
Mass Spectrometry-Based Proteomic Discovery of Prognostic Biomarkers in Adrenal Cortical Carcinoma

 , , , and
, , , and
Abstract
:Simple Summary
Abstract
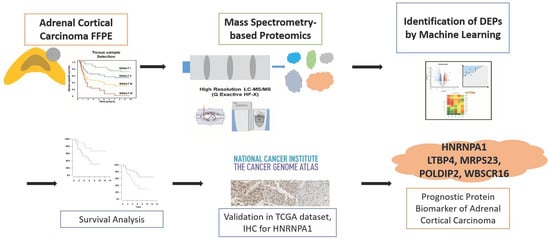
1. Introduction
2. Materials and Methods
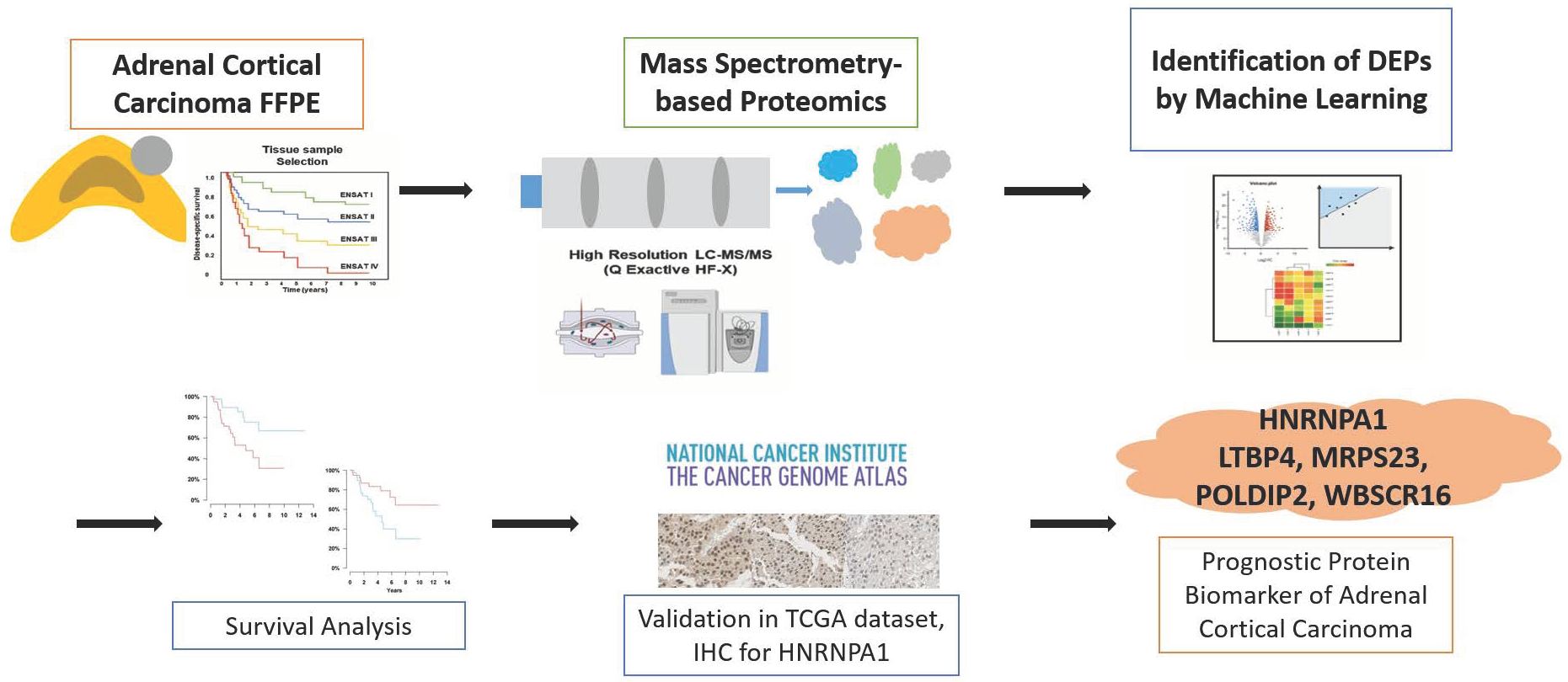
2.1. Study Design
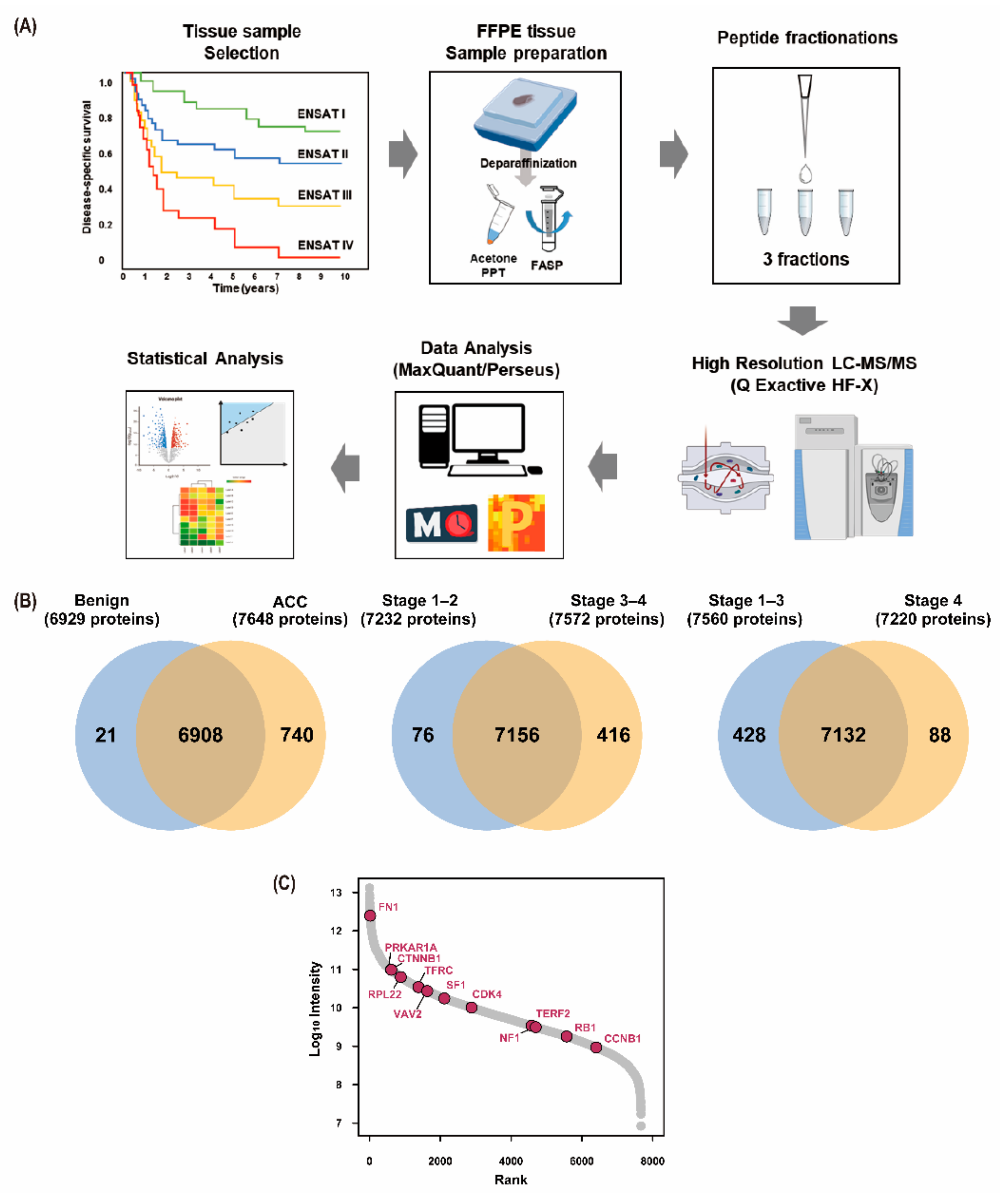
2.2. Sample Preparation for Proteomics Analysis
2.3. LC–MS/MS and MS Data Analysis
2.4. Label-Free Quantification and Bioinformatics Analysis
2.5. Immunohistochemistry Staining
2.6. Statistical Analysis
2.7. Ethical Statement
3. Results
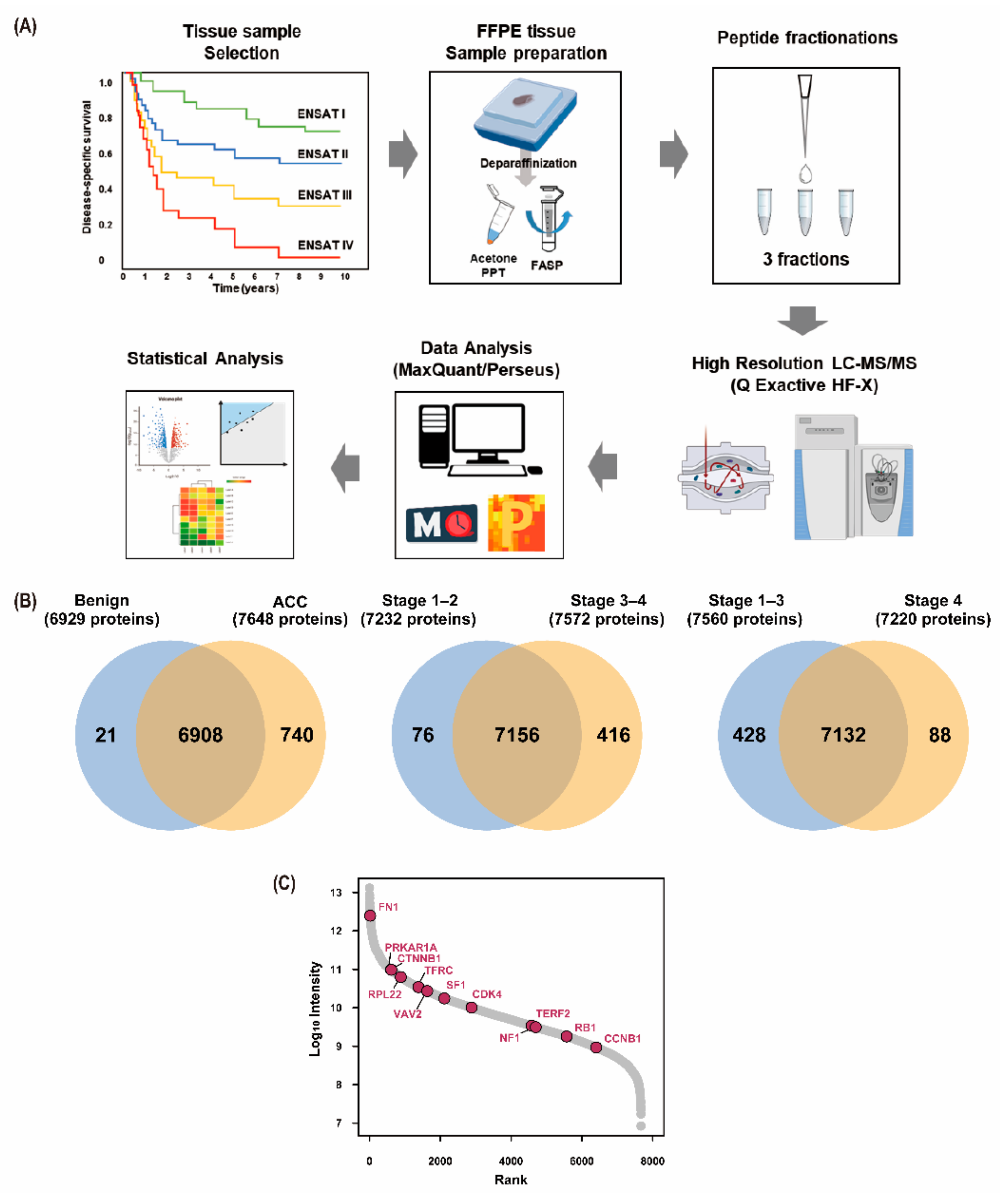
3.1. Baseline Characteristics of the Study Participants
3.2. Results of Proteomic Analysis
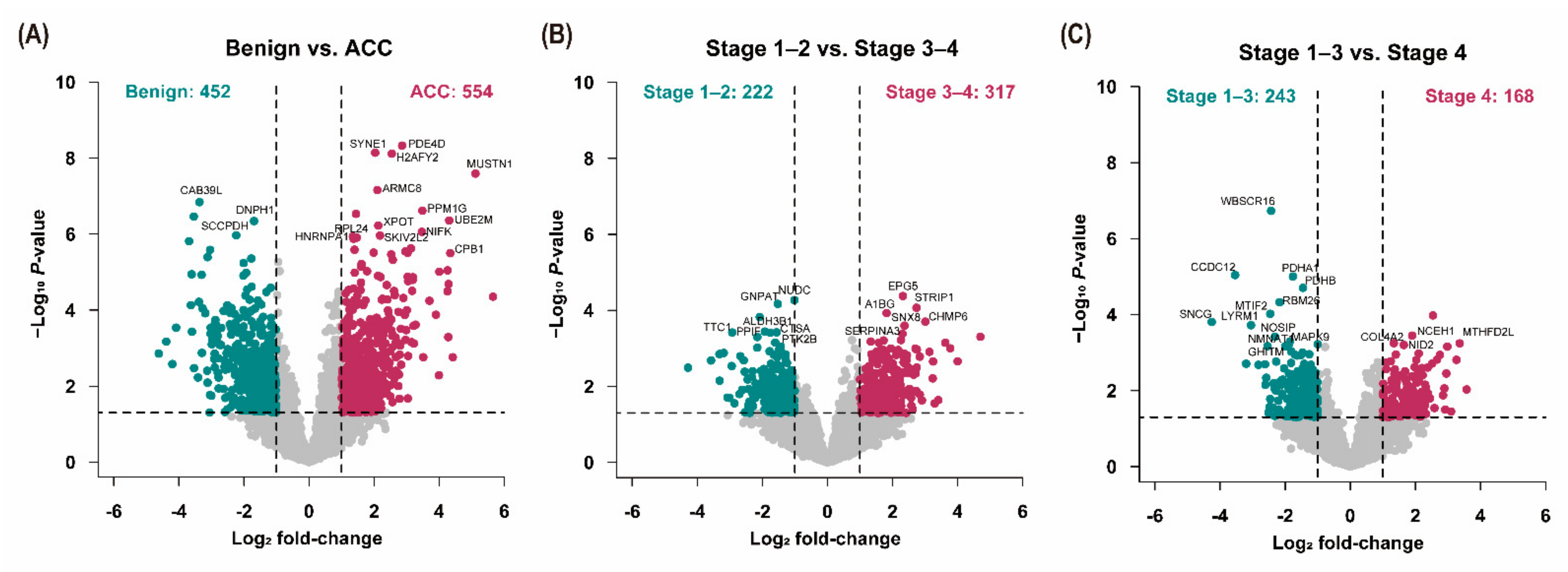
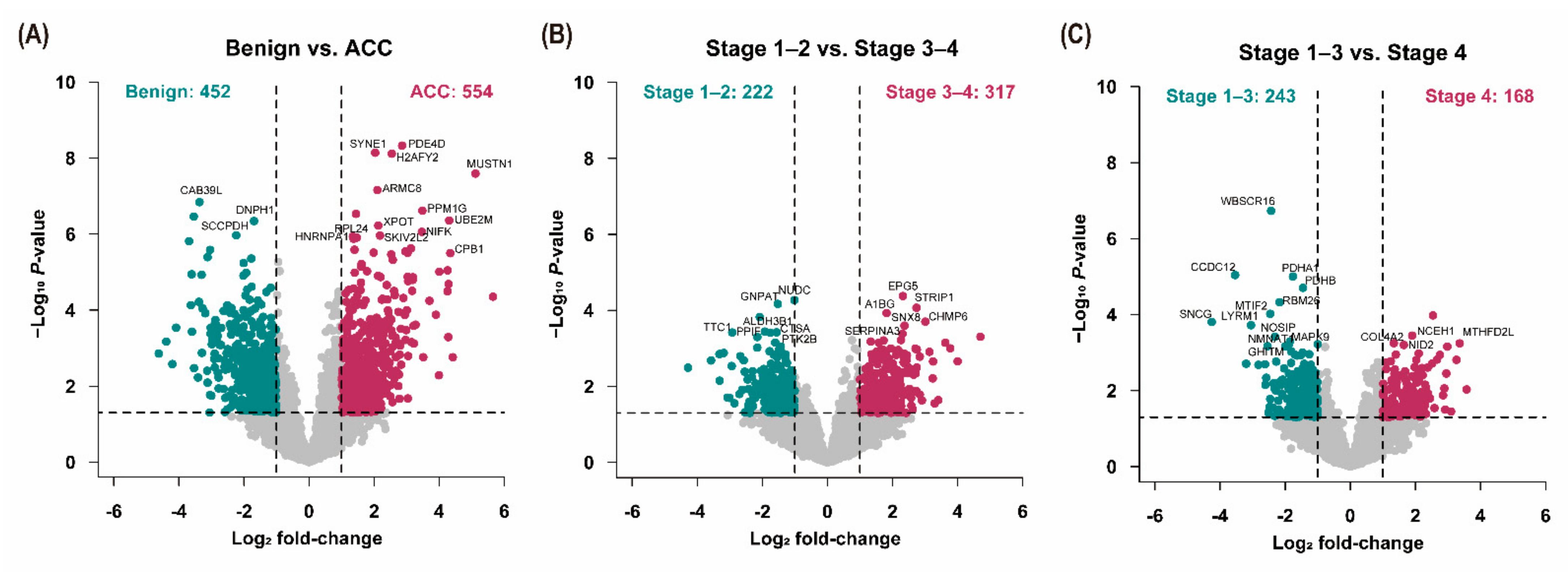
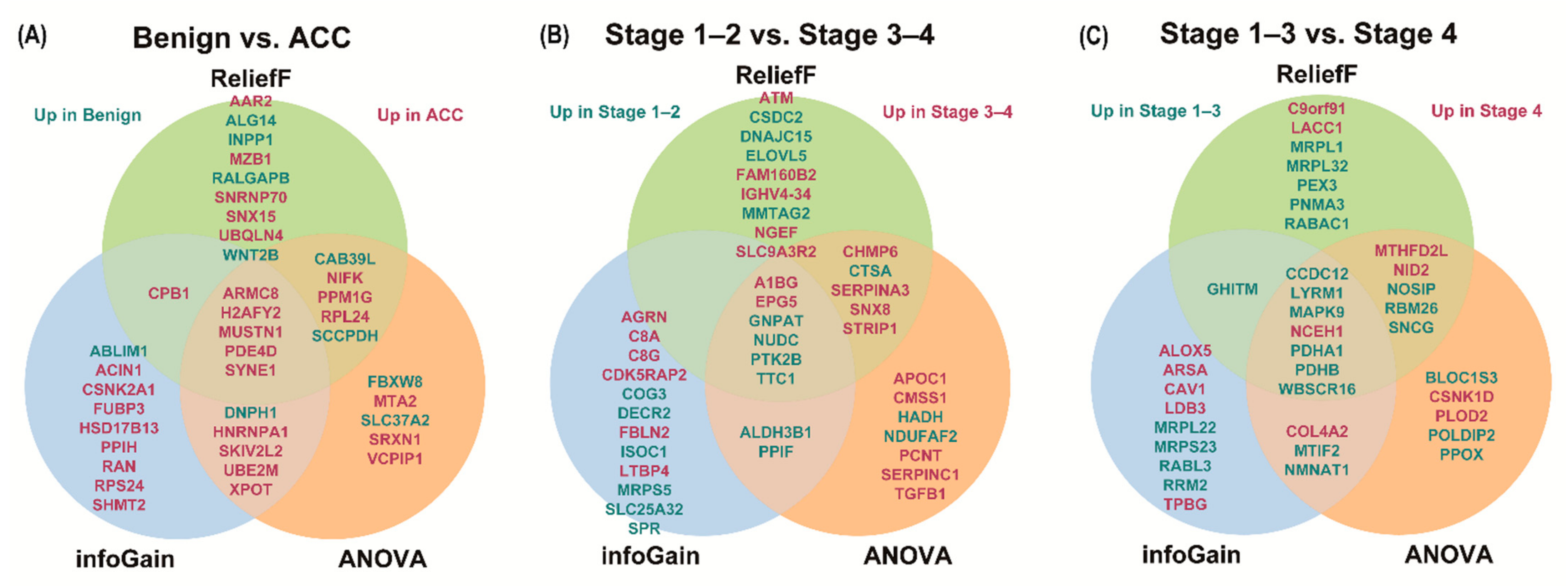
3.3. Analysis of Differentially Expressed Proteins
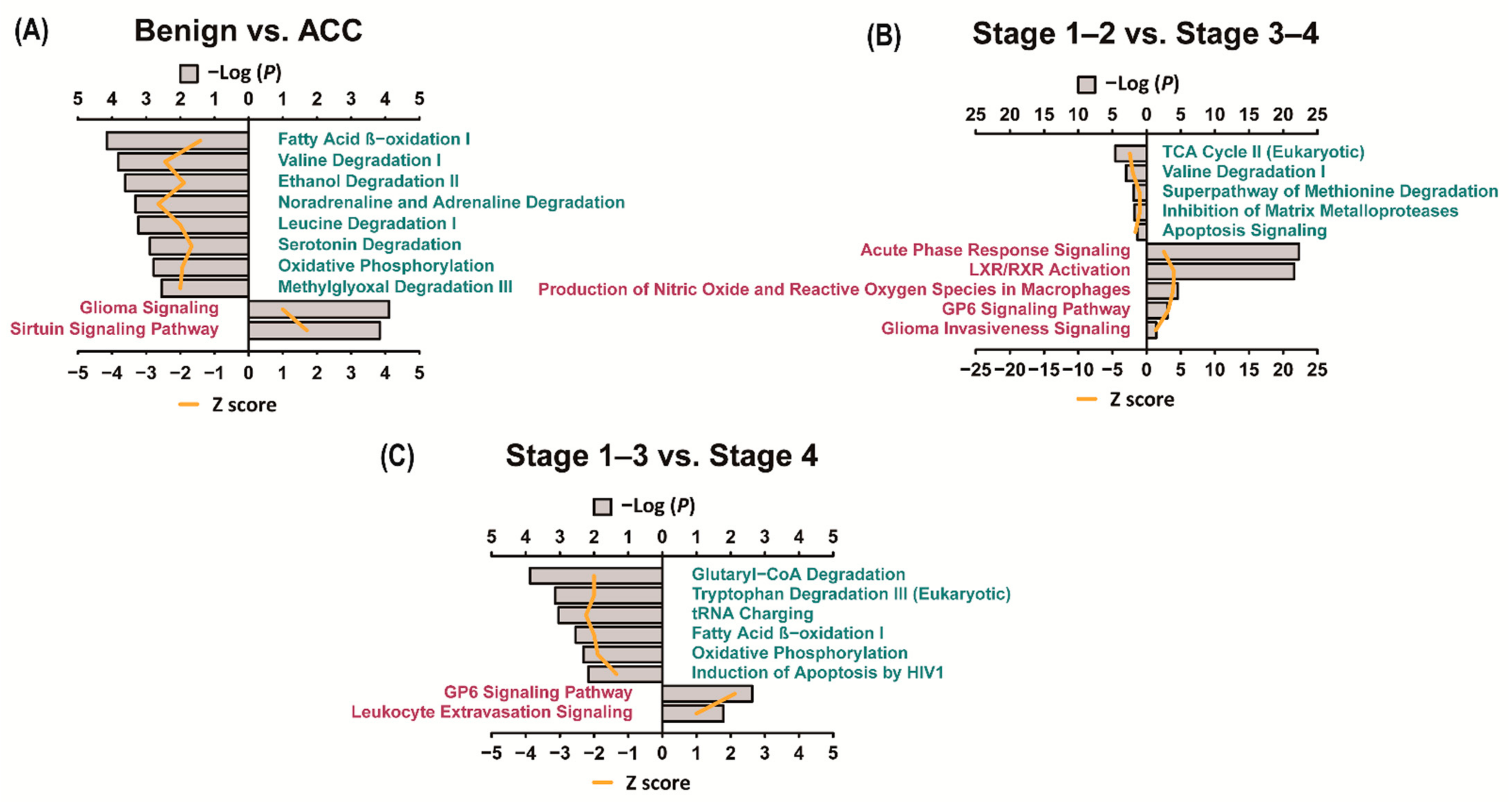
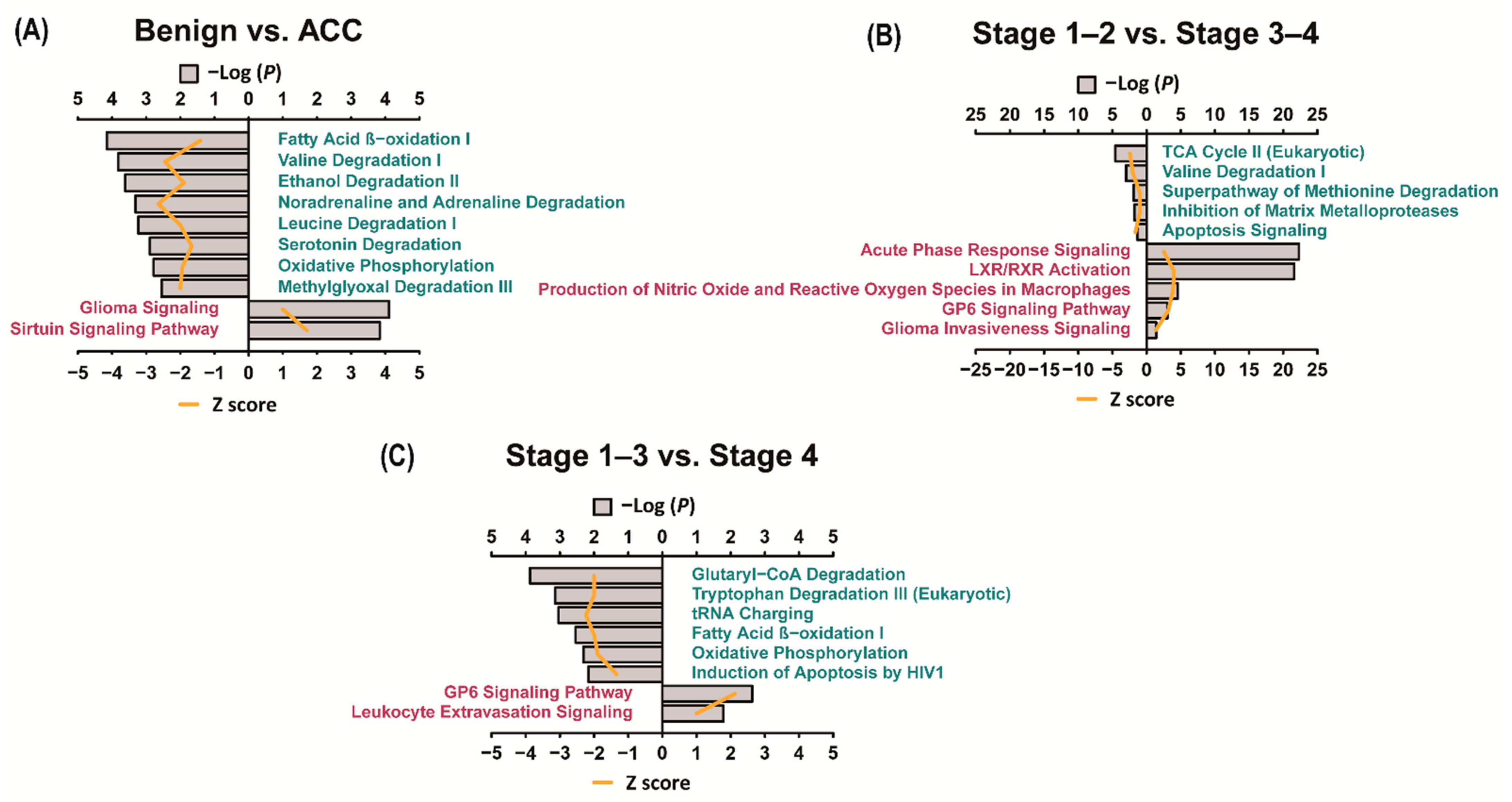
3.4. Ingenuity Pathway Analysis (IPA)
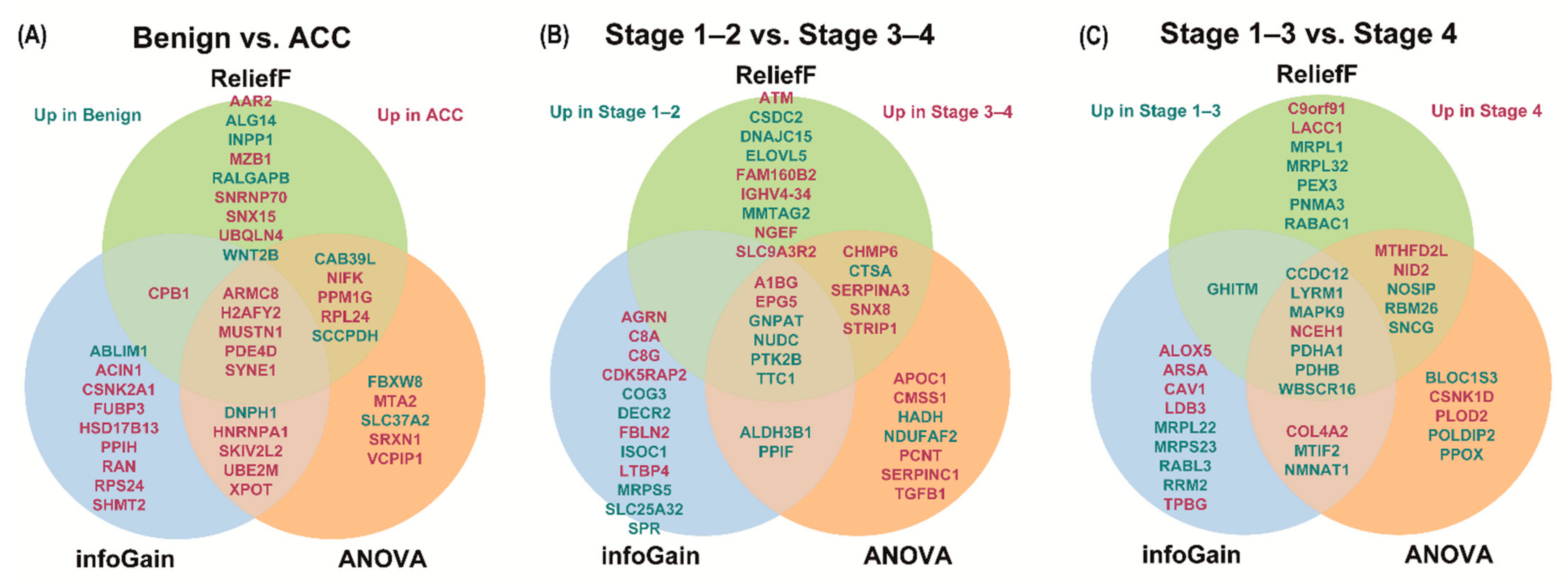
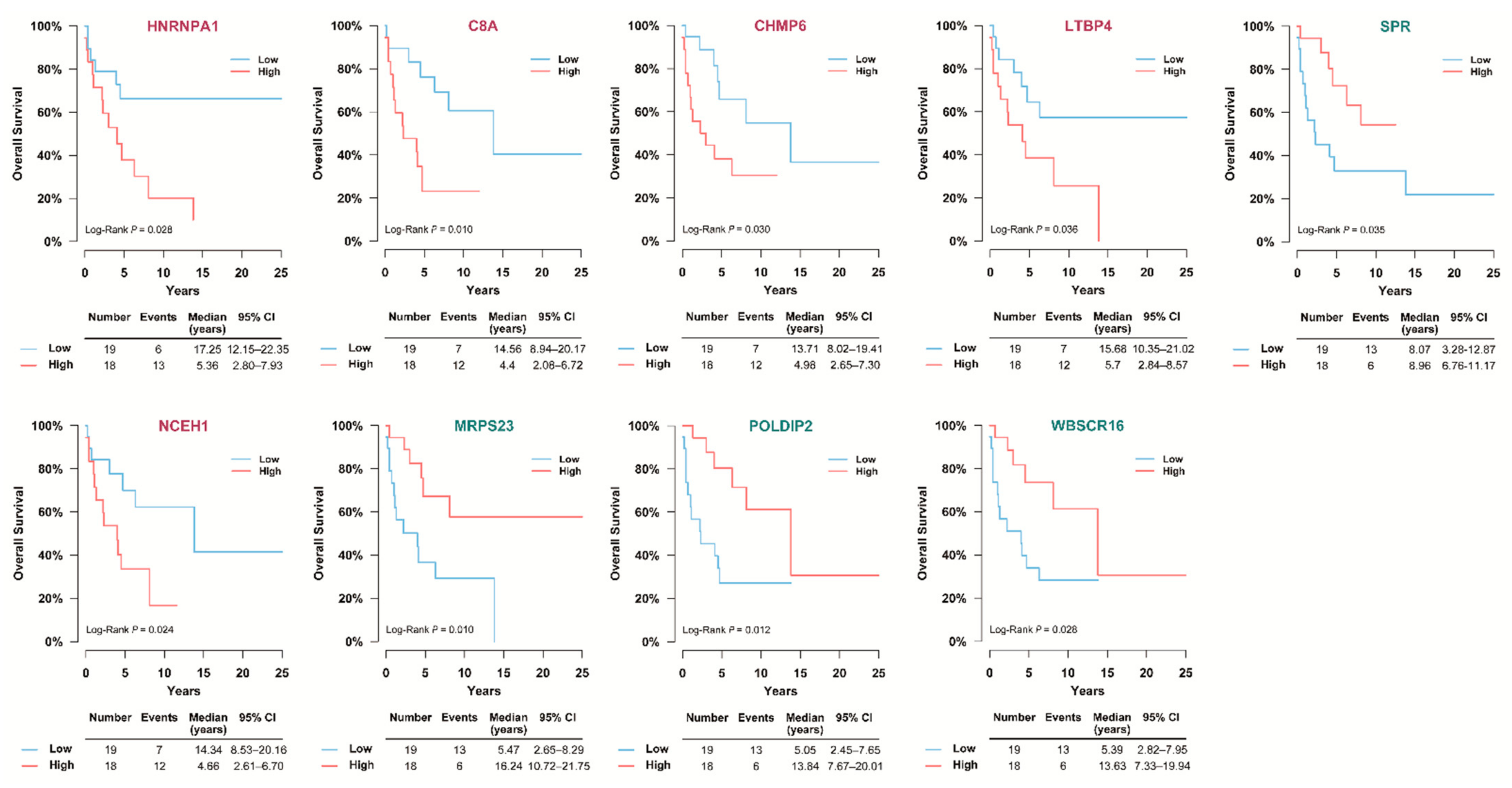
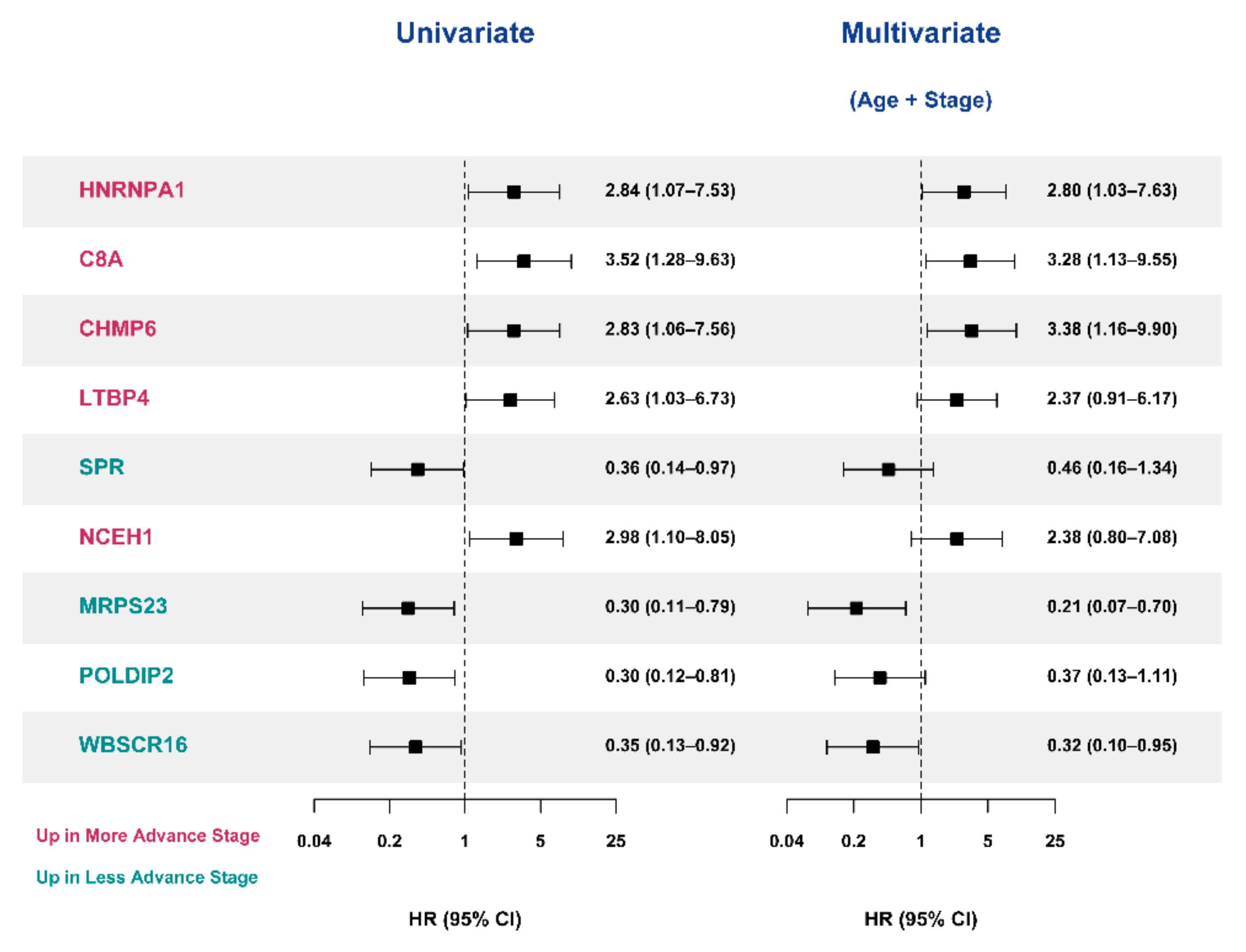
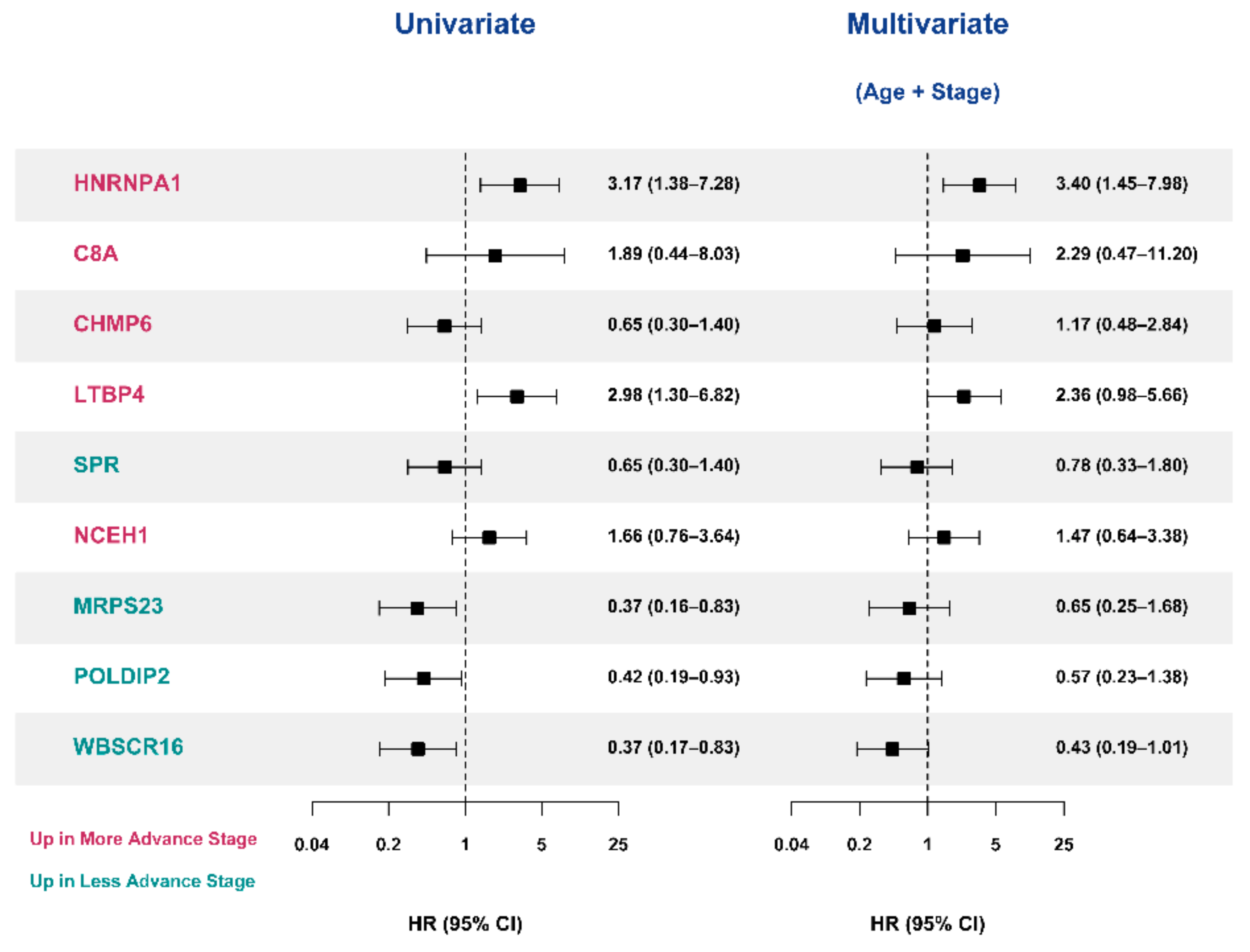
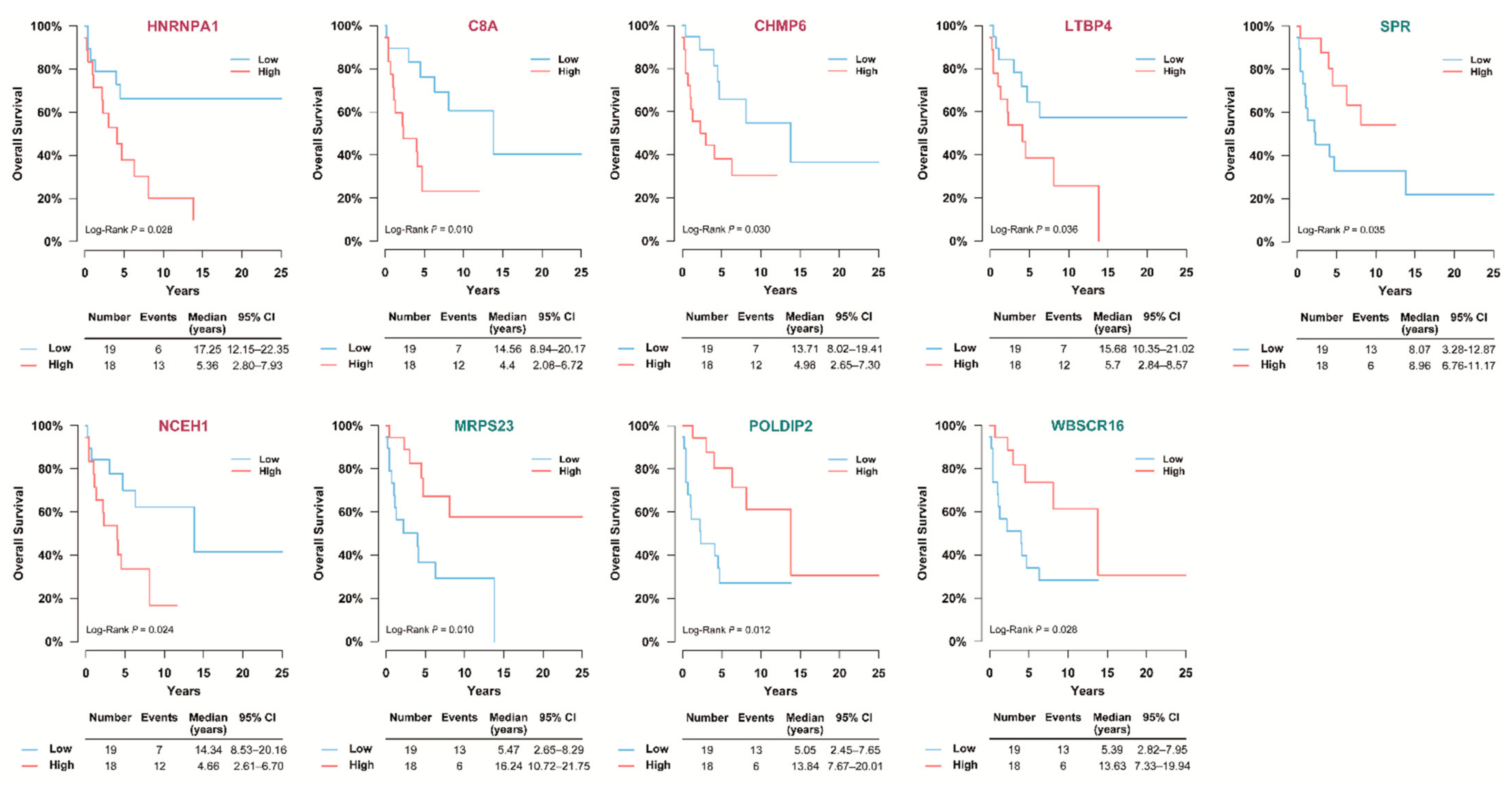
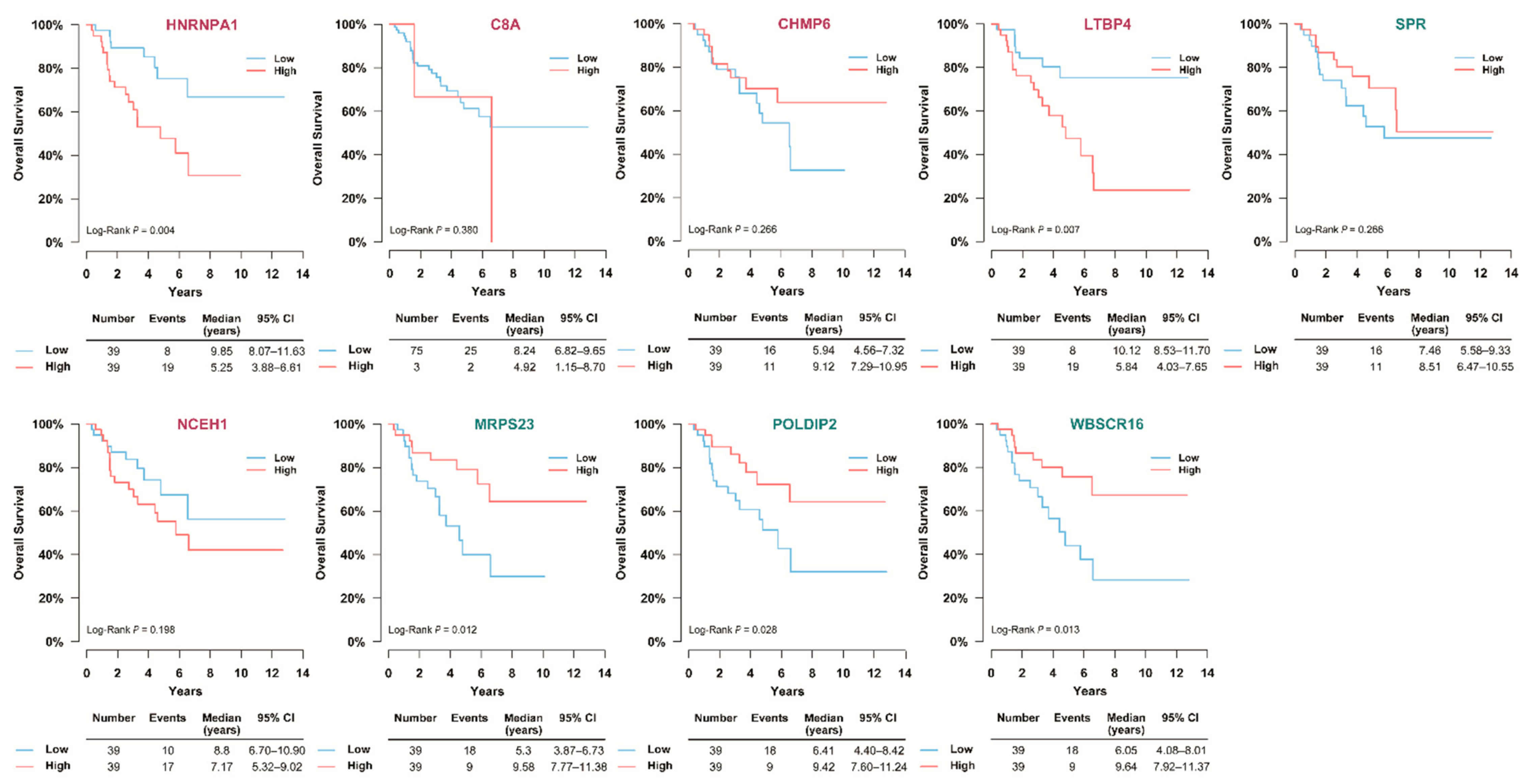
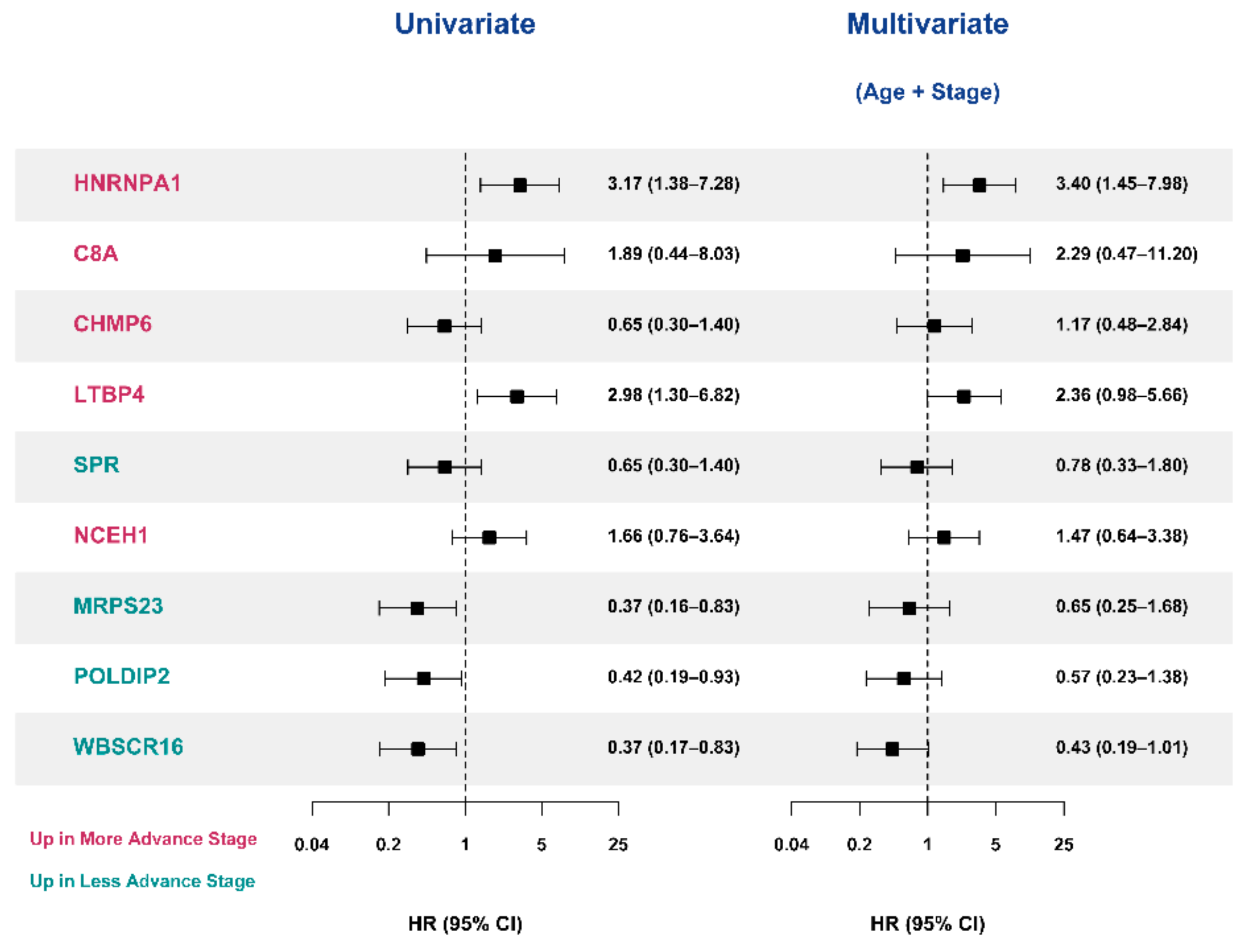
3.5. Selection of Prognostic Protein Biomarkers in the Seoul National University Hospital (SNUH) Cohort
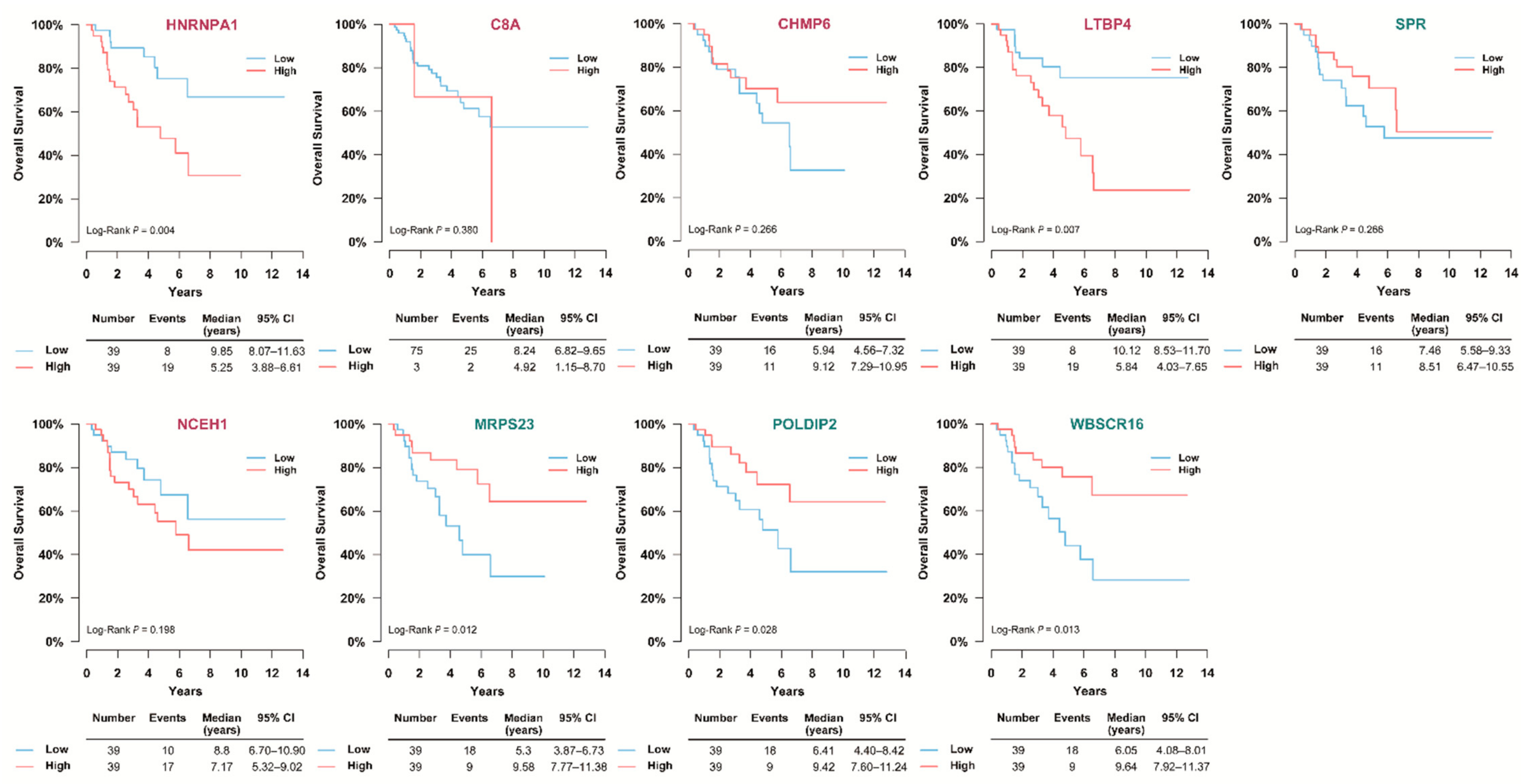
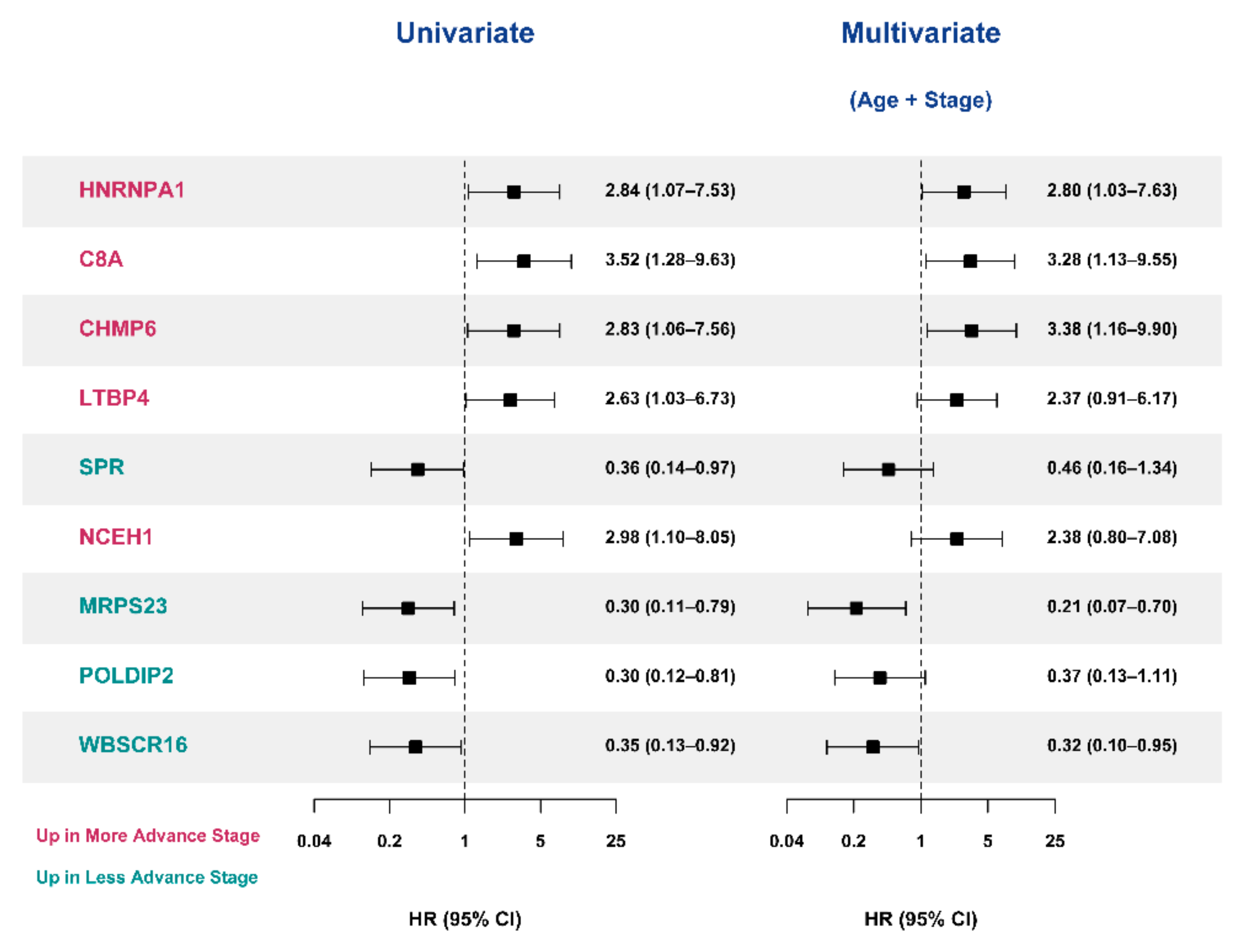
3.6. Validation of the Prognostic Value of Candidate Protein Biomarkers in the TCGA Cohort
3.7. Validation of the Prognostic Candidate Protein Biomarker by Immunohistochemistry Staining
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kebebew, E.; Reiff, E.; Duh, Q.Y.; Clark, O.H.; McMillan, A. Extent of disease at presentation and outcome for adrenocortical carcinoma: Have we made progress? World J. Surg. 2006, 30, 872–878. [Google Scholar] [CrossRef] [PubMed]
- Kerkhofs, T.M.; Verhoeven, R.H.; Van der Zwan, J.M.; Dieleman, J.; Kerstens, M.N.; Links, T.P.; Van de Poll-Franse, L.V.; Haak, H.R. Adrenocortical carcinoma: A population-based study on incidence and survival in the Netherlands since 1993. Eur. J. Cancer 2013, 49, 2579–2586. [Google Scholar] [CrossRef] [PubMed]
- Jouinot, A.; Bertherat, J. Adrenocortical carcinoma: Differentiating the good from the poor prognosis tumors. Eur. J. Endocrinol. 2018, 178, R215–R230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fassnacht, M.; Johanssen, S.; Quinkler, M.; Bucsky, P.; Willenberg, H.S.; Beuschlein, F.; Terzolo, M.; Mueller, H.-H.; Hahner, S.; Allolio, B.; et al. Limited prognostic value of the 2004 International Union Against Cancer staging classification for adrenocortical carcinoma: Proposal for a Revised TNM Classification. Cancer 2009, 115, 243–250. [Google Scholar] [CrossRef]
- Beuschlein, F.; Weigel, J.; Saeger, W.; Kroiss, M.; Wild, V.; Daffara, F.; Libe, R.; Ardito, A.; Al Ghuzlan, A.; Quinkler, M.; et al. Major prognostic role of ki67 in localized adrenocortical carcinoma after complete resection. J. Clin. Endocrinol. Metab. 2015, 100, 841–849. [Google Scholar] [CrossRef]
- Lim, J.S.; Lee, S.E.; Kim, J.H.; Kim, J.H. Characteristics of adrenocortical carcinoma in South Korea: A registry-based nationwide survey. Endocr. Connect. 2020, 9, 519–529. [Google Scholar] [CrossRef]
