
Proteins from the DNA Damage Response: Regulation, Dysfunction, and Anticancer Strategies


Abstract
Simple Summary
Abstract
1. Introduction
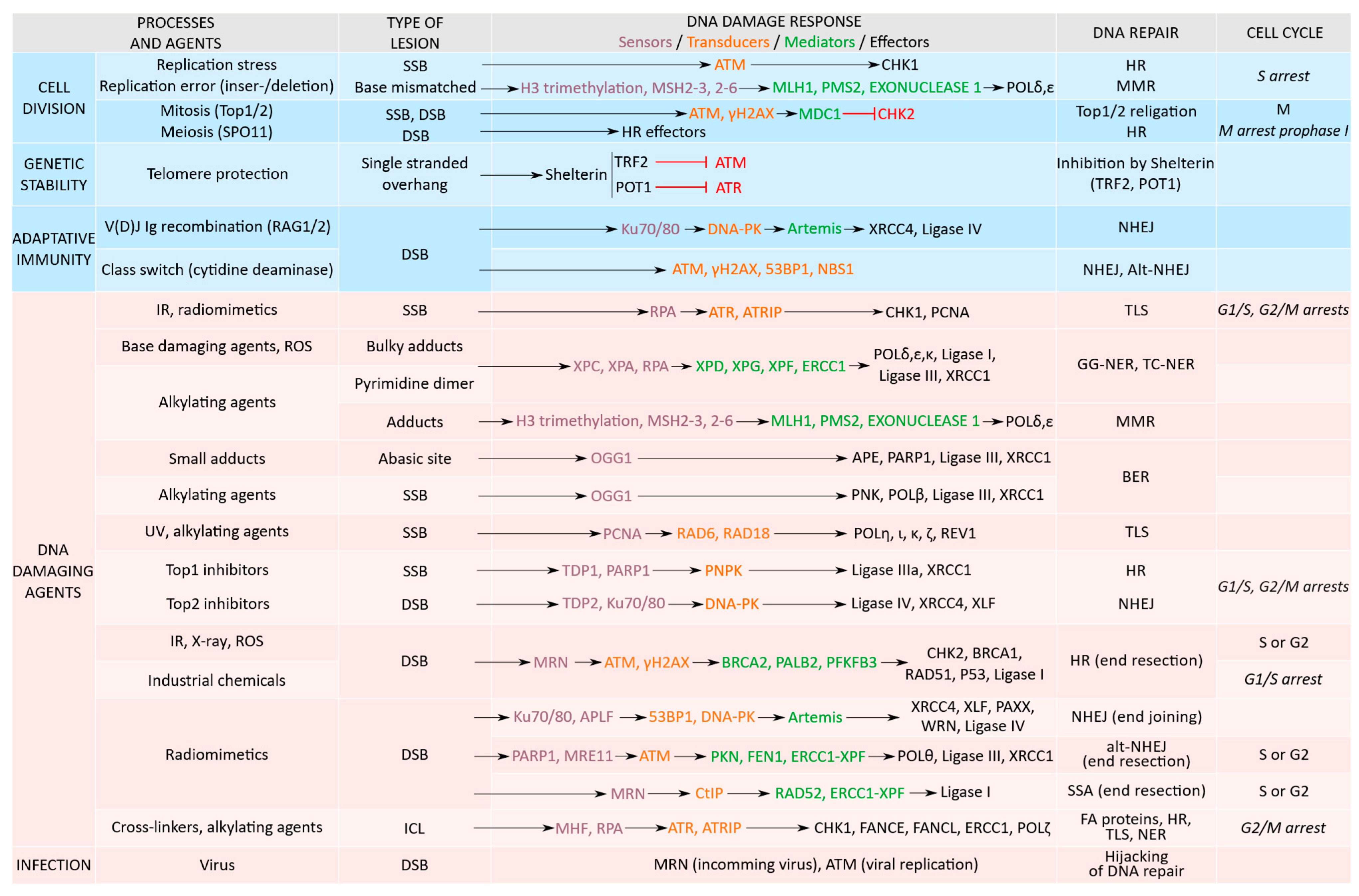
2. DDR Proteins Activation Is Function of the Type of DNA Damage
2.1. Replication Stress and DNA Damage
2.2. Physiological DNA Breaks Involved in Normal Cellular Processes
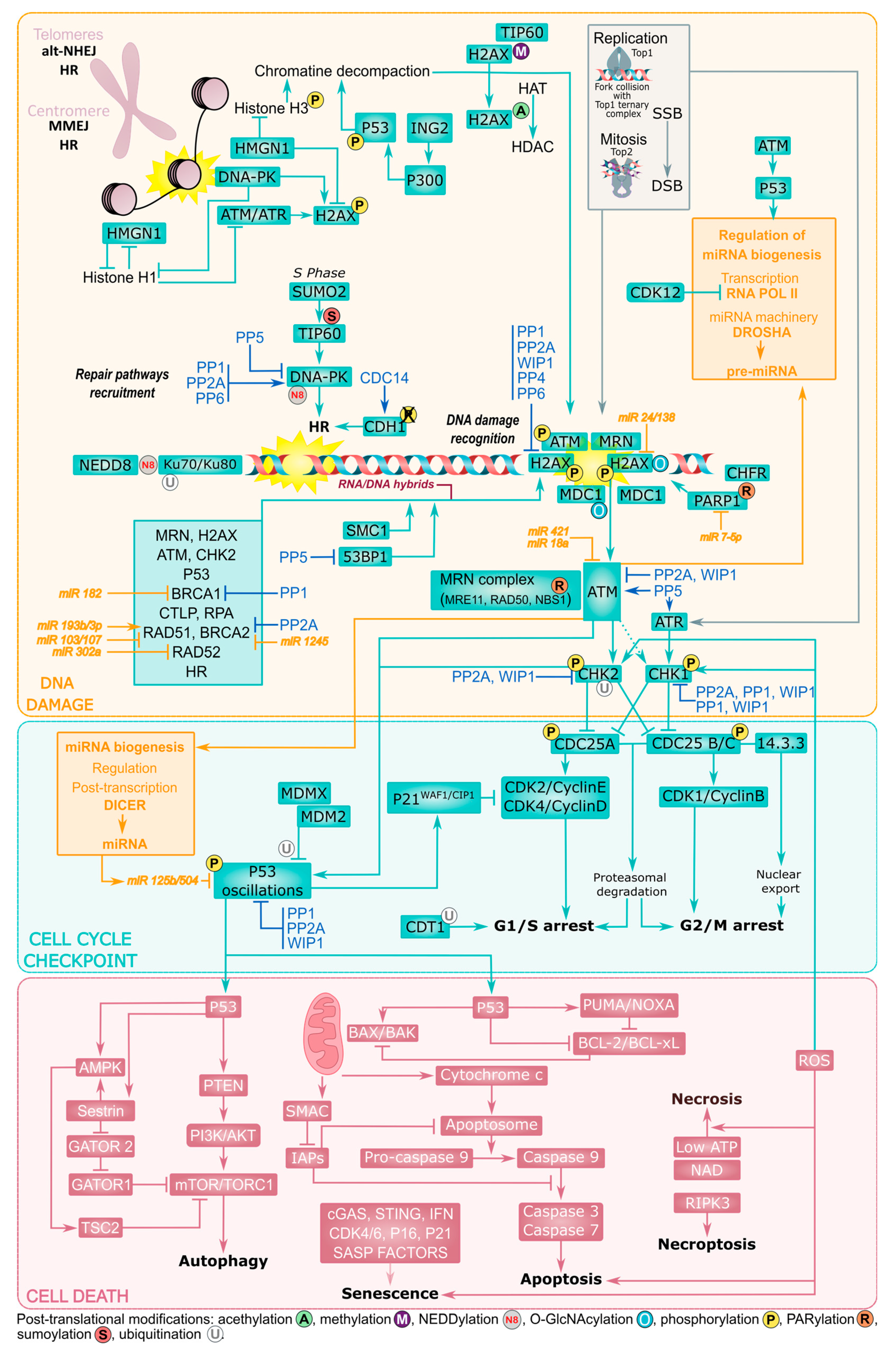
3. Structural Organization of the DDR
3.1. Selection of the Repair Mechanisms
3.2. Cell Cycle Checkpoints
3.3. Cell Death and P53 Signaling
3.4. Chromatin State and Chromosomal Damage Location
3.5. Post-Translational Modifications
3.6. ncRNAs as Emerging Regulators
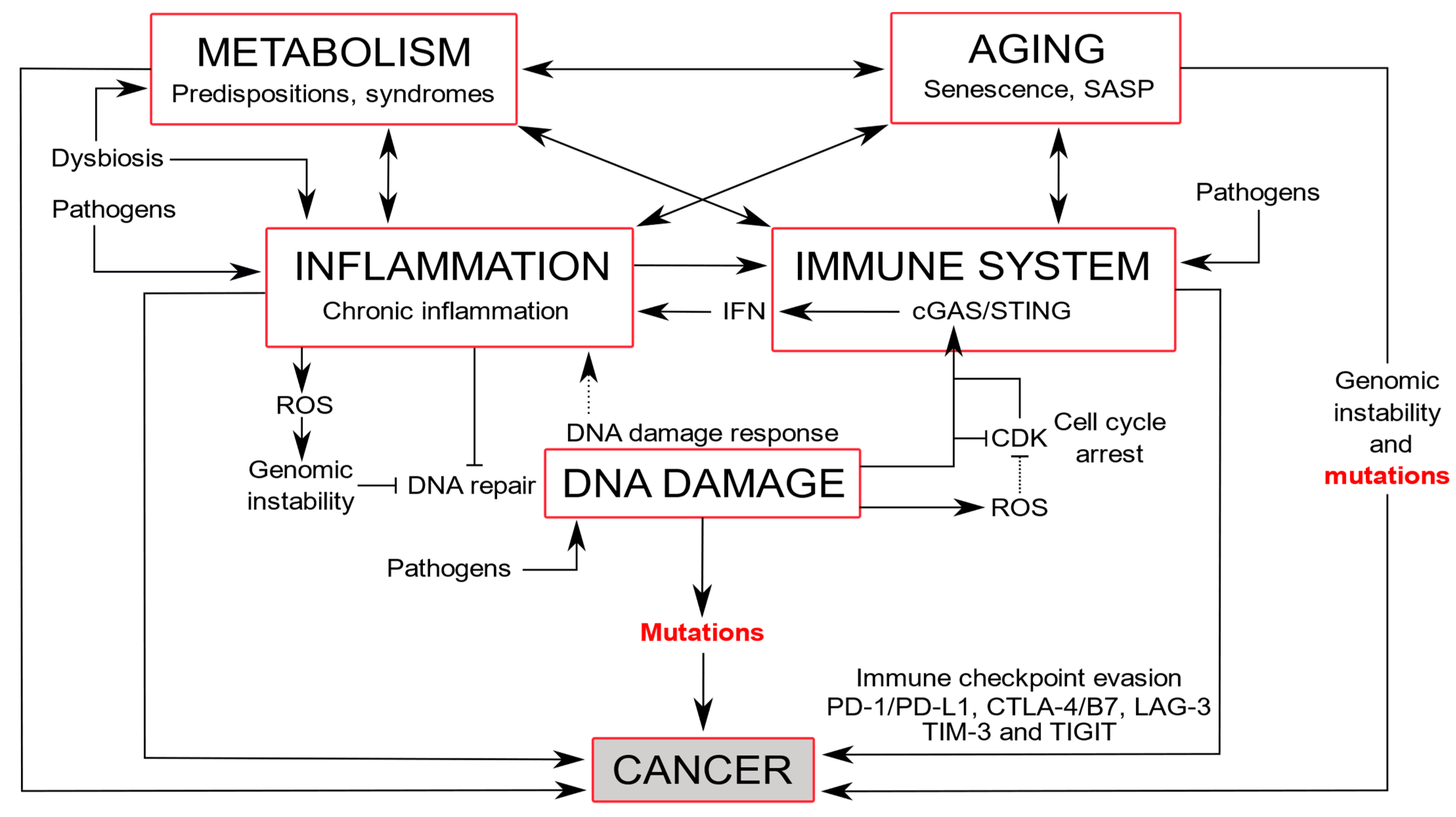
4. DDR Alterations and Associated Diseases
4.1. Immune Diseases and Inflammation

4.2. Neurodegeneration and Premature Aging
4.3. Aging
4.4. Cardiovascular and Metabolic Diseases
4.5. Cancers

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DDR Proteins | DDR Signaling Pathways | Protein Activity/Function | Cancer-Associated Mutations * |
|---|---|---|---|
| ATM | Cell cycle regulators | Kinases | Breast, colon, endometrial, leukemia, lung, lymphoma, pancreatic, prostate |
| ATR | Breast, colon, endometrial, gastric, lung, lymphoma | ||
| CHK2 | Cell cycle | Phosphatases | Bladder, breast, colon, endometrial, lung |
| APC-C/CDH1 | Breast, gastric, lung | ||
| PTEN | Cell death | Phosphatase | Breast, endometrial, gastric |
| P53 | Transcription factor | Found in 39.52% of all cancers (AACR) | |
| DNA repair proteins | DNA repair pathways | ||
| Ku70/Ku80 | NHEJ | Helicases | Breast, colorectal, lung, melanoma |
| DNA-PK | Kinase | Breast, colon, glioma, oesophagal, lung | |
| NBS1 | HR | MRN complex | Breast, colon, esophageal, head and neck, hepatoma, liver, lung, lymphoma, prostate |
| MRE11 | Breast | ||
| RAD50 | Colon, gastrointestinal, lung | ||
| BRCA1 | E3 ubiquitin-ligase | Breast, colon, gastrointestinal, haematological, lung, melanoma, ovarian, pancreatic, prostate | |
| BRCA2 | RAD51 binding to DNA | ||
| PALB2/FANCN | Recruitment of BRCA2 and RAD51 | Breast, colon, head and neck, lung, ovarian | |
| FANCA, FANCB | FA repair complex | Breast, colon, leukemia, liver, lung | |
| FANCO/RAD51C | DNA-dependent ATPase | Breast, colon, lung, ovarian, prostate | |
| RAD51D | Breast, lung, ovarian | ||
| BLM | Helicases | Breast, colon, endometrial, leukemia, lung, lymphoma, melanoma | |
| WRN | Colorectal, endometrial, lung, melanoma, pancreatic, thyroid | ||
| ERCC1 | NER | Nuclease | Colorectal, glioma, lung, skin |
| XPA, XPC | Scaffold protein | Bladder, colon, lung, skin | |
| XPD/ERCC2 | Helicase | ||
| XPG/ERCC5 | Endonuclease | ||
| OGG1 | BER | Glycosylase | Breast, lung, renal |
| PARP1 | ADP-ribosyltransferase | Breast, colon, endometrial, lung | |
| XRCC1 | Scaffold protein | Non-small cell lung | |
| MLH1 | MMR | ATPase | Brain, breast, colorectal, endometrial, hepatobiliary, lung, ovarian, pancreatic, skin, stomach, upper urinary |
| MSH2, MSH6 | Scaffold protein | ||
| MGMT | DR | Methyltransferase | Gliomas |
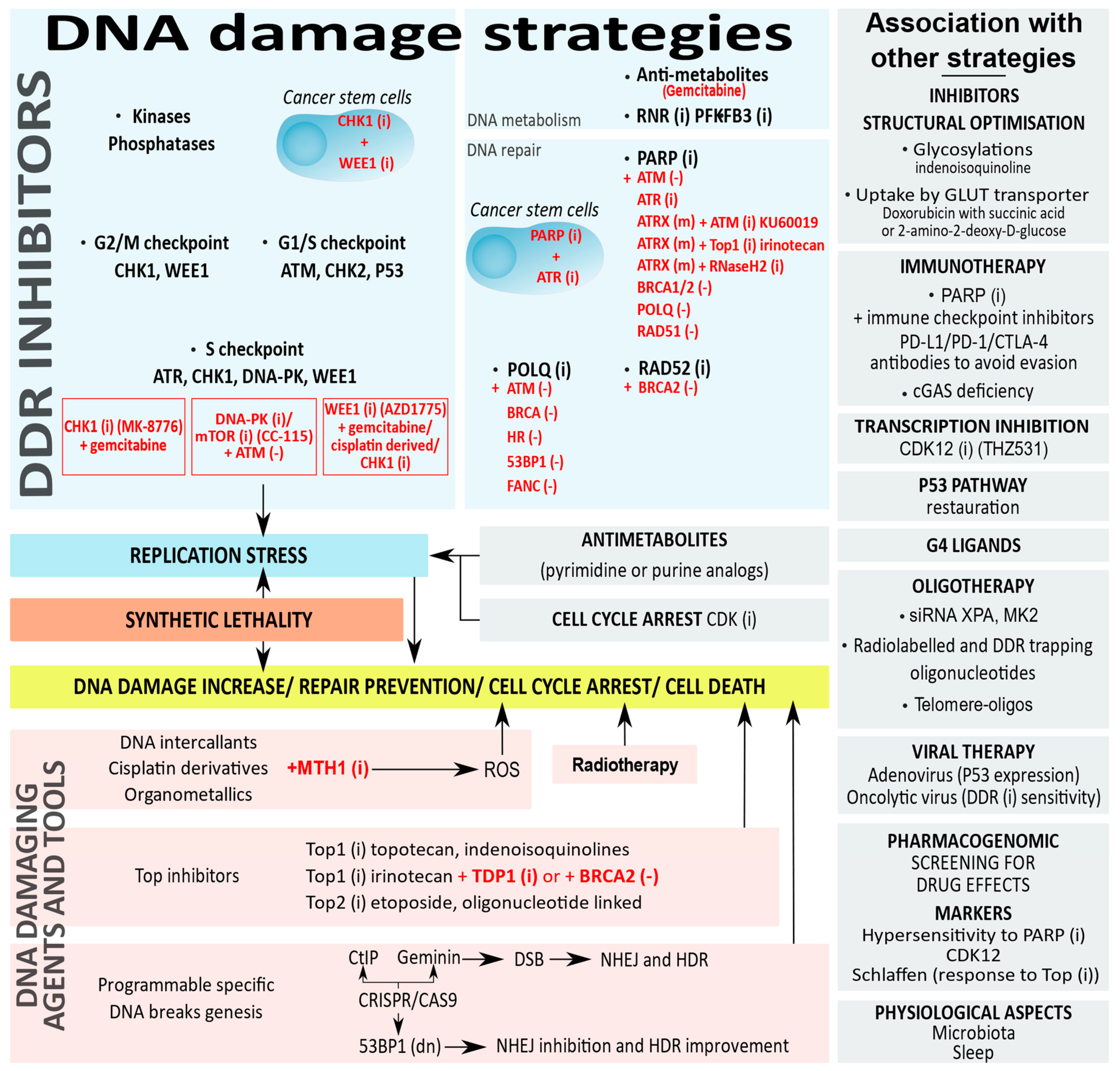
5. DDR Targets in Anticancer Therapies
5.1. Targeting DDR Proteins to Induce Deficiencies

5.2. Targeting the DDR Protein Network with Synthetic Lethality
5.3. Targeting Replication Stress
5.4. Replication Stress and Telomere Deprotection
5.5. Other Oligonucleotides, or Small Interfering RNA
5.6. Topoisomerase Inhibitors
5.7. DNA Damaging Drugs
5.8. DNA Damaging Tools
6. Drugging DDR Proteins in Association with Other Therapies
6.1. Inhibition of Cell Cycle Progression and Transcription
6.2. Restoration of the P53 Pathway
6.3. DDR Inhibition Strategies Associated to Cancer Immunotherapies
6.4. Pharmacogenomic and Predictive Markers in DDR Strategies
6.5. Viral Therapies to Boost the DDR
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Pearl, L.H.; Schierz, A.C.; Ward, S.E.; Al-Lazikani, B.; Pearl, F.M.G. Therapeutic opportunities within the DNA damage response. Nat. Rev. Cancer 2015, 15, 166–180. [Google Scholar] [CrossRef]
- Schlam-Babayov, S.; Bensimon, A.; Harel, M.; Geiger, T.; Aebersold, R.; Ziv, Y.; Shiloh, Y. Phosphoproteomics reveals novel modes of function and inter-relationships among PIKKs in response to genotoxic stress. EMBO J. 2021, 40, e104400. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.A.; Lin, Y.; Yan, S. Single-strand break end resection in genome integrity: Mechanism and regulation by APE2. Int. J. Mol. Sci. 2018, 19, 2389. [Google Scholar] [CrossRef] [PubMed]
- Ensminger, M.; Iloff, L.; Ebel, C.; Nikolova, T.; Kaina, B.; Löbrich, M. DNA breaks and chromosomal aberrations arise when replication meets base excision repair. J. Cell Biol. 2014, 206, 29–43. [Google Scholar] [CrossRef]
- Kuzminov, A. Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proc. Natl. Acad. Sci. USA 2001, 98, 8241–8246. [Google Scholar] [CrossRef]
- Jadoon, S.; Malik, A. DNA Damage by Heavy Metals in Animals and Human Beings: An Overview. Biochem. Pharmacol. Open Access 2017, 6. [Google Scholar] [CrossRef]
- Shah, G.A.; O’Shea, C.C. Viral and Cellular Genomes Activate Distinct DNA Damage Responses. Cell 2015, 162, 987–1002. [Google Scholar] [CrossRef]
- Adam, M.A.A.; Tabana, Y.M.; Musa, K.B.; Sandai, D.A. Effects of different mycotoxins on humans, cell genome and their involvement in cancer (Review). Oncol. Rep. 2017, 37, 1321–1336. [Google Scholar] [CrossRef]
- Virgilio, A.; Sinisi, A.; Russo, V.; Gerardo, S.; Santoro, A.; Galeone, A.; Taglialatela-Scafati, O.; Roperto, F. Ptaquiloside, the Major Carcinogen of Bracken Fern, in the Pooled Raw Milk of Healthy Sheep and Goats: An Underestimated, Global Concern of Food Safety. J. Agric. Food Chem. 2015, 63, 4886–4892. [Google Scholar] [CrossRef]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef]
- Muñoz, S.; Méndez, J. DNA replication stress: From molecular mechanisms to human disease. Chromosoma 2017, 126. [Google Scholar] [CrossRef]
- Petermann, E.; Orta, M.L.; Issaeva, N.; Schultz, N.; Helleday, T. Hydroxyurea-Stalled Replication Forks Become Progressively Inactivated and Require Two Different RAD51-Mediated Pathways for Restart and Repair. Mol. Cell 2010, 37, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Schorr, S.; Schneider, S.; Lammens, K.; Hopfner, K.; Carell, T. Mechanism of replication blocking and bypass of Y-family polymerase η by bulky acetylamino- fluorene DNA adducts. Proc. Natl. Acad. Sci. USA 2010, 107, 20720–20725. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Kim, J.M.; Shiotani, B.; Yang, K.; Zou, L.; D’Andrea, A.D. The FANCM/FAAP24 complex is required for the DNA interstrand crosslink-induced checkpoint response. Mol. Cell 2010, 39, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Castellano-Pozo, M.; García-Muse, T.; Aguilera, A. R-loops cause replication impairment and genome instability during meiosis. EMBO Rep. 2012, 13, 923–929. [Google Scholar] [CrossRef]
- Sollier, J.; Stork, C.T.; García-Rubio, M.L.; Paulsen, R.D.; Aguilera, A.; Cimprich, K.A. Transcription-Coupled Nucleotide Excision Repair Factors Promote R-Loop-Induced Genome Instability. Mol. Cell 2014, 56, 777–785. [Google Scholar] [CrossRef]
- Helmrich, A.; Ballarino, M.; Tora, L. Collisions between Replication and Transcription Complexes Cause Common Fragile Site Instability at the Longest Human Genes. Mol. Cell 2011, 44, 966–977. [Google Scholar] [CrossRef]
- Gómez-González, B.; García-Rubio, M.; Bermejo, R.; Gaillard, H.; Shirahige, K.; Marín, A.; Foiani, M.; Aguilera, A. Genome-wide function of THO/TREX in active genes prevents R-loop-dependent replication obstacles. EMBO J. 2011, 30, 3106–3119. [Google Scholar] [CrossRef] [PubMed]
- Burgess, R.C.; Misteli, T. Not All DDRs Are Created Equal: Non-Canonical DNA Damage Responses. Cell 2015, 162, 944–947. [Google Scholar] [CrossRef]
- Mailand, N.; Falck, J.; Lukas, C.; Syljuåsen, R.G.; Welcker, M.; Bartek, J.; Lukas, J. Rapid destruction of human Cdc25A in response to DNA damage. Science 2000, 288, 1425–1429. [Google Scholar] [CrossRef]
- Moiseeva, T.N.; Yin, Y.; Calderon, M.J.; Qian, C.; Schamus-Haynes, S.; Sugitani, N.; Osmanbeyoglu, H.U.; Rothenberg, E.; Watkins, S.C.; Bakkenist, C.J. An ATR and CHK1 kinase signaling mechanism that limits origin firing during unperturbed DNA replication. Proc. Natl. Acad. Sci. USA 2019, 116, 13374–13383. [Google Scholar] [CrossRef]
- Cortez, D. Replication-Coupled DNA Repair. Mol. Cell 2019, 74, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.; Gatenby, R.; Sidi, S. How Cells Handle DNA Breaks during Mitosis: Detection, signaling, repair, and fate choice. Cells 2019, 8, 1049. [Google Scholar] [CrossRef]
- Nielsen, C.F.; Zhang, T.; Barisic, M.; Kalitsis, P.; Hudson, D.F. Topoisomerase IIa is essential for maintenance of mitotic chromosome structure. Proc. Natl. Acad. Sci. USA 2020, 117, 12131–12142. [Google Scholar] [CrossRef]
- Lee, H.J.; Hwang, H.I.; Jang, Y.J. Mitotic DNA damage response: Polo-like kinase-1 is dephosphorylated through ATM-Chk1 pathway. Cell Cycle 2010, 9, 2389–2398. [Google Scholar] [CrossRef]
- Lee, K.Y.; Dutta, A. Chk1 promotes non-homologous end joining in G1 through direct phosphorylation of ASF1A. Cell Rep. 2021, 34, 108680. [Google Scholar] [CrossRef] [PubMed]
- Gentile, F.; Arcaro, A.; Pizzimenti, S.; Daga, M.; Paolo Cetrangolo, G.; Dianzani, C.; Lepore, A.; Graf, M.; Ames, P.R.J.; Barrera, G. DNA damage by lipid peroxidation products: Implications in cancer, inflammation and autoimmunity. AIMS Genet. 2017, 4, 103–137. [Google Scholar] [CrossRef]
- Cheng, E.Y.; Hunt, P.A.; Naluai-Cecchini, T.A.; Fligner, C.L.; Fujimoto, V.Y.; Pasternack, T.L.; Schwartz, J.M.; Steinauer, J.E.; Woodruff, T.J.; Cherry, S.M.; et al. Meiotic recombination in human oocytes. PLoS Genet. 2009, 5, e1000661. [Google Scholar] [CrossRef]
- Neale, M.J.; Pan, J.; Keeney, S. Endonucleolytic processing of covalent protein-linked DNA double-strand breaks. Nature 2005, 436, 1053–1057. [Google Scholar] [CrossRef]
- Hinch, A.G.; Zhang, G.; Becker, P.W.; Moralli, D.; Hinch, R.; Davies, B.; Bowden, R.; Donnelly, P. Factors influencing meiotic recombination revealed by whole-genome sequencing of single sperm. Science 2019, 363, eaau8861. [Google Scholar] [CrossRef]
- García-Rodríguez, A.; Gosálvez, J.; Agarwal, A.; Roy, R.; Johnston, S. DNA damage and repair in human reproductive cells. Int. J. Mol. Sci. 2019, 20, 31. [Google Scholar] [CrossRef]
- Har-Vardi, I.; Mali, R.; Breietman, M.; Sonin, Y.; Albotiano, S.; Levitas, E.; Potashnik, G.; Priel, E. DNA topoisomerases I and II in human mature sperm cells: Characterization and unique properties. Hum. Reprod. 2007, 22, 2183–2189. [Google Scholar] [CrossRef]
- Abe, H.; Alavattam, K.G.; Hu, Y.C.; Pang, Q.; Andreassen, P.R.; Hegde, R.S.; Namekawa, S.H. The Initiation of Meiotic Sex Chromosome Inactivation Sequesters DNA Damage Signaling from Autosomes in Mouse Spermatogenesis. Curr. Biol. 2020, 30, 408–420.e5. [Google Scholar] [CrossRef]
- Lorenz, M.; Jung, S.; Radbruch, A. Switch transcripts in immunoglobulin class switching. Science 1995, 67, 1825–1828. [Google Scholar] [CrossRef]
