
Transcription Factors, R-Loops and Deubiquitinating Enzymes: Emerging Targets in Myelodysplastic Syndromes and Acute Myeloid Leukemia


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
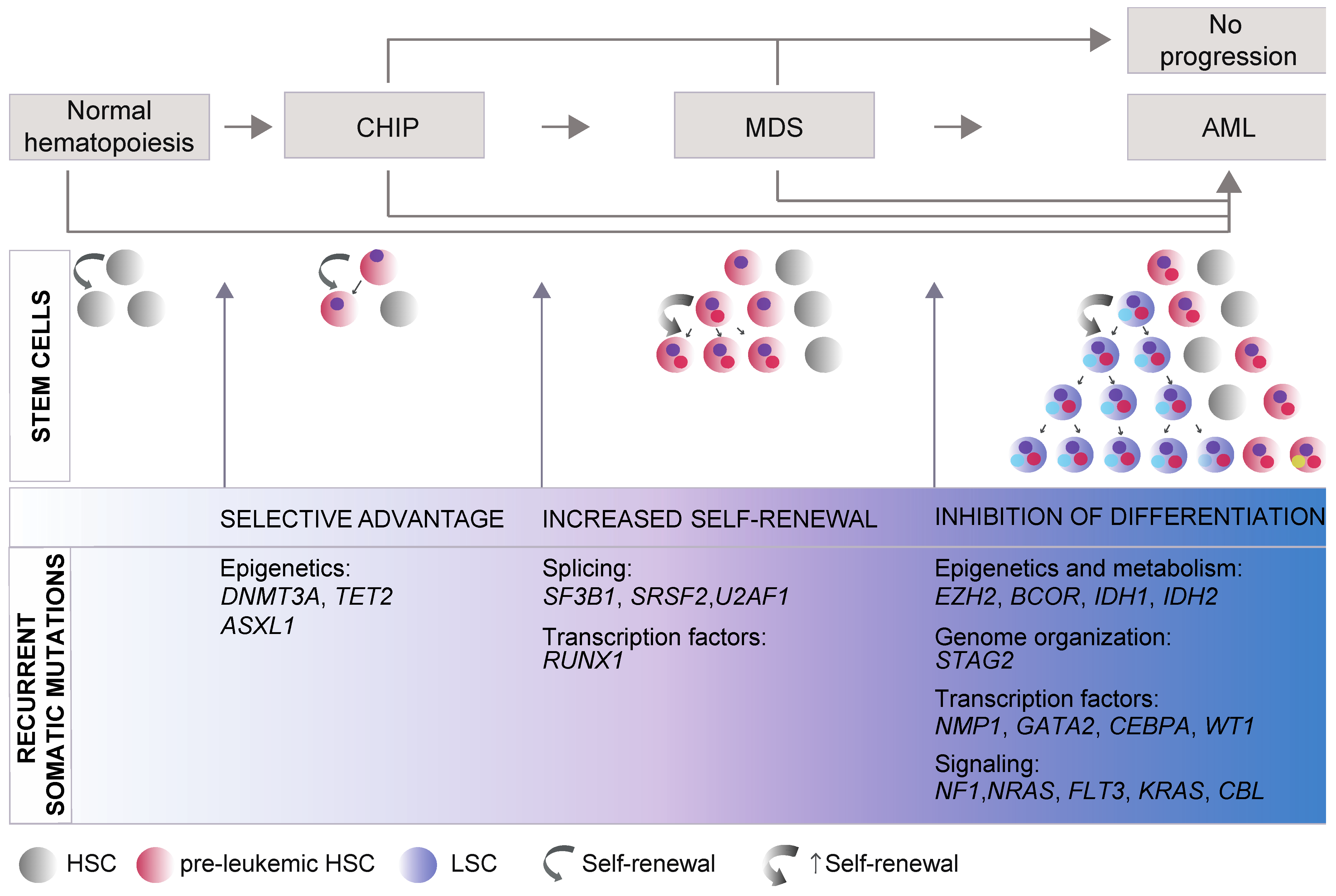
2. Genetics Underlying Evolutionary Trajectories of MDS Progression
2.1. Somatic Mutations
2.2. Familial Syndromes Predisposing to MDS
3. Transcription Factors in MDS and AML
3.1. Genome Rearrangements Involving TFs
3.2. Gene Mutations and Allelic Variants
3.3. Gene Dosage Effects
3.4. TFs Cross-Antagonism: The Paradigmatic Example of PU.I and GATA1
3.5. Mutations Leading to the Selective Expression of Specific TFs Isoforms
3.6. The Heterogeneous Spectrum of TFs and Mutation Involved in MDS/AML
4. Splicing Factors in MDS and AML
4.1. Splicing Factors’ Mutations in MDS and AML and Their Impact on Gene Expression
4.1.1. SF3B1 (Splicing Factor 3b Subunit 1)
4.1.2. U2AF1 (U2 Small Nuclear RNA Auxiliary Factor 1)
4.1.3. ZRSR2 (Zinc Finger CCCH-Type, RNA Binding Motif and Serine/Arginine Rich 2)
4.1.4. SRSF2 (Serine/Arginine-Rich Splicing Factor 2)
4.2. Common Themes in Alternative Splicing Alteration?
5. Genome Maintenance Pathways in HSCs and Their Implications in MDS/AML
5.1. R-Loops as a Source of DNA Damage
5.1.1. R-Loops and Mutations in Splicing Factors
5.1.2. Interplay between R-Loops and the Fanconi Anemia DNA Repair Pathway
5.2. Deubiquitinating Enzymes in HSCs Genome Stability
6. Emerging Therapeutic Targets
6.1. Transcription Factors’ Targeting
6.2. Targeting the Spliceosome
6.3. Are DUBs Possible Therapeutic Targets?
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Morrison, S.J.; Spradling, A.C. Stem cells and niches: Mechanisms that promote stem cell maintenance throughout life. Cell 2008, 132, 598–611. [Google Scholar] [CrossRef]
- De Haan, G.; Lazare, S.S. Aging of hematopoietic stem cells. Blood 2018, 131, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Laurenti, E.; Gottgens, B. From haematopoietic stem cells to complex differentiation landscapes. Nature 2018, 553, 418–426. [Google Scholar] [CrossRef]
- Cazzola, M. Myelodysplastic Syndromes. N. Engl. J. Med. 2020, 383, 1358–1374. [Google Scholar] [CrossRef]
- Lindsley, R.C.; Mar, B.G.; Mazzola, E.; Grauman, P.V.; Shareef, S.; Allen, S.L.; Pigneux, A.; Wetzler, M.; Stuart, R.K.; Erba, H.P.; et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 2015, 125, 1367–1376. [Google Scholar] [CrossRef] [PubMed]
- Sperling, A.S.; Gibson, C.J.; Ebert, B.L. The genetics of myelodysplastic syndrome: From clonal haematopoiesis to secondary leukaemia. Nat. Rev. Cancer 2017, 17, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Hellstrom-Lindberg, E.; Tobiasson, M.; Greenberg, P. Myelodysplastic syndromes: Moving towards personalized management. Haematologica 2020, 105, 1765–1779. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Corces-Zimmerman, M.R.; Hong, W.J.; Weissman, I.L.; Medeiros, B.C.; Majeti, R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc. Natl. Acad. Sci. USA 2014, 111, 2548–2553. [Google Scholar] [CrossRef]
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014, 506, 328–333. [Google Scholar] [CrossRef]
- Yamashita, M.; Dellorusso, P.V.; Olson, O.C.; Passegue, E. Dysregulated haematopoietic stem cell behaviour in myeloid leukaemogenesis. Nat. Rev. Cancer 2020, 20, 365–382. [Google Scholar] [CrossRef]
- Pollyea, D.A.; Jordan, C.T. Therapeutic targeting of acute myeloid leukemia stem cells. Blood 2017, 129, 1627–1635. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627. [Google Scholar] [CrossRef]
- Tyner, J.W.; Tognon, C.E.; Bottomly, D.; Wilmot, B.; Kurtz, S.E.; Savage, S.L.; Long, N.; Schultz, A.R.; Traer, E.; Abel, M.; et al. Functional genomic landscape of acute myeloid leukaemia. Nature 2018, 562, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Ogawa, S. Genetics of MDS. Blood 2019, 133, 1049–1059. [Google Scholar] [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P.; Ebert, B.L. Clonal hematopoiesis as a model for premalignant changes during aging. Exp. Hematol. 2020, 83, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Bick, A.G.; Weinstock, J.S.; Nandakumar, S.K.; Fulco, C.P.; Bao, E.L.; Zekavat, S.M.; Szeto, M.D.; Liao, X.; Leventhal, M.J.; Nasser, J.; et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature 2020, 586, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Gondek, L.P.; DeZern, A.E. Assessing clonal haematopoiesis: Clinical burdens and benefits of diagnosing myelodysplastic syndrome precursor states. Lancet Haematol. 2020, 7, e73–e81. [Google Scholar] [CrossRef]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef]
- Nagata, Y.; Makishima, H.; Kerr, C.M.; Przychodzen, B.P.; Aly, M.; Goyal, A.; Awada, H.; Asad, M.F.; Kuzmanovic, T.; Suzuki, H.; et al. Invariant patterns of clonal succession determine specific clinical features of myelodysplastic syndromes. Nat. Commun. 2019, 10, 5386. [Google Scholar] [CrossRef] [PubMed]
- Buscarlet, M.; Provost, S.; Zada, Y.F.; Barhdadi, A.; Bourgoin, V.; Lepine, G.; Mollica, L.; Szuber, N.; Dube, M.P.; Busque, L. DNMT3A and TET2 dominate clonal hematopoiesis and demonstrate benign phenotypes and different genetic predispositions. Blood 2017, 130, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef]
- Makishima, H.; Yoshizato, T.; Yoshida, K.; Sekeres, M.A.; Radivoyevitch, T.; Suzuki, H.; Przychodzen, B.; Nagata, Y.; Meggendorfer, M.; Sanada, M.; et al. Dynamics of clonal evolution in myelodysplastic syndromes. Nat. Genet. 2017, 49, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.F. Clinical and Therapeutic Implications of Cancer Stem Cells. N. Engl. J. Med. 2019, 380, 2237–2245. [Google Scholar] [CrossRef] [PubMed]
- Potter, N.; Miraki-Moud, F.; Ermini, L.; Titley, I.; Vijayaraghavan, G.; Papaemmanuil, E.; Campbell, P.; Gribben, J.; Taussig, D.; Greaves, M. Single cell analysis of clonal architecture in acute myeloid leukaemia. Leukemia 2019, 33, 1113–1123. [Google Scholar] [CrossRef]
- Arends, C.M.; Galan-Sousa, J.; Hoyer, K.; Chan, W.; Jager, M.; Yoshida, K.; Seemann, R.; Noerenberg, D.; Waldhueter, N.; Fleischer-Notter, H.; et al. Hematopoietic lineage distribution and evolutionary dynamics of clonal hematopoiesis. Leukemia 2018, 32, 1908–1919. [Google Scholar] [CrossRef] [PubMed]
- Buscarlet, M.; Provost, S.; Zada, Y.F.; Bourgoin, V.; Mollica, L.; Dube, M.P.; Busque, L. Lineage restriction analyses in CHIP indicate myeloid bias for TET2 and multipotent stem cell origin for DNMT3A. Blood 2018, 132, 277–280. [Google Scholar] [CrossRef]
- Von Bonin, M.; Jambor, H.K.; Teipel, R.; Stolzel, F.; Thiede, C.; Damm, F.; Kroschinsky, F.; Schetelig, J.; Chavakis, T.; Bornhauser, M. Clonal hematopoiesis and its emerging effects on cellular therapies. Leukemia 2021. [Google Scholar] [CrossRef]
- DeZern, A.E.; Gondek, L.P. Stem cell donors should be screened for CHIP. Blood Adv. 2020, 4, 784–788. [Google Scholar] [CrossRef]
- Frick, M.; Chan, W.; Arends, C.M.; Hablesreiter, R.; Halik, A.; Heuser, M.; Michonneau, D.; Blau, O.; Hoyer, K.; Christen, F.; et al. Role of Donor Clonal Hematopoiesis in Allogeneic Hematopoietic Stem-Cell Transplantation. J. Clin. Oncol. 2019, 37, 375–385. [Google Scholar] [CrossRef]
- Chen, J.; Kao, Y.R.; Sun, D.; Todorova, T.I.; Reynolds, D.; Narayanagari, S.R.; Montagna, C.; Will, B.; Verma, A.; Steidl, U. Myelodysplastic syndrome progression to acute myeloid leukemia at the stem cell level. Nat. Med. 2019, 25, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Menssen, A.J.; Walter, M.J. Genetics of progression from MDS to secondary leukemia. Blood 2020, 136, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Bolton, K.L.; Ptashkin, R.N.; Gao, T.; Braunstein, L.; Devlin, S.M.; Kelly, D.; Patel, M.; Berthon, A.; Syed, A.; Yabe, M.; et al. Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat. Genet. 2020, 52, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Bernard, E.; Nannya, Y.; Hasserjian, R.P.; Devlin, S.M.; Tuechler, H.; Medina-Martinez, J.S.; Yoshizato, T.; Shiozawa, Y.; Saiki, R.; Malcovati, L.; et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat. Med. 2020, 26, 1549–1556. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Ptashkin, R.; Bolton, K.L.; Sirenko, M.; Fong, C.; Spitzer, B.; Menghrajani, K.; Ossa, J.E.A.; Zhou, Y.; Bernard, E.; et al. Interplay between chromosomal alterations and gene mutations shapes the evolutionary trajectory of clonal hematopoiesis. Nat. Commun. 2021, 12, 338. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Parmar, K.; Mouly, E.; Delord, M.; Kim, J.M.; Regairaz, M.; Pla, M.; Vasquez, N.; Zhang, Q.S.; Pondarre, C.; et al. Bone marrow failure in Fanconi anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs hematopoietic stem and progenitor cells. Cell Stem Cell 2012, 11, 36–49. [Google Scholar] [CrossRef]
- Douglas, S.P.M.; Siipola, P.; Kovanen, P.E.; Pyorala, M.; Kakko, S.; Savolainen, E.R.; Salmenniemi, U.; Orte, K.; Kytola, S.; Pitkanen, E.; et al. ERCC6L2 defines a novel entity within inherited acute myeloid leukemia. Blood 2019, 133, 2724–2728. [Google Scholar] [CrossRef]
- Vulliamy, T.; Marrone, A.; Szydlo, R.; Walne, A.; Mason, P.J.; Dokal, I. Disease anticipation is associated with progressive telomere shortening in families with dyskeratosis congenita due to mutations in TERC. Nat. Genet. 2004, 36, 447–449. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Calado, R.T.; Ly, H.; Kajigaya, S.; Baerlocher, G.M.; Chanock, S.J.; Lansdorp, P.M.; Young, N.S. Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N. Engl. J. Med. 2005, 352, 1413–1424. [Google Scholar] [CrossRef]
- McBride, K.A.; Ballinger, M.L.; Killick, E.; Kirk, J.; Tattersall, M.H.; Eeles, R.A.; Thomas, D.M.; Mitchell, G. Li-Fraumeni syndrome: Cancer risk assessment and clinical management. Nat. Rev. Clin. Oncol. 2014, 11, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Hsu, A.P.; Sampaio, E.P.; Khan, J.; Calvo, K.R.; Lemieux, J.E.; Patel, S.Y.; Frucht, D.M.; Vinh, D.C.; Auth, R.D.; Freeman, A.F.; et al. Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood 2011, 118, 2653–2655. [Google Scholar] [CrossRef]
- Ostergaard, P.; Simpson, M.A.; Connell, F.C.; Steward, C.G.; Brice, G.; Woollard, W.J.; Dafou, D.; Kilo, T.; Smithson, S.; Lunt, P.; et al. Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome). Nat. Genet. 2011, 43, 929–931. [Google Scholar] [CrossRef] [PubMed]
- Tenen, D.G. Disruption of differentiation in human cancer: AML shows the way. Nat. Rev. Cancer 2003, 3, 89–101. [Google Scholar] [CrossRef]
- Graf, T.; Enver, T. Forcing cells to change lineages. Nature 2009, 462, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Okuno, Y.; Huang, G.; Rosenbauer, F.; Evans, E.K.; Radomska, H.S.; Iwasaki, H.; Akashi, K.; Moreau-Gachelin, F.; Li, Y.; Zhang, P.; et al. Potential autoregulation of transcription factor PU.1 by an upstream regulatory element. Mol. Cell Biol. 2005, 25, 2832–2845. [Google Scholar] [CrossRef]
- Schuetzmann, D.; Walter, C.; van Riel, B.; Kruse, S.; Konig, T.; Erdmann, T.; Tonges, A.; Bindels, E.; Weilemann, A.; Gebhard, C.; et al. Temporal autoregulation during human PU.1 locus SubTAD formation. Blood 2018, 132, 2643–2655. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, H.; Mizuno, S.; Arinobu, Y.; Ozawa, H.; Mori, Y.; Shigematsu, H.; Takatsu, K.; Tenen, D.G.; Akashi, K. The order of expression of transcription factors directs hierarchical specification of hematopoietic lineages. Genes Dev. 2006, 20, 3010–3021. [Google Scholar] [CrossRef]
