [Green Version]
- Nori, S.; Okada, Y.; Nishimura, S.; Sasaki, T.; Itakura, G.; Kobayashi, Y.; Renault-Mihara, F.; Shimizu, A.; Koya, I.; Yoshida, R.; et al. Long-term safety issues of IPSC-based cell therapy in a spinal cord injury model: Oncogenic transformation with epithelial-mesenchymal transition. Stem-Cell Rep. 2015, 4, 360–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sell, S.; Pierce, G.B. Maturation arrest of stem cell differentiation is a common pathway for the cellular origin of teratocarcinomas and epithelial cancers. Lab. Investig. 1994, 70, 6–22. [Google Scholar] [PubMed]
- Brastianos, P.K.; Carter, S.L.; Santagata, S.; Cahill, D.P.; Taylor-Weiner, A.; Jones, R.T.; Van Allen, E.M.; Lawrence, M.S.; Horowitz, P.M.; Cibulskis, K.; et al. Genomic characterization of brain metastases reveals branched evolution and potential therapeutic targets. Cancer Discov. 2015, 5, 1164–1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tu, S.-M.; Zhang, M.; Wood, C.G.; Pisters, L.L. Stem Cell Theory of Cancer: Origin of Tumor Heterogeneity and Plasticity. Cancers 2021, 13, 4006. https://doi.org/10.3390/cancers13164006
Tu S-M, Zhang M, Wood CG, Pisters LL. Stem Cell Theory of Cancer: Origin of Tumor Heterogeneity and Plasticity. Cancers. 2021; 13(16):4006. https://doi.org/10.3390/cancers13164006
Chicago/Turabian StyleTu, Shi-Ming, Miao Zhang, Christopher G. Wood, and Louis L. Pisters. 2021. "Stem Cell Theory of Cancer: Origin of Tumor Heterogeneity and Plasticity" Cancers 13, no. 16: 4006. https://doi.org/10.3390/cancers13164006
APA StyleTu, S.-M., Zhang, M., Wood, C. G., & Pisters, L. L. (2021). Stem Cell Theory of Cancer: Origin of Tumor Heterogeneity and Plasticity. Cancers, 13(16), 4006. https://doi.org/10.3390/cancers13164006