- Rivera-Munoz, P.; Soulas-Sprauel, P.; Le Guyader, G.; Abramowski, V.; Bruneau, S.; Fischer, A.; Pâques, F.; De Villartay, J.P. Reduced immunoglobulin class switch recombination in the absence of Artemis. Blood 2009, 114, 3601–3609. [Google Scholar] [CrossRef] [PubMed]
- Björkman, A.; Du, L.; Felgentreff, K.; Rosner, C.; Pankaj Kamdar, R.; Kokaraki, G.; Matsumoto, Y.; Davies, E.G.; van der Burg, M.; Notarangelo, L.D.; et al. DNA-PKcs Is Involved in Ig Class Switch Recombination in Human B Cells. J. Immunol. 2015, 195, 5608–5615. [Google Scholar] [CrossRef]
- Soulas-Sprauel, P.; Le Guyader, G.; Rivera-Munoz, P.; Abramowski, V.; Olivier-Martin, C.; Goujet-Zalc, C.; Charneau, P.; De Villartay, J.P. Role for DNA repair factor XRCC4 in immunoglobulin class switch recombination. J. Exp. Med. 2007, 204, 1717–1727. [Google Scholar] [CrossRef]
- Chi, X.; Li, Y.; Qiu, X. V(D)J recombination, somatic hypermutation and class switch recombination of immunoglobulins: Mechanism and regulation. Immunology 2020, 160, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, S.; Haddad, D.; Li, C.; Le, M.X.; Ling, A.K.; So, C.C.; Nepal, R.M.; Gommerman, J.L.; Yu, K.; Ketela, T.; et al. The SAGA Deubiquitination Module Promotes DNA Repair and Class Switch Recombination through ATM and DNAPK-Mediated γH2AX Formation. Cell Rep. 2016, 15, 1554–1565. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Zhang, X.; Pu, Q.; Huang, T.; Xie, Q.; Wang, Y.; Li, J.; Wang, Y.; Gu, H.; Huang, T.; et al. Immunoglobulin G Expression in Human Sperm and Possible Functional Significance. Sci. Rep. 2016, 6, 20166. [Google Scholar] [CrossRef] [PubMed]
- Bandaria, J.N.; Qin, P.; Berk, V.; Chu, S.; Yildiz, A. Shelterin protects chromosome ends by compacting telomeric chromatin. Cell 2016, 164, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Guo, X.; Ferguson, D.O.; Chang, S. Multiple roles for MRE11 at uncapped telomeres. Nature 2009, 460, 914–918. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef]
- Krenning, L.; van den Berg, J.; Medema, R.H. Life or Death after a Break: What Determines the Choice? Mol. Cell 2019, 76, 346–358. [Google Scholar] [CrossRef]
- Gutierrez, R.; O’Connor, T.R. DNA direct reversal repair and alkylating agent drug resistance. Cancer Drug Resist. 2021. [Google Scholar] [CrossRef]
- Dantuma, N.P.; Attikum, H. Spatiotemporal regulation of posttranslational modifications in the DNA damage response. EMBO J. 2016, 35, 6–23. [Google Scholar] [CrossRef] [PubMed]
- Carusillo, A.; Mussolino, C. DNA Damage: From Threat to Treatment. Cells 2020, 9, 1665. [Google Scholar] [CrossRef]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H.J. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef]
- Li, M.; Larsen, L.; Hedglin, M. Rad6/Rad18 Competes with DNA Polymerases η and δ for PCNA Encircling DNA. Biochemistry 2020, 59, 407–416. [Google Scholar] [CrossRef]
- Parameswaran, B.; Chiang, H.C.; Lu, Y.; Coates, J.; Deng, C.X.; Baer, R.; Lin, H.K.; Li, R.; Paull, T.T.; Hu, Y. Damage-induced BRCA1 phosphorylation by Chk2 contributes to the timing of end resection. Cell Cycle 2015, 14, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Wang, X.; Xue, X.; Li, L.; Hu, Y. A long noncoding RNA sensitizes genotoxic treatment by attenuating ATM activation and homologous recombination repair in cancers. PLoS Biol. 2020, 18, e3000666. [Google Scholar] [CrossRef]
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 2011, 11, 467–480. [Google Scholar] [CrossRef]
- Smith, T.B.; Dun, M.D.; Smith, N.D.; Curry, B.J.; Connaughton, H.S.; Aitken, R.J. The presence of a truncated base excision repair pathway in human spermatozoa that is mediated by OGG1. J. Cell Sci. 2013, 126, 1488–1497. [Google Scholar] [CrossRef]
- Khokhlova, E.V.; Fesenko, Z.S.; Sopova, J.V.; Leonova, E.I. Features of dna repair in the early stages of mammalian embryonic development. Genes 2020, 11, 1138. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.Y.; Yan, L.Y.; Wang, Z.B.; Luo, S.M.; Yeung, W.S.B.; Ou, X.H.; Sun, Q.Y.; Qiao, J. Meiotic chromatid recombination and segregation assessed with human single cell genome sequencing data. J. Med. Genet. 2019, 56, 156–163. [Google Scholar] [CrossRef]
- Cheung, V.; Yuen, V.M.; Wong, G.T.C.; Choi, S.W. The effect of sleep deprivation and disruption on DNA damage and health of doctors. Anaesthesia 2019, 74, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Zada, D.; Bronshtein, I.; Lerer-Goldshtein, T.; Garini, Y.; Appelbaum, L. Sleep increases chromosome dynamics to enable reduction of accumulating DNA damage in single neurons. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Sadoughi, F.; Hallajzadeh, J.; Asemi, Z.; Mansournia, M.A.; Alemi, F.; Yousefi, B. Signaling pathways involved in cell cycle arrest during the DNA breaks. DNA Repair 2021, 98, 103047. [Google Scholar] [CrossRef]
- Lee, D.H.; Chowdhury, D. What goes on must come off: Phosphatases gate-crash the DNA damage response. Trends Biochem. Sci. 2011, 36, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Ramos, F.; Villoria, M.T.; Alonso-Rodríguez, E.; Clemente-Blanco, A. Role of protein phosphatases PP1, PP2A, PP4 and Cdc14 in the DNA damage response. Cell Stress 2019, 3, 70–85. [Google Scholar] [CrossRef]
- Burdova, K.; Storchova, R.; Palek, M.; Macurek, L. WIP1 Promotes Homologous Recombination and Modulates Sensitivity to PARP Inhibitors. Cells 2019, 8, 1258. [Google Scholar] [CrossRef]
- Feringa, F.M.; Krenning, L.; Koch, A.; Van Den Berg, J.; Van Den Broek, B.; Jalink, K.; Medema, R.H. Hypersensitivity to DNA damage in antephase as a safeguard for genome stability. Nat. Commun. 2016, 7, 12618. [Google Scholar] [CrossRef]
- Hayashi, M.T.; Cesare, A.J.; Fitzpatrick, J.A.J.; Lazzerini-Denchi, E.; Karlseder, J. A telomere-dependent DNA damage checkpoint induced by prolonged mitotic arrest. Nat. Struct. Mol. Biol. 2012, 19, 387–394. [Google Scholar] [CrossRef]
- Kanellou, A.; Giakoumakis, N.N.; Panagopoulos, A.; Tsaniras, S.C.; Lygerou, Z. The licensing factor cdt1 links cell cycle progression to the DNA damage response. Anticancer Res. 2020, 40, 2449–2456. [Google Scholar] [CrossRef]
- Reyes, J.; Chen, J.Y.; Stewart-Ornstein, J.; Karhohs, K.W.; Mock, C.S.; Lahav, G. Fluctuations in p53 Signaling Allow Escape from Cell-Cycle Arrest. Mol. Cell 2018, 71, 581–591.e5. [Google Scholar] [CrossRef] [PubMed]
- Ngoi, N.Y.L.; Sundararajan, V.; Tan, D.S.P. Exploiting replicative stress in gynecological cancers as a therapeutic strategy. Int. J. Gynecol. Cancer 2020, 30, 1224–1238. [Google Scholar] [CrossRef] [PubMed]
- Matt, S.; Hofmann, T.G. The DNA damage-induced cell death response: A roadmap to kill cancer cells. Cell. Mol. Life Sci. 2016, 73, 2829–2850. [Google Scholar] [CrossRef] [PubMed]
- Oda, K.; Arakawa, H.; Tanaka, T.; Matsuda, K.; Tanikawa, C.; Mori, T.; Nishimori, H.; Tamai, K.; Tokino, T.; Nakamura, Y.; et al. p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by ser-46-phosphorylated p53. Cell 2000, 102, 849–862. [Google Scholar] [CrossRef]
- Cassina, A.; Silveira, P.; Cantu, L.; Montes, J.M.; Radi, R.; Sapiro, R. Defective human sperm cells are associated with mitochondrial dysfunction and oxidant production. Biol. Reprod. 2015, 93, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zeng, T.; Zhang, X.; Liu, C.; Wu, Z.; Yao, L.; Xie, C.; Xia, H.; Lin, Q.; Xie, L.; et al. ATR/Chk1 signaling induces autophagy through sumoylated RhoB-mediated lysosomal translocation of TSC2 after DNA damage. Nat. Commun. 2018, 9, 4139. [Google Scholar] [CrossRef]
- Zhou, X.; Liu, W.; Hu, X.; Dorrance, A.; Garzon, R.; Houghton, P.J.; Shen, C. Regulation of CHK1 by mTOR contributes to the evasion of DNA damage barrier of cancer cells. Sci. Rep. 2017, 7, 1535. [Google Scholar] [CrossRef]
- Shen, C.; Houghton, P.J. The mTOR pathway negatively controls ATM by up-regulating miRNAs. Proc. Natl. Acad. Sci. USA 2013, 110, 11869–11874. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.; Roelandt, R.; Bruggeman, I.; Estornes, Y.; Vandenabeele, P. Nuclear RIPK3 and MLKL contribute to cytosolic necrosome formation and necroptosis. Commun. Biol. 2018, 1, 6. [Google Scholar] [CrossRef] [PubMed]
- Toledo, L.; Neelsen, K.J.; Lukas, J. Replication Catastrophe: When a Checkpoint Fails because of Exhaustion. Mol. Cell 2017, 66, 735–749. [Google Scholar] [CrossRef] [PubMed]
- Stringer, J.M.; Winship, A.; Zerafa, N.; Wakefield, M.; Hutt, K. Oocytes can efficiently repair DNA double-strand breaks to restore genetic integrity and protect offspring health. Proc. Natl. Acad. Sci. USA 2020, 117, 11513–11522. [Google Scholar] [CrossRef] [PubMed]
- Stringer, J.M.; Winship, A.; Liew, S.H.; Hutt, K. The capacity of oocytes for DNA repair. Cell. Mol. Life Sci. 2018, 75, 2777–2792. [Google Scholar] [CrossRef] [PubMed]
- Gebel, J.; Tuppi, M.; Sänger, N.; Schumacher, B.; Dötsch, V. DNA Damaged Induced Cell Death in Oocytes. Molecules 2020, 25, 5714. [Google Scholar] [CrossRef] [PubMed]
- Rémillard-Labrosse, G.; Dean, N.L.; Allais, A.; Mihajlović, A.I.; Jin, S.G.; Son, W.Y.; Chung, J.T.; Pansera, M.; Henderson, S.; Mahfoudh, A.; et al. Human oocytes harboring damaged DNA can complete meiosis I. Fertil. Steril. 2020, 113, 1080–1089.e2. [Google Scholar] [CrossRef]
- Collins, J.K.; Jones, K.T. DNA damage responses in mammalian oocytes. Reproduction 2016, 152, R15–R22. [Google Scholar] [CrossRef]
- Collins, J.K.; Lane, S.I.R.; Merriman, J.A.; Jones, K.T. DNA damage induces a meiotic arrest in mouse oocytes mediated by the spindle assembly checkpoint. Nat. Commun. 2015, 6, 8553. [Google Scholar] [CrossRef]
- Archambeau, J.; Blondel, A.; Pedeux, R. Focus-ING on DNA integrity: Implication of ING proteins in cell cycle regulation and DNA repair modulation. Cancers 2020, 12, 58. [Google Scholar] [CrossRef]
- Aymard, F.; Aguirrebengoa, M.; Guillou, E.; Javierre, B.M.; Bugler, B.; Rocher, V.; Iacovoni, J.S.; Biernacka, A.; Skrzypczak, M.; Ginalski, K.; et al. Genome-wide mapping of long-range contacts unveils clustering of DNA double-strand breaks at damaged active genes. Nat. Struct. Mol. Biol. 2017, 24, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Aymard, F.; Bugler, B.; Schmidt, C.K.; Guillou, E.; Caron, P.; Briois, S.; Iacovoni, J.S.; Daburon, V.; Miller, K.M.; Jackson, S.P.; et al. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat. Struct. Mol. Biol. 2014, 21, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Lemaître, C.; Grabarz, A.; Tsouroula, K.; Andronov, L.; Furst, A.; Pankotai, T.; Heyer, V.; Rogier, M.; Attwood, K.M.; Kessler, P.; et al. Nuclear position dictates DNA repair pathway choice. Genes Dev. 2014, 28, 2450–2463. [Google Scholar] [CrossRef]
- van den Berg, J.; Joosten, S.E.; Kim, Y.; Manjón, A.G.; Krenning, L.; Koob, L.; Feringa, F.M.; Klompmaker, R.; van den Broek, B.; Jalink, K.; et al. DNA end-resection in highly accessible chromatin produces a toxic break. bioRxiv 2019. [Google Scholar] [CrossRef]
- Doksani, Y. The response to dna damage at telomeric repeats and its consequences for telomere function. Genes 2019, 10, 318. [Google Scholar] [CrossRef] [PubMed]