- Brown, A.L.; Arts, P.; Carmichael, C.L.; Babic, M.; Dobbins, J.; Chong, C.E.; Schreiber, A.W.; Feng, J.; Phillips, K.; Wang, P.P.S.; et al. RUNX1-mutated families show phenotype heterogeneity and a somatic mutation profile unique to germline predisposed AML. Blood Adv. 2020, 4, 1131–1144. [Google Scholar] [CrossRef]
- Haase, D.; Germing, U.; Schanz, J.; Pfeilstocker, M.; Nosslinger, T.; Hildebrandt, B.; Kundgen, A.; Lubbert, M.; Kunzmann, R.; Giagounidis, A.A.; et al. New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: Evidence from a core dataset of 2124 patients. Blood 2007, 110, 4385–4395. [Google Scholar] [CrossRef] [PubMed]
- Ebert, B.L.; Galili, N.; Tamayo, P.; Bosco, J.; Mak, R.; Pretz, J.; Tanguturi, S.; Ladd-Acosta, C.; Stone, R.; Golub, T.R.; et al. An erythroid differentiation signature predicts response to lenalidomide in myelodysplastic syndrome. PLoS Med. 2008, 5, e35. [Google Scholar] [CrossRef]
- Kronke, J.; Fink, E.C.; Hollenbach, P.W.; MacBeth, K.J.; Hurst, S.N.; Udeshi, N.D.; Chamberlain, P.P.; Mani, D.R.; Man, H.W.; Gandhi, A.K.; et al. Lenalidomide induces ubiquitination and degradation of CK1alpha in del(5q) MDS. Nature 2015, 523, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Aly, M.; Ramdzan, Z.M.; Nagata, Y.; Balasubramanian, S.K.; Hosono, N.; Makishima, H.; Visconte, V.; Kuzmanovic, T.; Adema, V.; Nazha, A.; et al. Distinct clinical and biological implications of CUX1 in myeloid neoplasms. Blood Adv. 2019, 3, 2164–2178. [Google Scholar] [CrossRef]
- Bochtler, T.; Frohling, S.; Kramer, A. Role of chromosomal aberrations in clonal diversity and progression of acute myeloid leukemia. Leukemia 2015, 29, 1243–1252. [Google Scholar] [CrossRef]
- Mitelman, F.; Johansson, B.; Mertens, F. The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer 2007, 7, 233–245. [Google Scholar] [CrossRef]
- Niki, M.; Okada, H.; Takano, H.; Kuno, J.; Tani, K.; Hibino, H.; Asano, S.; Ito, Y.; Satake, M.; Noda, T. Hematopoiesis in the fetal liver is impaired by targeted mutagenesis of a gene encoding a non-DNA binding subunit of the transcription factor, polyomavirus enhancer binding protein 2/core binding factor. Proc. Natl. Acad. Sci. USA 1997, 94, 5697–5702. [Google Scholar] [CrossRef] [PubMed]
- Okuda, T.; van Deursen, J.; Hiebert, S.W.; Grosveld, G.; Downing, J.R. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell 1996, 84, 321–330. [Google Scholar] [CrossRef]
- Sasaki, K.; Yagi, H.; Bronson, R.T.; Tominaga, K.; Matsunashi, T.; Deguchi, K.; Tani, Y.; Kishimoto, T.; Komori, T. Absence of fetal liver hematopoiesis in mice deficient in transcriptional coactivator core binding factor beta. Proc. Natl. Acad. Sci. USA 1996, 93, 12359–12363. [Google Scholar] [CrossRef]
- Beghini, A. Core Binding Factor Leukemia: Chromatin Remodeling Moves Towards Oncogenic Transcription. Cancers 2019, 11, 1973. [Google Scholar] [CrossRef]
- Wang, J.; Hoshino, T.; Redner, R.L.; Kajigaya, S.; Liu, J.M. ETO, fusion partner in t(8;21) acute myeloid leukemia, represses transcription by interaction with the human N-CoR/mSin3/HDAC1 complex. Proc. Natl. Acad. Sci. USA 1998, 95, 10860–10865. [Google Scholar] [CrossRef] [PubMed]
- Vangala, R.K.; Heiss-Neumann, M.S.; Rangatia, J.S.; Singh, S.M.; Schoch, C.; Tenen, D.G.; Hiddemann, W.; Behre, G. The myeloid master regulator transcription factor PU.1 is inactivated by AML1-ETO in t(8;21) myeloid leukemia. Blood 2003, 101, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Elagib, K.E.; Delehanty, L.L.; Goldfarb, A.N. Erythroid inhibition by the leukemic fusion AML1-ETO is associated with impaired acetylation of the major erythroid transcription factor GATA-1. Cancer Res. 2006, 66, 2990–2996. [Google Scholar] [CrossRef] [PubMed]
- Pabst, T.; Mueller, B.U.; Harakawa, N.; Schoch, C.; Haferlach, T.; Behre, G.; Hiddemann, W.; Zhang, D.E.; Tenen, D.G. AML1-ETO downregulates the granulocytic differentiation factor C/EBPalpha in t(8;21) myeloid leukemia. Nat. Med. 2001, 7, 444–451. [Google Scholar] [CrossRef]
- Stengel, K.R.; Ellis, J.D.; Spielman, C.L.; Bomber, M.L.; Hiebert, S.W. Definition of a small core transcriptional circuit regulated by AML1-ETO. Mol. Cell 2021, 81, 530–545.e535. [Google Scholar] [CrossRef] [PubMed]
- Junge, A.; Zandi, R.; Havgaard, J.H.; Gorodkin, J.; Cowland, J.B. Assessing the miRNA sponge potential of RUNX1T1 in t(8;21) acute myeloid leukemia. Gene 2017, 615, 35–40. [Google Scholar] [CrossRef]
- Grinev, V.V.; Barneh, F.; Ilyushonak, I.M.; Nakjang, S.; Smink, J.; van Oort, A.; Clough, R.; Seyani, M.; McNeill, H.; Reza, M.; et al. RUNX1/RUNX1T1 mediates alternative splicing and reorganises the transcriptional landscape in leukemia. Nat. Commun. 2021, 12, 520. [Google Scholar] [CrossRef]
- Krauth, M.T.; Eder, C.; Alpermann, T.; Bacher, U.; Nadarajah, N.; Kern, W.; Haferlach, C.; Haferlach, T.; Schnittger, S. High number of additional genetic lesions in acute myeloid leukemia with t(8;21)/RUNX1-RUNX1T1: Frequency and impact on clinical outcome. Leukemia 2014, 28, 1449–1458. [Google Scholar] [CrossRef]
- Duployez, N.; Marceau-Renaut, A.; Boissel, N.; Petit, A.; Bucci, M.; Geffroy, S.; Lapillonne, H.; Renneville, A.; Ragu, C.; Figeac, M.; et al. Comprehensive mutational profiling of core binding factor acute myeloid leukemia. Blood 2016, 127, 2451–2459. [Google Scholar] [CrossRef]
- Boissel, N.; Leroy, H.; Brethon, B.; Philippe, N.; de Botton, S.; Auvrignon, A.; Raffoux, E.; Leblanc, T.; Thomas, X.; Hermine, O.; et al. Incidence and prognostic impact of c-Kit, FLT3, and Ras gene mutations in core binding factor acute myeloid leukemia (CBF-AML). Leukemia 2006, 20, 965–970. [Google Scholar] [CrossRef]
- Christen, F.; Hoyer, K.; Yoshida, K.; Hou, H.A.; Waldhueter, N.; Heuser, M.; Hills, R.K.; Chan, W.; Hablesreiter, R.; Blau, O.; et al. Genomic landscape and clonal evolution of acute myeloid leukemia with t(8;21): An international study on 331 patients. Blood 2019, 133, 1140–1151. [Google Scholar] [CrossRef]
- Kovar, H. Dr. Jekyll and Mr. Hyde: The Two Faces of the FUS/EWS/TAF15 Protein Family. Sarcoma 2011, 2011, 837474. [Google Scholar] [CrossRef] [PubMed]
- Endo, A.; Tomizawa, D.; Aoki, Y.; Morio, T.; Mizutani, S.; Takagi, M. EWSR1/ELF5 induces acute myeloid leukemia by inhibiting p53/p21 pathway. Cancer Sci. 2016, 107, 1745–1754. [Google Scholar] [CrossRef]
- Ichikawa, H.; Shimizu, K.; Hayashi, Y.; Ohki, M. An RNA-binding protein gene, TLS/FUS, is fused to ERG in human myeloid leukemia with t(16;21) chromosomal translocation. Cancer Res. 1994, 54, 2865–2868. [Google Scholar] [PubMed]
- Martini, A.; La Starza, R.; Janssen, H.; Bilhou-Nabera, C.; Corveleyn, A.; Somers, R.; Aventin, A.; Foà, R.; Hagemeijer, A.; Mecucci, C.; et al. Recurrent rearrangement of the Ewing’s sarcoma gene, EWSR1, or its homologue, TAF15, with the transcription factor CIZ/NMP4 in acute leukemia. Cancer Res. 2002, 62, 5408–5412. [Google Scholar] [PubMed]
- Zerkalenkova, E.; Panfyorova, A.; Kazakova, A.; Baryshev, P.; Shelihova, L.; Kalinina, I.; Novichkova, G.; Maschan, M.; Maschan, A.; Olshanskaya, Y. Molecular characteristic of acute leukemias with t(16;21)/FUS-ERG. Ann. Hematol. 2018, 97, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Panagopoulos, I.; Lassen, C.; Isaksson, M.; Mitelman, F.; Mandahl, N.; Aman, P. Characteristic sequence motifs at the breakpoints of the hybrid genes FUS/CHOP, EWS/CHOP and FUS/ERG in myxoid liposarcoma and acute myeloid leukemia. Oncogene 1997, 15, 1357–1362. [Google Scholar] [CrossRef][Green Version]
- Torchia, E.C.; Boyd, K.; Rehg, J.E.; Qu, C.; Baker, S.J. EWS/FLI-1 Induces Rapid Onset of Myeloid/Erythroid Leukemia in Mice. Mol. Cell. Biol. 2007, 27, 7918–7934. [Google Scholar] [CrossRef] [PubMed]
- Svetoni, F.; Frisone, P.; Paronetto, M.P. Role of FET proteins in neurodegenerative disorders. RNA Biol. 2016, 13, 1089–1102. [Google Scholar] [CrossRef] [PubMed]
- Sotoca, A.M.; Prange, K.H.; Reijnders, B.; Mandoli, A.; Nguyen, L.N.; Stunnenberg, H.G.; Martens, J.H. The oncofusion protein FUS-ERG targets key hematopoietic regulators and modulates the all-trans retinoic acid signaling pathway in t(16;21) acute myeloid leukemia. Oncogene 2016, 35, 1965–1976. [Google Scholar] [CrossRef]
- Jaishankar, S.; Zhang, J.; Roussel, M.F.; Baker, S.J. Transforming activity of EWS/FLI is not strictly dependent upon DNA-binding activity. Oncogene 1999, 18, 5592–5597. [Google Scholar] [CrossRef]
- Welford, S.M.; Hebert, S.P.; Deneen, B.; Arvand, A.; Denny, C.T. DNA binding domain-independent pathways are involved in EWS/FLI1-mediated oncogenesis. J. Biol. Chem. 2001, 276, 41977–41984. [Google Scholar] [CrossRef]
- Kapeli, K.; Pratt, G.A.; Vu, A.Q.; Hutt, K.R.; Martinez, F.J.; Sundararaman, B.; Batra, R.; Freese, P.; Lambert, N.J.; Huelga, S.C.; et al. Distinct and shared functions of ALS-associated proteins TDP-43, FUS and TAF15 revealed by multisystem analyses. Nat. Commun. 2016, 7, 12143. [Google Scholar] [CrossRef] [PubMed]
- Paronetto, M.P.; Miñana, B.; Valcárcel, J. The Ewing Sarcoma Protein Regulates DNA Damage-Induced Alternative Splicing. Mol. Cell 2011, 43, 353–368. [Google Scholar] [CrossRef]
- Alberti, S.; Gladfelter, A.; Mittag, T. Considerations and Challenges in Studying Liquid-Liquid Phase Separation and Biomolecular Condensates. Cell 2019, 176, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, I.A.; Bartek, J.; Dundr, M. Phase separated microenvironments inside the cell nucleus are linked to disease and regulate epigenetic state, transcription and RNA processing. Semin. Cell Dev. Biol. 2019, 90, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Boulay, G.; Sandoval, G.J.; Riggi, N.; Iyer, S.; Buisson, R.; Naigles, B.; Awad, M.E.; Rengarajan, S.; Volorio, A.; McBride, M.J.; et al. Cancer-Specific Retargeting of BAF Complexes by a Prion-like Domain. Cell 2017, 171, 163–178.e119. [Google Scholar] [CrossRef] [PubMed]
- Kwon, I.; Kato, M.; Xiang, S.; Wu, L.; Theodoropoulos, P.; Mirzaei, H.; Han, T.; Xie, S.; Corden, J.L.; McKnight, S.L. Phosphorylation-Regulated Binding of RNA Polymerase II to Fibrous Polymers of Low-Complexity Domains. Cell 2013, 155, 1049–1060. [Google Scholar] [CrossRef]
- Schwartz, J.C.; Wang, X.; Podell, E.R.; Cech, T.R. RNA Seeds Higher-Order Assembly of FUS Protein. Cell Rep. 2013, 5, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Levone, B.R.; Lenzken, S.C.; Antonaci, M.; Maiser, A.; Rapp, A.; Conte, F.; Reber, S.; Mechtersheimer, J.; Ronchi, A.E.; Muhlemann, O.; et al. FUS-dependent liquid-liquid phase separation is important for DNA repair initiation. J. Cell Biol. 2021, 220. [Google Scholar] [CrossRef]
- Kon, A.; Shih, L.Y.; Minamino, M.; Sanada, M.; Shiraishi, Y.; Nagata, Y.; Yoshida, K.; Okuno, Y.; Bando, M.; Nakato, R.; et al. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat. Genet. 2013, 45, 1232–1237. [Google Scholar] [CrossRef]
- Ochi, Y.; Kon, A.; Sakata, T.; Nakagawa, M.M.; Nakazawa, N.; Kakuta, M.; Kataoka, K.; Koseki, H.; Nakayama, M.; Morishita, D.; et al. Combined Cohesin-RUNX1 Deficiency Synergistically Perturbs Chromatin Looping and Causes Myelodysplastic Syndromes. Cancer Discov. 2020, 10, 836–853. [Google Scholar] [CrossRef]
- Van der Meer, L.T.; Jansen, J.H.; van der Reijden, B.A. Gfi1 and Gfi1b: Key regulators of hematopoiesis. Leukemia 2010, 24, 1834–1843. [Google Scholar] [CrossRef] [PubMed]
- Khandanpour, C.; Thiede, C.; Valk, P.J.; Sharif-Askari, E.; Nuckel, H.; Lohmann, D.; Horsthemke, B.; Siffert, W.; Neubauer, A.; Grzeschik, K.H.; et al. A variant allele of Growth Factor Independence 1 (GFI1) is associated with acute myeloid leukemia. Blood 2010, 115, 2462–2472. [Google Scholar] [CrossRef] [PubMed]
