
Targeted Alpha-Particle Radiotherapy and Immune Checkpoint Inhibitors Induces Cooperative Inhibition on Tumor Growth of Malignant Melanoma

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines, Reagents, Materials, and Animals
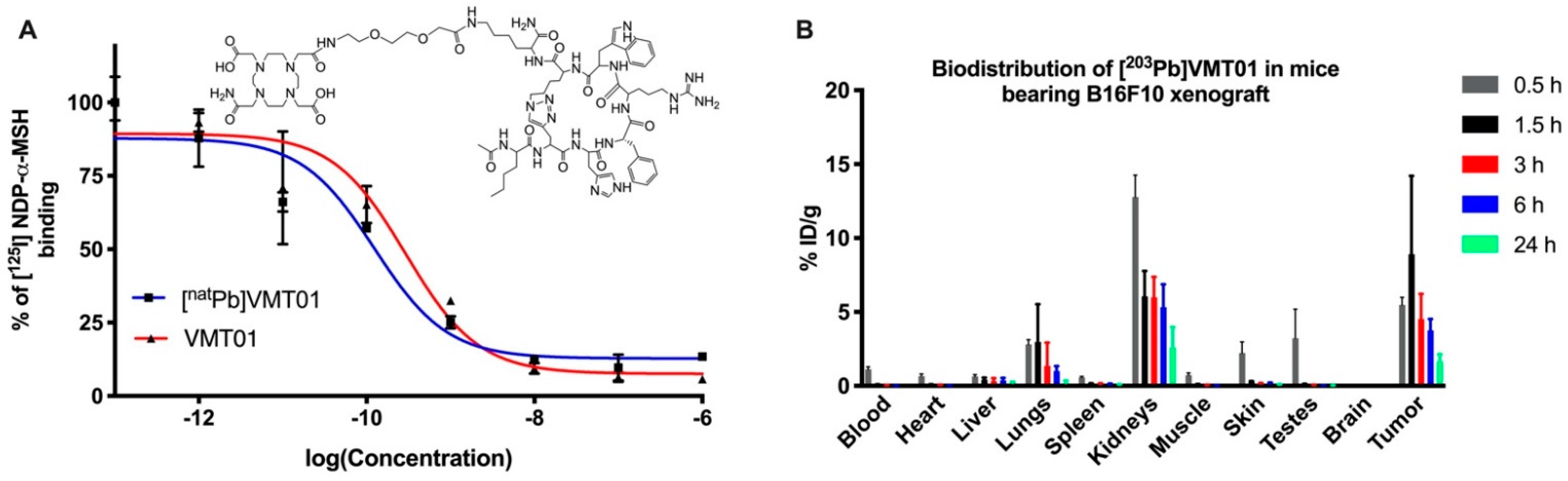
2.2. Radiolabeling, In Vivo Biodistribution and Kidney Dosimetry
2.3. Combination Therapy of Immune Checkpoint Inhibitors and [212Pb]VMT01
2.4. Combination of Immune Checkpoint Inhibitors and Single Dose of [212Pb]VMT01 in Rag1 KO Mice
2.5. Vaccination and Tumor Re-Challenge
2.6. Generation of Immunosensitized Syngeneic Melanoma Cells by [212Pb]VMT01
2.7. FACS Analysis of Tumor Infiltrating Lymphocytes
3. Results
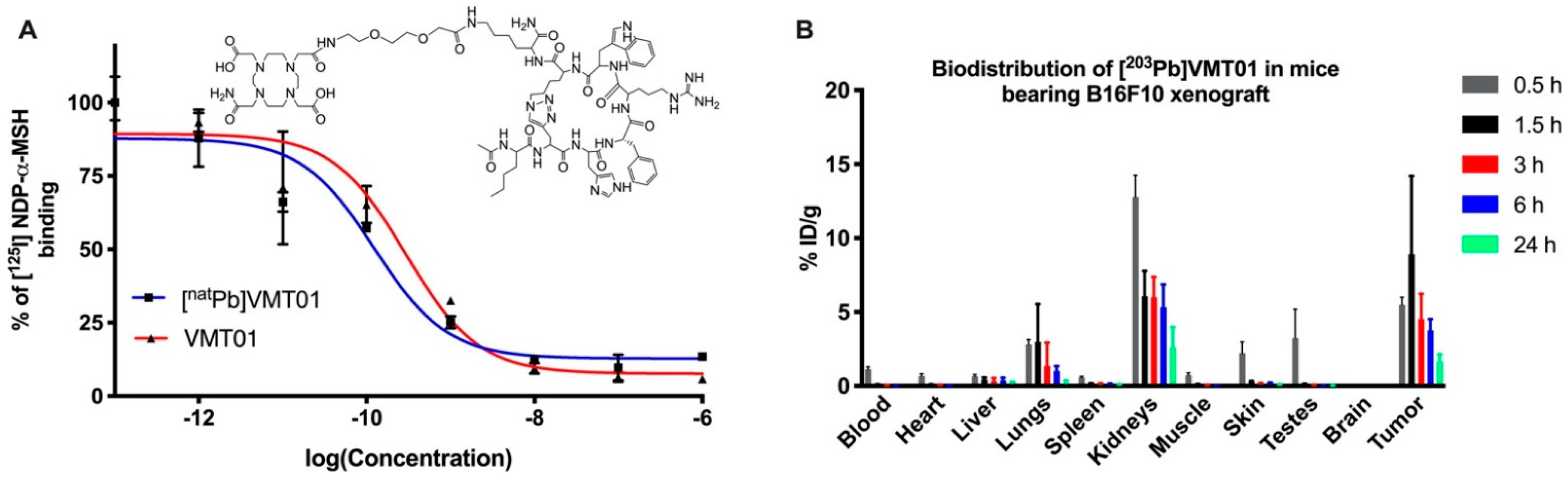
3.1. Radiolabeled Peptide VMT01 Delivers Ionizing Radiation to Melanoma Cells via Specific Binding to MC1R
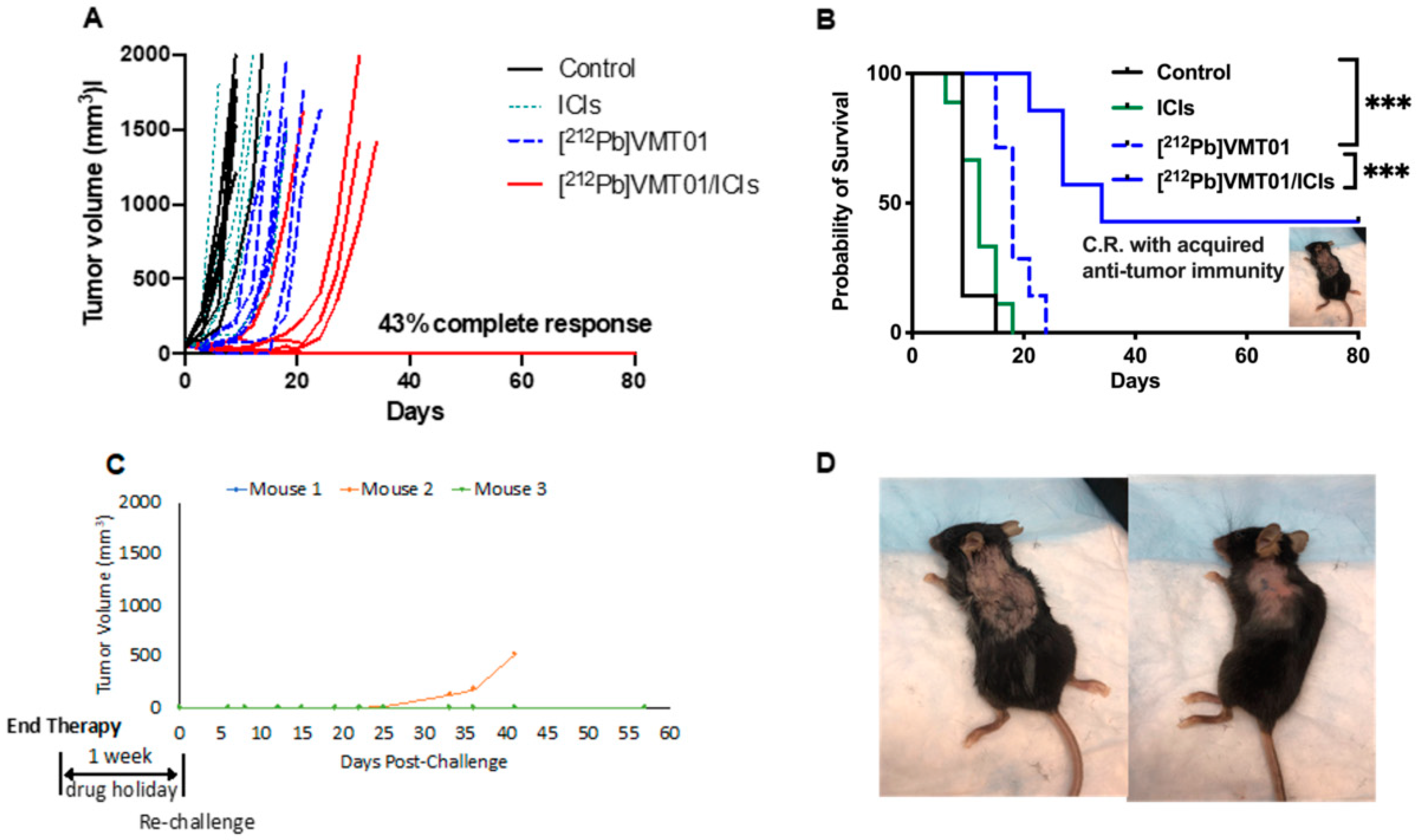
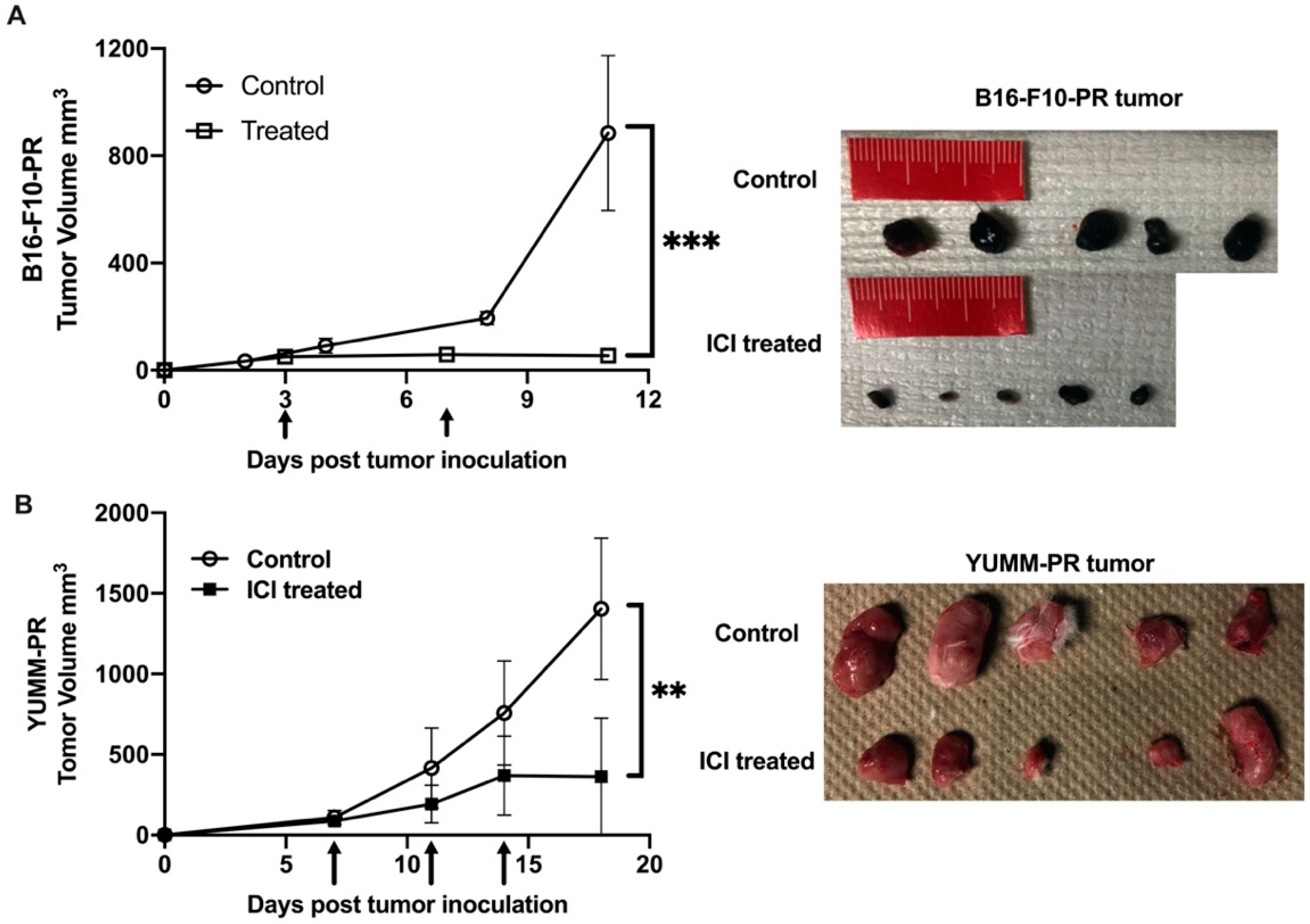
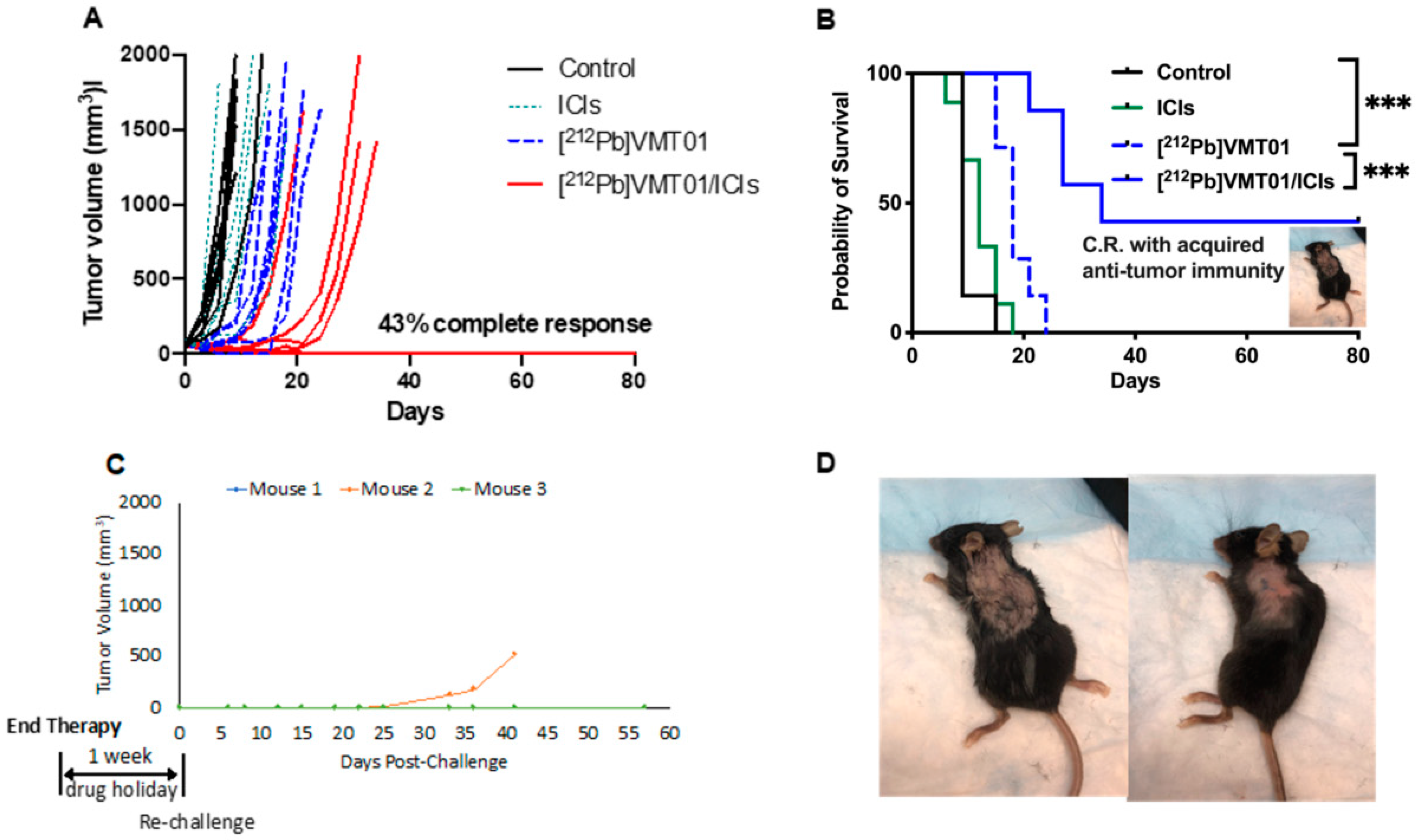
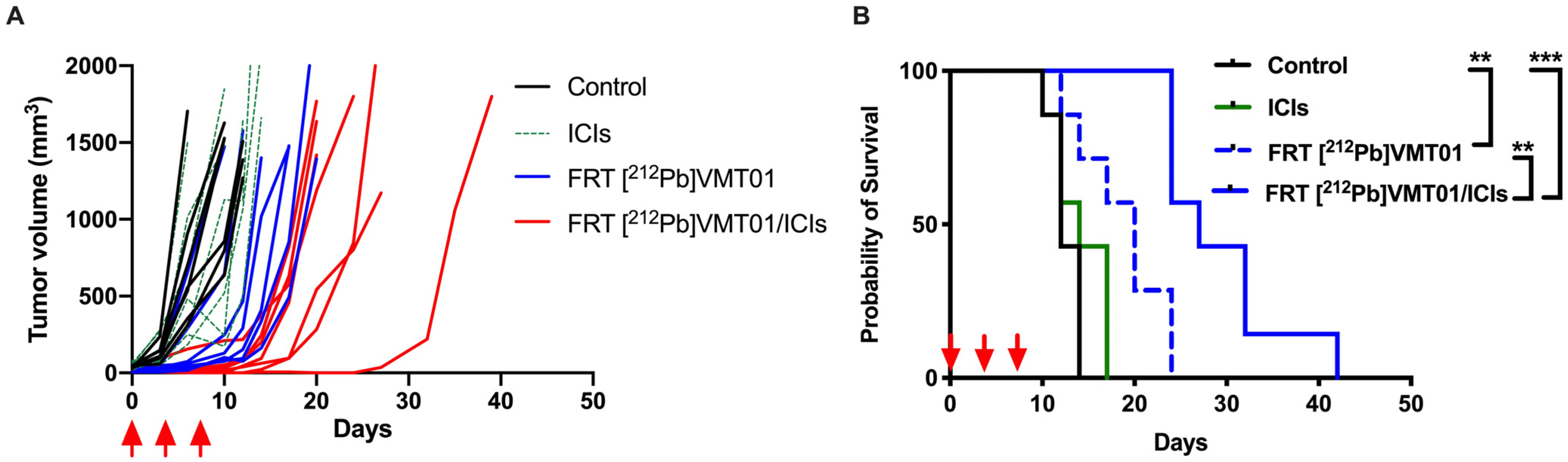
3.2. Combination of ICIs and [212Pb]VMT01 Induces Significant Tumor Inhibition and Lasting Anti-Tumor Immunity
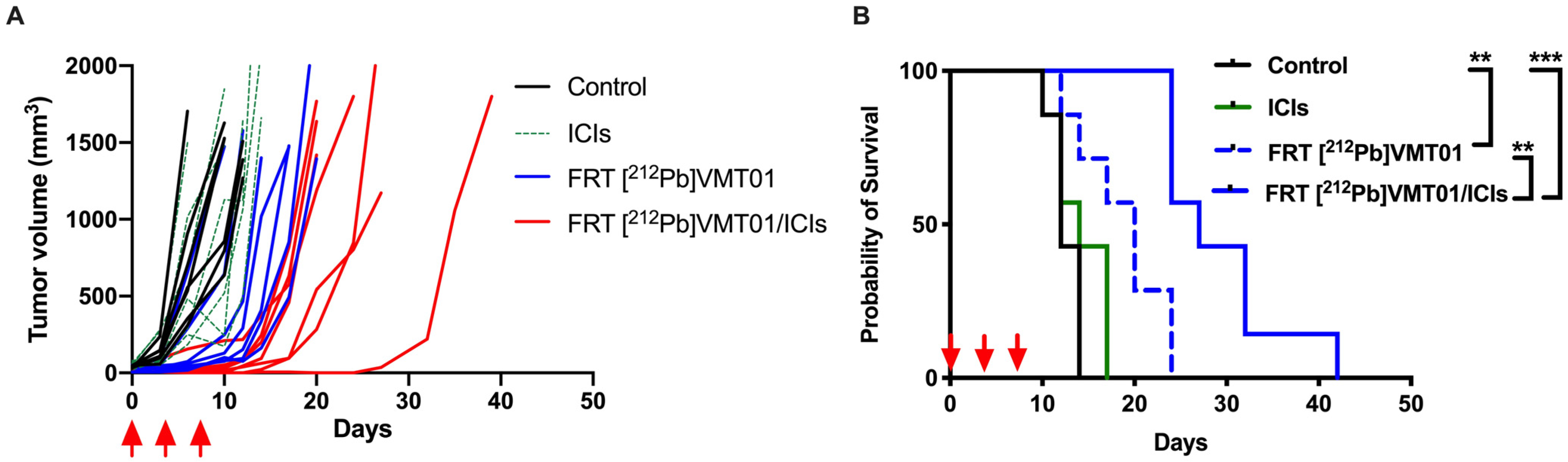
3.3. Combination of ICIs with Fractionated [212Pb]VMT01 Compromised the Cooperative Anti-Tumor Effects Observed for the Single-Dose α-TRT Plus ICIs Combination
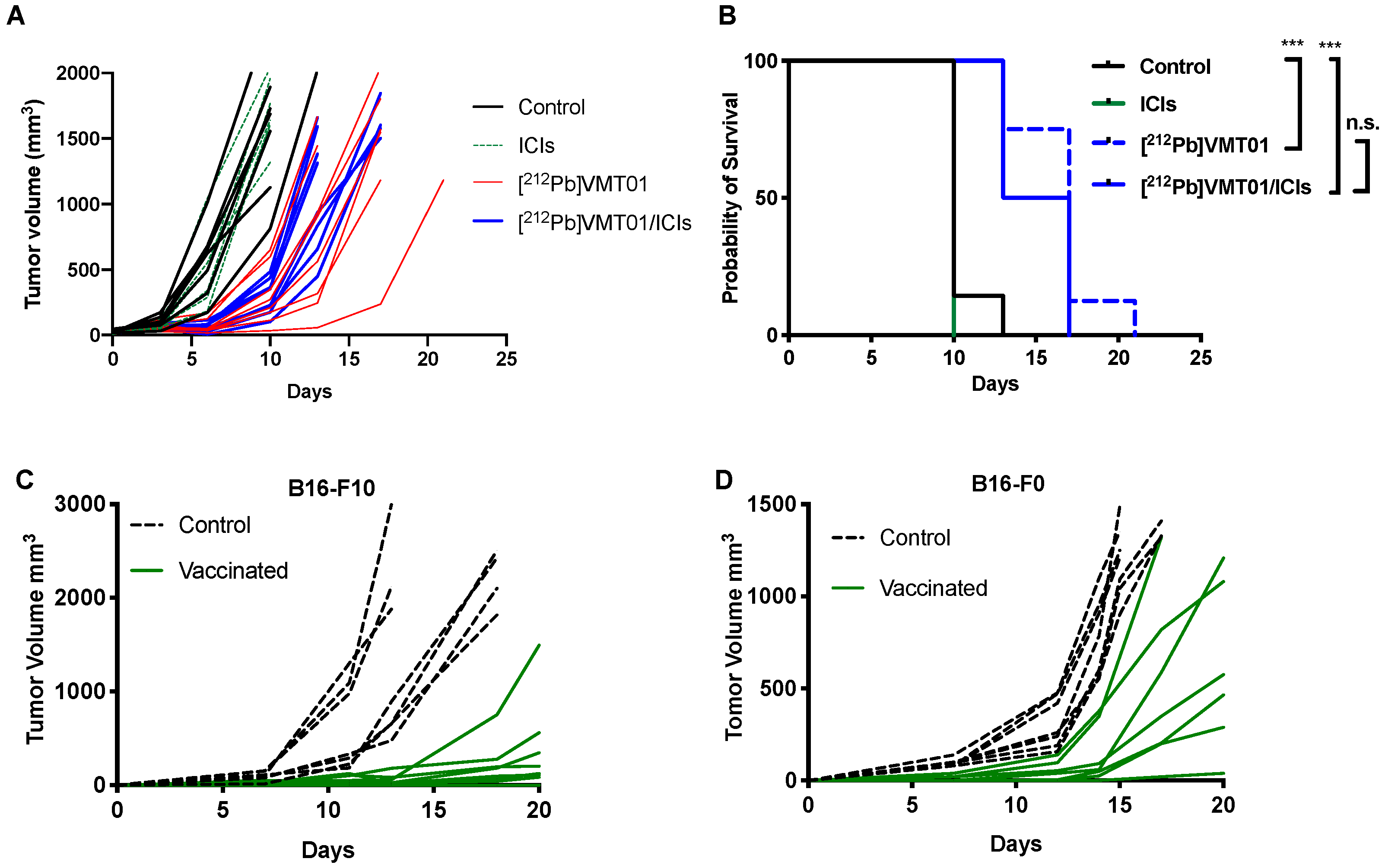
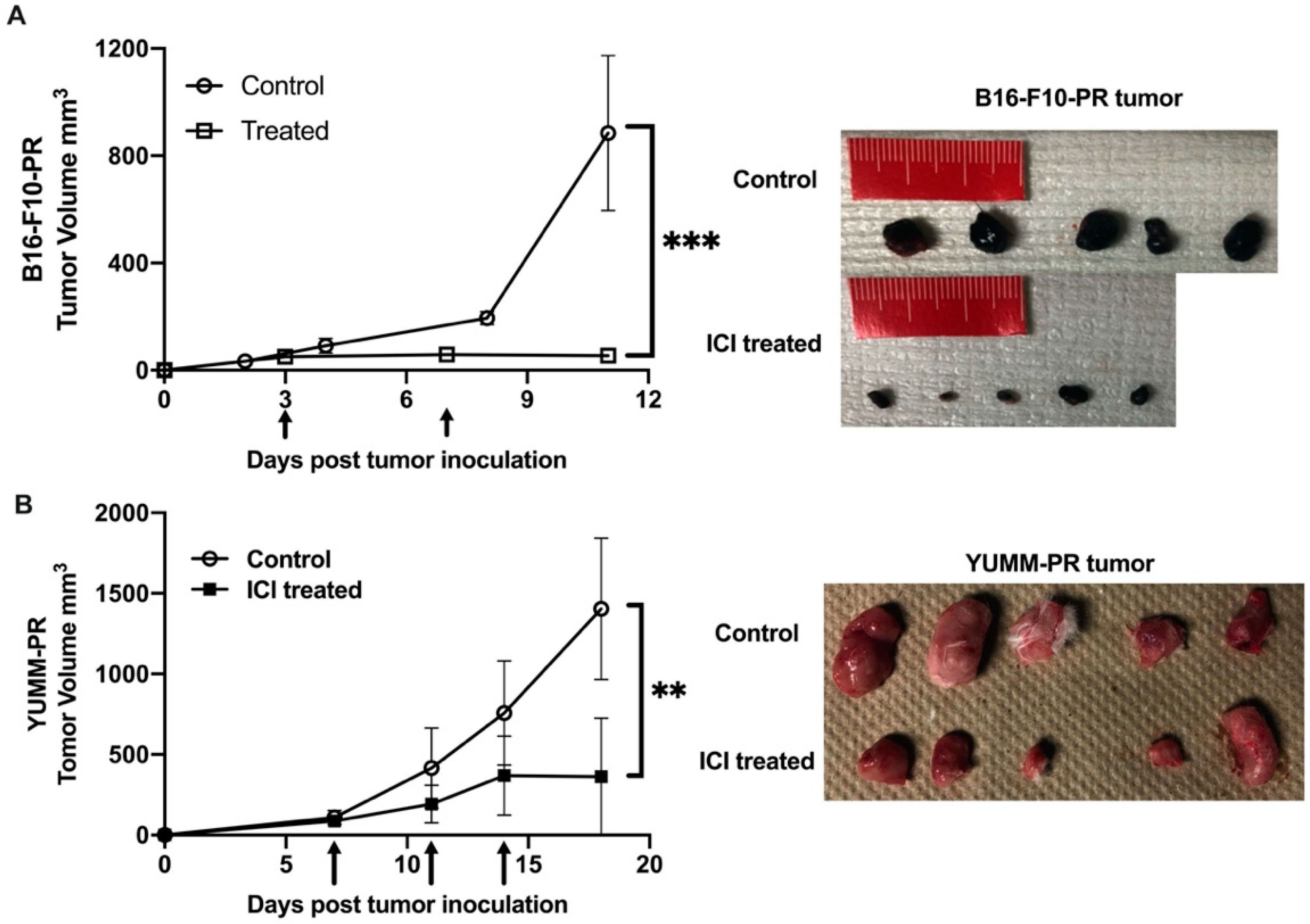
3.4. [212Pb]VMT01 Induces Anti-Tumor Immunity That Relies on the Involvement of Adaptive Immunity
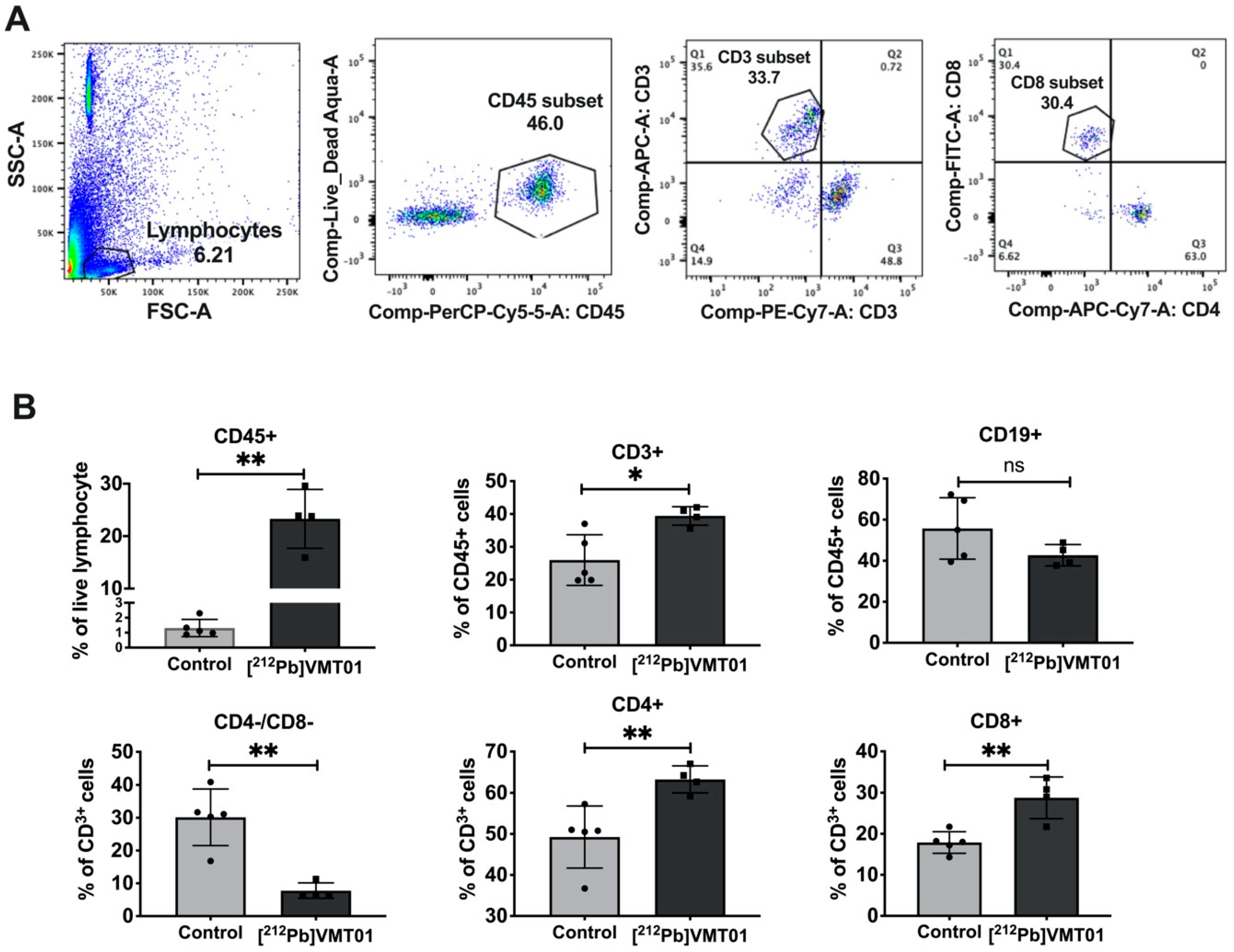
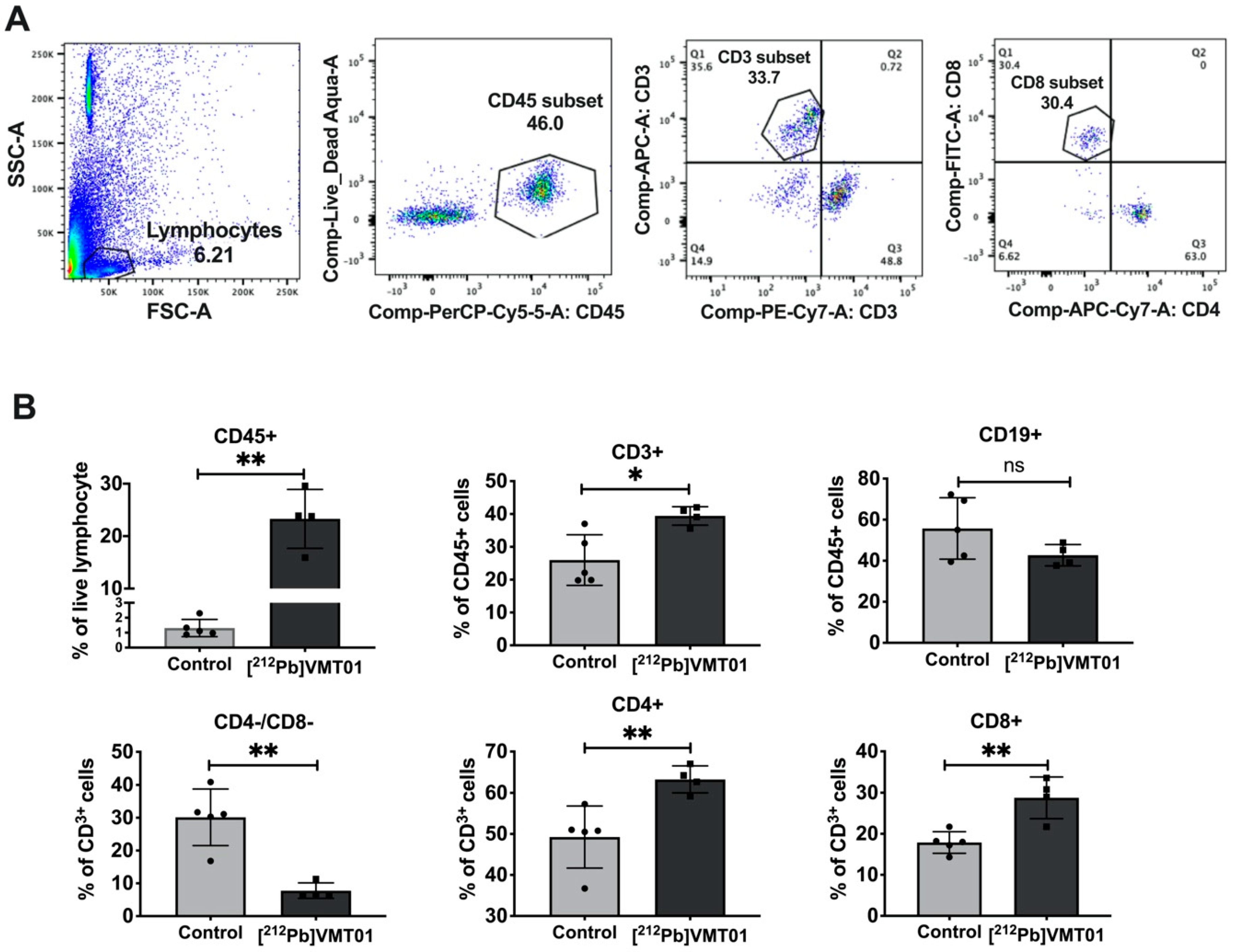
3.5. [212Pb]VMT01 Sensitizes Immunotolerant Melanoma Cells to ICIs and Induces Tumor-Infiltrating Lymphocytes
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Kim, G.; McKee, A.E.; Ning, Y.-M.; Hazarika, M.; Theoret, M.; Johnson, J.R.; Xu, Q.C.; Tang, S.; Sridhara, R.; Jiang, X. FDA approval summary: Vemurafenib for treatment of unresectable or metastatic melanoma with the BRAFV600E mutation. Clin. Cancer Res. 2014, 20, 4994–5000. [Google Scholar] [CrossRef] [Green Version]
- Barone, A.; Hazarika, M.; Theoret, M.R.; Mishra-Kalyani, P.; Chen, H.; He, K.; Sridhara, R.; Subramaniam, S.; Pfuma, E.; Wang, Y. FDA approval summary: Pembrolizumab for the treatment of patients with unresectable or metastatic melanoma. Clin. Cancer Res. 2017, 23, 5661–5665. [Google Scholar] [CrossRef] [Green Version]
- Beaver, J.A.; Theoret, M.R.; Mushti, S.; He, K.; Libeg, M.; Goldberg, K.; Sridhara, R.; McKee, A.E.; Keegan, P.; Pazdur, R. FDA approval of nivolumab for the first-line treatment of patients with BRAFV600 wild-type unresectable or metastatic melanoma. Clin. Cancer Res. 2017, 23, 3479–3483. [Google Scholar] [CrossRef] [Green Version]