- Campos, A.; Clemente-Blanco, A. Cell cycle and DNA repair regulation in the damage response: Protein phosphatases take over the reins. Int. J. Mol. Sci. 2020, 21, 446. [Google Scholar] [CrossRef]
- Goodarzi, A.A.; Jonnalagadda, J.C.; Douglas, P.; Young, D.; Ye, R.; Moorhead, G.B.G.; Lees-Miller, S.P.; Khanna, K.K. Autophosphorylation of ataxia-telangiectasia mutated is regulated by protein phosphatase 2A. EMBO J. 2004, 23, 4451–4461. [Google Scholar] [CrossRef]
- Zhang, J.; Bao, S.; Furumai, R.; Kucera, K.S.; Ali, A.; Dean, N.M.; Wang, X.-F. Protein Phosphatase 5 Is Required for ATR-Mediated Checkpoint Activation. Mol. Cell. Biol. 2005, 25, 9910–9919. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Gao, F.; Wang, T.; Flagg, T.; Deng, X. A nonhomologous end-joining pathway is required for protein phosphatase 2A promotion of DNA double-strand break repair. Neoplasia 2009, 11, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Wechsler, T.; Chen, B.P.C.; Harper, R.; Morotomi-Yano, K.; Huang, B.C.B.; Meek, K.; Cleaver, J.E.; Chen, D.J.; Wabl, M. DNA-PKcs function regulated specifically by protein phosphatase 5. Proc. Natl. Acad. Sci. USA 2004, 101, 1247–1252. [Google Scholar] [CrossRef]
- Mi, J.; Dziegielewski, J.; Bolesta, E.; Brautigan, D.L.; Larner, J.M. Activation of DNA-PK by ionizing radiation is mediated by protein phosphatase 6. PLoS ONE 2009, 4, e4395. [Google Scholar] [CrossRef]
- Lu, X.; Nannenga, B.; Donehower, L. PPM1D dephosphorylates Chk1 and p53 and Abrogates Cell Cycle Checkpoints. Genes Dev. 2005, 1162–1174. [Google Scholar] [CrossRef]
- Oliva-Trastoy, M.; Berthonaud, V.; Chevalier, A.; Ducrot, C.; Marsolier-Kergoat, M.C.; Mann, C.; Leteurtre, F. The Wip1 phosphatase (PPM1D) antagonizes activation of the Chk2 tumour suppressor kinase. Oncogene 2007, 26, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Landsverk, H.B.; Mora-Bermúdez, F.; Landsverk, O.J.B.; Hasvold, G.; Naderi, S.; Bakke, O.; Ellenberg, J.; Collas, P.; Syljuåsen, R.G.; Küntziger, T. The protein phosphatase 1 regulator PNUTS is a new component of the DNA damage response. EMBO Rep. 2010, 11, 868–875. [Google Scholar] [CrossRef]
- Macurek, L.; Lindqvist, A.; Voets, O.; Kool, J.; Vos, H.R.; Medema, R.H. Wip1 phosphatase is associated with chromatin and dephosphorylates γh2AX to promote checkpoint inhibition. Oncogene 2010, 29, 2281–2291. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.M.; Pace, S.M.; Allen, S.R.; Deng, C.X.; Hsu, L.C. A PP1-binding motif present in BRCA1 plays a role in its DNA repair function. Int. J. Biol. Sci. 2008, 4, 352–361. [Google Scholar] [CrossRef]
- Kang, Y.; Lee, J.H.; Hoan, N.N.; Sohn, H.M.; Chang, I.Y.; You, H.J. Protein phosphatase 5 regulates the function of 53BP1 after neocarzinostatin-induced DNA damage. J. Biol. Chem. 2009, 284, 9845–9853. [Google Scholar] [CrossRef]
- Eissler, C.L.; Mazón, G.; Powers, B.L.; Savinov, S.N.; Symington, L.S.; Hall, M.C. The Cdk/Cdc14 Module Controls Activation of the Yen1 Holliday Junction Resolvase to Promote Genome Stability. Mol. Cell 2014, 54, 80–93. [Google Scholar] [CrossRef]
- Batchelor, E.; Mock, C.S.; Bhan, I.; Loewer, A.; Lahav, G. A Mechanism for Triggering p53 Pulses in Response to DNA Damage. Mol Cell. 2008, 53, 277–289. [Google Scholar] [CrossRef]
- Smolka, M. Fine-tuning the DNA damage response: Protein phosphatase 2A checks on CHK2. Cell Cycle 2010, 9, 861–869. [Google Scholar] [CrossRef][Green Version]
- Freeman, A.K.; Monteiro, A.N.A. Phosphatases in the cellular response to DNA damage. Cell Commun. Signal. 2010, 8, 27. [Google Scholar] [CrossRef]
- Lee, D.H.; Pan, Y.; Kanner, S.; Sung, P.; Borowiec, J.A.; Chowdhury, D. A PP4 phosphatase complex dephosphorylates RPA2 to facilitate DNA repair via homologous recombination. Nat. Struct. Mol. Biol. 2010, 17, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Kvaratskhelia, M.; Hess, S.; Qu, Y.; Zou, Y. Modulation of replication protein A function by its hyperphosphorylation- induced conformational change involving DNA binding domain B. J. Biol. Chem. 2005, 280, 32775–32783. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.R.; Boutell, C.; Keppler, M.; Densham, R.; Weekes, D.; Alamshah, A.; Butler, L.; Galanty, Y.; Pangon, L.; Kiuchi, T.; et al. The SUMO modification pathway is involved in the BRCA1 response to genotoxic stress. Nature 2009, 462, 886–890. [Google Scholar] [CrossRef]
- Liu, C.; Vyas, A.; Kassab, M.A.; Singh, A.K.; Yu, X. The role of poly ADP-ribosylation in the first wave of DNA damage response. Nucleic Acids Res. 2017, 45, 8129–8141. [Google Scholar] [CrossRef]
- Brown, J.S.; Lukashchuk, N.; Sczaniecka-Clift, M.; Britton, S.; le Sage, C.; Calsou, P.; Beli, P.; Galanty, Y.; Jackson, S.P. Neddylation Promotes Ubiquitylation and Release of Ku from DNA-Damage Sites. Cell Rep. 2015, 11, 704–714. [Google Scholar] [CrossRef]
- Guo, Z.; Wang, S.; Xie, Y.; Han, Y.; Hu, S.; Guan, H.; Xie, D.; Bai, C.; Liu, X.; Gu, Y.; et al. HUWE1-dependent DNA-PKcs neddylation modulates its autophosphorylation in DNA damage response. Cell Death Dis. 2020, 11. [Google Scholar] [CrossRef]
- Gong, F.; Chiu, L.Y.; Miller, K.M. Acetylation Reader Proteins: Linking Acetylation Signaling to Genome Maintenance and Cancer. PLoS Genet. 2016, 12, e1006272. [Google Scholar] [CrossRef]
- Chen, Y.; Zhu, W.G. Biological function and regulation of histone and non-histone lysine methylation in response to DNA damage. Acta Biochim. Biophys. Sin. 2016, 48, 603–616. [Google Scholar] [CrossRef]
- Liu, C.; Li, J. O-GlcNAc: A Sweetheart of the Cell Cycle and DNA Damage Response. Front. Endocrinol. 2018, 9, 415. [Google Scholar] [CrossRef]
- Efimova, E.V.; Appelbe, O.K.; Ricco, N.; Lee, S.S.Y.; Liu, Y.; Wolfgeher, D.J.; Collins, T.N.; Flor, A.C.; Ramamurthy, A.; Warrington, S.; et al. O-GlcN acylation enhances double-strand break repair, promotes cancer cell proliferation, and prevents therapy-induced senescence in irradiated tumors. Mol. Cancer Res. 2019, 17, 1338–1350. [Google Scholar] [CrossRef]
- Burger, K.; Ketley, R.F.; Gullerova, M. Beyond the Trinity of ATM, ATR, and DNA-PK: Multiple Kinases Shape the DNA Damage Response in Concert With RNA Metabolism. Front. Mol. Biosci. 2019, 6, 61. [Google Scholar] [CrossRef]
- Anindya, R.; Aygün, O.; Svejstrup, J.Q. Damage-Induced Ubiquitylation of Human RNA Polymerase II by the Ubiquitin Ligase Nedd4, but Not Cockayne Syndrome Proteins or BRCA1. Mol. Cell 2007, 28, 386–397. [Google Scholar] [CrossRef] [PubMed]
- Naro, C.; Bielli, P.; Pagliarini, V.; Sette, C. The interplay between DNA damage response and RNA processing: The unexpected role of splicing factors as gatekeepers of genome stability. Front. Genet. 2015, 6, 142. [Google Scholar] [CrossRef] [PubMed]
- García-Muse, T.; Aguilera, A. R Loops: From Physiological to Pathological Roles. Cell 2019, 179, 604–618. [Google Scholar] [CrossRef] [PubMed]
- Olazabal-Herrero, A.; Green, A.M.; Chen, X.; Sung, P.; Pillai, M.M.; Kupfer, G.M. Binding of FANCD2 to SRSF1 Splicing Factor Prevents Genomic Instability through R Loop Regulation. Blood 2020, 136, 19. [Google Scholar] [CrossRef]
- Tuduri, S.; Crabbé, L.; Conti, C.; Tourrière, H.; Holtgreve-Grez, H.; Jauch, A.; Pantesco, V.; De Vos, J.; Thomas, A.; Theillet, C.; et al. Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat. Cell Biol. 2010, 11. Erratum in 2010, 12, 1122, doi:10.1038/ncb1110-1122. [Google Scholar] [CrossRef]
- Li, X.; Manley, J.L. Inactivation of the SR protein splicing factor ASF/SF2 results in genomic instability. Cell 2005, 122, 365–378. [Google Scholar] [CrossRef]
- Aguilera, A.; Huertas, P. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol. Cell 2003, 12, 711–721. [Google Scholar]
- Gan, W.; Guan, Z.; Liu, J.; Gui, T.; Shen, K.; Manley, J.L.; Li, X. R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev. 2011, 25, 2041–2056. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A.; Gómez-González, B. DNA-RNA hybrids: The risks of DNA breakage during transcription. Nat. Struct. Mol. Biol. 2017, 24, 439–443. [Google Scholar] [CrossRef] [PubMed]
- Ohle, C.; Tesorero, R.; Schermann, G.; Dobrev, N.; Sinning, I.; Fischer, T. Transient RNA-DNA Hybrids Are Required for Efficient Double-Strand Break Repair. Cell 2016, 167, 1001–1013.e7. [Google Scholar] [CrossRef]
- Lu, W.T.; Hawley, B.R.; Skalka, G.L.; Baldock, R.A.; Smith, E.M.; Bader, A.S.; Malewicz, M.; Watts, F.Z.; Wilczynska, A.; Bushell, M. Drosha drives the formation of DNA:RNA hybrids around DNA break sites to facilitate DNA repair. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Mikolaskova, B.; Jurcik, M.; Cipakova, I.; Kretova, M.; Chovanec, M.; Cipak, L. Maintenance of genome stability: The unifying role of interconnections between the DNA damage response and RNA-processing pathways. Curr. Genet. 2018, 64, 971–983. [Google Scholar] [CrossRef] [PubMed]
- Palancade, B.; Rothstein, R. The Ultimate (Mis)match: When DNA Meets RNA. Cells 2021, 10, 1433. [Google Scholar] [CrossRef]
- Hatchi, E.; Skourti-Stathaki, K.; Ventz, S.; Pinello, L.; Yen, A.; Kamieniarz-Gdula, K.; Dimitrov, S.; Pathania, S.; McKinney, K.M.; Eaton, M.L.; et al. BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol. Cell 2015, 57, 636–647. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, V.; Valdés-Sánchez, L.; Rodriguez-Martinez, D.; Bhattacharya, S.S. Formation of 53BP1 foci and ATM activation under oxidative stress is facilitated by RNA:DNA hybrids and loss of ATM-53BP1 expression promotes photoreceptor cell survival in mice [version 1; peer review: 1 approved, 3 approved with reservations]. F1000Research 2018, 7, 1233. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.D.; Yadav, T.; Giri, S.; Saez, B.; Graubert, T.A.; Zou, L. Functions of Replication Protein A as a Sensor of R Loops and a Regulator of RNaseH1. Mol. Cell 2017, 65, 832–847.e4. [Google Scholar] [CrossRef]
- Kabeche, L.; Nguyen, H.D.; Buisson, R.; Zou, L. A mitosis-specific and R loop-driven ATR pathway promotes faithful chromosome segregation. Science 2018, 359, 108–114. [Google Scholar] [CrossRef]
- Ketley, R.F.; Gullerova, M. Jack of all trades? The versatility of RNA in DNA double-strand break repair. Essays Biochem. 2020, 64, 721–735. [Google Scholar] [CrossRef]
- Gioia, U.; Francia, S.; Cabrini, M.; Brambillasca, S.; Michelini, F.; Jones-Weinert, C.W.; d’Adda di Fagagna, F. Pharmacological boost of DNA damage response and repair by enhanced biogenesis of DNA damage response RNAs. Sci. Rep. 2019, 9, 6460. [Google Scholar] [CrossRef]
- Yuan, M.; Zhao, S.; Chen, R.; Wang, G.; Bie, Y.; Wu, Q.; Cheng, J. MicroRNA-138 inhibits tumor growth and enhances chemosensitivity in human cervical cancer by targeting H2AX. Exp. Ther. Med. 2019, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Lal, A.; Pan, Y.; Navarro, F.; Dykxhoorn, D.M.; Moreau, L.; Meire, E.; Bentwich, Z.; Lieberman, J.; Chowdhury, D. MiR-24-mediated downregulation of H2AX suppresses DNA repair in terminally differentiated blood cells. Nat. Struct. Mol. Biol. 2009, 16, 492–498. [Google Scholar] [CrossRef]
- Lai, J.; Yang, H.; Zhu, Y.; Ruan, M.; Huang, Y.; Zhang, Q. MiR-7-5p-mediated downregulation of PARP1 impacts DNA homologous recombination repair and resistance to doxorubicin in small cell lung cancer. BMC Cancer 2019, 19, 602. [Google Scholar] [CrossRef]