- Lughezzani, G.; Sun, M.; Perrotte, P.; Jeldres, C.; Alasker, A.; Isbarn, H.; Budaus, L.; Shariat, S.F.; Guazzoni, G.; Montorsi, F.; et al. The European Network for the Study of Adrenal Tumors staging system is prognostically superior to the international union against cancer-staging system: A North American validation. Eur. J. Cancer 2010, 46, 713–719. [Google Scholar] [CrossRef]
- Else, T.; Kim, A.C.; Sabolch, A.; Raymond, V.M.; Kandathil, A.; Caoili, E.M.; Jolly, S.; Miller, B.S.; Giordano, T.J.; Hammer, G.D. Adrenocortical carcinoma. Endocr. Rev. 2014, 35, 282–326. [Google Scholar] [CrossRef] [Green Version]
- Kjellin, H.; Johansson, H.; Hoog, A.; Lehtio, J.; Jakobsson, P.J.; Kjellman, M. Differentially expressed proteins in malignant and benign adrenocortical tumors. PLoS ONE 2014, 9, e87951. [Google Scholar] [CrossRef]
- Romero Arenas, M.A.; Whitsett, T.G.; Aronova, A.; Henderson, S.A.; LoBello, J.; Habra, M.A.; Grubbs, E.G.; Lee, J.E.; Sircar, K.; Zarnegar, R.; et al. Protein Expression of PTTG1 as a Diagnostic Biomarker in Adrenocortical Carcinoma. Ann. Surg. Oncol. 2018, 25, 801–807. [Google Scholar] [CrossRef]
- Kim, H.M.; Koo, J.S. Expression of Glutamine Metabolism-Related and Amino Acid Transporter Proteins in Adrenal Cortical Neoplasms and Pheochromocytomas. Dis. Markers 2021, 2021, 8850990. [Google Scholar] [CrossRef]
- Moon, S.J.; Kim, J.H.; Kong, S.H.; Shin, C.S. Protein Expression of Cyclin B1, Transferrin Receptor, and Fibronectin Is Correlated with the Prognosis of Adrenal Cortical Carcinoma. Endocrinol. Metab. (Seoul) 2020, 35, 132–141. [Google Scholar] [CrossRef]
- Assie, G.; Letouze, E.; Fassnacht, M.; Jouinot, A.; Luscap, W.; Barreau, O.; Omeiri, H.; Rodriguez, S.; Perlemoine, K.; Rene-Corail, F.; et al. Integrated genomic characterization of adrenocortical carcinoma. Nat. Genet. 2014, 46, 607–612. [Google Scholar] [CrossRef]
- Zheng, S.; Cherniack, A.D.; Dewal, N.; Moffitt, R.A.; Danilova, L.; Murray, B.A.; Lerario, A.M.; Else, T.; Knijnenburg, T.A.; Ciriello, G.; et al. Comprehensive Pan-Genomic Characterization of Adrenocortical Carcinoma. Cancer Cell 2016, 29, 723–736. [Google Scholar] [CrossRef] [Green Version]
- Assie, G.; Jouinot, A.; Fassnacht, M.; Libe, R.; Garinet, S.; Jacob, L.; Hamzaoui, N.; Neou, M.; Sakat, J.; de La Villeon, B.; et al. Value of Molecular Classification for Prognostic Assessment of Adrenocortical Carcinoma. JAMA Oncol. 2019, 5, 1440–1447. [Google Scholar] [CrossRef]
- Li, J.; Lu, Y.; Akbani, R.; Ju, Z.; Roebuck, P.L.; Liu, W.; Yang, J.Y.; Broom, B.M.; Verhaak, R.G.; Kane, D.W.; et al. TCPA: A resource for cancer functional proteomics data. Nat. Methods 2013, 10, 1046–1047. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, T.; Mann, M.; Aebersold, R.; Yates, J.R., 3rd; Bairoch, A.; Bergeron, J.J. Mass spectrometry in high-throughput proteomics: Ready for the big time. Nat. Methods 2010, 7, 681–685. [Google Scholar] [CrossRef]
- Shabihkhani, M.; Lucey, G.M.; Wei, B.; Mareninov, S.; Lou, J.J.; Vinters, H.V.; Singer, E.J.; Cloughesy, T.F.; Yong, W.H. The procurement, storage, and quality assurance of frozen blood and tissue biospecimens in pathology, biorepository, and biobank settings. Clin. Biochem. 2014, 47, 258–266. [Google Scholar] [CrossRef] [Green Version]