- Hazarika, M.; Chuk, M.K.; Theoret, M.R.; Mushti, S.; He, K.; Weis, S.L.; Putman, A.H.; Helms, W.S.; Cao, X.; Li, H. US FDA approval summary: Nivolumab for treatment of unresectable or metastatic melanoma following progression on ipilimumab. Clin. Cancer Res. 2017, 23, 3484–3488. [Google Scholar] [CrossRef] [Green Version]
- Cameron, F.; Whiteside, G.; Perry, C. Ipilimumab. Drugs 2011, 71, 1093–1104. [Google Scholar] [CrossRef]
- Lanier, L.L.; O’Fallon, S.; Somoza, C.; Phillips, J.H.; Linsley, P.S.; Okumura, K.; Ito, D.; Azuma, M. CD80 (B7) and CD86 (B70) provide similar costimulatory signals for T cell proliferation, cytokine production, and generation of CTL. J. Immunol. 1995, 154, 97–105. [Google Scholar]
- van der Merwe, P.A.; Bodian, D.L.; Daenke, S.; Linsley, P.; Davis, S.J. CD80 (B7-1) binds both CD28 and CTLA-4 with a low affinity and very fast kinetics. J. Exp. Med. 1997, 185, 393–404. [Google Scholar] [CrossRef] [Green Version]
- Selby, M.J.; Engelhardt, J.J.; Quigley, M.; Henning, K.A.; Chen, T.; Srinivasan, M.; Korman, A.J. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol. Res. 2013, 1, 32–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agata, Y.; Kawasaki, A.; Nishimura, H.; Ishida, Y.; Tsubat, T.; Yagita, H.; Honjo, T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 1996, 8, 765–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keir, M.E.; Liang, S.C.; Guleria, I.; Latchman, Y.E.; Qipo, A.; Albacker, L.A.; Koulmanda, M.; Freeman, G.J.; Sayegh, M.H.; Sharpe, A.H. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J. Exp. Med. 2006, 203, 883–895. [Google Scholar] [CrossRef]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Kyi, C.; Postow, M.A. Immune checkpoint inhibitor combinations in solid tumors: Opportunities and challenges. Immunotherapy 2016, 8, 821–837. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Buque, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Golden, E.B.; Apetoh, L. Radiotherapy and immunogenic cell death. Semin. Radiat. Oncol. 2015, 25, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Maier, P.; Hartmann, L.; Wenz, F.; Herskind, C. Cellular pathways in response to ionizing radiation and their targetability for tumor radiosensitization. Int. J. Mol. Sci. 2016, 17, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garnett, C.T.; Palena, C.; Chakarborty, M.; Tsang, K.-Y.; Schlom, J.; Hodge, J.W. Sublethal irradiation of human tumor cells modulates phenotype resulting in enhanced killing by cytotoxic T lymphocytes. Cancer Res. 2004, 64, 7985–7994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lugade, A.A.; Moran, J.P.; Gerber, S.A.; Rose, R.C.; Frelinger, J.G.; Lord, E.M. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J. Immunol. 2005, 174, 7516–7523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalbasi, A.; June, C.H.; Haas, N.; Vapiwala, N. Radiation and immunotherapy: A synergistic combination. J. Clin. Invest. 2013, 123, 2756–2763. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.A.; Minn, A.J. Combination cancer therapy with immune checkpoint blockade: Mechanisms and strategies. Immunity 2018, 48, 417–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharabi, A.B.; Lim, M.; DeWeese, T.L.; Drake, C.G. Radiation and checkpoint blockade immunotherapy: Radiosensitisation and potential mechanisms of synergy. Lancet Oncol. 2015, 16, e498–e509. [Google Scholar] [CrossRef]
- Weichselbaum, R.R.; Liang, H.; Deng, L.; Fu, Y.X. Radiotherapy and immunotherapy: A beneficial liaison? Nat. Rev. Clin. Oncol. 2017, 14, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, M.B.; Krishnan, S.; Hodge, J.W.; Chang, J.Y. Immunotherapy and stereotactic ablative radiotherapy (ISABR): A curative approach? Nat. Rev. Clin. Oncol. 2016, 13, 516. [Google Scholar] [CrossRef] [PubMed]
- Mole, R. Whole body irradiation—Radiobiology or medicine? Br. J. Radiol. 1953, 26, 234–241. [Google Scholar] [CrossRef]
- Crittenden, M.; Kohrt, H.; Levy, R.; Jones, J.; Camphausen, K.; Dicker, A.; Demaria, S.; Formenti, S. Current clinical trials testing combinations of immunotherapy and radiation. Semin. Radiat. Oncol. 2015, 25, 54–64. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.; Demaria, S.; Formenti, S. Current clinical trials testing the combination of immunotherapy with radiotherapy. J. Immunother. Cancer 2016, 4, 51. [Google Scholar] [CrossRef] [Green Version]
- Abuodeh, Y.; Venkat, P.; Kim, S. Systematic review of case reports on the abscopal effect. Curr. Probl. Cancer. 2016, 40, 25–37. [Google Scholar] [CrossRef]
- Morris, Z.S.; Guy, E.I.; Werner, L.R.; Carlson, P.M.; Heinze, C.M.; Kler, J.S.; Busche, S.M.; Jaquish, A.A.; Sriramaneni, R.N.; Carmichael, L.L. Tumor-specific inhibition of in situ vaccination by distant untreated tumor sites. Cancer Immunol. Res. 2018, 6, 825–834. [Google Scholar] [CrossRef] [Green Version]
- Heppner, G.H. Tumor heterogeneity. Cancer Res. 1984, 44, 2259–2265. [Google Scholar]
