
Optimization of New Catalytic Topoisomerase II Inhibitors as an Anti-Cancer Therapy

 ,
,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. In Silico Experiments
2.2. Compounds
2.3. Solubility
2.4. Microsomal Stability Assays
2.5. Microscale Thermophoresis (MST) Assays
2.6. Fluorescence Polarization (FP) Assays
2.7. Other Routine Techniques
2.8. Statistics
3. Results
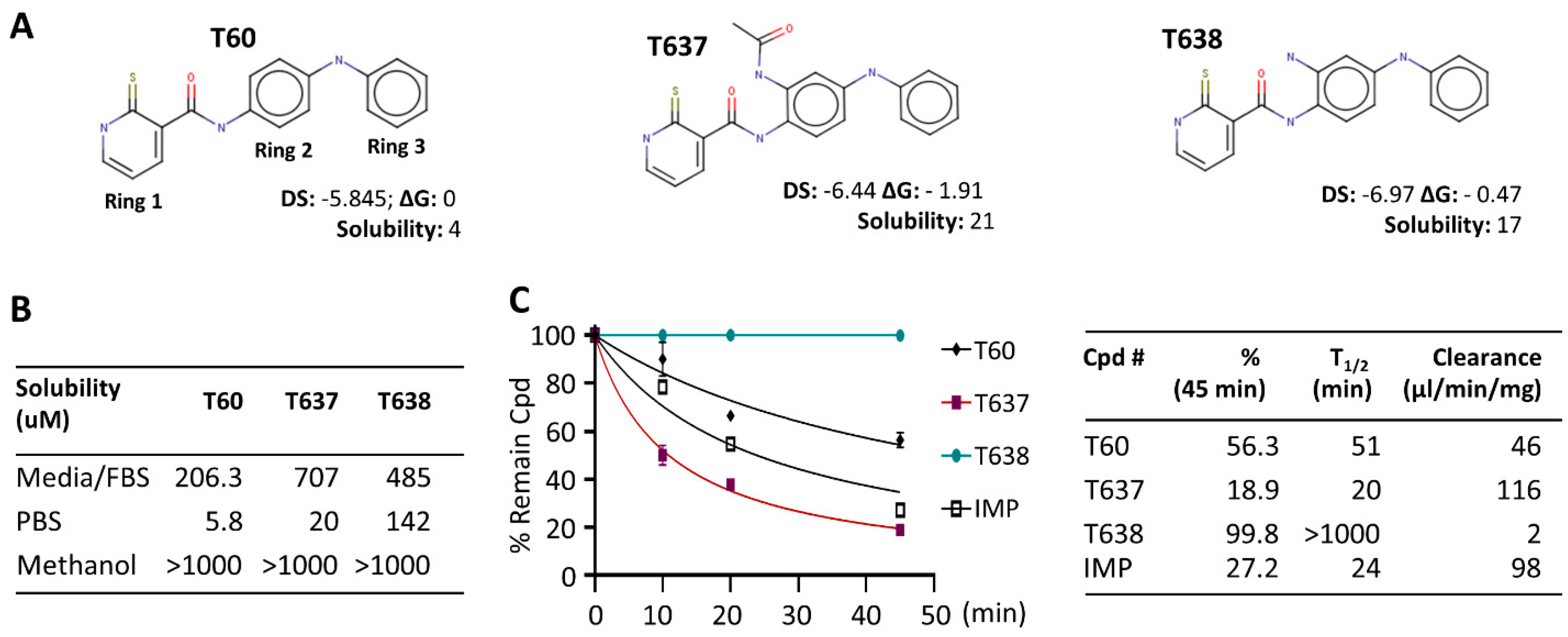
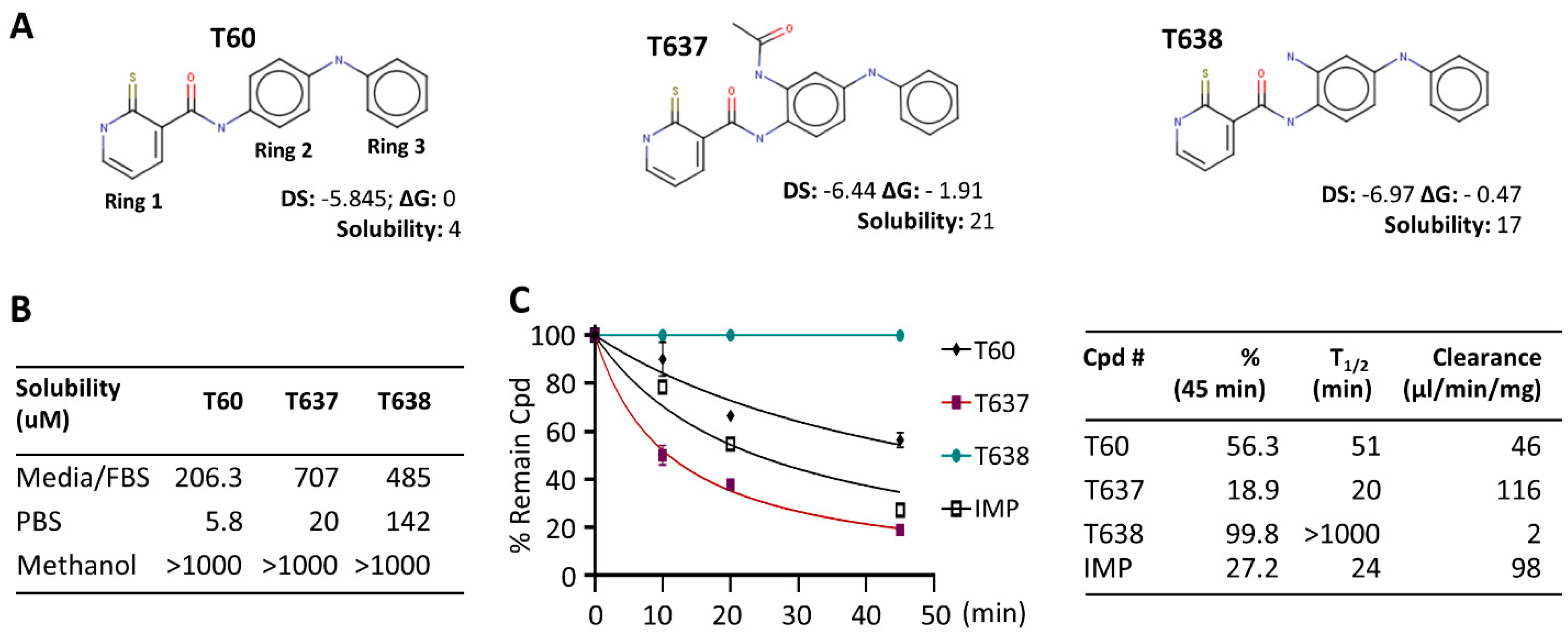
3.1. Modification of the Scaffold of T60 Using CADD
3.2. T638 Has Improved Solubility and Microsomal Stability in Comparison to T60
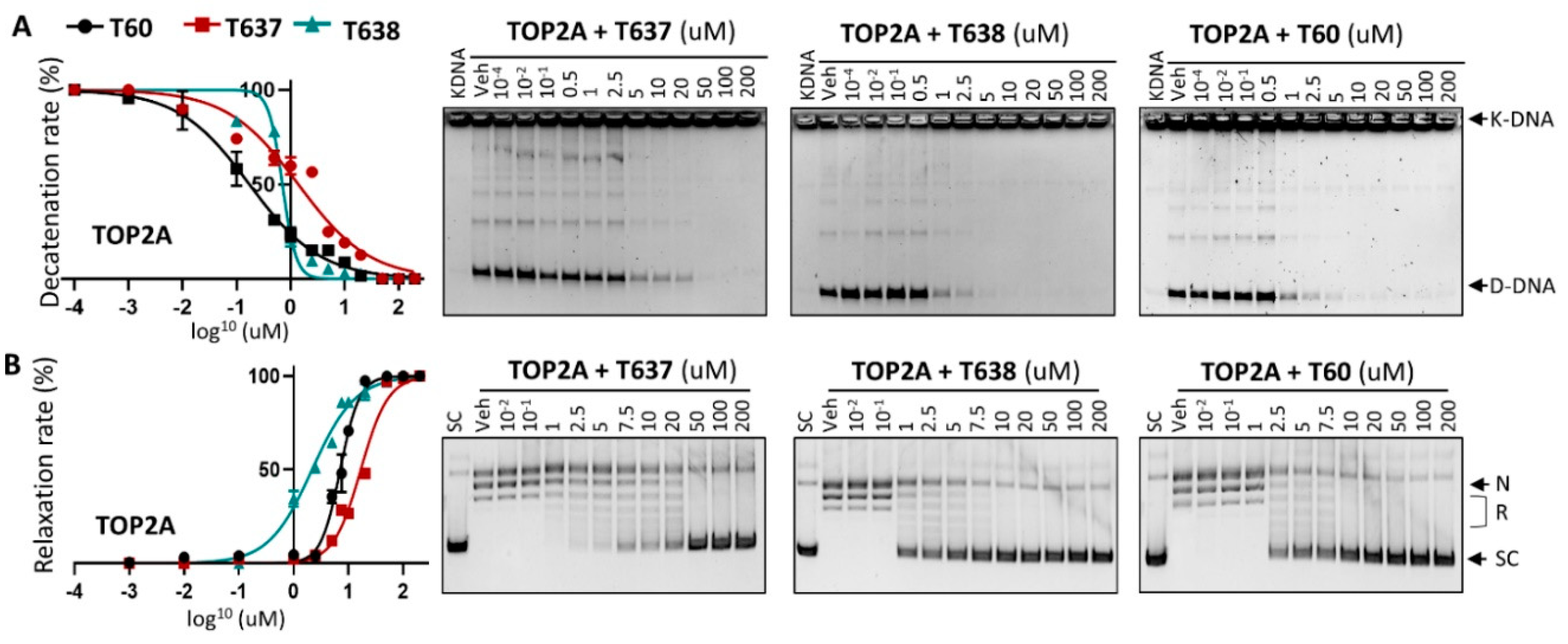
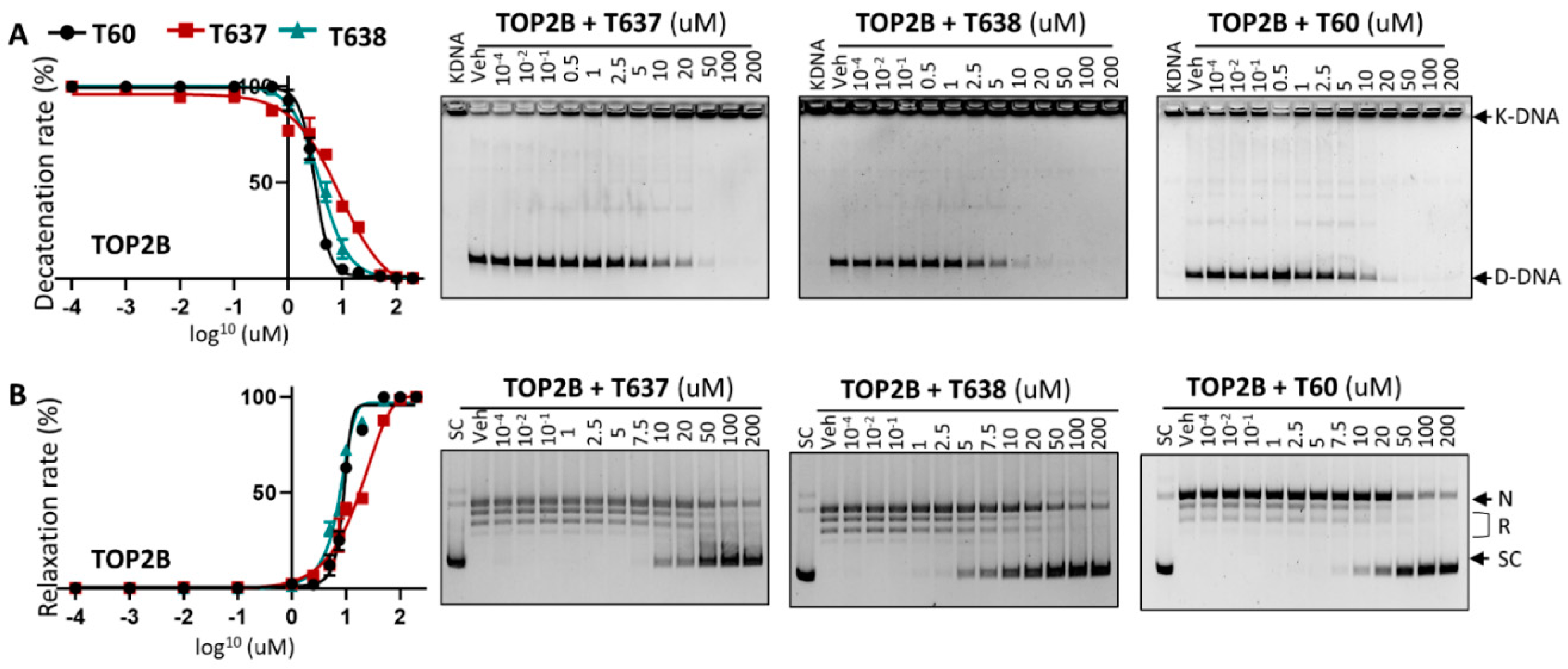
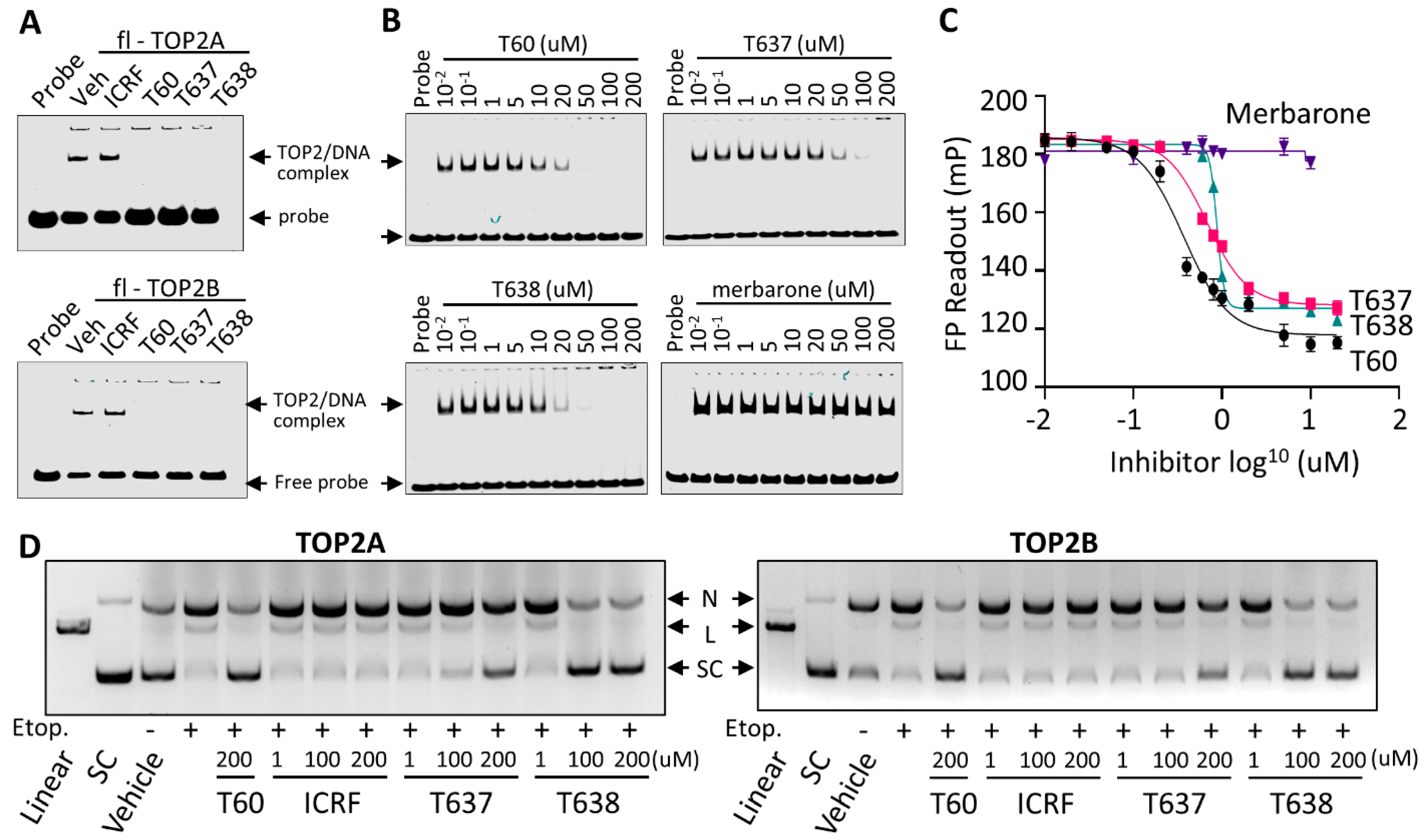
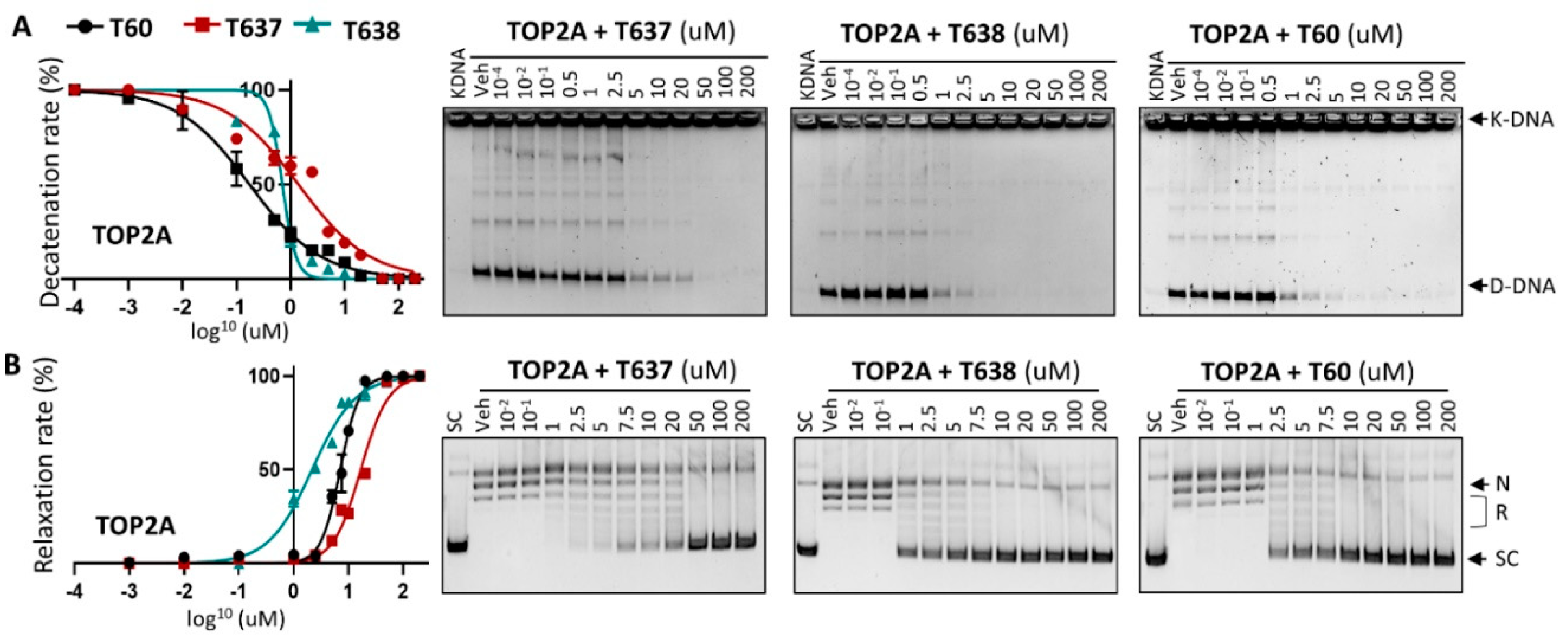
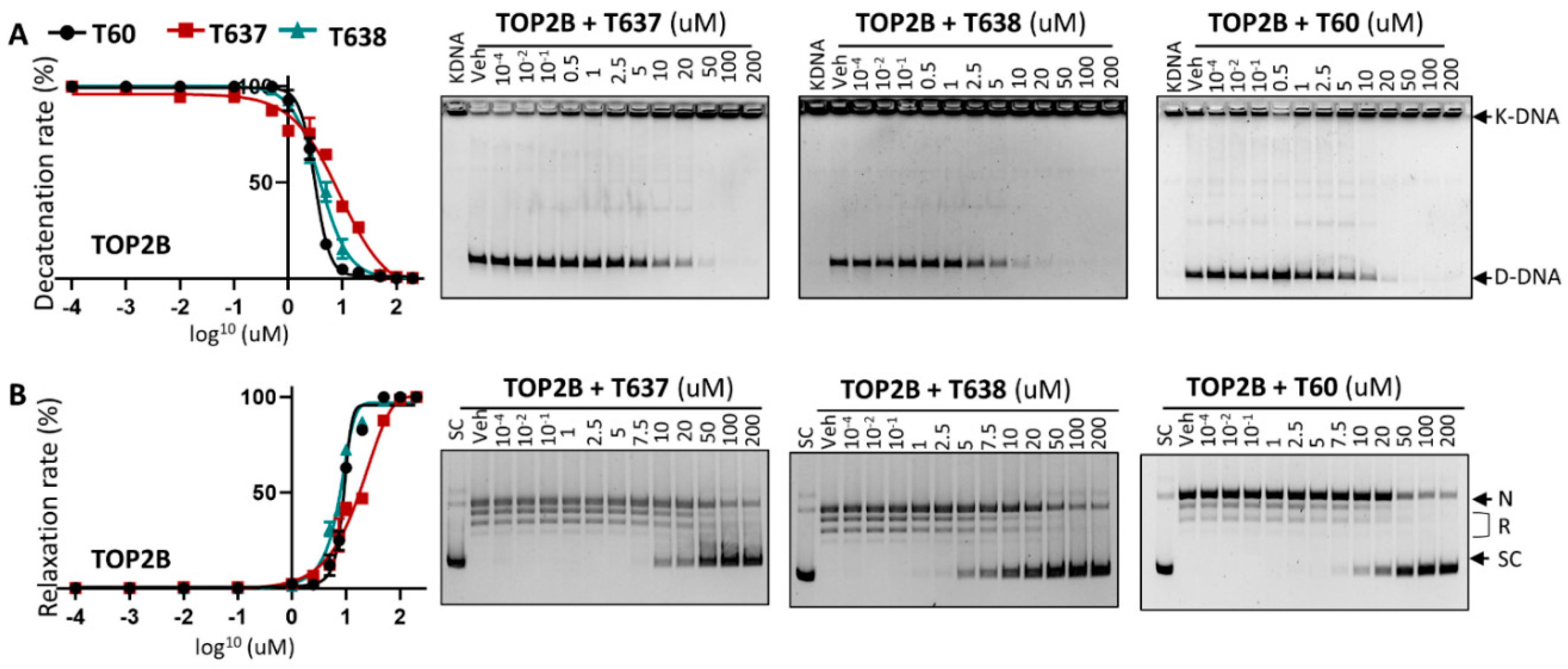
3.3. T638 Is an Inhibitor to Both TOP2A and TOP2B
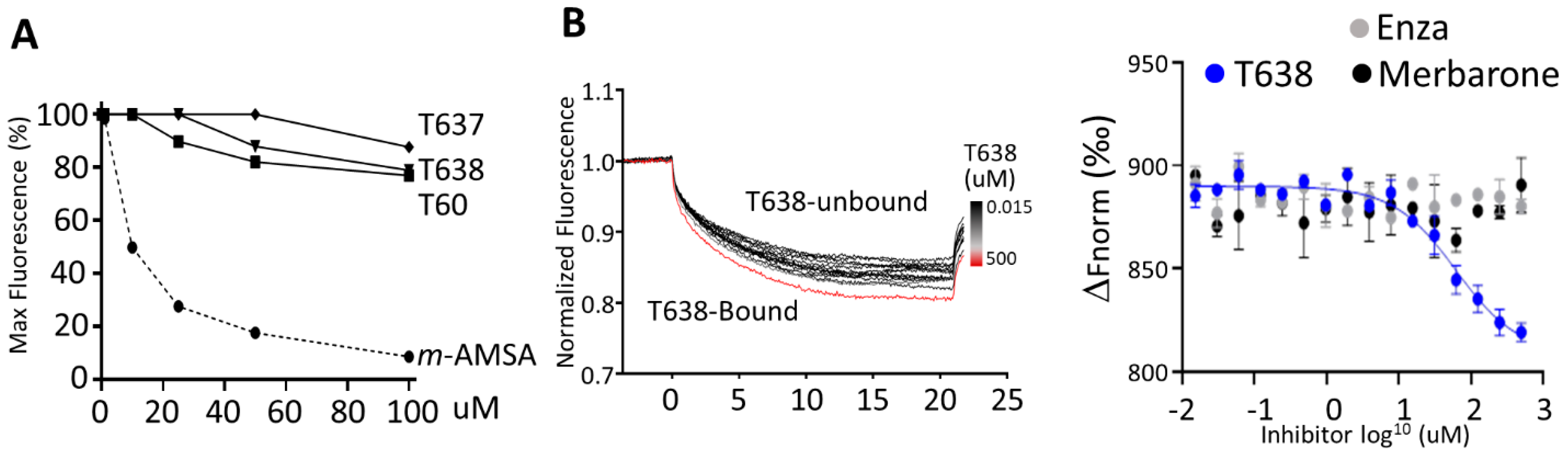
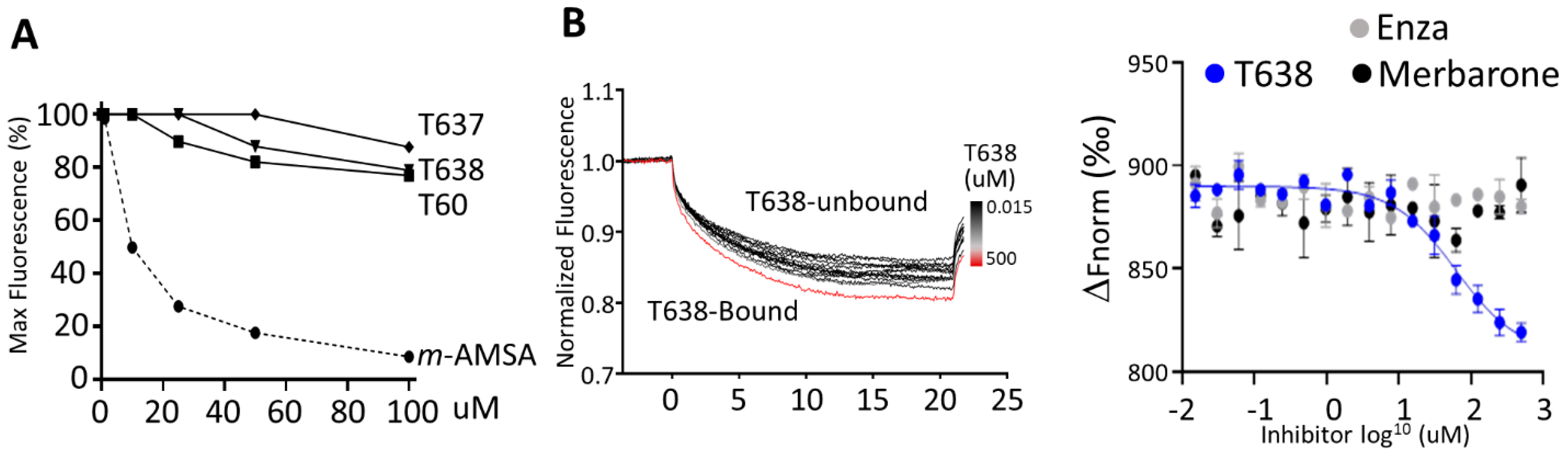
3.4. T638 Is Not a DNA Intercalator but a TOP2 Binder
3.5. T637 and T638 Block TOP2 Proteins from Interacting with DNA