- Botezatu, L.; Michel, L.C.; Helness, A.; Vadnais, C.; Makishima, H.; Hones, J.M.; Robert, F.; Vassen, L.; Thivakaran, A.; Al-Matary, Y.; et al. Epigenetic therapy as a novel approach for GFI136N-associated murine/human AML. Exp. Hematol. 2016, 44, 713–726.e714. [Google Scholar] [CrossRef]
- Vadnais, C.; Chen, R.; Fraszczak, J.; Yu, Z.; Boulais, J.; Pinder, J.; Frank, D.; Khandanpour, C.; Hebert, J.; Dellaire, G.; et al. GFI1 facilitates efficient DNA repair by regulating PRMT1 dependent methylation of MRE11 and 53BP1. Nat. Commun. 2018, 9, 1418. [Google Scholar] [CrossRef] [PubMed]
- Fraszczak, J.; Vadnais, C.; Rashkovan, M.; Ross, J.; Beauchemin, H.; Chen, R.; Grapton, D.; Khandanpour, C.; Moroy, T. Reduced expression but not deficiency of GFI1 causes a fatal myeloproliferative disease in mice. Leukemia 2019, 33, 110–121. [Google Scholar] [CrossRef]
- Kok, C.H.; Watkins, D.B.; Leclercq, T.; D’Andrea, R.J.; Hughes, T.P.; White, D.L. Low GFI1 expression in white blood cells of CP-CML patients at diagnosis is strongly associated with subsequent blastic transformation. Leukemia 2013, 27, 1427–1430. [Google Scholar] [CrossRef]
- Mori, N.; Morosetti, R.; Mizoguchi, H.; Koeffler, H.P. Progression of myelodysplastic syndrome: Allelic loss on chromosomal arm 1p. Br. J. Haematol. 2003, 122, 226–230. [Google Scholar] [CrossRef]
- Hones, J.M.; Botezatu, L.; Helness, A.; Vadnais, C.; Vassen, L.; Robert, F.; Hergenhan, S.M.; Thivakaran, A.; Schutte, J.; Al-Matary, Y.S.; et al. GFI1 as a novel prognostic and therapeutic factor for AML/MDS. Leukemia 2016, 30, 1237–1245. [Google Scholar] [CrossRef]
- Saleque, S.; Cameron, S.; Orkin, S.H. The zinc-finger proto-oncogene Gfi-1b is essential for development of the erythroid and megakaryocytic lineages. Genes Dev. 2002, 16, 301–306. [Google Scholar] [CrossRef]
- Garcon, L.; Lacout, C.; Svinartchouk, F.; Le Couedic, J.P.; Villeval, J.L.; Vainchenker, W.; Dumenil, D. Gfi-1B plays a critical role in terminal differentiation of normal and transformed erythroid progenitor cells. Blood 2005, 105, 1448–1455. [Google Scholar] [CrossRef]
- Thivakaran, A.; Botezatu, L.; Hones, J.M.; Schutte, J.; Vassen, L.; Al-Matary, Y.S.; Patnana, P.; Zeller, A.; Heuser, M.; Thol, F.; et al. Gfi1b: A key player in the genesis and maintenance of acute myeloid leukemia and myelodysplastic syndrome. Haematologica 2018, 103, 614–625. [Google Scholar] [CrossRef]
- Anguita, E.; Gupta, R.; Olariu, V.; Valk, P.J.; Peterson, C.; Delwel, R.; Enver, T. A somatic mutation of GFI1B identified in leukemia alters cell fate via a SPI1 (PU.1) centered genetic regulatory network. Dev. Biol. 2016, 411, 277–286. [Google Scholar] [CrossRef]
- Song, W.J.; Sullivan, M.G.; Legare, R.D.; Hutchings, S.; Tan, X.; Kufrin, D.; Ratajczak, J.; Resende, I.C.; Haworth, C.; Hock, R.; et al. Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat. Genet. 1999, 23, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Schnittger, S.; Dicker, F.; Kern, W.; Wendland, N.; Sundermann, J.; Alpermann, T.; Haferlach, C.; Haferlach, T. RUNX1 mutations are frequent in de novo AML with noncomplex karyotype and confer an unfavorable prognosis. Blood 2011, 117, 2348–2357. [Google Scholar] [CrossRef] [PubMed]
- Antony-Debre, I.; Manchev, V.T.; Balayn, N.; Bluteau, D.; Tomowiak, C.; Legrand, C.; Langlois, T.; Bawa, O.; Tosca, L.; Tachdjian, G.; et al. Level of RUNX1 activity is critical for leukemic predisposition but not for thrombocytopenia. Blood 2015, 125, 930–940. [Google Scholar] [CrossRef] [PubMed]
- Ling, K.W.; Ottersbach, K.; van Hamburg, J.P.; Oziemlak, A.; Tsai, F.Y.; Orkin, S.H.; Ploemacher, R.; Hendriks, R.W.; Dzierzak, E. GATA-2 plays two functionally distinct roles during the ontogeny of hematopoietic stem cells. J. Exp. Med. 2004, 200, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, R.; Yamamoto, M. Quantitative and qualitative impairments in GATA2 and myeloid neoplasms. IUBMB Life 2020, 72, 142–150. [Google Scholar] [CrossRef]
- Yamazaki, H.; Suzuki, M.; Otsuki, A.; Shimizu, R.; Bresnick, E.H.; Engel, J.D.; Yamamoto, M. A remote GATA2 hematopoietic enhancer drives leukemogenesis in inv(3)(q21;q26) by activating EVI1 expression. Cancer Cell 2014, 25, 415–427. [Google Scholar] [CrossRef]
- Groschel, S.; Sanders, M.A.; Hoogenboezem, R.; de Wit, E.; Bouwman, B.A.M.; Erpelinck, C.; van der Velden, V.H.J.; Havermans, M.; Avellino, R.; van Lom, K.; et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell 2014, 157, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Katayama, S.; Suzuki, M.; Yamaoka, A.; Keleku-Lukwete, N.; Katsuoka, F.; Otsuki, A.; Kure, S.; Engel, J.D.; Yamamoto, M. GATA2 haploinsufficiency accelerates EVI1-driven leukemogenesis. Blood 2017, 130, 908–919. [Google Scholar] [CrossRef]
- Kozyra, E.J.; Pastor, V.B.; Lefkopoulos, S.; Sahoo, S.S.; Busch, H.; Voss, R.K.; Erlacher, M.; Lebrecht, D.; Szvetnik, E.A.; Hirabayashi, S.; et al. Synonymous GATA2 mutations result in selective loss of mutated RNA and are common in patients with GATA2 deficiency. Leukemia 2020, 34, 2673–2687. [Google Scholar] [CrossRef]
- Burda, P.; Laslo, P.; Stopka, T. The role of PU.1 and GATA-1 transcription factors during normal and leukemogenic hematopoiesis. Leukemia 2010, 24, 1249–1257. [Google Scholar] [CrossRef]
- Nerlov, C.; Graf, T. PU.1 induces myeloid lineage commitment in multipotent hematopoietic progenitors. Genes Dev. 1998, 12, 2403–2412. [Google Scholar] [CrossRef]
- Rekhtman, N.; Radparvar, F.; Evans, T.; Skoultchi, A.I. Direct interaction of hematopoietic transcription factors PU.1 and GATA-1: Functional antagonism in erythroid cells. Genes Dev. 1999, 13, 1398–1411. [Google Scholar] [CrossRef]
- Anderson, K.L.; Smith, K.A.; Perkin, H.; Hermanson, G.; Anderson, C.G.; Jolly, D.J.; Maki, R.A.; Torbett, B.E. PU.1 and the granulocyte- and macrophage colony-stimulating factor receptors play distinct roles in late-stage myeloid cell differentiation. Blood 1999, 94, 2310–2318. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, L.; Caballero, N.; Fernandez-Calleja, L.; Karkoulia, E.; Strouboulis, J. Regulation of GATA1 levels in erythropoiesis. IUBMB Life 2020, 72, 89–105. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.A.; Iwama, A.; Iotzova, G.; Schulz, M.; Elsasser, A.; Vangala, R.K.; Tenen, D.G.; Hiddemann, W.; Behre, G. Granulocyte inducer C/EBPalpha inactivates the myeloid master regulator PU.1: Possible role in lineage commitment decisions. Blood 2002, 100, 483–490. [Google Scholar] [CrossRef]
- Radomska, H.S.; Huettner, C.S.; Zhang, P.; Cheng, T.; Scadden, D.T.; Tenen, D.G. CCAAT/enhancer binding protein alpha is a regulatory switch sufficient for induction of granulocytic development from bipotential myeloid progenitors. Mol. Cell Biol. 1998, 18, 4301–4314. [Google Scholar] [CrossRef] [PubMed]
- Rosenbauer, F.; Wagner, K.; Kutok, J.L.; Iwasaki, H.; Le Beau, M.M.; Okuno, Y.; Akashi, K.; Fiering, S.; Tenen, D.G. Acute myeloid leukemia induced by graded reduction of a lineage-specific transcription factor, PU.1. Nat. Genet. 2004, 36, 624–630. [Google Scholar] [CrossRef]
- Scott, E.W.; Simon, M.C.; Anastasi, J.; Singh, H. Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science 1994, 265, 1573–1577. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Okuno, Y.; Zhang, P.; Radomska, H.S.; Chen, H.; Iwasaki, H.; Akashi, K.; Klemsz, M.J.; McKercher, S.R.; Maki, R.A.; et al. Regulation of the PU.1 gene by distal elements. Blood 2001, 98, 2958–2965. [Google Scholar] [CrossRef]
- Will, B.; Vogler, T.O.; Narayanagari, S.; Bartholdy, B.; Todorova, T.I.; da Silva Ferreira, M.; Chen, J.; Yu, Y.; Mayer, J.; Barreyro, L.; et al. Minimal PU.1 reduction induces a preleukemic state and promotes development of acute myeloid leukemia. Nat. Med. 2015, 21, 1172–1181. [Google Scholar] [CrossRef]
- Mishra, M.; Thacker, G.; Sharma, A.; Singh, A.K.; Upadhyay, V.; Sanyal, S.; Verma, S.P.; Tripathi, A.K.; Bhatt, M.L.B.; Trivedi, A.K. FBW7 Inhibits Myeloid Differentiation in Acute Myeloid Leukemia via GSK3-Dependent Ubiquitination of PU.1. Mol. Cancer Res. 2021, 19, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Thacker, G.; Mishra, M.; Sharma, A.; Singh, A.K.; Sanyal, S.; Trivedi, A.K. E3 ligase SCF(SKP2) ubiquitinates and degrades tumor suppressor C/EBPalpha in acute myeloid leukemia. Life Sci. 2020, 257, 118041. [Google Scholar] [CrossRef] [PubMed]
- Pevny, L.; Simon, M.C.; Robertson, E.; Klein, W.H.; Tsai, S.F.; D’Agati, V.; Orkin, S.H.; Costantini, F. Erythroid differentiation in chimaeric mice blocked by a targeted mutation in the gene for transcription factor GATA-1. Nature 1991, 349, 257–260. [Google Scholar] [CrossRef]
- McDevitt, M.A.; Shivdasani, R.A.; Fujiwara, Y.; Yang, H.; Orkin, S.H. A “knockdown” mutation created by cis-element gene targeting reveals the dependence of erythroid cell maturation on the level of transcription factor GATA-1. Proc. Natl. Acad. Sci. USA 1997, 94, 6781–6785. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Onodera, K.; Motohashi, H.; Suwabe, N.; Hayashi, N.; Yanai, N.; Nabesima, Y.; Yamamoto, M. Arrest in primitive erythroid cell development caused by promoter-specific disruption of the GATA-1 gene. J. Biol. Chem. 1997, 272, 12611–12615. [Google Scholar] [CrossRef]
- Ling, T.; Crispino, J.D.; Zingariello, M.; Martelli, F.; Migliaccio, A.R. GATA1 insufficiencies in primary myelofibrosis and other hematopoietic disorders: Consequences for therapy. Expert Rev. Hematol. 2018, 11, 169–184. [Google Scholar] [CrossRef]
- Whyatt, D.; Lindeboom, F.; Karis, A.; Ferreira, R.; Milot, E.; Hendriks, R.; de Bruijn, M.; Langeveld, A.; Gribnau, J.; Grosveld, F.; et al. An intrinsic but cell-nonautonomous defect in GATA-1-overexpressing mouse erythroid cells. Nature 2000, 406, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, C.R.; Martinelli, V.; Rinaldi, P.; Ciancia, R.; del Vecchio, L. GATA1 is overexpressed in patients with essential thrombocythemia and polycythemia vera but not in patients with primary myelofibrosis or chronic myelogenous leukemia. Leuk. Lymphoma 2008, 49, 1416–1419. [Google Scholar] [CrossRef]
- Calligaris, R.; Bottardi, S.; Cogoi, S.; Apezteguia, I.; Santoro, C. Alternative translation initiation site usage results in two functionally distinct forms of the GATA-1 transcription factor. Proc. Natl. Acad. Sci. USA 1995, 92, 11598–11602. [Google Scholar] [CrossRef] [PubMed]
- Sankaran, V.G.; Ghazvinian, R.; Do, R.; Thiru, P.; Vergilio, J.A.; Beggs, A.H.; Sieff, C.A.; Orkin, S.H.; Nathan, D.G.; Lander, E.S.; et al. Exome sequencing identifies GATA1 mutations resulting in Diamond-Blackfan anemia. J. Clin. Invest. 2012, 122, 2439–2443. [Google Scholar] [CrossRef]
- Wechsler, J.; Greene, M.; McDevitt, M.A.; Anastasi, J.; Karp, J.E.; Le Beau, M.M.; Crispino, J.D. Acquired mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome. Nat. Genet. 2002, 32, 148–152. [Google Scholar] [CrossRef]
- Banno, K.; Omori, S.; Hirata, K.; Nawa, N.; Nakagawa, N.; Nishimura, K.; Ohtaka, M.; Nakanishi, M.; Sakuma, T.; Yamamoto, T.; et al. Systematic Cellular Disease Models Reveal Synergistic Interaction of Trisomy 21 and GATA1 Mutations in Hematopoietic Abnormalities. Cell Rep. 2016, 15, 1228–1241. [Google Scholar] [CrossRef]
- Labuhn, M.; Perkins, K.; Matzk, S.; Varghese, L.; Garnett, C.; Papaemmanuil, E.; Metzner, M.; Kennedy, A.; Amstislavskiy, V.; Risch, T.; et al. Mechanisms of Progression of Myeloid Preleukemia to Transformed Myeloid Leukemia in Children with Down Syndrome. Cancer Cell 2019, 36, 340. [Google Scholar] [CrossRef]
- Ohlsson, E.; Schuster, M.B.; Hasemann, M.; Porse, B.T. The multifaceted functions of C/EBPalpha in normal and malignant haematopoiesis. Leukemia 2016, 30, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Pulikkan, J.A.; Tenen, D.G.; Behre, G. C/EBPalpha deregulation as a paradigm for leukemogenesis. Leukemia 2017, 31, 2279–2285. [Google Scholar] [CrossRef]