- Heppner, G.H.; Shekhar, M. Tumor heterogeneity is fundamental to the tumor ecosystem. Oncology 2014, 28, 780. [Google Scholar]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Kwon, E.D.; Drake, C.G.; Scher, H.I.; Fizazi, K.; Bossi, A.; Van den Eertwegh, A.J.; Krainer, M.; Houede, N.; Santos, R.; Mahammedi, H. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): A multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014, 15, 700–712. [Google Scholar] [CrossRef] [Green Version]
- Brooks, E.D.; Chang, J.Y. Time to abandon single-site irradiation for inducing abscopal effects. Nat. Rev. Clin. Oncol. 2019, 16, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Arina, A.; Gutiontov, S.I.; Weichselbaum, R.R. Radiotherapy and immunotherapy for cancer: From “systemic” to “multisite”. Clin. Cancer Res. 2020, 26, 2777–2782. [Google Scholar] [CrossRef] [PubMed]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Yokoi, T.; Chiappori, A.; Lee, K.H.; de Wit, M. Durvalumab after chemoradiotherapy in stage III non–small-cell lung cancer. N. Engl. J. Med. 2017, 377, 1919–1929. [Google Scholar] [CrossRef] [Green Version]
- Luke, J.J.; Lemons, J.M.; Karrison, T.G.; Pitroda, S.P.; Melotek, J.M.; Zha, Y.; Al-Hallaq, H.A.; Arina, A.; Khodarev, N.N.; Janisch, L. Safety and clinical activity of pembrolizumab and multisite stereotactic body radiotherapy in patients with advanced solid tumors. J. Clin. Oncol. 2018, 36, 1611. [Google Scholar] [CrossRef]
- Jagodinsky, J.C.; Morris, Z.S. Priming and Propagating Anti-tumor Immunity: Focal Hypofractionated Radiation for in Situ Vaccination and Systemic Targeted Radionuclide Theranostics for Immunomodulation of Tumor Microenvironments. Semin. Radiat. Oncol. 2020, 30, 181–186. [Google Scholar] [CrossRef]
- Jagodinsky, J.C.; Arthur, I.S.; Castillo, J.S.; Chakravarty, I.; Zangl, L.M.; Brown, R.J.; Patel, R.B.; Jin, W.J.; Carlson, P.M.; Hernandez, R. Comparing type 1 interferon activation in tumor cells following external beam radiotherapy versus targeted radionuclide therapy. AACR 2020, 80, 16. [Google Scholar]
- Ersahin, D.; Doddamane, I.; Cheng, D. Targeted radionuclide therapy. Cancers 2011, 3, 3838–3855. [Google Scholar] [CrossRef] [PubMed]
- Dash, A.; F Russ Knapp, F.; Ra Pillai, M. Targeted radionuclide therapy-an overview. Curr. Radiopharm. 2013, 6, 152–180. [Google Scholar] [CrossRef]
- Gudkov, S.V.; Shilyagina, N.Y.; Vodeneev, V.A.; Zvyagin, A.V. Targeted radionuclide therapy of human tumors. Int. J. Mol. Sci. 2016, 17, 33. [Google Scholar] [CrossRef] [PubMed]