- Addis, M.F.; Tanca, A.; Pagnozzi, D.; Crobu, S.; Fanciulli, G.; Cossu-Rocca, P.; Uzzau, S. Generation of high-quality protein extracts from formalin-fixed, paraffin-embedded tissues. Proteomics 2009, 9, 3815–3823. [Google Scholar] [CrossRef]
- Gustafsson, O.J.; Arentz, G.; Hoffmann, P. Proteomic developments in the analysis of formalin-fixed tissue. Biochim. Biophys. Acta 2015, 1854, 559–580. [Google Scholar] [CrossRef]
- Piehowski, P.D.; Petyuk, V.A.; Sontag, R.L.; Gritsenko, M.A.; Weitz, K.K.; Fillmore, T.L.; Moon, J.; Makhlouf, H.; Chuaqui, R.F.; Boja, E.S.; et al. Residual tissue repositories as a resource for population-based cancer proteomic studies. Clin. Proteom. 2018, 15, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Kim, K.; Woo, J.; Park, J.; Kim, H.; Kim, Y.; Lee, K.E.; Moon, K.C.; Kim, J.Y.; Park, I.A.; et al. Quantitative Proteomic Analysis Identifies AHNAK (Neuroblast Differentiation-associated Protein AHNAK) as a Novel Candidate Biomarker for Bladder Urothelial Carcinoma Diagnosis by Liquid-based Cytology. Mol. Cell. Proteom. 2018, 17, 1788–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Lee, C.; Han, D.; Lee, J.S.; Lee, K.M.; Song, M.J.; Kim, K.; Lee, H.; Moon, K.C.; Kim, Y.; et al. Moesin (MSN) as a Novel Proteome-Based Diagnostic Marker for Early Detection of Invasive Bladder Urothelial Carcinoma in Liquid-Based Cytology. Cancers 2020, 12, 1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Kim, H.; Kim, S.Y.; Kim, Y.; Lee, J.-S.; Dan, K.; Seong, M.-W.; Han, D. In-depth blood proteome profiling analysis revealed distinct functional characteristics of plasma proteins between severe and non-severe COVID-19 patients. Sci. Rep. 2020, 10, 22418. [Google Scholar] [CrossRef]
- Rahnev, D. Entrainment of neural activity using transcranial magnetic stimulation. J. Neurosci. 2013, 33, 11325–11326. [Google Scholar] [CrossRef] [Green Version]
- Wichmann, C.; Meier, F.; Virreira Winter, S.; Brunner, A.-D.; Cox, J.; Mann, M. MaxQuant.Live Enables Global Targeting of More Than 25,000 Peptides. Mol. Cell. Proteom. 2019, 18, 982–994. [Google Scholar] [CrossRef] [Green Version]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Llinares, M.B.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- SchwanhAusser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [Green Version]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Urbanowicz, R.J.; Meeker, M.; La Cava, W.; Olson, R.S.; Moore, J.H. Relief-based feature selection: Introduction and review. J. Biomed. Inform. 2018, 85, 189–203. [Google Scholar] [CrossRef]
- Vergara, J.R.; Estévez, P.A. A review of feature selection methods based on mutual information. Neural Comput. Appl. 2013, 24, 175–186. [Google Scholar] [CrossRef]
- Jafari, P.; Azuaje, F. An assessment of recently published gene expression data analyses: Reporting experimental design and statistical factors. BMC Med. Inform. Decis. Mak. 2006, 6, 27. [Google Scholar] [CrossRef] [Green Version]
- Bi, N.; Suen, C.Y.; Nobile, N.; Tan, J. A multi-feature selection approach for gender identification of handwriting based on kernel mutual information. Pattern Recognit. Lett. 2019, 121, 123–132. [Google Scholar] [CrossRef]