- Ossipow, V.; Descombes, P.; Schibler, U. CCAAT/enhancer-binding protein mRNA is translated into multiple proteins with different transcription activation potentials. Proc. Natl. Acad. Sci. USA 1993, 90, 8219–8223. [Google Scholar] [CrossRef] [PubMed]
- Muller, C.; Bremer, A.; Schreiber, S.; Eichwald, S.; Calkhoven, C.F. Nucleolar retention of a translational C/EBPalpha isoform stimulates rDNA transcription and cell size. EMBO J. 2010, 29, 897–909. [Google Scholar] [CrossRef]
- Calkhoven, C.F.; Muller, C.; Leutz, A. Translational control of C/EBPalpha and C/EBPbeta isoform expression. Genes Dev. 2000, 14, 1920–1932. [Google Scholar]
- Greif, P.A.; Dufour, A.; Konstandin, N.P.; Ksienzyk, B.; Zellmeier, E.; Tizazu, B.; Sturm, J.; Benthaus, T.; Herold, T.; Yaghmaie, M.; et al. GATA2 zinc finger 1 mutations associated with biallelic CEBPA mutations define a unique genetic entity of acute myeloid leukemia. Blood 2012, 120, 395–403. [Google Scholar] [CrossRef]
- Fasan, A.; Eder, C.; Haferlach, C.; Grossmann, V.; Kohlmann, A.; Dicker, F.; Kern, W.; Haferlach, T.; Schnittger, S. GATA2 mutations are frequent in intermediate-risk karyotype AML with biallelic CEBPA mutations and are associated with favorable prognosis. Leukemia 2013, 27, 482–485. [Google Scholar] [CrossRef] [PubMed]
- Wilhelmson, A.S.; Porse, B.T. CCAAT enhancer binding protein alpha (CEBPA) biallelic acute myeloid leukaemia: Cooperating lesions, molecular mechanisms and clinical relevance. Br. J. Haematol. 2020, 190, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Mulet-Lazaro, R.; van Herk, S.; Erpelinck, C.A.J.; Bindels, E.; Sanders, M.A.; Vermeulen, C.; Renkens, I.; Valk, P.; Melnick, A.; de Ridder, J.; et al. Allele-specific expression of GATA2 due to epigenetic dysregulation in CEBPA double mutant AML. Blood 2021. [Google Scholar] [CrossRef] [PubMed]
- Ping, N.; Sun, A.; Song, Y.; Wang, Q.; Yin, J.; Cheng, W.; Xu, Y.; Wen, L.; Yao, H.; Ma, L.; et al. Exome sequencing identifies highly recurrent somatic GATA2 and CEBPA mutations in acute erythroid leukemia. Leukemia 2017, 31, 195–202. [Google Scholar] [CrossRef]
- Di Genua, C.; Valletta, S.; Buono, M.; Stoilova, B.; Sweeney, C.; Rodriguez-Meira, A.; Grover, A.; Drissen, R.; Meng, Y.; Beveridge, R.; et al. C/EBPalpha and GATA-2 Mutations Induce Bilineage Acute Erythroid Leukemia through Transformation of a Neomorphic Neutrophil-Erythroid Progenitor. Cancer Cell 2020, 37, 690–704.e698. [Google Scholar] [CrossRef] [PubMed]
- Perkins, A.; Xu, X.; Higgs, D.R.; Patrinos, G.P.; Arnaud, L.; Bieker, J.J.; Philipsen, S.; Workgroup, K.L.F.C. Kruppeling erythropoiesis: An unexpected broad spectrum of human red blood cell disorders due to KLF1 variants. Blood 2016, 127, 1856–1862. [Google Scholar] [CrossRef]
- Mansoor, A.; Mansoor, M.O.; Patel, J.L.; Zhao, S.; Natkunam, Y.; Bieker, J.J. KLF1/EKLF expression in acute leukemia is correlated with chromosomal abnormalities. Blood Cells Mol. Dis. 2020, 83, 102434. [Google Scholar] [CrossRef]
- Perdomo, J.; Fock, E.L.; Kaur, G.; Yan, F.; Khachigian, L.M.; Jans, D.A.; Chong, B.H. A monopartite sequence is essential for p45 NF-E2 nuclear translocation, transcriptional activity and platelet production. J. Thromb. Haemost. 2010, 8, 2542–2553. [Google Scholar] [CrossRef]
- Jutzi, J.S.; Basu, T.; Pellmann, M.; Kaiser, S.; Steinemann, D.; Sanders, M.A.; Hinai, A.S.A.; Zeilemaker, A.; Bojtine Kovacs, S.; Koellerer, C.; et al. Altered NFE2 activity predisposes to leukemic transformation and myelosarcoma with AML-specific aberrations. Blood 2019, 133, 1766–1777. [Google Scholar] [CrossRef] [PubMed]
- Dumitriu, B.; Bhattaram, P.; Dy, P.; Huang, Y.; Quayum, N.; Jensen, J.; Lefebvre, V. Sox6 is necessary for efficient erythropoiesis in adult mice under physiological and anemia-induced stress conditions. PLoS ONE 2010, 5, e12088. [Google Scholar] [CrossRef] [PubMed]
- Cantu, C.; Ierardi, R.; Alborelli, I.; Fugazza, C.; Cassinelli, L.; Piconese, S.; Bose, F.; Ottolenghi, S.; Ferrari, G.; Ronchi, A. Sox6 enhances erythroid differentiation in human erythroid progenitors. Blood 2011, 117, 3669–3679. [Google Scholar] [CrossRef] [PubMed]
- Barbarani, G.; Fugazza, C.; Barabino, S.M.L.; Ronchi, A.E. SOX6 blocks the proliferation of BCR-ABL1(+) and JAK2V617F(+) leukemic cells. Sci. Rep. 2019, 9, 3388. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Cazzola, M.; Boultwood, J.; Malcovati, L.; Vyas, P.; Bowen, D.; Pellagatti, A.; Wainscoat, J.S.; Hellstrom-Lindberg, E.; Gambacorti-Passerini, C.; et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N. Engl. J. Med. 2011, 365, 1384–1395. [Google Scholar] [CrossRef]
- Quesada, V.; Conde, L.; Villamor, N.; Ordóñez, G.R.; Jares, P.; Bassaganyas, L.; Ramsay, A.J.; Beà, S.; Pinyol, M.; Martínez-Trillos, A.; et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat. Genet. 2011, 44, 47–52. [Google Scholar] [CrossRef]
- Wang, L.; Lawrence, M.S.; Wan, Y.; Stojanov, P.; Sougnez, C.; Stevenson, K.; Werner, L.; Sivachenko, A.; DeLuca, D.S.; Zhang, L.; et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N. Engl. J. Med. 2011, 365, 2497–2506. [Google Scholar] [CrossRef]
- Haferlach, T. The Molecular Pathology of Myelodysplastic Syndrome. Pathobiol. J. Immunopathol. Mol. Cell. Biol. 2019, 86, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, M.; Drake, K.; Kanadia, R.N. An Integrated Model of Minor Intron Emergence and Conservation. Front. Genet. 2019, 10. [Google Scholar] [CrossRef]
- Cretu, C.; Schmitzová, J.; Ponce-Salvatierra, A.; Dybkov, O.; De Laurentiis, E.I.; Sharma, K.; Will, C.L.; Urlaub, H.; Lührmann, R.; Pena, V. Molecular Architecture of SF3b and Structural Consequences of Its Cancer-Related Mutations. Mol. Cell 2016, 64, 307–319. [Google Scholar] [CrossRef]
- Darman, R.B.; Seiler, M.; Agrawal, A.A.; Lim, K.H.; Peng, S.; Aird, D.; Bailey, S.L.; Bhavsar, E.B.; Chan, B.; Colla, S.; et al. Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3’ Splice Site Selection through Use of a Different Branch Point. Cell Rep. 2015, 13, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Park, S.-Y.; Sakashita, G.; Nariai, Y.; Kuwasako, K.; Muto, Y.; Urano, T.; Obayashi, E. Elucidation of the aberrant 3′ splice site selection by cancer-associated mutations on the U2AF1. Nat. Commun. 2020, 11, 4744. [Google Scholar] [CrossRef] [PubMed]
- Tronchère, H.; Wang, J.; Fu, X.D. A protein related to splicing factor U2AF35 that interacts with U2AF65 and SR proteins in splicing of pre-mRNA. Nature 1997, 388, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Zheng, X.; Luecke, S.; Green, M.R. The U2AF35-related protein Urp contacts the 3’ splice site to promote U12-type intron splicing and the second step of U2-type intron splicing. Genes Dev. 2010, 24, 2389–2394. [Google Scholar] [CrossRef] [PubMed]
- Madan, V.; Kanojia, D.; Li, J.; Okamoto, R.; Sato-Otsubo, A.; Kohlmann, A.; Sanada, M.; Grossmann, V.; Sundaresan, J.; Shiraishi, Y.; et al. Aberrant splicing of U12-type introns is the hallmark of ZRSR2 mutant myelodysplastic syndrome. Nat. Commun. 2015, 6, 6042. [Google Scholar] [CrossRef] [PubMed]
- Daubner, G.M.; Cléry, A.; Jayne, S.; Stevenin, J.; Allain, F.H. A syn-anti conformational difference allows SRSF2 to recognize guanines and cytosines equally well. EMBO J. 2012, 31, 162–174. [Google Scholar] [CrossRef]
- Saez, B.; Walter, M.J.; Graubert, T.A. Splicing factor gene mutations in hematologic malignancies. Blood 2017, 129, 1260–1269. [Google Scholar] [CrossRef]
- Hong, M.; He, G. The 2016 Revision to the World Health Organization Classification of Myelodysplastic Syndromes. J. Transl. Int. Med. 2017, 5, 139–143. [Google Scholar] [CrossRef]
- Malcovati, L.; Karimi, M.; Papaemmanuil, E.; Ambaglio, I.; Jädersten, M.; Jansson, M.; Elena, C.; Gallì, A.; Walldin, G.; Della Porta, M.G.; et al. SF3B1 mutation identifies a distinct subset of myelodysplastic syndrome with ring sideroblasts. Blood 2015, 126, 233–241. [Google Scholar] [CrossRef]
- Seiler, M.; Peng, S.; Agrawal, A.A.; Palacino, J.; Teng, T.; Zhu, P.; Smith, P.G.; Buonamici, S.; Yu, L. Somatic Mutational Landscape of Splicing Factor Genes and Their Functional Consequences across 33 Cancer Types. Cell Rep. 2018, 23, 282–296. [Google Scholar] [CrossRef] [PubMed]
- Mupo, A.; Seiler, M.; Sathiaseelan, V.; Pance, A.; Yang, Y.; Agrawal, A.A.; Iorio, F.; Bautista, R.; Pacharne, S.; Tzelepis, K.; et al. Hemopoietic-specific Sf3b1-K700E knock-in mice display the splicing defect seen in human MDS but develop anemia without ring sideroblasts. Leukemia 2017, 31, 720–727. [Google Scholar] [CrossRef]
- Obeng, E.A.; Chappell, R.J.; Seiler, M.; Chen, M.C.; Campagna, D.R.; Schmidt, P.J.; Schneider, R.K.; Lord, A.M.; Wang, L.; Gambe, R.G.; et al. Physiologic Expression of Sf3b1K700E Causes Impaired Erythropoiesis, Aberrant Splicing, and Sensitivity to Therapeutic Spliceosome Modulation. Cancer Cell 2016, 30, 404–417. [Google Scholar] [CrossRef] [PubMed]
- Will, C.L.; Schneider, C.; Hossbach, M.; Urlaub, H.; Rauhut, R.; Elbashir, S.; Tuschl, T.; Lührmann, R. The human 18S U11/U12 snRNP contains a set of novel proteins not found in the U2-dependent spliceosome. RNA 2004, 10, 929–941. [Google Scholar] [CrossRef]
- Yoshida, H.; Park, S.Y.; Oda, T.; Akiyoshi, T.; Sato, M.; Shirouzu, M.; Tsuda, K.; Kuwasako, K.; Unzai, S.; Muto, Y.; et al. A novel 3’ splice site recognition by the two zinc fingers in the U2AF small subunit. Genes Dev. 2015, 29, 1649–1660. [Google Scholar] [CrossRef]
- Okeyo-Owuor, T.; White, B.S.; Chatrikhi, R.; Mohan, D.R.; Kim, S.; Griffith, M.; Ding, L.; Ketkar-Kulkarni, S.; Hundal, J.; Laird, K.M.; et al. U2AF1 mutations alter sequence specificity of pre-mRNA binding and splicing. Leukemia 2015, 29, 909–917. [Google Scholar] [CrossRef]
- Ilagan, J.O.; Ramakrishnan, A.; Hayes, B.; Murphy, M.E.; Zebari, A.S.; Bradley, P.; Bradley, R.K. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res. 2015, 25, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, J.L.; Kielkopf, C.L. Splicing Factor Mutations in Myelodysplasias: Insights from Spliceosome Structures. Trends Genet. 2017, 33, 336–348. [Google Scholar] [CrossRef] [PubMed]
- Warnasooriya, C.; Feeney, C.F.; Laird, K.M.; Ermolenko, D.N.; Kielkopf, C.L. A splice site-sensing conformational switch in U2AF2 is modulated by U2AF1 and its recurrent myelodysplasia-associated mutation. Nucleic Acids Res. 2020, 48, 5695–5709. [Google Scholar] [CrossRef] [PubMed]
- Voith von Voithenberg, L.; Sánchez-Rico, C.; Kang, H.S.; Madl, T.; Zanier, K.; Barth, A.; Warner, L.R.; Sattler, M.; Lamb, D.C. Recognition of the 3’ splice site RNA by the U2AF heterodimer involves a dynamic population shift. Proc. Natl. Acad. Sci. USA 2016, 113, E7169–E7175. [Google Scholar] [CrossRef]
- Damm, F.; Kosmider, O.; Gelsi-Boyer, V.; Renneville, A.; Carbuccia, N.; Hidalgo-Curtis, C.; Della Valle, V.; Couronné, L.; Scourzic, L.; Chesnais, V.; et al. Mutations affecting mRNA splicing define distinct clinical phenotypes and correlate with patient outcome in myelodysplastic syndromes. Blood 2012, 119, 3211–3218. [Google Scholar] [CrossRef]
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011, 478, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Inoue, D.; Polaski, J.T.; Taylor, J.; Castel, P.; Chen, S.; Kobayashi, S.; Hogg, S.J.; Hayashi, Y.; Pineda, J.M.B.; El Marabti, E.; et al. Minor intron retention drives clonal hematopoietic disorders and diverse cancer predisposition. Nat. Genet. 2021, 53, 707–718. [Google Scholar] [CrossRef]