- Rezaeian, A.H.; Khanbabaei, H.; Calin, G.A. Therapeutic potential of the miRNA–ATM axis in the management of tumor radioresistance. Cancer Res. 2020, 80, 139–150. [Google Scholar] [CrossRef]
- Yan, D.; Ng, W.L.; Zhang, X.; Wang, P.; Zhang, Z.; Mo, Y.; Mao, H. Targeting DNA-PKcs and ATM with miR-101 Sensitizes Tumors to Radiation. Plos ONE 2010, 5, e11397. [Google Scholar] [CrossRef]
- Jia, X.Q.; Chen, Z.P.; Jiang, J.; Li, Y.; Kang, Z.; Wen, Z.P.; He, W.S.; Bin, L.W.; Yan, Y. MiR-182 regulates proliferation and apoptosis by targeting FBW7 in glioma cells. Int. J. Clin. Exp. Med. 2017, 10, 16085–16094. [Google Scholar]
- Huang, J.; Wang, Y.; Dhillon, K.K.; Calses, P.; Villegas, E. Systematic Screen Identi fi es miRNAs That Target RAD51 and RAD51D to Enhance Chemosensitivity. Mol. Cancer Res. 2013, 11, 1564–1574. [Google Scholar] [CrossRef]
- Song, L.; Dai, T.; Xie, Y.; Wang, C.; Lin, C.; Wu, Z.; Ying, Z.; Wu, J.; Li, M.; Li, J. Up-regulation of miR-1245 by c-myc targets BRCA2 and impairs DNA repair. J. Mol. Cell Biol. 2012, 4, 108–117. [Google Scholar] [CrossRef][Green Version]
- Liu, X.; Heng, C.; Li, Y.; Yu, L. MiR-302a sensitizes leukemia cells to etoposide by targeting Rad52. Oncotarget 2017, 8, 73884–73891. [Google Scholar] [CrossRef]
- Wang, Y.; Zeng, G.; Jiang, Y. The emerging roles of MiR-125b in cancers. Cancer Manag. Res. 2020, 12, 1079–1088. [Google Scholar] [CrossRef]
- Szpechcinski, A.; Florczuk, M.; Duk, K.; Zdral, A.; Rudzinski, S.; Bryl, M.; Czyzewicz, G.; Rudzinski, P.; Kupis, W.; Wojda, E.; et al. The expression of circulating miR-504 in plasma is associated with EGFR mutation status in non-small-cell lung carcinoma patients. Cell. Mol. Life Sci. 2019, 76, 3641–3656. [Google Scholar] [CrossRef]
- Cristini, A.; Groh, M.; Kristiansen, M.S.; Gromak, N. RNA/DNA Hybrid Interactome Identifies DXH9 as a Molecular Player in Transcriptional Termination and R-Loop-Associated DNA Damage. Cell Rep. 2018, 23, 1891–1905. [Google Scholar] [CrossRef]
- Bader, A.S.; Hawley, B.R.; Wilczynska, A.; Bushell, M. The roles of RNA in DNA double-strand break repair. Br. J. Cancer 2020, 122, 613–623. [Google Scholar] [CrossRef]
- Dimitrova, N.; Zamudio, J.R.; Jong, R.M.; Soukup, D.; Resnick, R.; Sarma, K.; Ward, A.J.; Raj, A.; Lee, J.T.; Sharp, P.A.; et al. LincRNA-p21 Activates p21 In cis to Promote Polycomb Target Gene Expression and to Enforce the G1/S Checkpoint. Mol. Cell 2014, 54, 777–790. [Google Scholar] [CrossRef]
- Santoni, G.; Morelli, M.B.; Nabissi, M.; Maggi, F.; Marinelli, O.; Santoni, M.; Amantini, C. Cross-talk between microRNAs, long non-coding RNAs and p21Cip1 in glioma: Diagnostic, prognostic and therapeutic roles. J. Cancer Metastasis Treat. 2020, 2020. [Google Scholar] [CrossRef]
- Chakraborty, A.; Tapryal, N.; Venkova, T.; Horikoshi, N.; Pandita, R.K.; Sarker, A.H.; Sarkar, P.S.; Pandita, T.K.; Hazra, T.K. Classical non-homologous end-joining pathway utilizes nascent RNA for error-free double-strand break repair of transcribed genes. Nat. Commun. 2016, 7, 13049. [Google Scholar] [CrossRef]
- Morio, T. Recent advances in the study of immunodeficiency and DNA damage response. Int. J. Hematol. 2017, 106, 357–365. [Google Scholar] [CrossRef]
- Nakad, R.; Schumacher, B. DNA damage response and immune defense: Links and mechanisms. Front. Genet. 2016, 7, 147. [Google Scholar] [CrossRef]
- Härtlova, A.; Erttmann, S.F.; Raffi, F.A.M.; Schmalz, A.M.; Resch, U.; Anugula, S.; Lienenklaus, S.; Nilsson, L.M.; Kröger, A.; Nilsson, J.A.; et al. DNA Damage Primes the Type I Interferon System via the Cytosolic DNA Sensor STING to Promote Anti-Microbial Innate Immunity. Immunity 2015, 42, 332–343. [Google Scholar] [CrossRef]
- Soriani, A.; Zingoni, A.; Cerboni, C.; Iannitto, M.L.; Ricciardi, M.R.; Di Gialleonardo, V.; Cippitelli, M.; Fionda, C.; Petrucci, M.T.; Guarini, A.; et al. ATM-ATR—dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood J. Am. Soc. Hematol. 2016, 113, 3503–3512. [Google Scholar] [CrossRef]
- Noble, P.W.; Bernatsky, S.; Clarke, A.E.; Isenberg, D.A.; Ramsey-Goldman, R.; Hansen, J.E. DNA-damaging autoantibodies and cancer: The lupus butterfly theory. Nat. Rev. Rheumatol. 2016, 12, 429–434. [Google Scholar] [CrossRef]
- Blatt, N.B.; Glick, G.D. Anti-DNA autoantibodies and systemic lupus erythematosus. Pharmacol. Ther. 1999, 83, 125–139. [Google Scholar] [CrossRef]
- Hansen, J.E.; Chan, G.; Liu, Y.; Hegan, D.C.; Dalal, S.; Dray, E.; Kwon, Y.; Xu, Y.; Xu, X.; Peterson-roth, E.; et al. Targeting Cancer with a Lupus Autoantibody. Sci. Transl. Med. 2012, 4, 157ra142. [Google Scholar] [CrossRef] [PubMed]
- Nastasi, C.; Mannarino, L.; D’Incalci, M. DNA Damage Response and Immune Defense. Int J Mol Sci. 2020, 21, 7504. [Google Scholar] [CrossRef] [PubMed]
- Erttmann, S.F.; Härtlova, A.; Sloniecka, M.; Raffi, F.A.; Hosseinzadeh, A.; Edgren, T.; Rofougaran, R.; Resch, U.; Fällman, M.; Ek, T.; et al. Loss of the DNA Damage Repair Kinase ATM Impairs Inflammasome-Dependent Anti-Bacterial Innate Immunity. Immunity 2016, 45, 106–118. [Google Scholar] [CrossRef]
- Sahan, A.Z.; Hazra, T.K.; Das, S. The Pivotal role of DNA repair in infection mediated-inflammation and cancer. Front. Microbiol. 2018, 9, 663. [Google Scholar] [CrossRef] [PubMed]
- Bode, C.; Fox, M.; Tewary, P.; Steinhagen, A.; Ellerkmann, R.K.; Klinman, D.; Baumgarten, G.; Hornung, V.; Steinhagen, F. Human plasmacytoid dentritic cells elicit a Type I Interferon response by sensing DNA via the cGAS-STING signaling pathway. Eur. J. Immunol. 2016, 46, 1615–1621. [Google Scholar] [CrossRef] [PubMed]
- Bennett, L.; Palucka, A.K.; Arce, E.; Cantrell, V.; Borvak, J.; Banchereau, J.; Pascual, V. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med. 2003, 197, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Hu, M.M.; Bian, L.J.; Liu, Y.; Chen, Q.; Shu, H.B. Phosphorylation of cGAS by CDK1 impairs self-DNA sensing in mitosis. Cell Discov. 2020, 6. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.S.; Lee, M.H.; Choi, M.H. Induction of pro-inflammatory cytokines by 29-kDa FN-f via cGAS/STING pathway. BMB Rep. 2019, 52, 336–341. [Google Scholar] [CrossRef]
- Rodriguez-iturbe, B.; Pons, H.; Johnson, R.J. Role of the immune system in hypertension. Physiol. Rev. 2017, 1127–1164. [Google Scholar] [CrossRef]
- Formanowicz, D.; Rybarczyk, A.; Radom, M. A Role of Inflammation and Immunity in Essential Hypertension—Modeled and Analyzed Using Petri Nets. Int. J. Mol. Sci. 2020, 21, 3348. [Google Scholar] [CrossRef]
- Czopek, A.; Moorhouse, R.; Farrah, T.; Lenoir, O.; Owen, E.; Van Bragt, J.; Costello, H.M.; Menolascina, F.; Webb, D.J.; Kluth, D.C.; et al. A novel role for myeloid endothelin-B receptors in hypertension. Eur. Heart J. 2019, 40, 768–784. [Google Scholar] [CrossRef]
- Kominsky, D.J.; Campbell, E.L.; Colgan, S.P. Metabolic Shifts in Immunity and Inflammation. J. Immunol. 2010, 184, 4062–4068. [Google Scholar] [CrossRef] [PubMed]
- Fulop, T.; Larbi, A.; Dupuis, G.; Le Page, A.; Frost, E.H.; Cohen, A.A.; Witkowski, J.M.; Franceschi, C. Immunosenescence and inflamm-aging as two sides of the same coin: Friends or Foes? Front. Immunol. 2018, 8, 1960. [Google Scholar] [CrossRef]
- Petr, M.A.; Tulika, T.; Carmona-Marin, L.M.; Scheibye-Knudsen, M. Protecting the Aging Genome. Trends Cell Biol. 2020, 30, 117–132. [Google Scholar] [CrossRef]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 155. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.E.; Ince, P.G.; Minett, T.; Matthews, F.E.; Heath, P.R.; Shaw, P.J.; Goodall, E.; Garwood, C.J.; Ratcliffe, L.E.; Brayne, C.; et al. Neuronal DNA damage response-associated dysregulation of signalling pathways and cholesterol metabolism at the earliest stages of Alzheimer-type pathology. Neuropathol. Appl. Neurobiol. 2016, 42, 167–179. [Google Scholar] [CrossRef]
- Abugable, A.A.; Morris, J.L.M.; Palminha, N.M.; Zaksauskaite, R.; Ray, S.; El-Khamisy, S.F. DNA repair and neurological disease: From molecular understanding to the development of diagnostics and model organisms. DNA Repair 2019, 81, 102669. [Google Scholar] [CrossRef]
- Kurihara, M.; Mano, T.; Saito, Y.; Murayama, S.; Toda, T.; Iwata, A. Colocalization of BRCA1 with Tau aggregates in human tauopathies. Brain Sci. 2020, 10, 7. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.M.R.; Rothblum-Oviatt, C.; Ellis, N.A.; Hickson, I.D.; Meyer, S.; Crawford, T.O.; Smogorzewska, A.; Pietrucha, B.; Weemaes, C.; Stewart, G.S. Chromosome instability syndromes. Nat. Rev. Dis. Prim. 2019, 5. [Google Scholar] [CrossRef]
- McKinnon, P.J. DNA repair deficiency and neurological disease. Nat. Rev. Neurosci. 2009, 10, 100–112. [Google Scholar] [CrossRef]
- Fang, E.F.; Scheibye-Knudsen, M.; Chua, K.F.; Mattson, M.P.; Croteau, D.L.; Bohr, V.A. Nuclear DNA damage signalling to mitochondria in ageing. Nat. Rev. Mol. Cell Biol. 2016, 17, 308–321. [Google Scholar] [CrossRef] [PubMed]
- Whittemore, K.; Vera, E.; Martínez-Nevado, E.; Sanpera, C.; Blasco, M.A. Telomere shortening rate predicts species life span. Proc. Natl. Acad. Sci. USA 2019, 116, 15122–15127. [Google Scholar] [CrossRef]
- Vizioli, M.G.; Liu, T.; Miller, K.N.; Robertson, N.A.; Gilroy, K.; Lagnado, A.B.; Perez-Garcia, A.; Kiourtis, C.; Dasgupta, N.; Lei, X.; et al. Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes Dev. 2020, 34, 428–445. [Google Scholar] [CrossRef]
- Tiwari, V.; Wilson, D.M. DNA Damage and Associated DNA Repair Defects in Disease and Premature Aging. Am. J. Hum. Genet. 2019, 105, 237–257. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.Y.; Heather, J.M.; Eisenhaure, T.; Garris, C.S.; Lieb, D.; Raychowdhury, R.; Hacohen, N. Extranuclear DNA accumulates in aged cells and contributes to senescence and inflammation. Aging Cell 2019, 18. [Google Scholar] [CrossRef]
- Tyrrell, D.J.; Blin, M.G.; Song, J.; Wood, S.C.; Goldstein, D.R. Aging impairs mitochondrial function and mitophagy and elevates interleukin 6 within the cerebral vasculature. J. Am. Heart Assoc. 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Scheibye-Knudsen, M.; Brace, L.E.; Kassahun, H.; Sengupta, T.; Nilsen, H.; Mitchell, J.R.; Croteau, D.L.; Bohr, V.A. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD +/SIRT1 reduction. Cell 2014, 157, 882–896. [Google Scholar] [CrossRef]
- Patel, J.; Baptiste, B.A.; Kim, E.; Hussain, M.; Croteau, D.L.; Bohr, V.A. DNA damage and mitochondria in cancer and aging. Carcinogenesis 2020, 41, 1625–1634. [Google Scholar] [CrossRef] [PubMed]
- Turan, V.; Oktay, K. BRCA-related ATM-mediated DNA double-strand break repair and ovarian aging. Hum. Reprod. Update 2020, 26, 43–57. [Google Scholar] [CrossRef]
- Lin, F.; Ma, X.S.; Wang, Z.B.; Wang, Z.W.; Luo, Y.B.; Huang, L.; Jiang, Z.Z.; Hu, M.W.; Schatten, H.; Sun, Q.Y. Different fates of oocytes with DNA double-strand breaks in vitro and in vivo. Cell Cycle 2014, 13, 2674–2680. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Hamatani, T.; Kamijo, S.; Iwai, M.; Kobanawa, M.; Ogawa, S.; Miyado, K.; Tanaka, M. Impact of Oxidative Stress on Age-Associated Decline in Oocyte Developmental Competence. Front. Endocrinol. 2019, 10, 811. [Google Scholar] [CrossRef]