- Ghanem, G.E.; Comunale, G.; Libert, A.; Vercammen-Grandjean, A.; Lejeune, F.J. Evidence for alpha-melanocyte-stimulating hormone (alpha-MSH) receptors on human malignant melanoma cells. Int. J. Cancer. 1988, 41, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Onfray, F.; Lopez, M.; Lundqvist, A.; Aguirre, A.; Escobar, A.; Serrano, A.; Korenblit, C.; Petersson, M.; Chhajlani, V.; Larsson, O.; et al. Tissue distribution and differential expression of melanocortin 1 receptor, a malignant melanoma marker. Br. J. Cancer. 2002, 87, 414–422. [Google Scholar] [CrossRef] [Green Version]
- Siegrist, W.; Solca, F.; Stutz, S.; Giuffre, L.; Carrel, S.; Girard, J.; Eberle, A.N. Characterization of receptors for alpha-melanocyte-stimulating hormone on human melanoma cells. Cancer Res. 1989, 49, 6352–6358. [Google Scholar] [PubMed]
- Tatro, J.B.; Atkins, M.; Mier, J.W.; Hardarson, S.; Wolfe, H.; Smith, T.; Entwistle, M.L.; Reichlin, S. Melanotropin receptors demonstrated in situ in human melanoma. J. Clin. Invest. 1990, 85, 1825–1832. [Google Scholar] [CrossRef]
- Martin, M.E.; Sue O’Dorisio, M.; Leverich, W.M.; Kloepping, K.C.; Walsh, S.A.; Schultz, M.K. “Click”-cyclized (68)Ga-labeled peptides for molecular imaging and therapy: Synthesis and preliminary in vitro and in vivo evaluation in a melanoma model system. Recent Results Cancer Res. 2013, 194, 149–175. [Google Scholar]
- Li, M.; Zhang, X.; Quinn, T.P.; Lee, D.; Liu, D.; Kunkel, F.; Zimmerman, B.E.; McAlister, D.; Olewein, K.; Menda, Y. Automated cassette-based production of high specific activity [203/212Pb] peptide-based theranostic radiopharmaceuticals for image-guided radionuclide therapy for cancer. Appl. Radiat. Isot. 2017, 127, 52–60. [Google Scholar] [CrossRef]
- Lee, D.; Li, M.; Bednarz, B.; Schultz, M.K. Modeling Cell and Tumor-Metastasis Dosimetry with the Particle and Heavy Ion Transport Code System (PHITS) Software for Targeted Alpha-Particle Radionuclide Therapy. Radiat. Res. 2018, 190, 236–247. [Google Scholar] [CrossRef] [PubMed]
- White, D.; Griffith, R.; Wilson, I. Report 46. J. Int. Comm. Radiat. Units Meas. 2016, os24. [Google Scholar] [CrossRef]
- Chan, H.S.; Konijnenberg, M.W.; Daniels, T.; Nysus, M.; Makvandi, M.; de Blois, E.; Breeman, W.A.; Atcher, R.W.; de Jong, M.; Norenberg, J.P. Improved safety and efficacy of 213 Bi-DOTATATE-targeted alpha therapy of somatostatin receptor-expressing neuroendocrine tumors in mice pre-treated with L-lysine. EJNMMI Res. 2016, 6, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Sagastume, E.E.; Lee, D.; McAlister, D.; DeGraffenreid, A.J.; Olewine, K.R.; Graves, S.; Copping, R.; Mirzadeh, S.; Zimmerman, B.E. 203/212Pb Theranostic Radiopharmaceuticals for Image-guided Radionuclide Therapy for Cancer. Curr. Med. Chem. 2020, 27, 7003–7031. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Perry, C.J.; Meeth, K.; Thakral, D.; Damsky, W.; Micevic, G.; Kaech, S.; Blenman, K.; Bosenberg, M. UV-induced somatic mutations elicit a functional T cell response in the YUMMER1.7 mouse melanoma model. Pigment. Cell Melanoma Res. 2017, 30, 428–435. [Google Scholar] [CrossRef]