- Urbanowicz, R.J.; Olson, R.S.; Schmitt, P.; Meeker, M.; Moore, J.H. Benchmarking relief-based feature selection methods for bioinformatics data mining. J. Biomed. Inform. 2018, 85, 168–188. [Google Scholar] [CrossRef]
- Giordano, T.J.; Thomas, D.G.; Kuick, R.; Lizyness, M.; Misek, D.E.; Smith, A.L.; Sanders, D.; Aljundi, R.T.; Gauger, P.G.; Thompson, N.W.; et al. Distinct Transcriptional Profiles of Adrenocortical Tumors Uncovered by DNA Microarray Analysis. Am. J. Pathol. 2003, 162, 521–531. [Google Scholar] [CrossRef] [Green Version]
- Giordano, T.J.; Kuick, R.; Else, T.; Gauger, P.G.; Vinco, M.; Bauersfeld, J.; Sanders, D.; Thomas, D.G.; Doherty, G.; Hammer, G. Molecular classification and prognostication of adrenocortical tumors by transcriptome profiling. Clin. Cancer Res. 2009, 15, 668–676. [Google Scholar] [CrossRef] [Green Version]
- Ragazzon, B.; Libe, R.; Gaujoux, S.; Assie, G.; Fratticci, A.; Launay, P.; Clauser, E.; Bertagna, X.; Tissier, F.; de Reynies, A.; et al. Transcriptome analysis reveals that p53 and β-catenin alterations occur in a group of aggressive adrenocortical Cancers. Cancer Res. 2010, 70, 8276–8281. [Google Scholar] [CrossRef] [Green Version]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227–232. [Google Scholar] [CrossRef]
- Yang, M.-S.; Wang, H.-S.; Wang, B.-S.; Li, W.-H.; Pang, Z.-F.; Zou, B.-K.; Zhang, X.; Shi, X.-T.; Mu, D.-B.; Zhang, D.-X.; et al. A comparative proteomic study identified calreticulin and prohibitin up-regulated in adrenocortical carcinomas. Diagn. Pathol. 2013, 8, 58. [Google Scholar] [CrossRef] [Green Version]
- Poli, G.; Ceni, E.; Armignacco, R.; Ercolino, T.; Canu, L.; Baroni, G.; Nesi, G.; Galli, A.; Mannelli, M.; Luconi, M. 2D-DIGE proteomic analysis identifies new potential therapeutic targets for adrenocortical carcinoma. Oncotarget 2015, 6, 5695–5706. [Google Scholar] [CrossRef] [Green Version]
- Roy, R.; Huang, Y.; Seckl, M.J.; Pardo, O.E. Emerging roles of hnRNPA1 in modulating malignant transformation. Wiley Interdiscip. Rev. RNA 2017, 8, e1431. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.-J.; Dai, Z.; Zhou, S.-L.; Fu, X.-T.; Zhao, Y.-M.; Shi, Y.-H.; Zhou, J.; Fan, J. Overexpression of HnRNP A1 promotes tumor invasion through regulating CD44v6 and indicates poor prognosis for hepatocellular carcinoma. Int. J. Cancer 2013, 132, 1080–1089. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, J.; Wang, W.; Xiang, L.; Wang, J.; Liu, S.; Zhou, H.; Guo, Z. High expression of hnRNPA1 promotes cell invasion by inducing EMT in gastric cancer. Oncol. Rep. 2018, 39, 1693–1701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talukdar, I.; Sen, S.; Urbano, R.; Thompson, J.; Yates, J.R.; Webster, N.J.G. hnRNP A1 and hnRNP F modulate the alternative splicing of exon 11 of the insulin receptor gene. PLoS ONE 2011, 6, e27869. [Google Scholar] [CrossRef] [Green Version]
- Mizdrak, M.; Kurir, T.T.; Bozic, J. The Role of Biomarkers in Adrenocortical Carcinoma: A Review of Current Evidence and Future Perspectives. Biomedicines 2021, 9, 174. [Google Scholar] [CrossRef]
- Koli, K.; Saharinen, J.; Hyytiäinen, M.; Penttinen, C.; Keski-Oja, J. Latency, activation, and binding proteins of TGF-beta. Microsc. Res. Tech. 2001, 52, 354–362. [Google Scholar] [CrossRef]