- Shukla, P.C.; Singh, K.K.; Yanagawa, B.; Teoh, H.; Verma, S. DNA damage repair and cardiovascular diseases. Can. J. Cardiol. 2010, 26, 13A–16A. [Google Scholar] [CrossRef]
- Shukla, P.C.; Singh, K.K.; Quan, A.; Al-Omran, M.; Teoh, H.; Lovren, F.; Cao, L.; Rovira, I.I.; Pan, Y.; Brezden-Masley, C.; et al. BRCA1 is an essential regulator of heart function and survival following myocardial infarction. Nat. Commun. 2011, 2. [Google Scholar] [CrossRef]
- Higo, T.; Naito, A.T.; Sumida, T.; Shibamoto, M.; Okada, K.; Nomura, S.; Nakagawa, A.; Yamaguchi, T.; Sakai, T.; Hashimoto, A.; et al. DNA single-strand break-induced DNA damage response causes heart failure. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Uryga, A.; Gray, K.; Bennett, M. DNA Damage and Repair in Vascular Disease. Annu. Rev. Physiol. 2016, 78, 45–66. [Google Scholar] [CrossRef]
- Shah, A.; Gray, K.; Figg, N.; Finigan, A.; Starks, L.; Bennett, M. Defective base excision repair of oxidative DNA damage in vascular smooth muscle cells promotes atherosclerosis. Circulation 2018, 138, 1446–1462. [Google Scholar] [CrossRef]
- Ishida, T.; Ishida, M.; Tashiro, S.; Yoshizumi, M.; Kihara, Y. Role of DNA damage in cardiovascular disease. Circ. J. 2014, 78, 42–50. [Google Scholar] [CrossRef]
- Kumar, V.; Agrawal, R.; Pandey, A.; Kopf, S.; Hoeffgen, M.; Kaymak, S.; Bandapalli, O.R.; Gorbunova, V.; Seluanov, A.; Mall, M.A.; et al. Compromised DNA repair is responsible for diabetes-associated fibrosis. EMBO J. 2020, 39, e103477. [Google Scholar] [CrossRef] [PubMed]
- Tay, V.S.Y.; Devaraj, S.; Koh, T.; Ke, G.; Crasta, K.C.; Ali, Y. Increased double strand breaks in diabetic β-cells with a p21 response that limits apoptosis. Sci. Rep. 2019, 9, 19341. [Google Scholar] [CrossRef]
- Włodarczyk, M.; Nowicka, G. Obesity, DNA damage, and development of obesity-related diseases. Int. J. Mol. Sci. 2019, 20, 1146. [Google Scholar] [CrossRef] [PubMed]
- Vergoni, B.; Cornejo, P.J.; Gilleron, J.; Djedaini, M.; Ceppo, F.; Jacquel, A.; Bouget, G.; Ginet, C.; Gonzalez, T.; Maillet, J.; et al. DNA damage and the activation of the p53 pathway mediate alterations in metabolic and secretory functions of adipocytes. Diabetes 2016, 65, 3062–3074. [Google Scholar] [CrossRef]
- Setayesh, T.; Mi, M.; Langie, S.A.S.; Godschalk, R.; Waldherr, M.; Bauer, T.; Leitner, S.; Bichler, C.; Prager, G.; Krupitza, G.; et al. Impact of Weight Loss Strategies on Obesity-Induced DNA Damage. Mol. Nutr. Food Res. 2019, 63, 1900045. [Google Scholar] [CrossRef] [PubMed]
- Fehrmann, R.S.N.; Karjalainen, J.M.; Krajewska, M.; Westra, H.J.; Maloney, D.; Simeonov, A.; Pers, T.H.; Hirschhorn, J.N.; Jansen, R.C.; Schultes, E.A.; et al. Gene expression analysis identifies global gene dosage sensitivity in cancer. Nat. Genet. 2015, 47, 115–125. [Google Scholar] [CrossRef]
- Caudron-Herger, M.; Diederichs, S. Mitochondrial mutations in human cancer: Curation of translation. RNA Biol. 2018, 15, 62–69. [Google Scholar] [CrossRef]
- Yuan, Y.; Ju, Y.S.; Kim, Y.; Li, J.; Wang, Y.; Yoon, C.J.; Yang, Y.; Martincorena, I.; Creighton, C.J.; Weinstein, J.N.; et al. Comprehensive molecular characterization of mitochondrial genomes in human cancers. Nat. Genet. 2020, 52, 342–352. [Google Scholar] [CrossRef]
- Kurian, A.W.; Hughes, E.; Handorf, E.A.; Gutin, A.; Allen, B.; Hartman, A.-R.; Hall, M.J. Breast and Ovarian Cancer Penetrance Estimates Derived From Germline Multiple-Gene Sequencing Results in Women. JCO Precis. Oncol. 2017. [Google Scholar] [CrossRef]
- Kurian, A.W.; Ward, K.C.; Howlader, N.; Deapen, D.; Hamilton, A.S. Genetic Testing and Results in a Population-Based Cohort of Breast Cancer Patients and Ovarian Cancer Patients. J. Clin. Oncol. 2021, 37, 1305–1316. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef]
- Cieslik, M.; Chinnaiyan, A.M. News & views Global cancer genomics project comes to fruition. Nature 2020, 578, 40. [Google Scholar]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Kay, J.; Thadhani, E.; Samson, L.; Engelward, B. Inflammation-induced DNA damage, mutations and cancer. DNA Repair 2019, 83. [Google Scholar] [CrossRef]
- Axelrad, J.E.; Lichtiger, S.; Yajnik, V. Inflammatory bowel disease and cancer: The role of inflammation, immunosuppression, and cancer treatment. World J. Gastroenterol. 2016, 22, 4794–4801. [Google Scholar] [CrossRef] [PubMed]
- Aiello, I.; Mul Fedele, M.L.; Román, F.; Marpegan, L.; Caldart, C.; Chiesa, J.J.; Golombek, D.A.; Finkielstein, C.V.; Paladino, N. Circadian disruption promotes tumor-immune microenvironment remodeling favoring tumor cell proliferation. Sci. Adv. 2020, 6. [Google Scholar] [CrossRef] [PubMed]
- Veena, M.S.; Raychaudhuri, S.; Basak, S.K.; Venkatesan, N.; Kumar, P.; Biswas, R.; Chakrabarti, R.; Lu, J.; Su, T.; Gallagher-Jones, M.; et al. Dysregulation of hsa-miR-34a and hsa-miR-449a leads to overexpression of PACS-1 and loss of DNA damage response (DDR) in cervical cancer. J. Biol. Chem. 2020, 295, 17169–17186. [Google Scholar] [CrossRef]
- Fell, V.L.; Schild-Poulter, C. Ku Regulates Signaling to DNA Damage Response Pathways through the Ku70 von Willebrand A Domain. Mol. Cell. Biol. 2012, 32, 76–87. [Google Scholar] [CrossRef]
- Bian, L.; Meng, Y.; Zhang, M.; Li, D. MRE11-RAD50-NBS1 complex alterations and DNA damage response: Implications for cancer treatment. Mol. Cancer 2019, 18, 169. [Google Scholar] [CrossRef] [PubMed]
- Gudmundsdottir, K.; Ashworth, A. The roles of BRCA1 and BRCA2 and associated proteins in the maintenance of genomic stability. Oncogene 2006, 25, 5864–5874. [Google Scholar] [CrossRef] [PubMed]
- Rajendra, E.; Oestergaard, V.H.; Langevin, F.; Wang, M.; Dornan, G.L.; Patel, K.J.; Passmore, L.A. The Genetic and Biochemical Basis of FANCD2 Monoubiquitination. Mol. Cell 2014, 54, 858–869. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.R.; Bernstein, K.A. RAD-ical new insights into RAD51 regulation. Genes 2018, 9, 629. [Google Scholar] [CrossRef]
- Chen, L.; Huang, S.; Lee, L.; Davalos, A.; Schiestl, R.H.; Campisi, J.; Oshima, J. WRN, the protein deficient in Werner syndrome, plays a critical structural role in optimizing DNA repair. Aging Cell 2003, 2, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Shell, S.M.; Hawkins, E.K.; Tsai, M.S.; Hlaing, A.S.; Rizzo, C.J.; Chazin, W.J. Xeroderma pigmentosum complementation group C protein (XPC) serves as a general sensor of damaged DNA. DNA Repair 2013, 12, 947–953. [Google Scholar] [CrossRef]
- Ba, X.; Boldogh, I. 8-Oxoguanine DNA glycosylase 1: Beyond repair of the oxidatively modified base lesions. Redox Biol. 2018, 14, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Leppard, J.B.; Dong, Z.; Mackey, Z.B.; Tomkinson, A.E. Physical and Functional Interaction between DNA Ligase IIIα and Poly(ADP-Ribose) Polymerase 1 in DNA Single-Strand Break Repair. Mol. Cell. Biol. 2003, 23, 5919–5927. [Google Scholar] [CrossRef]
- O’Brien, V.; Brown, R. Signalling cell cycle arrest and cell death through the MMR System. Carcinogenesis 2006, 27, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, S.M.; Cerami, E.; Baras, A.; Pugh, T.J.; Schultz, N.; Stricker, T.; Lindsay, J.; Del Vecchio Fitz, C.; Kumari, P.; Micheel, C.; et al. AACR project genie: Powering precision medicine through an international consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef]
- Yap, T.A.; Plummer, R.; Azad, N.S.; Helleday, T. The DNA Damaging Revolution: PARP Inhibitors and Beyond. Am. Soc. Clin. Oncol. Educ. B 2019, 185–195. [Google Scholar] [CrossRef]
- Reuvers, T.G.A.; Kanaar, R.; Nonnekens, J. DNA damage-inducing anticancer therapies: From global to precision damage. Cancers 2020, 12, 2098. [Google Scholar] [CrossRef] [PubMed]
- Pilié, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef]
- Zhou, P.; Wang, J.; Mishail, D.; Wang, C.-Y. Recent advancements in PARP inhibitors-based targeted cancer therapy. Precis. Clin. Med. 2020, 3, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Gourley, C.; Balmaña, J.; Ledermann, J.A.; Serra, V.; Dent, R.; Loibl, S.; Pujade-Lauraine, E.; Boulton, S.J. Moving from poly (ADP-ribose) polymerase inhibition to targeting DNA repair and DNA damage response in cancer therapy. J. Clin. Oncol. 2019, 37, 2257–2269. [Google Scholar] [CrossRef] [PubMed]
- Laurini, E.; Marson, D.; Fermeglia, A.; Aulic, S.; Fermeglia, M.; Pricl, S. Role of Rad51 and DNA repair in cancer: A molecular perspective. Pharmacol. Ther. 2020, 208, 107492. [Google Scholar] [CrossRef]
- Yin, Y.; Chen, F. Targeting human MutT homolog 1 (MTH1) for cancer eradication: Current progress and perspectives. Acta Pharm. Sin. B 2020, 10, 2259–2271. [Google Scholar] [CrossRef]
- Schrempf, A.; Slyskova, J.; Loizou, J.I. Targeting the DNA Repair Enzyme Polymerase θ in Cancer Therapy. Trends Cancer 2021, 7, 98–111. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, H.; Lord, C.J.; Tutt, A.H.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Foskolou, I.P.; Jorgensen, C.; Leszczynska, K.B.; Olcina, M.M.; Tarhonskaya, H.; Haisma, B.; D’Angiolella, V.; Myers, W.K.; Domene, C.; Flashman, E.; et al. Ribonucleotide Reductase Requires Subunit Switching in Hypoxia to Maintain DNA Replication. Mol. Cell 2017, 66, 206–220.e9. [Google Scholar] [CrossRef]
- Xiao, Y.; Jin, L.; Deng, C.; Guan, Y.; Kalogera, E.; Ray, U.; Thirusangu, P.; Staub, J.; Sarkar Bhattacharya, S.; Xu, H.; et al. Inhibition of PFKFB3 induces cell death and synergistically enhances chemosensitivity in endometrial cancer. Oncogene 2021, 40, 1409–1424. [Google Scholar] [CrossRef]
- Karakaidos, P.; Karagiannis, D.; Rampias, T. Resolving DNA damage: Epigenetic regulation of DNA repair. Molecules 2020, 25, 2496. [Google Scholar] [CrossRef]
- Thurn, K.T.; Thomas, S.; Raha, P.; Qureshi, I.; Munster, P.N. Histone deacetylase regulation of ATM-mediated DNA damage signaling. Mol. Cancer Ther. 2013, 12, 2078–2087. [Google Scholar] [CrossRef]
- Loughery, J.E.P.; Dunne, P.D.; O’Neill, K.M.; Meehan, R.R.; McDaid, J.R.; Walsh, C.P. DNMT1 deficiency triggers mismatch repair defects in human cells through depletion of repair protein levels in a process involving the DNA damage response. Hum. Mol. Genet. 2011, 20, 3241–3255. [Google Scholar] [CrossRef]
- Topatana, W.; Juengpanich, S.; Li, S.; Cao, J.; Hu, J.; Lee, J.; Suliyanto, K.; Ma, D.; Zhang, B.; Chen, M.; et al. Advances in synthetic lethality for cancer therapy: Cellular mechanism and clinical translation. J. Hematol. Oncol. 2020, 13, 118. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Sachdev, E.; Tabatabai, R.; Roy, V.; Rimel, B.J.; Mita, M.M. PARP Inhibition in Cancer: An Update on Clinical Development. Target. Oncol. 2019, 14, 657–679. [Google Scholar] [CrossRef]
- Daud, A.I.; Ashworth, M.T.; Strosberg, J.; Goldman, J.W.; Mendelson, D.; Springett, G.; Venook, A.P.; Loechner, S.; Rosen, L.S.; Shanahan, F.; et al. Phase I dose-escalation trial of checkpoint kinase 1 inhibitor MK-8776 as monotherapy and in combination with gemcitabine in patients with advanced solid tumors. J. Clin. Oncol. 2015, 33, 1060–1066. [Google Scholar] [CrossRef] [PubMed]