- Miao, Y.; Hylarides, M.; Fisher, D.R.; Shelton, T.; Moore, H.; Wester, D.W.; Fritzberg, A.R.; Winkelmann, C.T.; Hoffman, T.; Quinn, T.P. Melanoma therapy via peptide-targeted α-radiation. Clin. Cancer Res. 2005, 11, 5616–5621. [Google Scholar] [CrossRef] [Green Version]
- Weichselbaum, R.R. The 46th David A. Karnofsky Memorial Award lecture: Oligometastasis—from conception to treatment. J. Clin. Oncol. 2018, 36, 3240–3250. [Google Scholar] [CrossRef]
- Sgouros, G.; Roeske, J.C.; McDevitt, M.R.; Palm, S.; Allen, B.J.; Fisher, D.R.; Brill, A.B.; Song, H.; Howell, R.W.; Akabani, G. MIRD Pamphlet No. 22 (abridged): Radiobiology and dosimetry of α-particle emitters for targeted radionuclide therapy. J. Nucl. Med. 2010, 51, 311–328. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, T.; Mitsuhashi, N.; Furuta, M.; Hasegawa, M.; Ohno, T.; Saito, Y.; Sakurai, H.; Nakano, T.; Niibe, H. Apoptosis induced by heavy ion (carbon) irradiation of two human tumours with different radiosensitivities in vivo: Relative biological effectiveness (RBE) of carbon beam. Anticancer Res. 1998, 18, 253–256. [Google Scholar]
- Chauhan, V.; Howland, M.; Chen, J.; Kutzner, B.; Wilkins, R.C. Differential effects of alpha-particle radiation and X-irradiation on genes associated with apoptosis. Radiol. Res. Pract. 2011, 2011, 679806. [Google Scholar] [CrossRef] [Green Version]
- Vanpouille-Box, C.; Alard, A.; Aryankalayil, M.J.; Sarfraz, Y.; Diamond, J.M.; Schneider, R.J.; Inghirami, G.; Coleman, C.N.; Formenti, S.C.; Demaria, S. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun. 2017, 8, 15618. [Google Scholar] [CrossRef]
- Huang, A.C.; Postow, M.A.; Orlowski, R.J.; Mick, R.; Bengsch, B.; Manne, S.; Xu, W.; Harmon, S.; Giles, J.R.; Wenz, B. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 2017, 545, 60–65. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Ribas, A.; Hamid, O.; Daud, A.; Wolchok, J.D.; Joshua, A.M.; Hwu, W.-J.; Weber, J.S.; Gangadhar, T.C.; Joseph, R.W. Durable complete response after discontinuation of pembrolizumab in patients with metastatic melanoma. J. Clin. Oncol. 2018, 36, 1668–1674. [Google Scholar] [CrossRef]
- Umeshappa, C.S.; Zhu, Y.; Bhanumathy, K.K.; Omabe, M.; Chibbar, R.; Xiang, J. Innate and adoptive immune cells contribute to natural resistance to systemic metastasis of B16 melanoma. Cancer Biother. Radiopharm. 2015, 30, 72–78. [Google Scholar] [CrossRef]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef]