- Yeh, H.-W.; Lee, S.-S.; Chang, C.-Y.; Lang, Y.-D.; Jou, Y.-S. A New Switch for TGFβ in Cancer. Cancer Res. 2019, 79, 3797–3805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sterner-Kock, A.; Thorey, I.S.; Koli, K.; Wempe, F.; Otte, J.; Bangsow, T.; Kuhlmeier, K.; Kirchner, T.; Jin, S.; Keski-Oja, J.; et al. Disruption of the gene encoding the latent transforming growth factor-beta binding protein 4 (LTBP-4) causes abnormal lung development, cardiomyopathy, and colorectal cancer. Genes Dev. 2002, 16, 2264–2273. [Google Scholar] [CrossRef] [Green Version]
- Bultmann, I.; Conradi, A.; Kretschmer, C.; Sterner-Kock, A. Latent transforming growth factor β-binding protein 4 is downregulated in esophageal cancer via promoter methylation. PLoS ONE 2013, 8, e65614. [Google Scholar] [CrossRef] [Green Version]
- Kretschmer, C.; Conradi, A.; Kemmner, W.; Sterner-Kock, A. Latent transforming growth factor binding protein 4 (LTBP4) is downregulated in mouse and human DCIS and mammary carcinomas. Cell. Oncol. 2011, 34, 419–434. [Google Scholar] [CrossRef] [Green Version]
- Pu, M.; Wang, J.; Huang, Q.; Zhao, G.; Xia, C.; Shang, R.; Zhang, Z.; Bian, Z.; Yang, X.; Tao, K. High MRPS23 expression contributes to hepatocellular carcinoma proliferation and indicates poor survival outcomes. Tumor Biol. 2017, 39. [Google Scholar] [CrossRef] [Green Version]
- Lyng, H.; Brovig, R.S.; Svendsrud, D.H.; Holm, R.; Kaalhus, O.; Knutstad, K.; Oksefjell, H.; Sundfor, K.; Kristensen, G.B.; Stokke, T. Gene expressions and copy numbers associated with metastatic phenotypes of uterine cervical cancer. BMC Genom. 2006, 7, 268. [Google Scholar] [CrossRef] [Green Version]
- Gatza, M.L.; Silva, G.O.; Parker, J.S.; Fan, C.; Perou, C.M. An integrated genomics approach identifies drivers of proliferation in luminal-subtype human breast cancer. Nat. Genet. 2014, 46, 1051–1059. [Google Scholar] [CrossRef] [Green Version]
- Tissier, A.; Janel-Bintz, R.; Coulon, S.; Klaile, E.; Kannouche, P.; Fuchs, R.P.; Cordonnier, A.M. Crosstalk between replicative and translesional DNA polymerases: PDIP38 interacts directly with Poleta. DNA Repair 2010, 9, 922–928. [Google Scholar] [CrossRef]
- Klaile, E.; Kukalev, A.; Obrink, B.; Muller, M.M. PDIP38 is a novel mitotic spindle-associated protein that affects spindle organization and chromosome segregation. Cell Cycle 2008, 7, 3180–3186. [Google Scholar] [CrossRef]
- Cheng, X.; Kanki, T.; Fukuoh, A.; Ohgaki, K.; Takeya, R.; Aoki, Y.; Hamasaki, N.; Kang, D. PDIP38 associates with proteins constituting the mitochondrial DNA nucleoid. J. Biochem. 2005, 138, 673–678. [Google Scholar] [CrossRef]
- Brown, D.I.; Lassegue, B.; Lee, M.; Zafari, R.; Long, J.S.; Saavedra, H.I.; Griendling, K.K. Poldip2 knockout results in perinatal lethality, reduced cellular growth and increased autophagy of mouse embryonic fibroblasts. PLoS ONE 2014, 9, e96657. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.C.; Kuo, C.C.; Chian, C.F.; Tzao, C.; Chang, S.Y.; Shih, Y.L.; Lin, Y.W.; Yu, M.H.; Su, H.Y. Knockdown of POLDIP2 suppresses tumor growth and invasion capacity and is linked to unfavorable transformation ability and metastatic feature in non-small cell lung cancer. Exp. Cell Res. 2018, 368, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Chian, C.F.; Hwang, Y.T.; Terng, H.J.; Lee, S.C.; Chao, T.Y.; Chang, H.; Ho, C.L.; Wu, Y.Y.; Perng, W.C. Panels of tumor-derived RNA markers in peripheral blood of patients with non-small cell lung cancer: Their dependence on age, gender and clinical stages. Oncotarget 2016, 7, 50582–50595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, G.; Massoudi, D.; Muir, A.M.; Joshi, D.C.; Zhang, C.L.; Chiu, S.Y.; Greenspan, D.S. WBSCR16 Is a Guanine Nucleotide Exchange Factor Important for Mitochondrial Fusion. Cell Rep. 2017, 20, 923–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sessions, D.T.; Kashatus, D.F. Mitochondrial dynamics in cancer stem cells. Cell. Mol. Life Sci. 2021, 78, 3803–3816. [Google Scholar] [CrossRef] [PubMed]
- de Reynies, A.; Assie, G.; Rickman, D.S.; Tissier, F.; Groussin, L.; Rene-Corail, F.; Dousset, B.; Bertagna, X.; Clauser, E.; Bertherat, J. Gene Expression profiling reveals a new classification of adrenocortical tumors and identifies molecular predictors of malignancy and survival. J. Clin. Oncol. 2009, 27, 1108–1115. [Google Scholar] [CrossRef]
- Fragoso, M.C.; Almeida, M.Q.; Mazzuco, T.L.; Mariani, B.M.; Brito, L.P.; Goncalves, T.C.; Alencar, G.A.; Lima, L.D.O.; Faria, A.M.; Bourdeau, I.; et al. Combined expression of BUB1B, DLGAP5, and PINK1 as predictors of poor outcome in adrenocortical tumors: Validation in a Brazilian cohort of adult and pediatric patients. Eur. J. Endocrinol. 2012, 166, 61–67. [Google Scholar] [CrossRef]
- Drelon, C.; Berthon, A.; Mathieu, M.; Ragazzon, B.; Kuick, R.; Tabbal, H.; Septier, A.; Rodriguez, S.; Batisse-Lignier, M.; Sahut-Barnola, I.; et al. EZH2 is overexpressed in adrenocortical carcinoma and is associated with disease progression. Hum. Mol. Genet. 2016, 25, 2789–2800. [Google Scholar] [CrossRef]
- Ruggiero, C.; Doghman-Bouguerra, M.; Sbiera, S.; Sbiera, I.; Parsons, M.; Ragazzon, B.; Morin, A.; Robidel, E.; Favier, J.; Bertherat, J.; et al. Dosage-dependent regulation ofVAV2expression by steroidogenic factor-1 drives adrenocortical carcinoma cell invasion. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef]
- Sbiera, S.; Sbiera, I.; Ruggiero, C.; Doghman-Bouguerra, M.; Korpershoek, E.; de Krijger, R.R.; Ettaieb, H.; Haak, H.; Volante, M.; Papotti, M.; et al. Assessment of VAV2 Expression Refines Prognostic Prediction in Adrenocortical Carcinoma. J. Clin. Endocrinol. Metab. 2017, 102, 3491–3498. [Google Scholar] [CrossRef] [Green Version]
- Mete, O.; Gucer, H.; Kefeli, M.; Asa, S.L. Diagnostic and Prognostic Biomarkers of Adrenal Cortical Carci-noma. Am. J. Surg. Pathol. 2018, 42, 201–213. [Google Scholar] [CrossRef]
- Tian, X.; Xu, W.H.; Anwaier, A.; Wang, H.K.; Wan, F.N.; Cao, D.L.; Luo, W.J.; Shi, G.H.; Qu, Y.Y.; Zhang, H.L.; et al. Construction of a robust prognostic model for adult adrenocortical carcinoma: Results from bioinformatics and real-world data. J. Cell. Mol. Med. 2021, 25, 3898–3911. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | ACC (n = 37) | Benign (n = 8) |
|---|---|---|
| Age | 48.5 ± 12.9 | 51.9 ± 10.5 |
| Male | 15 (40.5) | 4 (50.0) |
| Initial Stage (ENSAT) | ||
| I | 2 (5.4) | - |