- Leijen, S.; Van Geel, R.M.J.M.; Pavlick, A.C.; Tibes, R.; Rosen, L.; Razak, A.R.A.; Lam, R.; Demuth, T.; Rose, S.; Lee, M.A.; et al. Phase I study evaluating WEE1 inhibitor AZD1775 as monotherapy and in combination with gemcitabine, cisplatin, or carboplatin in patients with advanced solid tumors. J. Clin. Oncol. 2016, 34, 4371–4380. [Google Scholar] [CrossRef] [PubMed]
- Di Rorà, A.G.L.; Bocconcelli, M.; Ferrari, A.; Terragna, C.; Bruno, S.; Imbrogno, E.; Beeharry, N.; Robustelli, V.; Ghetti, M.; Napolitano, R.; et al. Synergism through WEE1 and CHK1 inhibition in acute lymphoblastic leukemia. Cancers 2019, 11, 1654. [Google Scholar] [CrossRef]
- Munster, P.; Mita, M.; Mahipal, A.; Nemunaitis, J.; Massard, C.; Mikkelsen, T.; Cruz, C.; Paz-Ares, L.; Hidalgo, M.; Rathkopf, D.; et al. First-in-human phase i study of a dual mTOR kinase and DNA-PK inhibitor (CC-115) in advanced malignancy. Cancer Manag. Res. 2019, 11, 10463–10476. [Google Scholar] [CrossRef]
- Jette, N.R.; Kumar, M.; Radhamani, S.; Arthur, G.; Goutam, S.; Yip, S.; Kolinsky, M.; Williams, G.J.; Bose, P.; Lees-Miller, S.P. ATM-deficient cancers provide new opportunities for precision oncology. Cancers 2020, 12, 687. [Google Scholar] [CrossRef]
- Neeb, A.; Herranz, N.; Arce-Gallego, S.; Miranda, S.; Buroni, L.; Yuan, W.; Athie, A.; Casals, T.; Carmichael, J.; Rodrigues, D.N.; et al. Advanced Prostate Cancer with ATM Loss: PARP and ATR Inhibitors. Eur. Urol. 2021, 79, 200–211. [Google Scholar] [CrossRef]
- Southgate, H.E.D.; Chen, L.; Tweddle, D.A.; Curtin, N.J. ATR Inhibition Potentiates PARP Inhibitor Cytotoxicity in High Risk Neuroblastoma Cell Lines by Multiple Mechanisms. Cancers 2020, 12, 1095. [Google Scholar] [CrossRef]
- Higgins, G.S.; Prevo, R.; Lee, Y.F.; Helleday, T.; Muschel, R.J.; Taylor, S.; Yoshimura, M.; Hickson, I.D.; Bernhard, E.J.; McKenna, W.G. A small interfering RNA screen of genes involved in DNA repair identifies tumor-specific radiosensitization by POLQ knockdown. Cancer Res. 2010, 70, 2984–2993. [Google Scholar] [CrossRef] [PubMed]
- George, S.L.; Lorenzi, F.; King, D.; Hartlieb, S.; Campbell, J.; Pemberton, H.; Toprak, U.H.; Barker, K.; Tall, J.; da Costa, B.M.; et al. Therapeutic vulnerabilities in the DNA damage response for the treatment of ATRX mutant neuroblastoma. EBioMedicine 2020, 59, 102971. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, A.; Fernandes, M.; Catarino, R.; Medeiros, R. RAD52 functions in homologous recombination and its importance on genomic integrity maintenance and cancer therapy. Cancers 2019, 11, 1622. [Google Scholar] [CrossRef] [PubMed]
- Ciccarone, F.; Zampieri, M.; Caiafa, P. PARP1 orchestrates epigenetic events setting up chromatin domains. Semin. Cell Dev. Biol. 2017, 63, 123–134. [Google Scholar] [CrossRef]
- Muvarak, N.E.; Chowdhury, K.; Xia, L.; Robert, C.; Choi, E.Y.; Cai, Y.; Bellani, M.; Zou, Y.; Singh, Z.N.; Duong, V.H.; et al. Enhancing the Cytotoxic Effects of PARP Inhibitors with DNA Demethylating Agents—A Potential Therapy for Cancer. Cancer Cell 2016, 30, 637–650. [Google Scholar] [CrossRef]
- Marijon, H.; Lee, D.H.; Ding, L.W.; Sun, H.; Gery, S.; de Gramont, A.; Koeffler, H.P. Co-targeting poly(ADP-ribose) polymerase (PARP) and histone deacetylase (HDAC) in triple-negative breast cancer: Higher synergism in BRCA mutated cells. Biomed. Pharmacother. 2018, 99, 543–551. [Google Scholar] [CrossRef]
- Park, Y.; Chui, M.H.; Rahmanto, Y.S.; Yu, Z.C.; Shamanna, R.A.; Bellani, M.A.; Gaillard, S.; Ayhan, A.; Viswanathan, A.; Seidman, M.M.; et al. Loss of ARID1A in tumor cells renders selective vulnerability to combined ionizing radiation and PARP inhibitor therapy. Clin. Cancer Res. 2019, 25, 5584–5593. [Google Scholar] [CrossRef]
- Abad, E.; Graifer, D.; Lyakhovich, A. DNA damage response and resistance of cancer stem cells. Cancer Lett. 2020, 474, 106–117. [Google Scholar] [CrossRef]
- Mir, S.E.; Hamer, P.C.D.W.; Krawczyk, P.M.; Balaj, L.; Claes, A.; Niers, J.M.; Van Tilborg, A.A.G.; Zwinderman, A.H.; Geerts, D.; Kaspers, G.J.L.; et al. Article In Silico Analysis of Kinase Expression Identifies WEE1 as a Gatekeeper against Mitotic Catastrophe in Glioblastoma. Cancer Cell 2010, 244–257. [Google Scholar] [CrossRef]
- Ahmed, S.U.; Carruthers, R.; Gilmour, L.; Yildirim, S.; Watts, C.; Chalmers, A.J. Selective Inhibition of Parallel DNA Damage Response Pathways Optimizes Radiosensitization of Glioblastoma Stem-like Cells. Cancer Res. 2015, 75, 4416–4428. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhang, Y.; Chen, S.; Kmieciak, M.; Leng, Y.; Lin, H.; Rizzo, K.A.; Dumur, C.I.; Ferreira-Gonzalez, A.; Dai, Y.; et al. A regimen combining the Wee1 inhibitor AZD1775 with HDAC inhibitors targets human acute myeloid leukemia cells harboring various genetic mutations. Leukemia 2015, 29, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Jeggo, P.A.; Pearl, L.H.; Carr, A.M. DNA repair, genome stability and cancer: A historical perspective. Nat. Rev. Cancer 2016, 16, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Marabitti, V.; Lillo, G.; Malacaria, E.; Palermo, V.; Pichierri, P.; Franchitto, A. Checkpoint defects elicit a WRNIP1-mediated response to counteract R-loop-associated genomic instability. Cancers 2020, 12, 389. [Google Scholar] [CrossRef]
- Park, S.R.; Namkoong, S.; Friesen, L.; Cho, C.S.; Zhang, Z.Z.; Chen, Y.C.; Yoon, E.; Kim, C.H.; Kwak, H.; Kang, H.M.; et al. Single-Cell Transcriptome Analysis of Colon Cancer Cell Response to 5-Fluorouracil-Induced DNA Damage. Cell Rep. 2020, 32, 108077. [Google Scholar] [CrossRef]
- Sfeir, A.; Kosiyatrakul, S.T.; Hockemeyer, D.; MacRae, S.L.; Karlseder, J.; Schildkraut, C.L.; de Lange, T. Mammalian Telomeres Resemble Fragile Sites and Require TRF1 for Efficient Replication. Cell 2009, 138, 90–103. [Google Scholar] [CrossRef]
- Parkinson, G.N.; Lee, M.P.H.; Neidle, S. Crystal structure of parallel quadruplexes from human telomeric DNA. Nature 2002, 417, 876–880. [Google Scholar] [CrossRef]
- Masamsetti, V.P.; Low, R.R.J.; Mak, K.S.; O’Connor, A.; Riffkin, C.D.; Lamm, N.; Crabbe, L.; Karlseder, J.; Huang, D.C.S.; Hayashi, M.T.; et al. Replication stress induces mitotic death through parallel pathways regulated by WAPL and telomere deprotection. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Van Ly, D.; Low, R.R.J.; Frölich, S.; Bartolec, T.K.; Kafer, G.R.; Pickett, H.A.; Gaus, K.; Cesare, A.J. Telomere Loop Dynamics in Chromosome End Protection. Mol. Cell 2018, 71, 510–525.e6. [Google Scholar] [CrossRef] [PubMed]
- Cesare, A.J.; Kaul, Z.; Cohen, S.B.; Napier, C.E.; Pickett, H.A.; Neumann, A.A.; Reddel, R.R. Spontaneous occurrence of telomeric DNA damage response in the absence of chromosome fusions. Nat. Struct. Mol. Biol. 2009, 16, 1244–1251. [Google Scholar] [CrossRef]
- Cesare, A.J.; Hayashi, M.T.; Crabbe, L.; Karlseder, J. The Telomere deprotection response is functionally distinct from the Genomic DNA damage response. Mol. Cell 2013, 51, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Rossiello, F.; Aguado, J.; Sepe, S.; Iannelli, F.; Nguyen, Q.; Pitchiaya, S.; Carninci, P.; Di Fagagna, F.D.A. DNA damage response inhibition at dysfunctional telomeres by modulation of telomeric DNA damage response RNAs. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Rankin, A.M.; Forman, L.; Sarkar, S.; Faller, D.V. Enhanced cytotoxicity from deoxyguanosine-enriched T-oligo in prostate cancer cells. Nucleic Acid Ther. 2013, 23, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Puri, N.; Pitman, R.T.; Mulnix, R.E.; Erickson, T.; Iness, A.N.; Vitali, C.; Zhao, Y.; Salgia, R. Non-small cell lung cancer is susceptible to induction of DNA damage responses and inhibition of angiogenesis by telomere overhang oligonucleotides. Cancer Lett. 2014, 343, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Weng, D.; Cunin, M.C.; Song, B.; Price, B.D.; Eller, M.S.; Gilchrest, B.A.; Calderwood, S.K.; Gong, J. Radiosensitization of mammary carcinoma cells by telomere homolog oligonucleotide pretreatment. Breast Cancer Res. 2010, 12. [Google Scholar] [CrossRef]
- Martínez, P.; Thanasoula, M.; Muñoz, P.; Liao, C.; Tejera, A.; McNees, C.; Flores, J.M.; Fernández-Capetillo, O.; Tarsounas, M.; Blasco, M.A. Increased telomere fragility and fusions resulting from TRF1 deficiency lead to degenerative pathologies and increased cancer in mice. Genes Dev. 2009, 23, 2060–2075. [Google Scholar] [CrossRef] [PubMed]
- Casagrande, V.; Salvati, E.; Alvino, A.; Bianco, A.; Ciammaichella, A.; D’Angelo, C.; Ginnari-Satriani, L.; Serrilli, A.M.; Iachettini, S.; Leonetti, C.; et al. N-cyclic bay-substituted perylene g-quadruplex ligands have selective antiproliferative effects on cancer cells and induce telomere damage. J. Med. Chem. 2011, 54, 1140–1156. [Google Scholar] [CrossRef] [PubMed]
- Kosiol, N.; Juranek, S.; Brossart, P.; Heine, A.; Paeschke, K. G-quadruplexes: A promising target for cancer therapy. Mol. Cancer 2021, 20, 40. [Google Scholar] [CrossRef]
- Beniaminov, A.D.; Novikov, R.A.; Mamaeva, O.K.; Mitkevich, V.A.; Smirnov, I.P.; Livshits, M.A.; Shchyolkina, A.K.; Kaluzhny, D.N. Light-induced oxidation of the telomeric G4 DNA in complex with Zn(II) tetracarboxymethyl porphyrin. Nucleic Acids Res. 2016, 44, 10031–10041. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.N.; Zhao, S.J.; Wu, B.; Li, X.Z.; Kong, D.M. A new cationic porphyrin derivative (TMPipEOPP) with large side arm substituents: A highly selective G-quadruplex optical probe. PLoS ONE 2012, 7, e35586. [Google Scholar] [CrossRef]
- Salvati, E.; Leonetti, C.; Rizzo, A.; Scarsella, M.; Mottolese, M.; Galati, R.; Sperduti, I.; Stevens, M.F.G.; D’Incalci, M.; Blasco, M.; et al. Telomere damage induced by the G-quadruplex ligand RHPS4 has an antitumor effect. J. Clin. Investig. 2007, 117, 3236–3247. [Google Scholar] [CrossRef] [PubMed]
- Salvati, E.; Rizzo, A.; Iachettini, S.; Zizza, P.; Cingolani, C.; D’Angelo, C.; Porru, M.; Mondello, C.; Aiello, A.; Farsetti, A.; et al. A basal level of DNA damage and telomere deprotection increases the sensitivity of cancer cells to G-quadruplex interactive compounds. Nucleic Acids Res. 2015, 43, 1759–1769. [Google Scholar] [CrossRef]
- Haddach, M.; Schwaebe, M.K.; Michaux, J.; Nagasawa, J.; O’Brien, S.E.; Whitten, J.P.; Pierre, F.; Kerdoncuff, P.; Darjania, L.; Stansfield, R.; et al. Discovery of CX-5461, the first direct and selective inhibitor of RNA polymerase I, for cancer therapeutics. ACS Med. Chem. Lett. 2012, 3, 602–606. [Google Scholar] [CrossRef]
- Bruno, P.M.; Lu, M.; Dennis, K.A.; Inam, H.; Moore, C.J.; Sheehe, J.; Elledge, S.J.; Hemann, M.T.; Pritchard, J.R. The primary mechanism of cytotoxicity of the chemotherapeutic agent CX-5461 is topoisomerase II poisoning. Proc. Natl. Acad. Sci. USA 2020, 117, 4053–4060. [Google Scholar] [CrossRef] [PubMed]
- Bossaert, M.; Pipier, A.; Riou, J.F.; Noirot, C.; Nguyên, L.T.; Serre, R.F.; Bouchez, O.; Defrancq, E.; Calsou, P.; Britton, S.; et al. Transcription-associated topoisomerase 2α (TOP2A) activity is a major effector of cytotoxicity induced by G-quadruplex ligands. Elife 2021, 10, e65184. [Google Scholar] [CrossRef]