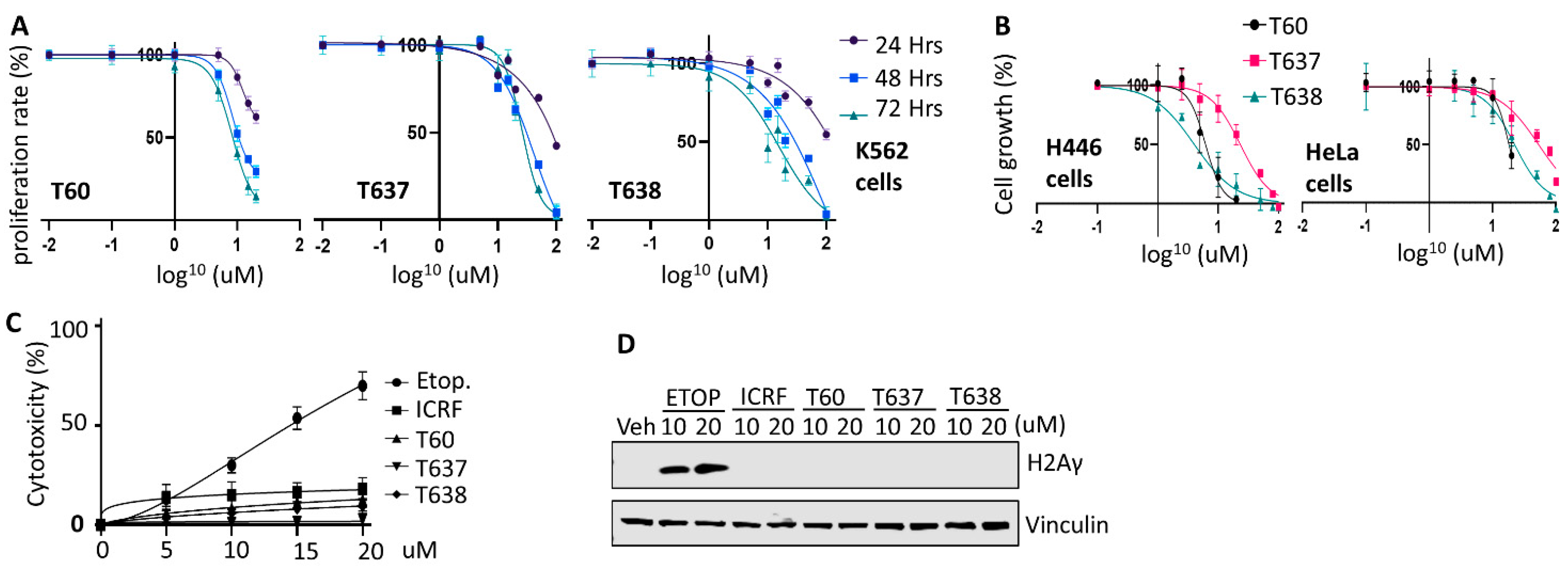
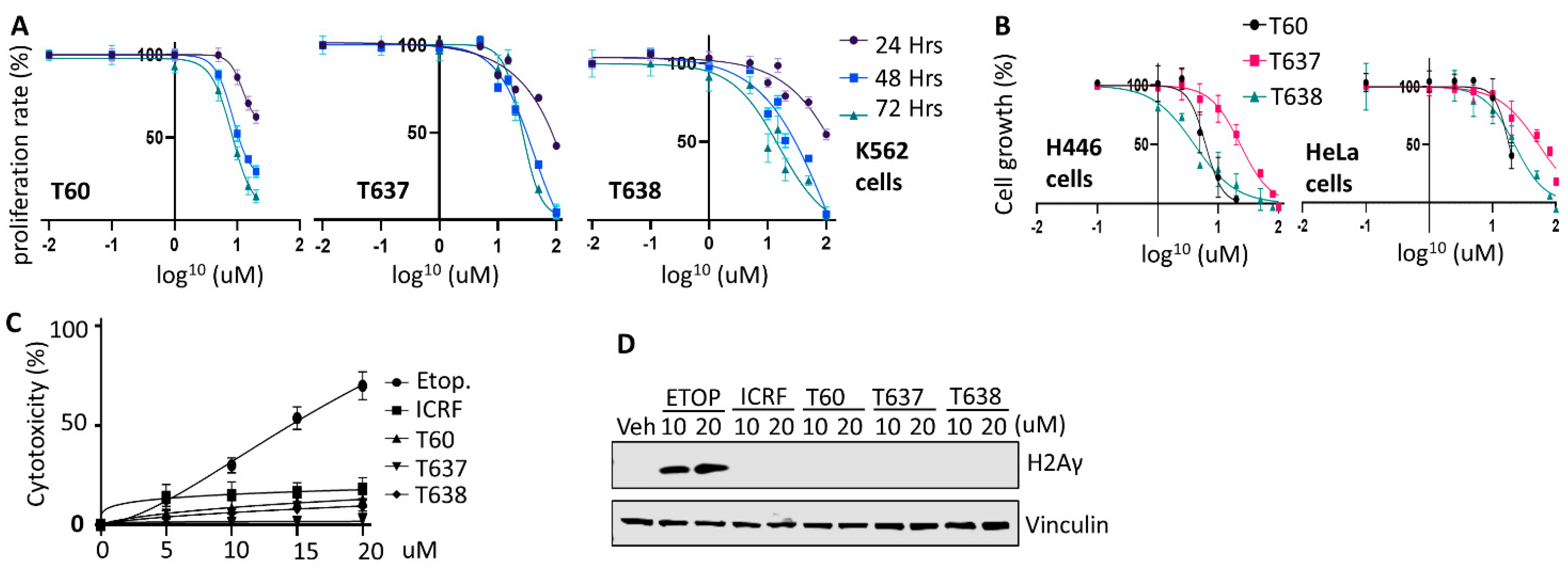
3.6. T637 and T638 Inhibit Cancer Cell Growth and Have Limited Genotoxicity
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nitiss, J.L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer 2009, 9, 327–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokiniwa, H.; Horiguchi, J.; Takata, D.; Kikuchi, M.; Rokutanda, N.; Nagaoka, R.; Sato, A.; Odawara, H.; Tozuka, K.; Oyama, T.; et al. Topoisomerase II alpha expression and the Ki-67 labeling index correlate with prognostic factors in estrogen receptor-positive and human epidermal growth factor type-2-negative breast cancer. Breast Cancer 2012, 19, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Jarvinen, T.A.; Kononen, J.; Pelto-Huikko, M.; Isola, J. Expression of topoisomerase IIalpha is associated with rapid cell proliferation, aneuploidy, and c-erbB2 overexpression in breast cancer. Am. J. Pathol. 1996, 148, 2073–2082. [Google Scholar] [PubMed]