- Lee, S.C.; North, K.; Kim, E.; Jang, E.; Obeng, E.; Lu, S.X.; Liu, B.; Inoue, D.; Yoshimi, A.; Ki, M.; et al. Synthetic Lethal and Convergent Biological Effects of Cancer-Associated Spliceosomal Gene Mutations. Cancer Cell 2018, 34, 225–241.e228. [Google Scholar] [CrossRef]
- Madan, V.; Cao, Z.; Teoh, W.W.; Dakle, P.; Han, L.; Shyamsunder, P.; Jeitany, M.; Zhou, S.; Li, J.; Nordin, H.B.M.; et al. ZRSR1 cooperates with ZRSR2 in regulating splicing of U12-type introns in murine hematopoietic cells. Haematologica 2021. [Google Scholar] [CrossRef]
- Kim, E.; Ilagan, J.O.; Liang, Y.; Daubner, G.M.; Lee, S.C.; Ramakrishnan, A.; Li, Y.; Chung, Y.R.; Micol, J.B.; Murphy, M.E.; et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 2015, 27, 617–630. [Google Scholar] [CrossRef]
- Zhang, J.; Lieu, Y.K.; Ali, A.M.; Penson, A.; Reggio, K.S.; Rabadan, R.; Raza, A.; Mukherjee, S.; Manley, J.L. Disease-associated mutation in SRSF2 misregulates splicing by altering RNA-binding affinities. Proc. Natl. Acad. Sci. USA 2015, 112, E4726–E4734. [Google Scholar] [CrossRef] [PubMed]
- Pellagatti, A.; Armstrong, R.N.; Steeples, V.; Sharma, E.; Repapi, E.; Singh, S.; Sanchi, A.; Radujkovic, A.; Horn, P.; Dolatshad, H.; et al. Impact of spliceosome mutations on RNA splicing in myelodysplasia: Dysregulated genes/pathways and clinical associations. Blood 2018, 132, 1225–1240. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, Y.; Malcovati, L.; Gallì, A.; Sato-Otsubo, A.; Kataoka, K.; Sato, Y.; Watatani, Y.; Suzuki, H.; Yoshizato, T.; Yoshida, K.; et al. Aberrant splicing and defective mRNA production induced by somatic spliceosome mutations in myelodysplasia. Nat. Commun. 2018, 9, 3649. [Google Scholar] [CrossRef] [PubMed]
- Dolatshad, H.; Pellagatti, A.; Liberante, F.G.; Llorian, M.; Repapi, E.; Steeples, V.; Roy, S.; Scifo, L.; Armstrong, R.N.; Shaw, J.; et al. Cryptic splicing events in the iron transporter ABCB7 and other key target genes in SF3B1-mutant myelodysplastic syndromes. Leukemia 2016, 30, 2322–2331. [Google Scholar] [CrossRef] [PubMed]
- Bakker, S.T.; Passegue, E. Resilient and resourceful: Genome maintenance strategies in hematopoietic stem cells. Exp. Hematol. 2013, 41, 915–923. [Google Scholar] [CrossRef]
- Biechonski, S.; Yassin, M.; Milyavsky, M. DNA-damage response in hematopoietic stem cells: An evolutionary trade-off between blood regeneration and leukemia suppression. Carcinogenesis 2017, 38, 367–377. [Google Scholar] [CrossRef]
- Niedernhofer, L.J. DNA repair is crucial for maintaining hematopoietic stem cell function. DNA Repair 2008, 7, 523–529. [Google Scholar] [CrossRef]
- Mgbemena, V.E.; Signer, R.A.J.; Wijayatunge, R.; Laxson, T.; Morrison, S.J.; Ross, T.S. Distinct Brca1 Mutations Differentially Reduce Hematopoietic Stem Cell Function. Cell Rep. 2017, 18, 947–960. [Google Scholar] [CrossRef] [PubMed]
- Vasanthakumar, A.; Arnovitz, S.; Marquez, R.; Lepore, J.; Rafidi, G.; Asom, A.; Weatherly, M.; Davis, E.M.; Neistadt, B.; Duszynski, R.; et al. Brca1 deficiency causes bone marrow failure and spontaneous hematologic malignancies in mice. Blood 2016, 127, 310–313. [Google Scholar] [CrossRef] [PubMed]
- Lancini, C.; van den Berk, P.C.; Vissers, J.H.; Gargiulo, G.; Song, J.Y.; Hulsman, D.; Serresi, M.; Tanger, E.; Blom, M.; Vens, C.; et al. Tight regulation of ubiquitin-mediated DNA damage response by USP3 preserves the functional integrity of hematopoietic stem cells. J. Exp. Med. 2014, 211, 1759–1777. [Google Scholar] [CrossRef] [PubMed]
- Pilzecker, B.; Buoninfante, O.A.; van den Berk, P.; Lancini, C.; Song, J.Y.; Citterio, E.; Jacobs, H. DNA damage tolerance in hematopoietic stem and progenitor cells in mice. Proc. Natl. Acad. Sci. USA 2017, 114, E6875–E6883. [Google Scholar] [CrossRef]
- Niraj, J.; Farkkila, A.; D’Andrea, A.D. The Fanconi Anemia Pathway in Cancer. Annu. Rev. Cancer Biol. 2019, 3, 457–478. [Google Scholar] [CrossRef]
- Rossi, D.J.; Bryder, D.; Seita, J.; Nussenzweig, A.; Hoeijmakers, J.; Weissman, I.L. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature 2007, 447, 725–729. [Google Scholar] [CrossRef] [PubMed]
- Rübe, C.; Fricke, A.; Widmann, T.; Fürst, T.; Madry, H.; Pfreundschuh, M.; Rübe, C.; Freitag, M. Accumulation of DNA Damage in Hematopoietic Stem and Progenitor Cells during Human Aging. PLoS ONE 2011, 6, e17487. [Google Scholar] [CrossRef]
- Beerman, I.; Seita, J.; Inlay, M.A.; Weissman, I.L.; Rossi, D.J. Quiescent hematopoietic stem cells accumulate DNA damage during aging that is repaired upon entry into cell cycle. Cell Stem Cell 2014, 15, 37–50. [Google Scholar] [CrossRef]
- Milyavsky, M.; Gan, O.I.; Trottier, M.; Komosa, M.; Tabach, O.; Notta, F.; Lechman, E.; Hermans, K.G.; Eppert, K.; Konovalova, Z.; et al. A Distinctive DNA Damage Response in Human Hematopoietic Stem Cells Reveals an Apoptosis-Independent Role for p53 in Self-Renewal. Stem Cell 2010, 7, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Welch, J.S.; Ley, T.J.; Link, D.C.; Miller, C.A.; Larson, D.E.; Koboldt, D.C.; Wartman, L.D.; Lamprecht, T.L.; Liu, F.; Xia, J.; et al. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012, 150, 264–278. [Google Scholar] [CrossRef] [PubMed]
- Osorio, F.G.; Rosendahl Huber, A.; Oka, R.; Verheul, M.; Patel, S.H.; Hasaart, K.; de la Fonteijne, L.; Varela, I.; Camargo, F.D.; van Boxtel, R. Somatic Mutations Reveal Lineage Relationships and Age-Related Mutagenesis in Human Hematopoiesis. Cell Rep. 2018, 25, 2308–2316. [Google Scholar] [CrossRef]
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D.; et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Shlush, L.I.; Mitchell, A.; Heisler, L.; Abelson, S.; Ng, S.W.K.; Trotman-Grant, A.; Medeiros, J.J.F.; Rao-Bhatia, A.; Jaciw-Zurakowsky, I.; Marke, R.; et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature 2017, 547, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Mohrin, M.; Bourke, E.; Alexander, D.; Warr, M.R.; Barry-Holson, K.; Le Beau, M.M.; Morrison, C.G.; Passegue, E. Hematopoietic stem cell quiescence promotes error-prone DNA repair and mutagenesis. Cell Stem Cell 2010, 7, 174–185. [Google Scholar] [CrossRef]
- Greaves, M.F.; Wiemels, J. Origins of chromosome translocations in childhood leukaemia. Nat. Rev. Cancer 2003, 3, 639–649. [Google Scholar] [CrossRef]
- Abdel-Wahab, O.; Mullally, A.; Hedvat, C.; Garcia-Manero, G.; Patel, J.; Wadleigh, M.; Malinge, S.; Yao, J.; Kilpivaara, O.; Bhat, R.; et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood 2009, 114, 144–147. [Google Scholar] [CrossRef]
- Flach, J.; Bakker, S.T.; Mohrin, M.; Conroy, P.C.; Pietras, E.M.; Reynaud, D.; Alvarez, S.; Diolaiti, M.E.; Ugarte, F.; Forsberg, E.C.; et al. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature 2014, 512, 198–202. [Google Scholar] [CrossRef]
- Sperka, T.; Wang, J.; Rudolph, K.L. DNA damage checkpoints in stem cells, ageing and cancer. Nat. Rev. Mol. Cell Biol. 2012, 13, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Sung, P.A.; Libura, J.; Richardson, C. Etoposide and illegitimate DNA double-strand break repair in the generation of MLL translocations: New insights and new questions. DNA Repair 2006, 5, 1109–1118. [Google Scholar] [CrossRef]
- Pui, C.H.; Relling, M.V. Topoisomerase II inhibitor-related acute myeloid leukaemia. Br. J. Haematol. 2000, 109, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Belotserkovskii, B.P.; Tornaletti, S.; D’Souza, A.D.; Hanawalt, P.C. R-loop generation during transcription: Formation, processing and cellular outcomes. DNA Repair 2018, 71, 69–81. [Google Scholar] [CrossRef]
- Garcia-Muse, T.; Aguilera, A. R Loops: From Physiological to Pathological Roles. Cell 2019, 179, 604–618. [Google Scholar] [CrossRef] [PubMed]
- Barroso, S.; Herrera-Moyano, E.; Munoz, S.; Garcia-Rubio, M.; Gomez-Gonzalez, B.; Aguilera, A. The DNA damage response acts as a safeguard against harmful DNA-RNA hybrids of different origins. EMBO Rep. 2019, 20, e47250. [Google Scholar] [CrossRef]
- Crossley, M.P.; Bocek, M.; Cimprich, K.A. R-Loops as Cellular Regulators and Genomic Threats. Mol. Cell 2019, 73, 398–411. [Google Scholar] [CrossRef] [PubMed]
- Flach, J.; Jann, J.C.; Knaflic, A.; Riabov, V.; Streuer, A.; Altrock, E.; Xu, Q.; Schmitt, N.; Oblander, J.; Nowak, V.; et al. Replication stress signaling is a therapeutic target in myelodysplastic syndromes with splicing factor mutations. Haematologica 2020. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, J.Y.; Huang, Y.J.; Gu, Y.; Qiu, J.; Qian, H.; Shao, C.; Zhang, X.; Hu, J.; Li, H.; et al. The Augmented R-Loop Is a Unifying Mechanism for Myelodysplastic Syndromes Induced by High-Risk Splicing Factor Mutations. Mol. Cell 2018, 69, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.D.; Leong, W.Y.; Li, W.; Reddy, P.N.G.; Sullivan, J.D.; Walter, M.J.; Zou, L.; Graubert, T.A. Spliceosome Mutations Induce R Loop-Associated Sensitivity to ATR Inhibition in Myelodysplastic Syndromes. Cancer Res. 2018, 78, 5363–5374. [Google Scholar] [CrossRef] [PubMed]
- Tam, A.S.; Stirling, P.C. Splicing, genome stability and disease: Splice like your genome depends on it! Curr. Genet. 2019, 65, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, J.; Manley, J.L. Loss of splicing factor ASF/SF2 induces G2 cell cycle arrest and apoptosis, but inhibits internucleosomal DNA fragmentation. Genes Dev. 2005, 19, 2705–2714. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Ahmed, D.; Dolatshad, H.; Tatwavedi, D.; Schulze, U.; Sanchi, A.; Ryley, S.; Dhir, A.; Carpenter, L.; Watt, S.M.; et al. SF3B1 mutations induce R-loop accumulation and DNA damage in MDS and leukemia cells with therapeutic implications. Leukemia 2020, 34, 2525–2530. [Google Scholar] [CrossRef]
- Skourti-Stathaki, K.; Proudfoot, N.J.; Gromak, N. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol. Cell 2011, 42, 794–805. [Google Scholar] [CrossRef]
- Garcia-Rubio, M.L.; Perez-Calero, C.; Barroso, S.I.; Tumini, E.; Herrera-Moyano, E.; Rosado, I.V.; Aguilera, A. The Fanconi Anemia Pathway Protects Genome Integrity from R-loops. PLoS Genet. 2015, 11, e1005674. [Google Scholar] [CrossRef]
- Schwab, R.A.; Nieminuszczy, J.; Shah, F.; Langton, J.; Lopez Martinez, D.; Liang, C.C.; Cohn, M.A.; Gibbons, R.J.; Deans, A.J.; Niedzwiedz, W. The Fanconi Anemia Pathway Maintains Genome Stability by Coordinating Replication and Transcription. Mol. Cell 2015, 60, 351–361. [Google Scholar] [CrossRef]
- Liang, Z.; Liang, F.; Teng, Y.; Chen, X.; Liu, J.; Longerich, S.; Rao, T.; Green, A.M.; Collins, N.B.; Xiong, Y.; et al. Binding of FANCI-FANCD2 Complex to RNA and R-Loops Stimulates Robust FANCD2 Monoubiquitination. Cell Rep. 2019, 26, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Madireddy, A.; Kosiyatrakul, S.T.; Boisvert, R.A.; Herrera-Moyano, E.; Garcia-Rubio, M.L.; Gerhardt, J.; Vuono, E.A.; Owen, N.; Yan, Z.; Olson, S.; et al. FANCD2 Facilitates Replication through Common Fragile Sites. Mol. Cell 2016, 64, 388–404. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Hejna, J.; Takata, M. Regulation of R-loops and genome instability in Fanconi anemia. J. Biochem. 2019, 165, 465–470. [Google Scholar] [CrossRef]
- Moriel-Carretero, M.; Ovejero, S.; Gerus-Durand, M.; Vryzas, D.; Constantinou, A. Fanconi anemia FANCD2 and FANCI proteins regulate the nuclear dynamics of splicing factors. J. Cell Biol. 2017, 216, 4007–4026. [Google Scholar] [CrossRef]