- Xu, H.; Di Antonio, M.; McKinney, S.; Mathew, V.; Ho, B.; O’Neil, N.J.; Santos, N.D.; Silvester, J.; Wei, V.; Garcia, J.; et al. CX-5461 is a DNA G-quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Khot, A.; Brajanovski, N.; Cameron, D.P.; Hein, N.; Maclachlan, K.H.; Sanij, E.; Lim, J.; Soong, J.; Link, E.; Blombery, P.; et al. First-in-human RNA polymerase I transcription inhibitor CX-5461 in patients with advanced hematologic cancers: Results of a phase I dose-escalation study. Cancer Discov. 2019, 9, 1036–1049. [Google Scholar] [CrossRef]
- Liang, X.; Li, D.; Leng, S.; Zhu, X. RNA-based pharmacotherapy for tumors: From bench to clinic and back. Biomed. Pharmacother. 2020, 125, 109997. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.W.; Dreaden, E.C.; Morandell, S.; Zhou, W.; Dhara, S.S.; Sriram, G.; Lam, F.C.; Patterson, J.C.; Quadir, M.; Dinh, A.; et al. Enhancing chemotherapy response through augmented synthetic lethality by co-targeting nucleotide excision repair and cell-cycle checkpoints. Nat. Commun. 2020, 11, 4124. [Google Scholar] [CrossRef]
- Peaucellier, G.; Shartzer, K.; Jiang, W.; Maggio, K.; Kinsey, W.H. Anti-peptide Antibody Identifies a 57 kDa Protein Tyrosine Kinase in the Sea Urchin Egg Cortex: Tyrosine kinase/fertilization/src/egg/antibody. Dev. Growth Differ. 1993, 35. [Google Scholar] [CrossRef]
- Jackson, M.R.; Bavelaar, B.M.; Waghorn, P.A.; Gill, M.R.; El-Sagheer, A.H.; Brown, T.; Tarsounas, M.; Vallis, K.A. Radiolabeled oligonucleotides targeting the RNA subunit of telomerase inhibit telomerase and induce DNA damage in telomerase-positive cancer cells. Cancer Res. 2019, 79, 4627–4637. [Google Scholar] [CrossRef]
- Girard, P.M.; Berthault, N.; Kozlac, M.; Ferreira, S.; Jdey, W.; Bhaskara, S.; Alekseev, S.; Thomas, F.; Dutreix, M. Evolution of tumor cells during AsiDNA treatment results in energy exhaustion, decrease in responsiveness to signal, and higher sensitivity to the drug. Evol. Appl. 2020, 13, 1673–1680. [Google Scholar] [CrossRef] [PubMed]
- Dragomir, M.P.; Kopetz, S.; Ajani, J.A.; Calin, G.A. Non-coding RNAs in GI cancers: From cancer hallmarks to clinical utility. Gut 2020, 69, 748–763. [Google Scholar] [CrossRef]
- Heestand, G.M.; Schwaederle, M.; Gatalica, Z.; Arguello, D.; Kurzrock, R. Topoisomerase expression and amplification in solid tumours: Analysis of 24,262 patients. Eur. J. Cancer 2017, 83, 80–87. [Google Scholar] [CrossRef]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef]
- Pommier, Y.; Cushman, M.; Doroshow, J.H. Novel clinical indenoisoquinoline topoisomerase I inhibitors: A twist around the camptothecins. Oncotarget 2018, 9, 37286–37288. [Google Scholar] [CrossRef]
- Thomas, A.; Pommier, Y. Targeting topoisomerase I in the era of precision medicine. Clin. Cancer Res. 2019, 25, 6581–6589. [Google Scholar] [CrossRef]
- Skok, Ž.; Zidar, N.; Kikelj, D.; Ilaš, J. Dual Inhibitors of Human DNA Topoisomerase II and Other Cancer-Related Targets. J. Med. Chem. 2020, 63, 884–904. [Google Scholar] [CrossRef]
- Wang, W.; Rodriguez-Silva, M.; Acanda de la Rocha, A.M.; Wolf, A.L.; Lai, Y.; Liu, Y.; Reinhold, W.C.; Pommier, Y.; Chambers, J.W.; Tse-Dinh, Y.C. Tyrosyl-DNA phosphodiesterase 1 and topoisomerase I activities as predictive indicators for Glioblastoma susceptibility to genotoxic agents. Cancers 2019, 11, 1416. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Min, A.; Im, S.A.; Jang, H.; Kim, Y.J.; Kim, H.J.; Lee, K.H.; Kim, T.Y.; Lee, K.W.; Oh, D.Y.; et al. TDP1 and TOP1 modulation in Olaparib-resistant cancer determines the efficacy of subsequent chemotherapy. Cancers 2020, 12, 334. [Google Scholar] [CrossRef]
- Infante Lara, L.; Fenner, S.; Ratcliffe, S.; Isidro-Llobet, A.; Hann, M.; Bax, B.; Osheroff, N. Coupling the core of the anticancer drug etoposide to an oligonucleotide induces topoisomerase II-mediated cleavage at specific DNA sequences. Nucleic Acids Res. 2018, 46, 2218–2233. [Google Scholar] [CrossRef]
- Molejon, M.I.; Weiz, G.; Breccia, J.D.; Vaccaro, M.I. Glycoconjugation: An approach to cancer therapeutics. World J. Clin. Oncol. 2020, 11, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Beck, D.E.; Agama, K.; Marchand, C.; Chergui, A.; Pommier, Y.; Cushman, M. Synthesis and biological evaluation of new carbohydrate-substituted indenoisoquinoline topoisomerase I inhibitors and improved syntheses of the experimental anticancer agents indotecan (LMP400) and indimitecan (LMP776). J. Med. Chem. 2014, 57, 1495–1512. [Google Scholar] [CrossRef]
- Ma, Y.; Chen, H.; Su, S.; Wang, T.; Zhang, C.; Fida, G.; Cui, S.; Zhao, J.; Gu, Y. Galactose as broad ligand for multiple tumor imaging and therapy. J. Cancer 2015, 6, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Cui, S.; Li, S.; Du, C.; Tian, J.; Wan, S.; Qian, Z.; Gu, Y.; Chen, W.R.; Wang, G. Targeted cancer therapy with a 2-deoxyglucose-based adriamycin complex. Cancer Res. 2013, 73, 1362–1373. [Google Scholar] [CrossRef]
- Puyo, S.; Montaudon, D.; Pourquier, P. From old alkylating agents to new minor groove binders. Crit. Rev. Oncol. Hematol. 2014, 89, 43–61. [Google Scholar] [CrossRef]
- Spitaleri, G.; Matei, D.V.; Curigliano, G.; Detti, S.; Verweij, F.; Zambito, S.; Scardino, E.; Rocco, B.; Nolè, F.; Ariu, L.; et al. Phase II trial of estramustine phosphate and oral etoposide in patients with hormone-refractory prostate cancer. Ann. Oncol. 2009, 20, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, A.; Herkommer, K.; Glück, M.; Speit, G. DNA-damaging effect of cyclophosphamide on human blood cells in vivo and in vitro studied with the single-cell gel test (comet assay). Environ. Mol. Mutagen. 1995, 25, 180–187. [Google Scholar] [CrossRef]
- Yuan, W.X.; Gui, Y.X.; Na, W.N.; Chao, J.; Yang, X. Circulating microRNA-125b and microRNA-130a expression profiles predict chemoresistance to R-CHOP in diffuse large B-cell lymphoma patients. Oncol. Lett. 2016, 11, 423–432. [Google Scholar] [CrossRef]
- Hu, J.; Lieb, J.D.; Sancar, A.; Adar, S. Cisplatin DNA damage and repair maps of the human genome at single-nucleotide resolution. Proc. Natl. Acad. Sci. USA 2016, 113, 11507–11512. [Google Scholar] [CrossRef]
- Woods, D.; Turchi, J.J. Chemotherapy induced DNA damage response Convergence of drugs and pathways. Cancer Biol. Ther. 2013, 14, 379–389. [Google Scholar] [CrossRef]
- Zhang, P.; Sadler, P.J. Advances in the design of organometallic anticancer complexes. J. Organomet. Chem. 2017, 839, 5–14. [Google Scholar] [CrossRef]
- Molinaro, C.; Martoriati, A.; Pelinski, L.; Cailliau, K. Copper complexes as anticancer agents targeting topoisomerases i and ii. Cancers 2020, 12, 2863. [Google Scholar] [CrossRef]
- Fast, O.G.; Gentry, B.; Strouth, L.; Niece, M.B.; Beckford, F.A.; Shell, S.M. Polynuclear ruthenium organometallic compounds induce DNA damage in human cells identified by the nucleotide excision repair factor XPC. Biosci. Rep. 2019, 39, BSR20190378. [Google Scholar] [CrossRef]
- Wambang, N.; Schifano-Faux, N.; Martoriati, A.; Henry, N.; Baldeyrou, B.; Bal-Mahieu, C.; Bousquet, T.; Pellegrini, S.; Meignan, S.; Cailliau, K.; et al. Synthesis, Structure, and Antiproliferative Activity of Ruthenium(II) Arene Complexes of Indenoisoquinoline Derivatives. Organometallics 2016, 35. [Google Scholar] [CrossRef]
- Murray, V.; Chen, J.K.; Chung, L.H. The interaction of the metallo-glycopeptide anti-tumour drug bleomycin with DNA. Int. J. Mol. Sci. 2018, 19, 1372. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Kim, H.S.; Seo, Y.R. Understanding of ROS-Inducing Strategy in Anticancer Therapy. Oxid. Med. Cell. Longev. 2019, 2019. [Google Scholar] [CrossRef]
- Magkouta, S.F.; Pappas, A.G.; Vaitsi, P.C.; Agioutantis, P.C.; Pateras, I.S.; Moschos, C.A.; Iliopoulou, M.P.; Kosti, C.N.; Loutrari, H.V.; Gorgoulis, V.G.; et al. MTH1 favors mesothelioma progression and mediates paracrine rescue of bystander endothelium from oxidative damage. JCI Insight 2020, 5, e134885. [Google Scholar] [CrossRef]
- Doudna, J.A.; Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.D.; Richardson, C.D.; Corn, J.E. Advances in genome editing through control of DNA repair pathways. Nat. Cell Biol. 2019, 21, 1468–1478. [Google Scholar] [CrossRef]
- Charpentier, M.; Khedher, A.H.Y.; Menoret, S.; Brion, A.; Lamribet, K.; Dardillac, E.; Boix, C.; Perrouault, L.; Tesson, L.; Geny, S.; et al. CtIP fusion to Cas9 enhances transgene integration by homology-dependent repair. Nat. Commun. 2018, 9, 1133. [Google Scholar] [CrossRef] [PubMed]
- Jayavaradhan, R.; Pillis, D.M.; Goodman, M.; Zhang, F.; Zhang, Y.; Andreassen, P.R.; Malik, P. CRISPR-Cas9 fusion to dominant-negative 53BP1 enhances HDR and inhibits NHEJ specifically at Cas9 target sites. Nat. Commun. 2019, 10, 2866. [Google Scholar] [CrossRef]
- Benitez, E.K.; Lomova Kaufman, A.; Cervantes, L.; Clark, D.N.; Ayoub, P.G.; Senadheera, S.; Osborne, K.; Sanchez, J.M.; Crisostomo, R.V.; Wang, X.; et al. Global and Local Manipulation of DNA Repair Mechanisms to Alter Site-Specific Gene Editing Outcomes in Hematopoietic Stem Cells. Front. Genome Ed. 2020, 2. [Google Scholar] [CrossRef]
- Deutsch, E.; Chargari, C.; Galluzzi, L.; Kroemer, G. Optimising efficacy and reducing toxicity of anticancer radioimmunotherapy. Lancet Oncol. 2019, 20, e452–e463. [Google Scholar] [CrossRef]
- Huang, R.X.; Zhou, P.K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct. Target. Ther. 2020, 5. [Google Scholar] [CrossRef]
- Biau, J.; Chautard, E.; Verrelle, P.; Dutreix, M. Altering DNA repair to improve radiation therapy: Specific and multiple pathway targeting. Front. Oncol. 2019, 9, 1009. [Google Scholar] [CrossRef] [PubMed]
- de Jong, M.; Breeman, W.A.; Valkema, R.; Bernard, B.F.; Krenning, E.P. Combination radionuclide therapy using 177Lu- and 90Y-labeled somatostatin analogs. J. Nucl. Med. 2005, 46 (Suppl. 1), 13S–17S. [Google Scholar]
- Hofman, M.S.; Violet, J.; Hicks, R.J.; Ferdinandus, J.; Thang, S.P.; Akhurst, T.; Iravani, A.; Kong, G. Articles [177Lu] -PSMA-617 radionuclide treatment in patients with metastatic castration-resistant prostate cancer (LuPSMA trial): A single-centre, single-arm, phase 2 study. Lancet Oncol. 2018, 19, 825–833. [Google Scholar] [CrossRef]
- Pokrovska, T.D.; Jacobus, E.J.; Puliyadi, R.; Prevo, R.; Frost, S.; Dyer, A.; Baugh, R.; Rodriguez-Berriguete, G.; Fisher, K.; Granata, G.; et al. External beam radiation therapy and enadenotucirev: Inhibition of the DDR and mechanisms of radiation-mediated virus increase. Cancers 2020, 12, 798. [Google Scholar] [CrossRef]
- Maude, S.L.; Enders, G.H. Cdk inhibition in human cells compromises Chk1 function and activates a DNA damage response. Cancer Res. 2005, 65, 780–786. [Google Scholar] [PubMed]
- Militello, A.M.; Zielli, T.; Boggiani, D.; Michiara, M.; Naldi, N.; Bortesi, B.; Zanelli, P.; Uliana, V.; Giuliotti, S.; Musolino, A. Mechanism of action and clinical efficacy of CDK4/6 inhibitors in BRCA-mutated, estrogen receptor-positive breast cancers: Case report and literature review. Front. Oncol. 2019, 9, 759. [Google Scholar] [CrossRef]