- Labbe, D.P.; Sweeney, C.J.; Brown, M.; Galbo, P.; Rosario, S.; Wadosky, K.M.; Ku, S.Y.; Sjostrom, M.; Alshalalfa, M.; Erho, N.; et al. TOP2A and EZH2 Provide Early Detection of an Aggressive Prostate Cancer Subgroup. Clin. Cancer Res. 2017, 23, 7072–7083. [Google Scholar] [CrossRef] [Green Version]
- Dingemans, A.M.; Witlox, M.A.; Stallaert, R.A.; van der Valk, P.; Postmus, P.E.; Giaccone, G. Expression of DNA topoisomerase IIalpha and topoisomerase IIbeta genes predicts survival and response to chemotherapy in patients with small cell lung cancer. Clin. Cancer Res. 1999, 5, 2048–2058. [Google Scholar] [PubMed]
- Woessner, R.D.; Mattern, M.R.; Mirabelli, C.K.; Johnson, R.K.; Drake, F.H. Proliferation- and cell cycle-dependent differences in expression of the 170 kilodalton and 180 kilodalton forms of topoisomerase II in NIH-3T3 cells. Cell Growth Differ. 1991, 2, 209–214. [Google Scholar]
- Kimura, K.; Saijo, M.; Ui, M.; Enomoto, T. Growth state- and cell cycle-dependent fluctuation in the expression of two forms of DNA topoisomerase II and possible specific modification of the higher molecular weight form in the M phase. J. Biol. Chem. 1994, 269, 1173–1176. [Google Scholar] [CrossRef]
- Wang, J.C. DNA topoisomerases. Annu. Rev. Biochem 1996, 65, 635–692. [Google Scholar] [CrossRef] [PubMed]
- Grue, P.; Grasser, A.; Sehested, M.; Jensen, P.B.; Uhse, A.; Straub, T.; Ness, W.; Boege, F. Essential mitotic functions of DNA topoisomerase IIalpha are not adopted by topoisomerase IIbeta in human H69 cells. J. Biol. Chem. 1998, 273, 33660–33666. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Beck, W.T. DNA topoisomerase II expression, stability, and phosphorylation in two VM-26-resistant human leukemic CEM sublines. Oncol. Res. 1995, 7, 103–111. [Google Scholar]
- Yang, X.; Li, W.; Prescott, E.D.; Burden, S.J.; Wang, J.C. DNA topoisomerase IIbeta and neural development. Science 2000, 287, 131–134. [Google Scholar] [CrossRef] [Green Version]