- Arora, S.; Velichinskii, R.; Lesh, R.W.; Ali, U.; Kubiak, M.; Bansal, P.; Borghaei, H.; Edelman, M.J.; Boumber, Y. Existing and emerging biomarkers for immune checkpoint immunotherapy in solid tumors. Adv. Ther. 2019, 36, 2638–2678. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Xu, L.; Li, L.; Liu, X.; Gao, J.; Bai, Y. Inhibiting the CD8+ T cell infiltration in the tumor microenvironment after radiotherapy is an important mechanism of radioresistance. Sci. Rep. 2018, 8, 11934. [Google Scholar] [CrossRef]
- Cooper, A.; Robinson, S.J.; Pickard, C.; Jackson, C.L.; Friedmann, P.S.; Healy, E. α-Melanocyte-stimulating hormone suppresses antigen-induced lymphocyte proliferation in humans independently of melanocortin 1 receptor gene status. J. Immunol. 2005, 175, 4806–4813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, S.J.; Healy, E. Human melanocortin 1 receptor (MC1R) gene variants alter melanoma cell growth and adhesion to extracellular matrix. Oncogene 2002, 21, 8037–8046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kepp, O.; Senovilla, L.; Vitale, I.; Vacchelli, E.; Adjemian, S.; Agostinis, P.; Apetoh, L.; Aranda, F.; Barnaba, V.; Bloy, N. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 2014, 3, e955691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaipl, U.S.; Multhoff, G.; Scheithauer, H.; Lauber, K.; Hehlgans, S.; Frey, B.; Rödel, F. Kill and spread the word: Stimulation of antitumor immune responses in the context of radiotherapy. Immunotherapy 2014, 6, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Schaue, D.; McBride, W.H. Links between innate immunity and normal tissue radiobiology. Radiat. Res. 2010, 173, 406–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.-D.; Mauceri, H.; Beckett, M.; Darga, T. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef] [Green Version]
- Gameiro, S.R.; Jammed, M.L.; Wattenberg, M.M.; Tsang, K.Y.; Ferrone, S.; Hodge, J.W. Radiation-induced immunogenic modulation of tumor enhances antigen processing and calreticulin exposure, resulting in enhanced T-cell killing. Oncotarget 2014, 5, 403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golden, E.; Pellicciotta, I.; Demaria, S.; Barcellos-Hoff, M.H.; Formenti, S.C. The convergence of radiation and immunogenic cell death signaling pathways. Front. Oncol. 2012, 2, 88. [Google Scholar] [CrossRef] [Green Version]