- Howlett, N.G.; Taniguchi, T.; Olson, S.; Cox, B.; Waisfisz, Q.; De Die-Smulders, C.; Persky, N.; Grompe, M.; Joenje, H.; Pals, G.; et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science 2002, 297, 606–609. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, S.L.; Tian, L.; Kahkonen, M.; Schwartzentruber, J.; Kircher, M.; Consortium, F.C.; Majewski, J.; Dyment, D.A.; Innes, A.M.; University of Washington Centers for Mendelian Genomics; et al. Biallelic mutations in BRCA1 cause a new Fanconi anemia subtype. Cancer Discov. 2015, 5, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Navarro, S.; Meza, N.W.; Quintana-Bustamante, O.; Casado, J.A.; Jacome, A.; McAllister, K.; Puerto, S.; Surralles, J.; Segovia, J.C.; Bueren, J.A. Hematopoietic dysfunction in a mouse model for Fanconi anemia group D1. Mol. Ther. 2006, 14, 525–535. [Google Scholar] [CrossRef] [PubMed]
- San Martin Alonso, M.; Noordermeer, S.M. Untangling the crosstalk between BRCA1 and R-loops during DNA repair. Nucleic Acids Res. 2021. [Google Scholar] [CrossRef]
- Hatchi, E.; Skourti-Stathaki, K.; Ventz, S.; Pinello, L.; Yen, A.; Kamieniarz-Gdula, K.; Dimitrov, S.; Pathania, S.; McKinney, K.M.; Eaton, M.L.; et al. BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol. Cell 2015, 57, 636–647. [Google Scholar] [CrossRef]
- Bhatia, V.; Barroso, S.I.; Garcia-Rubio, M.L.; Tumini, E.; Herrera-Moyano, E.; Aguilera, A. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 2014, 511, 362–365. [Google Scholar] [CrossRef]
- Tan, S.L.W.; Chadha, S.; Liu, Y.; Gabasova, E.; Perera, D.; Ahmed, K.; Constantinou, S.; Renaudin, X.; Lee, M.; Aebersold, R.; et al. A Class of Environmental and Endogenous Toxins Induces BRCA2 Haploinsufficiency and Genome Instability. Cell 2017, 169, 1105–1118.e1115. [Google Scholar] [CrossRef]
- Shivji, M.K.K.; Renaudin, X.; Williams, C.H.; Venkitaraman, A.R. BRCA2 Regulates Transcription Elongation by RNA Polymerase II to Prevent R-Loop Accumulation. Cell Rep. 2018, 22, 1031–1039. [Google Scholar] [CrossRef]
- D’Alessandro, G.; Whelan, D.R.; Howard, S.M.; Vitelli, V.; Renaudin, X.; Adamowicz, M.; Iannelli, F.; Jones-Weinert, C.W.; Lee, M.; Matti, V.; et al. BRCA2 controls DNA:RNA hybrid level at DSBs by mediating RNase H2 recruitment. Nat. Commun. 2018, 9, 5376. [Google Scholar] [CrossRef]
- Gorthi, A.; Romero, J.C.; Loranc, E.; Cao, L.; Lawrence, L.A.; Goodale, E.; Iniguez, A.B.; Bernard, X.; Masamsetti, V.P.; Roston, S.; et al. EWS-FLI1 increases transcription to cause R-loops and block BRCA1 repair in Ewing sarcoma. Nature 2018, 555, 387–391. [Google Scholar] [CrossRef]
- Garaycoechea, J.I.; Crossan, G.P.; Langevin, F.; Mulderrig, L.; Louzada, S.; Yang, F.; Guilbaud, G.; Park, N.; Roerink, S.; Nik-Zainal, S.; et al. Alcohol and endogenous aldehydes damage chromosomes and mutate stem cells. Nature 2018, 553, 171–177. [Google Scholar] [CrossRef]
- Liao, W.; Du, C.; Wang, J. The cGAS-STING Pathway in Hematopoiesis and Its Physiopathological Significance. Front. Immunol. 2020, 11, 573915. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, K.J.; Carroll, P.; Martin, C.A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017, 548, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Walter, D.; Lier, A.; Geiselhart, A.; Thalheimer, F.B.; Huntscha, S.; Sobotta, M.C.; Moehrle, B.; Brocks, D.; Bayindir, I.; Kaschutnig, P.; et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature 2015, 520, 549–552. [Google Scholar] [CrossRef] [PubMed]
- Mankan, A.K.; Schmidt, T.; Chauhan, D.; Goldeck, M.; Honing, K.; Gaidt, M.; Kubarenko, A.V.; Andreeva, L.; Hopfner, K.P.; Hornung, V. Cytosolic RNA:DNA hybrids activate the cGAS-STING axis. EMBO J. 2014, 33, 2937–2946. [Google Scholar] [CrossRef]
- Weinreb, J.T.; Ghazale, N.; Pradhan, K.; Gupta, V.; Potts, K.S.; Tricomi, B.; Daniels, N.J.; Padgett, R.A.; De Oliveira, S.; Verma, A.; et al. Excessive R-loops trigger an inflammatory cascade leading to increased HSPC production. Dev. Cell 2021, 56, 627–640.e625. [Google Scholar] [CrossRef] [PubMed]
- Pilger, D.; Seymour, L.W.; Jackson, S.P. Interfaces between cellular responses to DNA damage and cancer immunotherapy. Genes Dev. 2021. [Google Scholar] [CrossRef] [PubMed]
- Yau, R.; Rape, M. The increasing complexity of the ubiquitin code. Nat. Cell Biol. 2016, 18, 579–586. [Google Scholar] [CrossRef]
- Mevissen, T.E.T.; Komander, D. Mechanisms of Deubiquitinase Specificity and Regulation. Annu. Rev. Biochem. 2017, 86, 159–192. [Google Scholar] [CrossRef] [PubMed]
- Coleman, K.E.; Huang, T.T. In a Class of Its Own: A New Family of Deubiquitinases Promotes Genome Stability. Mol. Cell 2018, 70, 1–3. [Google Scholar] [CrossRef]
- Schwertman, P.; Bekker-Jensen, S.; Mailand, N. Regulation of DNA double-strand break repair by ubiquitin and ubiquitin-like modifiers. Nat. Rev. Mol. Cell Biol. 2016, 17, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Heideker, J.; Wertz, I.E. DUBs, the regulation of cell identity and disease. Biochem. J. 2015, 467, 191. [Google Scholar] [CrossRef] [PubMed]
- Moran-Crusio, K.; Reavie, L.B.; Aifantis, I. Regulation of hematopoietic stem cell fate by the ubiquitin proteasome system. Trends Immunol. 2012, 33, 357–363. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sarodaya, N.; Karapurkar, J.; Kim, K.S.; Hong, S.H.; Ramakrishna, S. The Role of Deubiquitinating Enzymes in Hematopoiesis and Hematological Malignancies. Cancers 2020, 12, 1103. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Seshasayee, D.; Noubade, R.; French, D.M.; Liu, J.; Chaurushiya, M.S.; Kirkpatrick, D.S.; Pham, V.C.; Lill, J.R.; Bakalarski, C.E.; et al. Loss of the tumor suppressor BAP1 causes myeloid transformation. Science 2012, 337, 1541–1546. [Google Scholar] [CrossRef]
- Abdel-Wahab, O.; Dey, A. The ASXL-BAP1 axis: New factors in myelopoiesis, cancer and epigenetics. Leukemia 2013, 27, 10–15. [Google Scholar] [CrossRef]
- Asada, S.; Goyama, S.; Inoue, D.; Shikata, S.; Takeda, R.; Fukushima, T.; Yonezawa, T.; Fujino, T.; Hayashi, Y.; Kawabata, K.C.; et al. Mutant ASXL1 cooperates with BAP1 to promote myeloid leukaemogenesis. Nat. Commun. 2018, 9, 2733. [Google Scholar] [CrossRef]
- Muto, T.; Walker, C.S.; Choi, K.; Hueneman, K.; Smith, M.A.; Gul, Z.; Garcia-Manero, G.; Ma, A.; Zheng, Y.; Starczynowski, D.T. Adaptive response to inflammation contributes to sustained myelopoiesis and confers a competitive advantage in myelodysplastic syndrome HSCs. Nat. Immunol. 2020, 21, 535–545. [Google Scholar] [CrossRef]
- Nakagawa, M.M.; Rathinam, C.V. A20 deficiency in hematopoietic stem cells causes lymphopenia and myeloproliferation due to elevated Interferon-gamma signals. Sci. Rep. 2019, 9, 12658. [Google Scholar] [CrossRef] [PubMed]
- Meyer, T.; Jahn, N.; Lindner, S.; Rohner, L.; Dolnik, A.; Weber, D.; Scheffold, A.; Kopff, S.; Paschka, P.; Gaidzik, V.I.; et al. Functional characterization of BRCC3 mutations in acute myeloid leukemia with t(8;21)(q22;q22.1). Leukemia 2020, 34, 404–415. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Nagata, Y.; Grossmann, V.; Radivoyevitch, T.; Okuno, Y.; Nagae, G.; Hosono, N.; Schnittger, S.; Sanada, M.; Przychodzen, B.; et al. BRCC3 mutations in myeloid neoplasms. Haematologica 2015, 100, 1051–1057. [Google Scholar] [CrossRef]
- Donaghy, R.; Han, X.; Rozenova, K.; Lv, K.; Jiang, Q.; Doepner, M.; Greenberg, R.A.; Tong, W. The BRISC deubiquitinating enzyme complex limits hematopoietic stem cell expansion by regulating JAK2 K63-ubiquitination. Blood 2019, 133, 1560–1571. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Liang, L.; Yi, H.; Su, T.; Yang, Z.; Nie, L.; Liu, J. USP7 inhibition inhibits proliferation and induces megakaryocytic differentiation in MDS cells by upregulating gelsolin. Br. J. Haematol. 2020, 190, 418–429. [Google Scholar] [CrossRef]
- Cartel, M.; Mouchel, P.L.; Gotanegre, M.; David, L.; Bertoli, S.; Mansat-De Mas, V.; Besson, A.; Sarry, J.E.; Manenti, S.; Didier, C. Inhibition of ubiquitin-specific protease 7 sensitizes acute myeloid leukemia to chemotherapy. Leukemia 2021, 35, 417–432. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, E.L.; Schauer, N.J.; Yang, J.; Lamberto, I.; Doherty, L.; Bhatt, S.; Nonami, A.; Meng, C.; Letai, A.; Wright, R.; et al. Inhibition of USP10 induces degradation of oncogenic FLT3. Nat. Chem. Biol. 2017, 13, 1207–1215. [Google Scholar] [CrossRef]
- Zerkalenkova, E.; Lebedeva, S.; Kazakova, A.; Baryshev, P.; Meyer, C.; Marschalek, R.; Novichkova, G.; Maschan, M.; Maschan, A.; Olshanskaya, Y. A case of pediatric acute myeloid leukemia with t(11;16)(q23;q24) leading to a novel KMT2A-USP10 fusion gene. Genes Chromosomes Cancer 2018, 57, 522–524. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, J.; Shiba, N.; Tsujimoto, S.I.; Yoshida, M.; Nakabayashi, K.; Ogata-Kawata, H.; Okamura, K.; Takeuchi, M.; Osumi, T.; Tomizawa, D.; et al. Whole transcriptome sequencing reveals a KMT2A-USP2 fusion in infant acute myeloid leukemia. Genes Chromosomes Cancer 2019, 58, 669–672. [Google Scholar] [CrossRef]
- Blackburn, P.R.; Huang, L.; Dalovisio, A.; Pitel, B.A.; Chen, D.; Oliveira, J.L.; Wood, A.J.; Smadbeck, J.B.; Johnson, S.H.; Vasmatzis, G.; et al. Secondary acquisition of BCR-ABL1 fusion in de novo GATA2-MECOM positive acute myeloid leukemia with subsequent emergence of a rare KMT2A-ASXL2 fusion. Cancer Genet. 2020, 241, 67–71. [Google Scholar] [CrossRef]
- Madan, V.; Li, J.; Zhou, S.; Teoh, W.W.; Han, L.; Meggendorfer, M.; Malcovati, L.; Cazzola, M.; Ogawa, S.; Haferlach, T.; et al. Distinct and convergent consequences of splice factor mutations in myelodysplastic syndromes. Am. J. Hematol. 2020, 95, 133–143. [Google Scholar] [CrossRef]
- Nijnik, A.; Woodbine, L.; Marchetti, C.; Dawson, S.; Lambe, T.; Liu, C.; Rodrigues, N.P.; Crockford, T.L.; Cabuy, E.; Vindigni, A.; et al. DNA repair is limiting for haematopoietic stem cells during ageing. Nature 2007, 447, 686–690. [Google Scholar] [CrossRef]
- Citterio, E. Fine-tuning the ubiquitin code at DNA double-strand breaks: Deubiquitinating enzymes at work. Front. Genet. 2015, 6, 282. [Google Scholar] [CrossRef]
- Parmar, K.; Kim, J.; Sykes, S.; Shimamura, A.; Stuckert, P.; Zhu, K.; Hamilton, A.; Deloach, M.; Kutok, J.; Akashi, K.; et al. Hematopoietic Stem Cell Defects in Mice with Deficiency of Fancd2 or Usp1. Stem Cells 2010, 28, 1186–1195. [Google Scholar] [CrossRef]
- Van den Berk, P.; Lancini, C.; Company, C.; Serresi, M.; Sanchez-Bailon, M.P.; Hulsman, D.; Pritchard, C.; Song, J.Y.; Schmitt, M.J.; Tanger, E.; et al. USP15 Deubiquitinase Safeguards Hematopoiesis and Genome Integrity in Hematopoietic Stem Cells and Leukemia Cells. Cell Rep. 2020, 33, 108533. [Google Scholar] [CrossRef] [PubMed]
- Adorno, M.; Sikandar, S.; Mitra, S.S.; Kuo, A.; Nicolis Di Robilant, B.; Haro-Acosta, V.; Ouadah, Y.; Quarta, M.; Rodriguez, J.; Qian, D.; et al. Usp16 contributes to somatic stem-cell defects in Down’s syndrome. Nature 2013, 501, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Jones, A.E.; Yang, W.; Liu, S.; Dai, Q.; Liu, Y.; Swindle, C.S.; Zhou, D.; Zhang, Z.; Ryan, T.M.; et al. The histone H2A deubiquitinase Usp16 regulates hematopoiesis and hematopoietic stem cell function. Proc. Natl. Acad. Sci. USA 2016, 113, E51–E60. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Nandakumar, V.; Jiang, X.X.; Jones, L.; Yang, A.G.; Huang, X.F.; Chen, S.Y. The control of hematopoietic stem cell maintenance, self-renewal, and differentiation by Mysm1-mediated epigenetic regulation. Blood 2013, 122, 2812–2822. [Google Scholar] [CrossRef] [PubMed]
- Fiore, A.; Liang, Y.; Lin, Y.H.; Tung, J.; Wang, H.; Langlais, D.; Nijnik, A. Deubiquitinase MYSM1 in the Hematopoietic System and beyond: A Current Review. Int. J. Mol. Sci. 2020, 21, 3007. [Google Scholar] [CrossRef] [PubMed]
- Sacco, J.J.; Coulson, J.M.; Clague, M.J.; Urbe, S. Emerging roles of deubiquitinases in cancer-associated pathways. IUBMB Life 2010, 62, 140–157. [Google Scholar] [CrossRef]