- Haffner, M.C.; Aryee, M.J.; Toubaji, A.; Esopi, D.M.; Albadine, R.; Gurel, B.; Isaacs, W.B.; Bova, G.S.; Liu, W.; Xu, J.; et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat. Genet. 2010, 42, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Ju, B.G.; Lunyak, V.V.; Perissi, V.; Garcia-Bassets, I.; Rose, D.W.; Glass, C.K.; Rosenfeld, M.G. A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science 2006, 312, 1798–1802. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Yang, L.; Tanasa, B.; Hutt, K.; Ju, B.G.; Ohgi, K.; Zhang, J.; Rose, D.W.; Fu, X.D.; Glass, C.K.; et al. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell 2009, 139, 1069–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pendleton, M.; Lindsey, R.H., Jr.; Felix, C.A.; Grimwade, D.; Osheroff, N. Topoisomerase II and leukemia. Ann. N. Y. Acad. Sci. 2014, 1310, 98–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef] [Green Version]
- Schroeder, P.E.; Jensen, P.B.; Sehested, M.; Hofland, K.F.; Langer, S.W.; Hasinoff, B.B. Metabolism of dexrazoxane (ICRF-187) used as a rescue agent in cancer patients treated with high-dose etoposide. Cancer Chemother. Pharm. 2003, 52, 167–174. [Google Scholar] [CrossRef]