- Krajewska, M.; Dries, R.; Grassetti, A.V.; Dust, S.; Gao, Y.; Huang, H.; Sharma, B.; Day, D.S.; Kwiatkowski, N.; Pomaville, M.; et al. CDK12 loss in cancer cells affects DNA damage response genes through premature cleavage and polyadenylation. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Quereda, V.; Bayle, S.; Vena, F.; Frydman, S.M.; Monastyrskyi, A.; Roush, W.R.; Duckett, D.R. Therapeutic Targeting of CDK12/CDK13 in Triple-Negative Breast Cancer. Cancer Cell 2019, 36, 545–558.e7. [Google Scholar] [CrossRef]
- Jiang, L.; Zawacka-Pankau, J. The p53/MDM2/MDMX-targeted therapies—A clinical synopsis. Cell Death Dis. 2020, 11, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Blandino, G.; Di Agostino, S. New therapeutic strategies to treat human cancers expressing mutant p53 proteins. J. Exp. Clin. Cancer Res. 2018, 37, 30. [Google Scholar] [CrossRef] [PubMed]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef]
- Lambert, J.M.R.; Gorzov, P.; Veprintsev, D.B.; Söderqvist, M.; Segerbäck, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J.N. PRIMA-1 Reactivates Mutant p53 by Covalent Binding to the Core Domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef]
- Aryee, D.N.T.; Niedan, S.; Ban, J.; Schwentner, R.; Muehlbacher, K.; Kauer, M.; Kofler, R.; Kovar, H. Variability in functional p53 reactivation by PRIMA-1 Met/APR-246 in Ewing sarcoma. Br. J. Cancer 2013, 109, 2696–2704. [Google Scholar] [CrossRef]
- Liu, X.; Wilcken, R.; Joerger, A.C.; Chuckowree, I.S.; Amin, J.; Spencer, J.; Fersht, A.R. Small molecule induced reactivation of mutant p53 in cancer cells. Nucleic Acids Res. 2013, 41, 6034–6044. [Google Scholar] [CrossRef]
- Bauer, M.R.; Jones, R.N.; Baud, M.G.J.; Wilcken, R.; Boeckler, F.M.; Fersht, A.R.; Joerger, A.C.; Spencer, J. Harnessing fluorine-sulfur contacts and multipolar interactions for the design of p53 mutant Y220C rescue drugs Harnessing Fluorine-Sulfur Contacts and Multipolar Interactions for the Design of p53 Mutant Y220C Rescue Drugs. ACS Chem. Biol. 2016, 11, 2265–2274. [Google Scholar] [CrossRef]
- Gupta, A.; Shah, K.; Oza, M.J.; Behl, T. Reactivation of p53 gene by MDM2 inhibitors: A novel therapy for cancer treatment. Biomed. Pharmacother. 2019, 109, 484–492. [Google Scholar] [CrossRef]
- Macurek, L.; Benada, J.; Müllers, E.; Halim, V.A.; Krejčíková, K.; Burdová, K.; Pecháčková, S.; Hodný, Z.; Lindqvist, A.; Medema, R.H.; et al. Downregulation of Wip1 phosphatase modulates the cellular threshold of DNA damage signaling in mitosis. Cell Cycle 2013, 12, 251–262. [Google Scholar] [CrossRef]
- Kong, W.; Jiang, X.; Mercer, W.E. Downregulation of Wip-1 phosphatase expression in MCF-7 breast cancer cells enhances doxorubicin-induced apoptosis through p53-mediated transcriptional activation of Bax. Cancer Biol. Ther. 2009, 8, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wang, L.; Yao, D.; Yang, T.; Cao, W.M.; Dou, J.; Pang, J.C.; Guan, S.; Zhang, H.; Yu, Y.; et al. Wip1 inhibitor GSK2830371 inhibits neuroblastoma growth by inducing Chk2/p53-mediated apoptosis. Sci. Rep. 2016, 6, 38011. [Google Scholar] [CrossRef]
- Hirai, H.; Iwasawa, Y.; Okada, M.; Arai, T.; Nishibata, T.; Kobayashi, M.; Kimura, T.; Kaneko, N.; Ohtani, J.; Yamanaka, K.; et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol. Cancer Ther. 2009, 8, 2992–3000. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, J.; Booher, R.N.; Kraker, A.; Lawrence, T.; Leopold, W.R.; Sun, Y. Radiosensitization of p53 mutant cells by PD0166285, a novel G2 checkpoint abrogator. Cancer Res. 2001, 61, 8211–8217. [Google Scholar] [PubMed]
- Yin, Y.; Shen, Q.; Tao, R.; Chang, W.; Li, R.; Xie, G.; Liu, W.; Zhang, P.; Tao, K. Wee1 inhibition can suppress tumor proliferation and sensitize p53 mutant colonic cancer cells to the anticancer effect of irinotecan. Mol. Med. Rep. 2018, 17, 3344–3349. [Google Scholar] [CrossRef] [PubMed]
- Reisländer, T.; Groelly, F.J.; Tarsounas, M. DNA Damage and Cancer Immunotherapy: A STING in the Tale. Mol. Cell 2020, 80, 21–28. [Google Scholar] [CrossRef]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.K.; Hsu, J.M.; Hsu, J.L.; Yu, W.H.; Du, Y.; Lee, H.H.; et al. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef] [PubMed]
- Shaverdian, N.; Lisberg, A.E.; Bornazyan, K.; Veruttipong, D.; Goldman, J.W.; Formenti, S.C.; Garon, E.B.; Lee, P. Articles Previous radiotherapy and the clinical activity and toxicity of pembrolizumab in the treatment of non-small-cell lung cancer: A secondary analysis of the KEYNOTE-001 phase 1 trial. Lancet Oncol. 2017, 18, 895–903. [Google Scholar] [CrossRef]
- Le, D.T.; Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, K.; Lu, S.; Kemberling, H.; Wilt, C.; et al. Mismatch-repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Brown, J.S.; Sundar, R.; Lopez, J. Combining DNA damaging therapeutics with immunotherapy: More haste, less speed. Br. J. Cancer 2018, 118, 312–324. [Google Scholar] [CrossRef]
- Chen, H.; Chen, H.; Zhang, J.; Wang, Y.; Simoneau, A.; Yang, H.; Levine, A.S.; Zou, L.; Chen, Z.; Lan, L. Molecular biology cGAS suppresses genomic instability as a decelerator of replication forks. Sci. Adv. 2020, 6. [Google Scholar] [CrossRef]
- Roden, D.M.; McLeod, H.L.; Relling, M.V.; Williams, M.S.; Mensah, G.A.; Peterson, J.F.; Van Driest, S.L. Pharmacogenomics. Lancet 2019, 394, 521–532. [Google Scholar] [CrossRef]
- Khanna, K.K.; Duijf, P.H.G. Complexities of pharmacogenomic interactions in cancer. Mol. Cell. Oncol. 2020, 7. [Google Scholar] [CrossRef]
- Pang, B.; De Jong, J.; Qiao, X.; Wessels, L.F.A.; Neefjes, J. Chemical profiling of the genome with anti-cancer drugs defines target specificities. Nat. Chem. Biol. 2015, 11, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Meng, J.; Zhang, Y.; Gu, J.; Han, Z.; Wang, X.; Gao, S. Development and validation of a six-RNA binding proteins prognostic signature and candidate drugs for prostate cancer. Genomics 2020, 112, 4980–4992. [Google Scholar] [CrossRef]
- Zimmermann, M.; Murina, O.; Reijns, M.A.M.; Agathanggelou, A.; Challis, R.; Tarnauskaite, Ž.; Muir, M.; Fluteau, A.; Aregger, M.; McEwan, A.; et al. CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature 2018, 559, 285–289. [Google Scholar] [CrossRef]
- Hamilton, G.; Rath, B. Pharmacogenetics of platinum-based chemotherapy in non-small cell lung cancer: Predictive validity of polymorphisms of ERCC1. Expert Opin. Drug Metab. Toxicol. 2018, 14, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Guffanti, F.; Fruscio, R.; Rulli, E.; Damia, G. The impact of DNA damage response gene polymorphisms on therapeutic outcomes in late stage ovarian cancer. Sci. Rep. 2016, 6, 38142. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Thomas, A.; Miettinen, M.; Pommier, Y. Schlafen 11 (SLFN11), a restriction factor for replicative stress induced by DNA-targeting anti-cancer therapies. Pharmacol. Ther. 2019, 201, 94–102. [Google Scholar] [CrossRef]
- Abbosh, C.; Birkbak, N.J.; Wilson, G.A.; Jamal-Hanjani, M.; Constantin, T.; Salari, R.; Le Quesne, J.; Moore, D.A.; Veeriah, S.; Rosenthal, R.; et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 2017, 545, 446–451. [Google Scholar] [CrossRef]
- Christensen, E.; Birkenkamp-Demtröder, K.; Sethi, H.; Shchegrova, S.; Salari, R.; Nordentoft, I.; Wu, H.T.; Knudsen, M.; Lamy, P.; Lindskrog, S.V.; et al. Early detection of metastatic relapse and monitoring of therapeutic efficacy by ultra-deep sequencing of plasma cell-free DNA in patients with urothelial bladder carcinoma. J. Clin. Oncol. 2019, 37, 1547–1557. [Google Scholar] [CrossRef]
- Mithraprabhu, S.; Hocking, J.; Ramachandran, M.; Choi, K.; Klarica, D.; Khong, T.; Reynolds, J.; Spencer, A. DNA-repair gene mutations are highly prevalent in circulating tumour dna from multiple myeloma patients. Cancers 2019, 11, 917. [Google Scholar] [CrossRef]
- Wilsker, D.F.; Barrett, A.M.; Dull, A.B.; Lawrence, S.M.; Hollingshead, M.G.; Chen, A.; Kummar, S.; Parchment, R.E.; Doroshow, J.H.; Kinders, R.J. Evaluation of pharmacodynamic responses to cancer therapeutic agents using DNA damage markers. Clin. Cancer Res. 2019, 25, 3084–3095. [Google Scholar] [CrossRef]
- Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; Biedrzycki, B.; Donehower, R.C.; De Chapelle, A.; Koshiji, M.; Bhaijee, F.; Huebner, T.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Sun, L.-L.; Yang, R.-Y.; Li, C.-W.; Chen, M.-K.; Shao, B.; Hsu, J.-M.; Chan, L.-C.; Yang, Y.; Hsu, J.L.; Lai, Y.-J.; et al. Inhibition of ATR downregulates PD-L1 and sensitizes tumor cells to T cell-mediated killing. Am. J. Cancer Res. 2018, 8, 1307–1316. [Google Scholar]
- Sato, H.; Niimi, A.; Yasuhara, T.; Bunga, T.; Permata, M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat Commun. 2017, 8, 1751. [Google Scholar] [CrossRef]
- Luftig, M.A. Viruses and the DNA Damage Response: Activation and Antagonism. Annu. Rev. Virol. 2014, 1, 605–625. [Google Scholar] [CrossRef] [PubMed]
- Stracker, T.H.; Carson, C.T.; Weitzman, M.D. Adenovirus oncoproteins inactivate the Mre11—Rad50—NBS1 DNA repair complex. Nature 2002, 418, 348–352. [Google Scholar] [CrossRef]
- Zhang, W.W.; Li, L.; Li, D.; Liu, J.; Li, X.; Li, W.; Xu, X.; Zhang, M.J.; Chandler, L.A.; Lin, H.; et al. The First Approved Gene Therapy Product for Cancer Ad-p53 (Gendicine): 12 Years in the Clinic. Hum. Gene Ther. 2018, 29, 160–179. [Google Scholar] [CrossRef]
- Ganesh, S.; Gibbons, D.; Ge, Y.; Vanroey, M.; Robinson, M.; Jooss, K. Combination therapy with radiation or cisplatin enhances the potency of Ad5/35 chimeric oncolytic adenovirus in a preclinical model of head and neck cancer. Cancer Gene Ther. 2009, 16, 383–392. [Google Scholar] [CrossRef]
- Kuhn, I.; Harden, P.; Bauzon, M.; Chartier, C.; Nye, J.; Thorne, S.; Reid, T.; Ni, S.; Lieber, A.; Fisher, K.; et al. Directed Evolution Generates a Novel Oncolytic Virus for the Treatment of Colon Cancer. PLoS ONE 2008, 3, e2409. [Google Scholar] [CrossRef]
- Xiao, X.; Liang, J.; Huang, C.; Li, K.; Xing, F.; Zhu, W.; Lin, Z.; Xu, W.; Wu, G.; Zhang, J.; et al. DNA-PK inhibition synergizes with oncolytic virus M1 by inhibiting antiviral response and potentiating DNA damage. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Ning, J.; Wakimoto, H.; Peters, C.; Martuza, R.L.; Rabkin, S.D. Rad51 degradation: Role in oncolytic virus-poly (ADP Ribose) polymerase inhibitor combination therapy in glioblastoma. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Molinaro, C.; Martoriati, A.; Cailliau, K. Proteins from the DNA Damage Response: Regulation, Dysfunction, and Anticancer Strategies. Cancers 2021, 13, 3819. https://doi.org/10.3390/cancers13153819
Molinaro C, Martoriati A, Cailliau K. Proteins from the DNA Damage Response: Regulation, Dysfunction, and Anticancer Strategies. Cancers. 2021; 13(15):3819. https://doi.org/10.3390/cancers13153819
Chicago/Turabian StyleMolinaro, Caroline, Alain Martoriati, and Katia Cailliau. 2021. "Proteins from the DNA Damage Response: Regulation, Dysfunction, and Anticancer Strategies" Cancers 13, no. 15: 3819. https://doi.org/10.3390/cancers13153819
APA StyleMolinaro, C., Martoriati, A., & Cailliau, K. (2021). Proteins from the DNA Damage Response: Regulation, Dysfunction, and Anticancer Strategies. Cancers, 13(15), 3819. https://doi.org/10.3390/cancers13153819