- Clague, M.J.; Coulson, J.M.; Urbe, S. Cellular functions of the DUBs. J. Cell Sci. 2012, 125, 277–286. [Google Scholar] [CrossRef]
- Cohn, M.A.; Kowal, P.; Yang, K.; Haas, W.; Huang, T.T.; Gygi, S.P.; D’Andrea, A.D. A UAF1-containing multisubunit protein complex regulates the Fanconi anemia pathway. Mol. Cell 2007, 28, 786–797. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Parmar, K.; Huang, M.; Weinstock, D.M.; Ruit, C.A.; Kutok, J.L.; D’Andrea, A.D. Inactivation of murine Usp1 results in genomic instability and a Fanconi anemia phenotype. Dev. Cell 2009, 16, 314–320. [Google Scholar] [CrossRef]
- Ulrich, H.D.; Walden, H. Ubiquitin signalling in DNA replication and repair. Nat. Rev. Mol. Cell Biol. 2010, 11, 479–489. [Google Scholar] [CrossRef]
- Dexheimer, T.S.; Rosenthal, A.S.; Luci, D.K.; Liang, Q.; Villamil, M.A.; Chen, J.; Sun, H.; Kerns, E.H.; Simeonov, A.; Jadhav, A.; et al. Synthesis and structure-activity relationship studies of N-benzyl-2-phenylpyrimidin-4-amine derivatives as potent USP1/UAF1 deubiquitinase inhibitors with anticancer activity against nonsmall cell lung cancer. J. Med. Chem. 2014, 57, 8099–8110. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.S.; Li, H.; Roberts, E.A.; Gaudiano, E.F.; Clairmont, C.; Sambel, L.A.; Ponnienselvan, K.; Liu, J.C.; Yang, C.; Kozono, D.; et al. USP1 Is Required for Replication Fork Protection in BRCA1-Deficient Tumors. Mol. Cell 2018, 72, 925–941.e924. [Google Scholar] [CrossRef] [PubMed]
- Lasorella, A.; Uo, T.; Iavarone, A. Id proteins at the cross-road of development and cancer. Oncogene 2001, 20, 8326–8333. [Google Scholar] [CrossRef] [PubMed]
- Suh, H.C.; Leeanansaksiri, W.; Ji, M.; Klarmann, K.D.; Renn, K.; Gooya, J.; Smith, D.; McNiece, I.; Lugthart, S.; Valk, P.J.; et al. Id1 immortalizes hematopoietic progenitors in vitro and promotes a myeloproliferative disease in vivo. Oncogene 2008, 27, 5612–5623. [Google Scholar] [CrossRef]
- Mistry, H.; Hsieh, G.; Buhrlage, S.J.; Huang, M.; Park, E.; Cuny, G.D.; Galinsky, I.; Stone, R.M.; Gray, N.S.; D’Andrea, A.D.; et al. Small-molecule inhibitors of USP1 target ID1 degradation in leukemic cells. Mol. Cancer 2013, 12, 2651–2662. [Google Scholar] [CrossRef] [PubMed]
- Das, D.S.; Das, A.; Ray, A.; Song, Y.; Samur, M.K.; Munshi, N.C.; Chauhan, D.; Anderson, K.C. Blockade of Deubiquitylating Enzyme USP1 Inhibits DNA Repair and Triggers Apoptosis in Multiple Myeloma Cells. Clin. Cancer Res. 2017, 23, 4280–4289. [Google Scholar] [CrossRef]
- Li, M.; Brooks, C.L.; Kon, N.; Gu, W. A dynamic role of HAUSP in the p53-Mdm2 pathway. Mol. Cell 2004, 13, 879–886. [Google Scholar] [CrossRef]
- Cummins, J.M.; Rago, C.; Kohli, M.; Kinzler, K.W.; Lengauer, C.; Vogelstein, B. Tumour suppression: Disruption of HAUSP gene stabilizes p53. Nature 2004, 428, 1–2. [Google Scholar] [CrossRef]
- Valles, G.J.; Bezsonova, I.; Woodgate, R.; Ashton, N.W. USP7 Is a Master Regulator of Genome Stability. Front. Cell Dev. Biol. 2020, 8, 717. [Google Scholar] [CrossRef] [PubMed]
- Su, D.; Ma, S.; Shan, L.; Wang, Y.; Wang, Y.; Cao, C.; Liu, B.; Yang, C.; Wang, L.; Tian, S.; et al. Ubiquitin-specific protease 7 sustains DNA damage response and promotes cervical carcinogenesis. J. Clin. Invest. 2018, 128, 4280–4296. [Google Scholar] [CrossRef]
- Wertz, I.E.; Wang, X. From Discovery to Bedside: Targeting the Ubiquitin System. Cell Chem. Biol. 2019, 26, 156–177. [Google Scholar] [CrossRef] [PubMed]
- Galarreta, A.; Valledor, P.; Ubieto-Capella, P.; Lafarga, V.; Zarzuela, E.; Munoz, J.; Malumbres, M.; Lecona, E.; Fernandez-Capetillo, O. USP7 limits CDK1 activity throughout the cell cycle. EMBO J. 2021, e99692. [Google Scholar] [CrossRef]
- Harrigan, J.A.; Jacq, X.; Martin, N.M.; Jackson, S.P. Deubiquitylating enzymes and drug discovery: Emerging opportunities. Nat. Rev. Drug Discov. 2018, 17, 57–78. [Google Scholar] [CrossRef]
- Maat, H.; Atsma, T.; Hogeling, S.; Rodríguez López, A.; Jaques, J.; Olthuis, M.; de Vries, M.; Gravesteijn, C.; Brouwers-Vos, A.; van der Meer, N.; et al. The USP7-TRIM27 axis mediates non-canonical PRC1.1 function and is a druggable target in leukemia. iScience 2021, 24. [Google Scholar] [CrossRef]
- Liang, L.; Peng, Y.; Zhang, J.; Zhang, Y.; Roy, M.; Han, X.; Xiao, X.; Sun, S.; Liu, H.; Nie, L.; et al. Deubiquitylase USP7 regulates human terminal erythroid differentiation by stabilizing GATA1. Haematologica 2019, 104, 2178–2187. [Google Scholar] [CrossRef]
- Frisan, E.; Vandekerckhove, J.; de Thonel, A.; Pierre-Eugene, C.; Sternberg, A.; Arlet, J.B.; Floquet, C.; Gyan, E.; Kosmider, O.; Dreyfus, F.; et al. Defective nuclear localization of Hsp70 is associated with dyserythropoiesis and GATA-1 cleavage in myelodysplastic syndromes. Blood 2012, 119, 1532–1542. [Google Scholar] [CrossRef]
- Hopfer, O.; Nolte, F.; Mossner, M.; Komor, M.; Kmetsch, A.; Benslasfer, O.; Reissmann, M.; Nowak, D.; Hoelzer, D.; Thiel, E.; et al. Epigenetic dysregulation of GATA1 is involved in myelodysplastic syndromes dyserythropoiesis. Eur. J. Haematol. 2012, 88, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Nishi, R.; Wijnhoven, P.; le Sage, C.; Tjeertes, J.; Galanty, Y.; Forment, J.V.; Clague, M.J.; Urbe, S.; Jackson, S.P. Systematic characterization of deubiquitylating enzymes for roles in maintaining genome integrity. Nat. Cell Biol. 2014, 16, 1016–1026. [Google Scholar] [CrossRef] [PubMed]
- Orthwein, A.; Noordermeer, S.M.; Wilson, M.D.; Landry, S.; Enchev, R.I.; Sherker, A.; Munro, M.; Pinder, J.; Salsman, J.; Dellaire, G.; et al. A mechanism for the suppression of homologous recombination in G1 cells. Nature 2015, 528, 422–426. [Google Scholar] [CrossRef]
- Wijnhoven, P.; Konietzny, R.; Blackford, A.N.; Travers, J.; Kessler, B.M.; Nishi, R.; Jackson, S.P. USP4 Auto-Deubiquitylation Promotes Homologous Recombination. Mol. Cell 2015, 60, 362–373. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, H.; Wang, X.; Tian, Q.; Hu, Z.; Peng, C.; Jiang, P.; Wang, T.; Guo, W.; Chen, Y.; et al. The Deubiquitylating Enzyme USP4 Cooperates with CtIP in DNA Double-Strand Break End Resection. Cell Rep. 2015, 13, 93–107. [Google Scholar] [CrossRef]
- Mu, J.J.; Wang, Y.; Luo, H.; Leng, M.; Zhang, J.; Yang, T.; Besusso, D.; Jung, S.Y.; Qin, J. A proteomic analysis of ataxia telangiectasia-mutated (ATM)/ATM-Rad3-related (ATR) substrates identifies the ubiquitin-proteasome system as a regulator for DNA damage checkpoints. J. Biol. Chem. 2007, 282, 17330–17334. [Google Scholar] [CrossRef] [PubMed]
- Fielding, A.B.; Concannon, M.; Darling, S.; Rusilowicz-Jones, E.V.; Sacco, J.J.; Prior, I.A.; Clague, M.J.; Urbe, S.; Coulson, J.M. The deubiquitylase USP15 regulates topoisomerase II alpha to maintain genome integrity. Oncogene 2018, 37, 2326–2342. [Google Scholar] [CrossRef]
- Peng, Y.; Liao, Q.; Tan, W.; Peng, C.; Hu, Z.; Chen, Y.; Li, Z.; Li, J.; Zhen, B.; Zhu, W.; et al. The deubiquitylating enzyme USP15 regulates homologous recombination repair and cancer cell response to PARP inhibitors. Nat. Commun. 2019, 10, 1224. [Google Scholar] [CrossRef] [PubMed]
- Eichhorn, P.J.; Rodon, L.; Gonzalez-Junca, A.; Dirac, A.; Gili, M.; Martinez-Saez, E.; Aura, C.; Barba, I.; Peg, V.; Prat, A.; et al. USP15 stabilizes TGF-beta receptor I and promotes oncogenesis through the activation of TGF-beta signaling in glioblastoma. Nat. Med. 2012, 18, 429–435. [Google Scholar] [CrossRef]
- Padmanabhan, A.; Candelaria, N.; Wong, K.K.; Nikolai, B.C.; Lonard, D.M.; O’Malley, B.W.; Richards, J.S. USP15-dependent lysosomal pathway controls p53-R175H turnover in ovarian cancer cells. Nat. Commun. 2018, 9, 1270. [Google Scholar] [CrossRef]
- Niederkorn, M.; Hueneman, K.; Choi, K.; Varney, M.E.; Romano, L.; Pujato, M.A.; Greis, K.D.; Inoue, J.I.; Meetei, R.; Starczynowski, D.T. TIFAB Regulates USP15-Mediated p53 Signaling during Stressed and Malignant Hematopoiesis. Cell Rep. 2020, 30, 2776–2790.e2776. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Jin, J.; Hu, H.; Li, H.S.; Romano, S.; Xiao, Y.; Nakaya, M.; Zhou, X.; Cheng, X.; Yang, P.; et al. USP15 stabilizes MDM2 to mediate cancer-cell survival and inhibit antitumor T cell responses. Nat. Immunol. 2014, 15, 562–570. [Google Scholar] [CrossRef]
- Liu, W.T.; Huang, K.Y.; Lu, M.C.; Huang, H.L.; Chen, C.Y.; Cheng, Y.L.; Yu, H.C.; Liu, S.Q.; Lai, N.S.; Huang, H.B. TGF-beta upregulates the translation of USP15 via the PI3K/AKT pathway to promote p53 stability. Oncogene 2017, 36, 2715–2723. [Google Scholar] [CrossRef]
- Sugawara, T.; Oguro, H.; Negishi, M.; Morita, Y.; Ichikawa, H.; Iseki, T.; Yokosuka, O.; Nakauchi, H.; Iwama, A. FET family proto-oncogene Fus contributes to self-renewal of hematopoietic stem cells. Exp. Hematol. 2010, 38, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Das, T.; Park, J.K.; Park, J.; Kim, E.; Rape, M.; Kim, E.E.; Song, E.J. USP15 regulates dynamic protein-protein interactions of the spliceosome through deubiquitination of PRP31. Nucleic Acids Res. 2017, 45, 5010–5011. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Moran-Crusio, K.; Reavie, L.; Shih, A.; Abdel-Wahab, O.; Ndiaye-Lobry, D.; Lobry, C.; Figueroa, M.E.; Vasanthakumar, A.; Patel, J.; Zhao, X.; et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 2011, 20, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.L.; Smith, M.D.; Lv, L.; Nakagawa, T.; Li, Z.; Sun, S.C.; Brown, N.G.; Xiong, Y.; Xu, Y.P. USP15 suppresses tumor immunity via deubiquitylation and inactivation of TET2. Sci. Adv. 2020, 6. [Google Scholar] [CrossRef] [PubMed]
- Georges, A.; Gros, P.; Fodil, N. USP15: A review of its implication in immune and inflammatory processes and tumor progression. Genes Immun. 2021. [Google Scholar] [CrossRef]
- Teyra, J.; Singer, A.U.; Schmitges, F.W.; Jaynes, P.; Kit Leng Lui, S.; Polyak, M.J.; Fodil, N.; Krieger, J.R.; Tong, J.; Schwerdtfeger, C.; et al. Structural and Functional Characterization of Ubiquitin Variant Inhibitors of USP15. Structure 2019, 27, 590–605.e595. [Google Scholar] [CrossRef]
- Huang, M.E.; Ye, Y.C.; Chen, S.R.; Chai, J.R.; Lu, J.X.; Zhoa, L.; Gu, L.J.; Wang, Z.Y. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood 1988, 72, 567–572. [Google Scholar] [CrossRef]
- Lo-Coco, F.; Avvisati, G.; Vignetti, M.; Thiede, C.; Orlando, S.M.; Iacobelli, S.; Ferrara, F.; Fazi, P.; Cicconi, L.; Di Bona, E.; et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N. Engl. J. Med. 2013, 369, 111–121. [Google Scholar] [CrossRef]
- Schanda, J.; Lee, C.W.; Wohlan, K.; Muller-Kuller, U.; Kunkel, H.; Coco, I.Q.; Stein, S.; Metz, A.; Koch, J.; Lausen, J.; et al. Suppression of RUNX1/ETO oncogenic activity by a small molecule inhibitor of tetramerization. Haematologica 2017, 102, e170–e174. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Johnson, D.T.; Davis, A.G.; Zhou, J.H.; Ball, E.D.; Zhang, D.E. MicroRNA let-7b downregulates AML1-ETO oncogene expression in t(8;21) AML by targeting its 3’UTR. Exp. Hematol. Oncol. 2021, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Bondeson, D.P.; Mares, A.; Smith, I.E.; Ko, E.; Campos, S.; Miah, A.H.; Mulholland, K.E.; Routly, N.; Buckley, D.L.; Gustafson, J.L.; et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Cui, D.; Chen, X.; Xiong, X.; Zhao, Y. PROTACs: An Emerging Targeting Technique for Protein Degradation in Drug Discovery. Bioessays 2018, 40, e1700247. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Milazzo, J.P.; Somerville, T.D.D.; Tarumoto, Y.; Huang, Y.H.; Ostrander, E.L.; Wilkinson, J.E.; Challen, G.A.; Vakoc, C.R. A TFIID-SAGA Perturbation that Targets MYB and Suppresses Acute Myeloid Leukemia. Cancer Cell 2018, 33, 13–28.e18. [Google Scholar] [CrossRef] [PubMed]