- Azarova, A.M.; Lyu, Y.L.; Lin, C.P.; Tsai, Y.C.; Lau, J.Y.; Wang, J.C.; Liu, L.F. Roles of DNA topoisomerase II isozymes in chemotherapy and secondary malignancies. Proc. Natl. Acad. Sci. USA 2007, 104, 11014–11019. [Google Scholar] [CrossRef] [Green Version]
- Nichols, C.R.; Breeden, E.S.; Loehrer, P.J.; Williams, S.D.; Einhorn, L.H. Secondary leukemia associated with a conventional dose of etoposide: Review of serial germ cell tumor protocols. J. Natl. Cancer Inst. 1993, 85, 36–40. [Google Scholar] [CrossRef]
- Sorensen, B.S.; Sinding, J.; Andersen, A.H.; Alsner, J.; Jensen, P.B.; Westergaard, O. Mode of action of topoisomerase II-targeting agents at a specific DNA sequence. Uncoupling the DNA binding, cleavage and religation events. J. Mol. Biol 1992, 228, 778–786. [Google Scholar] [CrossRef]
- Gormley, N.A.; Orphanides, G.; Meyer, A.; Cullis, P.M.; Maxwell, A. The interaction of coumarin antibiotics with fragments of DNA gyrase B protein. Biochemistry 1996, 35, 5083–5092. [Google Scholar] [CrossRef]