| II | 7 (18.9) | - |
| III | 17 (45.9) | - |
| IV | 11 (29.7) | - |
| Death | 19 (51.4) | - |
| Follow-up, years (IQR) | 4.0 (1.3–8.1) | - |
| Cortisol Secretion a | ||
| Yes | 18 (48.6) | - |
| No | 9 (24.3) | - |
| Mitosis Count b | ||
| ≥20/HPF | 7 (18.9) | - |
| <20/HPF | 22 (59.5) | - |
| Ki67 c | ||
| ≥20% | 11 (29.7) | - |
| 10–19% | 4 (10.8) | - |
| <10% | 7 (18.9) | - |
| Rank | Benign vs. ACC | Stage 1–2 vs. 3–4 | Stage 1–3 vs. 4 | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| InfoGain | ANOVA | ReliefF | InfoGain | ANOVA | ReliefF | InfoGain | ANOVA | ReliefF | ||||||||||
| Gene Name | Score | Gene Name | Score | Gene Name | Score | Gene Name | Score | Gene Name | Score | Gene Name | Score | Gene Name | Score | Gene Name | Score | Gene Name | Score | |
| 1 | H2AFY2 | 0.787 | PDE4D | 62.944 | MUSTN1 | 0.321 | EPG5 | 0.681 | EPG5 | 24.961 | NUDC | 0.239 | WBSCR16 | 0.658 | WBSCR16 | 52.070 | WBSCR16 | 0.303 |
| 2 | ABLIM1 | 0.654 | SYNE1 | 60.441 | ARMC8 | 0.298 | GNPAT | 0.531 | NUDC | 24.001 | GNPAT | 0.226 | CCDC12 | 0.658 | CCDC12 | 31.394 | NOSIP | 0.234 |
| 3 | DNPH1 | 0.654 | H2AFY2 | 60.114 | SNX15 | 0.296 | DECR2 | 0.531 | GNPAT | 23.111 | FAM160B2 | 0.217 | PDHA1 | 0.658 | PDHA1 | 31.002 | CCDC12 | 0.196 |
| 4 | ACIN1 | 0.531 | MUSTN1 | 53.536 | SYNE1 | 0.291 | PPIF | 0.531 | STRIP1 | 22.252 | SNX8 | 0.191 | LYRM1 | 0.581 | PDHB | 28.034 | GHITM | 0.188 |
| 5 | RPS24 | 0.531 | ARMC8 | 48.449 | PDE4D | 0.276 | SPR | 0.531 | A1BG | 21.073 | NGEF | 0.182 | RRM2 | 0.581 | RBM26 | 24.492 | MRPL32 | 0.184 |
| 6 | FUBP3 | 0.526 | CAB39L | 44.931 | CAB39L | 0.275 | SLC25A32 | 0.531 | NDUFAF2 | 20.221 | CTSA | 0.170 | NCEH1 | 0.519 | MTIF2 | 21.829 | PDHB | 0.179 |
| 7 | SYNE1 | 0.526 | PPM1G | 42.526 | RALGAPB | 0.270 | COG3 | 0.531 | CHMP6 | 19.245 | A1BG | 0.166 | ARSA | 0.519 | CSNK1D | 21.478 | SNCG | 0.171 |
| 8 | ARMC8 | 0.526 | MTA2 | 41.666 | SNRNP70 | 0.251 | ISOC1 | 0.531 | SNX8 | 18.413 | ATM | 0.164 | CAV1 | 0.519 | SNCG | 20.043 | PDHA1 | 0.170 |
| 9 | HSD17B13 | 0.526 | SLC37A2 | 40.937 | AAR2 | 0.245 | TTC1 | 0.522 | ALDH3B1 | 17.218 | PTK2B | 0.163 | RABL3 | 0.519 | LYRM1 | 19.369 | RBM26 | 0.167 |
| 10 | PDE4D | 0.526 | UBE2M | 39.877 | MZB1 | 0.239 | MRPS5 | 0.522 | CTSA | 17.069 | STRIP1 | 0.160 | MRPS23 | 0.510 | NCEH1 | 17.250 | NCEH1 | 0.153 |
| 11 | UBE2M | 0.526 | DNPH1 | 39.713 | H2AFY2 | 0.237 | AGRN | 0.478 | TTC1 | 17.034 | CHMP6 | 0.158 | TPBG | 0.499 | NOSIP | 17.024 | PNMA3 | 0.152 |
| 12 | SKIV2L2 | 0.526 | XPOT | 38.513 | NIFK | 0.230 | FBLN2 | 0.478 | HADH | 17.000 | MMTAG2 | 0.157 | PDHB | 0.475 | NMNAT1 | 16.105 | LYRM1 | 0.146 |
| 13 | SHMT2 | 0.526 | NIFK | 36.911 | ALG14 | 0.225 | CDK5RAP2 | 0.478 | CMSS1 | 16.775 | DNAJC15 | 0.156 | MAPK9 | 0.475 | COL4A2 | 15.781 | NID2 | 0.142 |
| 14 | CPB1 | 0.526 | SCCPDH | 36.042 | CPB1 | 0.220 | A1BG | 0.458 | APOC1 | 16.197 | EPG5 | 0.153 | MTIF2 | 0.475 | MTHFD2L | 15.740 | RABAC1 | 0.140 |
| 15 | XPOT | 0.526 | SKIV2L2 | 35.957 | WNT2B | 0.210 | C8G | 0.458 | PPIF | 16.157 | TTC1 | 0.153 | NMNAT1 | 0.475 | POLDIP2 | 15.577 | C9orf91 | 0.138 |
| 16 | HNRNPA1 | 0.526 | HNRNPA1 | 35.727 | SCCPDH | 0.201 | LTBP4 | 0.458 | PCNT | 15.578 | SERPINA3 | 0.151 | MRPL22 | 0.475 | MAPK9 | 15.468 | MTHFD2L | 0.138 |