- Golden, E.B.; Frances, D.; Pellicciotta, I.; Demaria, S.; Helen Barcellos-Hoff, M.; Formenti, S.C. Radiation fosters dose-dependent and chemotherapy-induced immunogenic cell death. Oncoimmunology 2014, 3, e28518. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 0.5 h | 1.5 h | 3 h | 6 h | 24 h | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Average | Std. Dev | Average | Std. Dev | Average | Std. Dev | Average | Std. Dev | Average | Std. Dev | |
| Blood | 1.12 | 0.34 | 0.08 | 0.02 | 0.03 | 0.01 | 0.02 | 0.01 | 0.01 | 0.00 |
| Heart | 0.66 | 0.30 | 0.08 | 0.03 | 0.05 | 0.01 | 0.03 | 0.01 | 0.02 | 0.00 |
| Liver | 0.64 | 0.25 | 0.43 | 0.12 | 0.31 | 0.19 | 0.37 | 0.17 | 0.22 | 0.06 |
| Spleen | 0.58 | 0.14 | 0.17 | 0.01 | 0.12 | 0.03 | 0.11 | 0.04 | 0.09 | 0.01 |
| Lungs | 2.80 | 0.66 | 2.98 | 2.55 | 1.34 | 1.57 | 1.00 | 0.34 | 0.20 | 0.16 |
| Kidneys | 12.78 | 2.97 | 6.06 | 1.72 | 5.98 | 1.38 | 5.32 | 1.55 | 2.59 | 1.39 |
| Tumor | 5.47 | 1.05 | 8.90 | 6.30 | 4.50 | 1.71 | 3.76 | 0.77 | 1.68 | 0.46 |
| Muscle | 0.73 | 0.29 | 0.09 | 0.04 | 0.04 | 0.02 | 0.03 | 0.01 | 0.02 | 0.00 |
| Skin | 2.23 | 1.49 | 0.27 | 0.05 | 0.15 | 0.03 | 0.17 | 0.06 | 0.09 | 0.01 |
| Brain | 0.06 | 0.02 | 0.02 | 0.01 | 0.01 | 0.00 | 0.01 | 0.00 | 0.01 | 0.00 |
| Testes | 3.23 | 3.93 | 0.10 | 0.04 | 0.04 | 0.01 | 0.04 | 0.01 | 0.03 | 0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, M.; Liu, D.; Lee, D.; Cheng, Y.; Baumhover, N.J.; Marks, B.M.; Sagastume, E.A.; Ballas, Z.K.; Johnson, F.L.; Morris, Z.S.; et al. Targeted Alpha-Particle Radiotherapy and Immune Checkpoint Inhibitors Induces Cooperative Inhibition on Tumor Growth of Malignant Melanoma. Cancers 2021, 13, 3676. https://doi.org/10.3390/cancers13153676
Li M, Liu D, Lee D, Cheng Y, Baumhover NJ, Marks BM, Sagastume EA, Ballas ZK, Johnson FL, Morris ZS, et al. Targeted Alpha-Particle Radiotherapy and Immune Checkpoint Inhibitors Induces Cooperative Inhibition on Tumor Growth of Malignant Melanoma. Cancers. 2021; 13(15):3676. https://doi.org/10.3390/cancers13153676
Chicago/Turabian StyleLi, Mengshi, Dijie Liu, Dongyoul Lee, Yinwen Cheng, Nicholas J. Baumhover, Brenna M. Marks, Edwin A. Sagastume, Zuhair K. Ballas, Frances L. Johnson, Zachary S. Morris, and et al. 2021. "Targeted Alpha-Particle Radiotherapy and Immune Checkpoint Inhibitors Induces Cooperative Inhibition on Tumor Growth of Malignant Melanoma" Cancers 13, no. 15: 3676. https://doi.org/10.3390/cancers13153676
APA StyleLi, M., Liu, D., Lee, D., Cheng, Y., Baumhover, N. J., Marks, B. M., Sagastume, E. A., Ballas, Z. K., Johnson, F. L., Morris, Z. S., & Schultz, M. K. (2021). Targeted Alpha-Particle Radiotherapy and Immune Checkpoint Inhibitors Induces Cooperative Inhibition on Tumor Growth of Malignant Melanoma. Cancers, 13(15), 3676. https://doi.org/10.3390/cancers13153676