- Oo, Z.M.; Illendula, A.; Grembecka, J.; Schmidt, C.; Zhou, Y.; Esain, V.; Kwan, W.; Frost, I.; North, T.E.; Rajewski, R.A.; et al. A tool compound targeting the core binding factor Runt domain to disrupt binding to CBFbeta in leukemic cells. Leuk. Lymphoma 2018, 59, 2188–2200. [Google Scholar] [CrossRef]
- Fennell, K.A.; Bell, C.C.; Dawson, M.A. Epigenetic therapies in acute myeloid leukemia: Where to from here? Blood 2019, 134, 1891–1901. [Google Scholar] [CrossRef]
- Bots, M.; Verbrugge, I.; Martin, B.P.; Salmon, J.M.; Ghisi, M.; Baker, A.; Stanley, K.; Shortt, J.; Ossenkoppele, G.J.; Zuber, J.; et al. Differentiation therapy for the treatment of t(8;21) acute myeloid leukemia using histone deacetylase inhibitors. Blood 2014, 123, 1341–1352. [Google Scholar] [CrossRef]
- McGrath, J.P.; Williamson, K.E.; Balasubramanian, S.; Odate, S.; Arora, S.; Hatton, C.; Edwards, T.M.; O’Brien, T.; Magnuson, S.; Stokoe, D.; et al. Pharmacological Inhibition of the Histone Lysine Demethylase KDM1A Suppresses the Growth of Multiple Acute Myeloid Leukemia Subtypes. Cancer Res. 2016, 76, 1975–1988. [Google Scholar] [CrossRef] [PubMed]
- Ravasio, R.; Ceccacci, E.; Nicosia, L.; Hosseini, A.; Rossi, P.L.; Barozzi, I.; Fornasari, L.; Zuffo, R.D.; Valente, S.; Fioravanti, R.; et al. Targeting the scaffolding role of LSD1 (KDM1A) poises acute myeloid leukemia cells for retinoic acid-induced differentiation. Sci. Adv. 2020, 6, eaax2746. [Google Scholar] [CrossRef]
- Maiques-Diaz, A.; Spencer, G.J.; Lynch, J.T.; Ciceri, F.; Williams, E.L.; Amaral, F.M.R.; Wiseman, D.H.; Harris, W.J.; Li, Y.; Sahoo, S.; et al. Enhancer Activation by Pharmacologic Displacement of LSD1 from GFI1 Induces Differentiation in Acute Myeloid Leukemia. Cell Rep. 2018, 22, 3641–3659. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.; Heyes, E.; Scheiblecker, L.; Eder, T.; Volpe, G.; Frampton, J.; Nerlov, C.; Valent, P.; Grembecka, J.; Grebien, F. CEBPA-mutated leukemia is sensitive to genetic and pharmacological targeting of the MLL1 complex. Leukemia 2019, 33, 1608–1619. [Google Scholar] [CrossRef]
- Morita, K.; Suzuki, K.; Maeda, S.; Matsuo, A.; Mitsuda, Y.; Tokushige, C.; Kashiwazaki, G.; Taniguchi, J.; Maeda, R.; Noura, M.; et al. Genetic regulation of the RUNX transcription factor family has antitumor effects. J. Clin. Invest. 2017, 127, 2815–2828. [Google Scholar] [CrossRef] [PubMed]
- Antony-Debre, I.; Paul, A.; Leite, J.; Mitchell, K.; Kim, H.M.; Carvajal, L.A.; Todorova, T.I.; Huang, K.; Kumar, A.; Farahat, A.A.; et al. Pharmacological inhibition of the transcription factor PU.1 in leukemia. J. Clin. Invest. 2017, 127, 4297–4313. [Google Scholar] [CrossRef]
- Sridhar, R.; Takei, H.; Syed, R.; Kobayashi, I.S.; Hui, L.B.; Kamal, A.; Tenen, D.G.; Kobayashi, S.S. Styryl Quinazolinones as Potential Inducers of Myeloid Differentiation via Upregulation of C/EBPalpha. Molecules 2018, 23, 1938. [Google Scholar] [CrossRef] [PubMed]
- Kwok, A.; Raulf, N.; Habib, N. Developing small activating RNA as a therapeutic: Current challenges and promises. Ther. Deliv. 2019, 10, 151–164. [Google Scholar] [CrossRef]
- Sarker, D.; Plummer, R.; Meyer, T.; Sodergren, M.H.; Basu, B.; Chee, C.E.; Huang, K.W.; Palmer, D.H.; Ma, Y.T.; Evans, T.R.J.; et al. MTL-CEBPA, a Small Activating RNA Therapeutic Upregulating C/EBP-alpha, in Patients with Advanced Liver Cancer: A First-in-Human, Multicenter, Open-Label, Phase I Trial. Clin. Cancer Res. 2020, 26, 3936–3946. [Google Scholar] [CrossRef]
- Namasu, C.Y.; Katzerke, C.; Brauer-Hartmann, D.; Wurm, A.A.; Gerloff, D.; Hartmann, J.U.; Schwind, S.; Muller-Tidow, C.; Hilger, N.; Fricke, S.; et al. ABR, a novel inducer of transcription factor C/EBPalpha, contributes to myeloid differentiation and is a favorable prognostic factor in acute myeloid leukemia. Oncotarget 2017, 8, 103626–103639. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, J.S.; Laursen, L.G.; Schuster, M.B.; Pundhir, S.; Schoof, E.; Ge, Y.; d’Altri, T.; Vitting-Seerup, K.; Rapin, N.; Gentil, C.; et al. Mutant CEBPA directly drives the expression of the targetable tumor-promoting factor CD73 in AML. Sci. Adv. 2019, 5, eaaw4304. [Google Scholar] [CrossRef] [PubMed]
- Bonnal, S.C.; López-Oreja, I.; Valcárcel, J. Roles and mechanisms of alternative splicing in cancer—Implications for care. Nat. Rev. Clin. Oncol. 2020, 17, 457–474. [Google Scholar] [CrossRef]
- Webb, T.R.; Joyner, A.S.; Potter, P.M. The development and application of small molecule modulators of SF3b as therapeutic agents for cancer. Drug Discov. Today 2013, 18, 43–49. [Google Scholar] [CrossRef]
- Yokoi, A.; Kotake, Y.; Takahashi, K.; Kadowaki, T.; Matsumoto, Y.; Minoshima, Y.; Sugi, N.H.; Sagane, K.; Hamaguchi, M.; Iwata, M.; et al. Biological validation that SF3b is a target of the antitumor macrolide pladienolide. FEBS J. 2011, 278, 4870–4880. [Google Scholar] [CrossRef] [PubMed]
- Teng, T.; Tsai, J.H.C.; Puyang, X.; Seiler, M.; Peng, S.; Prajapati, S.; Aird, D.; Buonamici, S.; Caleb, B.; Chan, B.; et al. Splicing modulators act at the branch point adenosine binding pocket defined by the PHF5A–SF3b complex. Nat. Commun. 2017, 8, 15522. [Google Scholar] [CrossRef] [PubMed]
- Finci, L.I.; Zhang, X.; Huang, X.; Zhou, Q.; Tsai, J.; Teng, T.; Agrawal, A.; Chan, B.; Irwin, S.; Karr, C.; et al. The cryo-EM structure of the SF3b spliceosome complex bound to a splicing modulator reveals a pre-mRNA substrate competitive mechanism of action. Genes Dev. 2018, 32, 309–320. [Google Scholar] [CrossRef]
- Shirai, C.L.; White, B.S.; Tripathi, M.; Tapia, R.; Ley, J.N.; Ndonwi, M.; Kim, S.; Shao, J.; Carver, A.; Saez, B.; et al. Mutant U2AF1-expressing cells are sensitive to pharmacological modulation of the spliceosome. Nat. Commun. 2017, 8, 14060. [Google Scholar] [CrossRef]
- Fan, L.; Lagisetti, C.; Edwards, C.C.; Webb, T.R.; Potter, P.M. Sudemycins, novel small molecule analogues of FR901464, induce alternative gene splicing. ACS Chem. Biol. 2011, 6, 582–589. [Google Scholar] [CrossRef]
- Convertini, P.; Shen, M.; Potter, P.M.; Palacios, G.; Lagisetti, C.; de la Grange, P.; Horbinski, C.; Fondufe-Mittendorf, Y.N.; Webb, T.R.; Stamm, S. Sudemycin E influences alternative splicing and changes chromatin modifications. Nucleic Acids Res. 2014, 42, 4947–4961. [Google Scholar] [CrossRef]
- Shirai, C.L.; Tripathi, M.; Ley, J.N.; Ndonwi, M.; White, B.S.; Tapia, R.; Saez, B.; Bertino, A.; Shao, J.; Kim, S.; et al. Preclinical Activity of Splicing Modulators in U2AF1 Mutant MDS/AML. Blood 2015, 126, 1653. [Google Scholar] [CrossRef]
- Steensma, D.P.; Wermke, M.; Klimek, V.M.; Greenberg, P.L.; Font, P.; Komrokji, R.S.; Yang, J.; Brunner, A.M.; Carraway, H.E.; Ades, L.; et al. Results of a Clinical Trial of H3B-8800, a Splicing Modulator, in Patients with Myelodysplastic Syndromes (MDS), Acute Myeloid Leukemia (AML) or Chronic Myelomonocytic Leukemia (CMML). Blood 2019, 134, 673. [Google Scholar] [CrossRef]
- Steensma, D.P.; Wermke, M.; Klimek, V.M.; Greenberg, P.L.; Font, P.; Komrokji, R.S.; Yang, J.; Brunner, A.M.; Carraway, H.E.; Ades, L.; et al. Phase I First-in-Human Dose Escalation Study of the oral SF3B1 modulator H3B-8800 in myeloid neoplasms. Leukemia 2021. [Google Scholar] [CrossRef]
- Jagtap, P.K.A.; Kubelka, T.; Soni, K.; Will, C.L.; Garg, D.; Sippel, C.; Kapp, T.G.; Potukuchi, H.K.; Schorpp, K.; Hadian, K.; et al. Identification of phenothiazine derivatives as UHM-binding inhibitors of early spliceosome assembly. Nat. Commun. 2020, 11, 5621. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Kurzrock, R.; Naing, A.; Wheler, J.J.; Falchook, G.S.; Schiffman, J.S.; Faulkner, N.; Pilat, M.J.; O’Brien, J.; LoRusso, P. A phase I, open-label, single-arm, dose-escalation study of E7107, a precursor messenger ribonucleic acid (pre-mRNA) splicesome inhibitor administered intravenously on days 1 and 8 every 21 days to patients with solid tumors. Investig. New Drugs 2014, 32, 436–444. [Google Scholar] [CrossRef]
- Shibata, N.; Ohoka, N.; Tsuji, G.; Demizu, Y.; Miyawaza, K.; Ui-Tei, K.; Akiyama, T.; Naito, M. Deubiquitylase USP25 prevents degradation of BCR-ABL protein and ensures proliferation of Ph-positive leukemia cells. Oncogene 2020, 39, 3867–3878. [Google Scholar] [CrossRef]
- Weinstock, J.; Wu, J.; Cao, P.; Kingsbury, W.D.; McDermott, J.L.; Kodrasov, M.P.; McKelvey, D.M.; Suresh Kumar, K.G.; Goldenberg, S.J.; Mattern, M.R.; et al. Selective Dual Inhibitors of the Cancer-Related Deubiquitylating Proteases USP7 and USP47. ACS Med. Chem. Lett. 2012, 3, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; Xu, H.Z.; Shan, H.Z.; Liu, M.; Lu, Y.; Fang, Z.X.; Jin, J.; Jing, B.; Xiao, X.H.; Gao, S.M.; et al. Targeting USP47 overcomes tyrosine kinase inhibitor resistance and eradicates leukemia stem/progenitor cells in chronic myelogenous leukemia. Nat. Commun. 2021, 12, 51. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Tian, Z.; Nicholson, B.; Kumar, K.G.; Zhou, B.; Carrasco, R.; McDermott, J.L.; Leach, C.A.; Fulcinniti, M.; Kodrasov, M.P.; et al. A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell 2012, 22, 345–358. [Google Scholar] [CrossRef]
- Peterson, L.F.; Sun, H.; Liu, Y.; Potu, H.; Kandarpa, M.; Ermann, M.; Courtney, S.M.; Young, M.; Showalter, H.D.; Sun, D.; et al. Targeting deubiquitinase activity with a novel small-molecule inhibitor as therapy for B-cell malignancies. Blood 2015, 125, 3588–3597. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Meng, C.; Weisberg, E.; Case, A.; Lamberto, I.; Magin, R.S.; Adamia, S.; Wang, J.; Gray, N.; Liu, S.; et al. Inhibition of the deubiquitinase USP10 induces degradation of SYK. Br. J. Cancer 2020, 122, 1175–1184. [Google Scholar] [CrossRef]
- Akiyama, H.; Umezawa, Y.; Ishida, S.; Okada, K.; Nogami, A.; Miura, O. Inhibition of USP9X induces apoptosis in FLT3-ITD-positive AML cells cooperatively by inhibiting the mutant kinase through aggresomal translocation and inducing oxidative stress. Cancer Lett. 2019, 453, 84–94. [Google Scholar] [CrossRef]
- Altun, M.; Kramer, H.B.; Willems, L.I.; McDermott, J.L.; Leach, C.A.; Goldenberg, S.J.; Kumar, K.G.; Konietzny, R.; Fischer, R.; Kogan, E.; et al. Activity-based chemical proteomics accelerates inhibitor development for deubiquitylating enzymes. Chem. Biol. 2011, 18, 1401–1412. [Google Scholar] [CrossRef] [PubMed]
- Ernst, A.; Avvakumov, G.; Tong, J.; Fan, Y.; Zhao, Y.; Alberts, P.; Persaud, A.; Walker, J.R.; Neculai, A.M.; Neculai, D.; et al. A strategy for modulation of enzymes in the ubiquitin system. Science 2013, 339, 590–595. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barabino, S.M.L.; Citterio, E.; Ronchi, A.E. Transcription Factors, R-Loops and Deubiquitinating Enzymes: Emerging Targets in Myelodysplastic Syndromes and Acute Myeloid Leukemia. Cancers 2021, 13, 3753. https://doi.org/10.3390/cancers13153753
Barabino SML, Citterio E, Ronchi AE. Transcription Factors, R-Loops and Deubiquitinating Enzymes: Emerging Targets in Myelodysplastic Syndromes and Acute Myeloid Leukemia. Cancers. 2021; 13(15):3753. https://doi.org/10.3390/cancers13153753
Chicago/Turabian StyleBarabino, Silvia M. L., Elisabetta Citterio, and Antonella Ellena Ronchi. 2021. "Transcription Factors, R-Loops and Deubiquitinating Enzymes: Emerging Targets in Myelodysplastic Syndromes and Acute Myeloid Leukemia" Cancers 13, no. 15: 3753. https://doi.org/10.3390/cancers13153753
APA StyleBarabino, S. M. L., Citterio, E., & Ronchi, A. E. (2021). Transcription Factors, R-Loops and Deubiquitinating Enzymes: Emerging Targets in Myelodysplastic Syndromes and Acute Myeloid Leukemia. Cancers, 13(15), 3753. https://doi.org/10.3390/cancers13153753