| 17 | CSNK2A1 | 0.526 | VCPIP1 | 35.452 | PPM1G | 0.200 | C8A | 0.458 | SERPINA3 | 15.499 | IGHV4-34 | 0.146 | LDB3 | 0.475 | NID2 | 15.440 | MAPK9 | 0.137 |
| 18 | MUSTN1 | 0.526 | RPL24 | 35.172 | RPL24 | 0.199 | NUDC | 0.396 | SERPINC1 | 15.308 | CSDC2 | 0.144 | ALOX5 | 0.475 | BLOC1S3 | 15.217 | MRPL1 | 0.137 |
| 19 | PPIH | 0.526 | FBXW8 | 34.574 | UBQLN4 | 0.198 | PTK2B | 0.396 | PTK2B | 15.098 | ELOVL5 | 0.144 | COL4A2 | 0.431 | PPOX | 15.193 | LACC1 | 0.136 |
| 20 | RAN | 0.526 | SRXN1 | 32.834 | INPP1 | 0.196 | ALDH3B1 | 0.396 | TGFB1 | 15.072 | SLC9A3R2 | 0.142 | GHITM | 0.427 | PLOD2 | 15.117 | PEX3 | 0.135 |
| Variables | SNUH Cohort | TCGA Cohort | ||||
|---|---|---|---|---|---|---|
| C-Index (95% CI) | NRI (95% CI) | p-Value for NRI | C-Index (95% CI) | NRI (95% CI) | p-Value for NRI | |
| Age+Stage | 0.690 (0.551—0.829) | - | - | 0.674 (0.552—0.796) | - | - |
| Age+Stage+HNRNPA1 | 0.704 (0.563—0.845) | 0.813 (0.225—1.401) | 0.007 | 0.757 (0.662—0.852) | 0.625 (0.179—1.070) | 0.006 |
| Age+Stage C8A | 0.727 (0.612—0.841) | 0.152 (−0.480—0.784) | 0.637 | 0.674 (0.550—0.798) | 0.228 (−0.070—0.526) | 0.134 |
| Age+Stage+CHMP6 | 0.756 (0.645—0.867) | 0.152 (−0.480—0.784) | 0.637 | 0.683 (0.568—0.797) | −0.234 (−0.704—0.236) | 0.329 |
| Age+Stage+LTBP4 | 0.702 (0.561—0.842) | 0.596 (−0.018—1.211) | 0.057 | 0.752 (0.639—0.865) | 0.585 (0.138—1.031) | 0.010 |
| Age+Stage+SPR | 0.718 (0.583—0.852) | 0.503 (−0.088—1.094) | 0.095 | 0.681 (0.559—0.803) | 0.311 (−0.154—0.776) | 0.190 |
| Age+Stage+NCEH1 | 0.695 (0.550—0.839) | 0.485 (−0.140—1.111) | 0.128 | 0.689 (0.575—0.803) | 0.428 (−0.030—0.885) | 0.067 |
| Age+Stage+MRPS23 | 0.740 (0.618—0.862) | 0.819 (0.239—1.398) | 0.006 | 0.729 (0.632—0.825) | 0.508 (0.052—0.963) | 0.029 |
| Age+Stage+POLDIP2 | 0.733 (0.612—0.855) | 0.591 (−0.024—1.205) | 0.060 | 0.741 (0.666—0.816) | 0.508 (0.052—0.963) | 0.029 |
| Age+Stage+WBSCR16 | 0.740 (0.606—0.874) | 0.702 (0.098—1.305) | 0.023 | 0.743 (0.668—0.818) | 0.508 (0.052—0.963) | 0.029 |
| Age+Stage+HNRNPA1+MRPS23+WBSCR16 a | 0.708 (0.579—0.838) | 0.175 (−0.447—0.797) | 0.58 | 0.752 (0.659—0.845) | 0.492 (0.091—0.894) | 0.016 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jang, H.N.; Moon, S.J.; Jung, K.C.; Kim, S.W.; Kim, H.; Han, D.; Kim, J.H. Mass Spectrometry-Based Proteomic Discovery of Prognostic Biomarkers in Adrenal Cortical Carcinoma. Cancers 2021, 13, 3890. https://doi.org/10.3390/cancers13153890
Jang HN, Moon SJ, Jung KC, Kim SW, Kim H, Han D, Kim JH. Mass Spectrometry-Based Proteomic Discovery of Prognostic Biomarkers in Adrenal Cortical Carcinoma. Cancers. 2021; 13(15):3890. https://doi.org/10.3390/cancers13153890
Chicago/Turabian StyleJang, Han Na, Sun Joon Moon, Kyeong Cheon Jung, Sang Wan Kim, Hyeyoon Kim, Dohyun Han, and Jung Hee Kim. 2021. "Mass Spectrometry-Based Proteomic Discovery of Prognostic Biomarkers in Adrenal Cortical Carcinoma" Cancers 13, no. 15: 3890. https://doi.org/10.3390/cancers13153890
APA StyleJang, H. N., Moon, S. J., Jung, K. C., Kim, S. W., Kim, H., Han, D., & Kim, J. H. (2021). Mass Spectrometry-Based Proteomic Discovery of Prognostic Biomarkers in Adrenal Cortical Carcinoma. Cancers, 13(15), 3890. https://doi.org/10.3390/cancers13153890