- Roca, J.; Ishida, R.; Berger, J.M.; Andoh, T.; Wang, J.C. Antitumor bisdioxopiperazines inhibit yeast DNA topoisomerase II by trapping the enzyme in the form of a closed protein clamp. Proc. Natl. Acad. Sci. USA 1994, 91, 1781–1785. [Google Scholar] [CrossRef] [Green Version]
- Larsen, A.K.; Escargueil, A.E.; Skladanowski, A. Catalytic topoisomerase II inhibitors in cancer therapy. Pharmacol. Ther. 2003, 99, 167–181. [Google Scholar] [CrossRef]
- Matias-Barrios, V.M.; Radaeva, M.; Song, Y.; Alperstein, Z.; Lee, A.R.; Schmitt, V.; Lee, J.; Ban, F.; Xie, N.; Qi, J.; et al. Discovery of New Catalytic Topoisomerase II Inhibitors for Anticancer Therapeutics. Front. Oncol. 2020, 10, 633142. [Google Scholar] [CrossRef]
- Chemical Computing Group (ULC). Molecular Operating Environment (MOE); Chemical Computing Group (ULC): Montreal, QC, Canada, 2019. [Google Scholar]
- He, B.; Kemppainen, J.A.; Wilson, E.M. FXXLF and WXXLF sequences mediate the NH2-terminal interaction with the ligand binding domain of the androgen receptor. J. Biol. Chem. 2000, 275, 22986–22994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delaney, J.S. ESOL: Estimating aqueous solubility directly from molecular structure. J. Chem. Inf. Comput. Sci. 2004, 44, 1000–1005. [Google Scholar] [CrossRef] [PubMed]
- Ali, J.; Camilleri, P.; Brown, M.B.; Hutt, A.J.; Kirton, S.B. In silico prediction of aqueous solubility using simple QSPR models: The importance of phenol and phenol-like moieties. J. Chem. Inf. Modeling 2012, 52, 2950–2957. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fratev, F.; Sirimulla, S. An improved free energy perturbation fep+ Sampling protocol for flexible Ligand-Binding Domains. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.S.; Deng, Y.; Wu, Y.; Sindhikara, D.; Rask, A.R.; Kimura, T.; Abel, R.; Wang, L. Accurate and reliable prediction of the binding affinities of macrocycles to their protein targets. J. Chem. Theory Comput. 2017, 13, 6290–6300. [Google Scholar] [CrossRef]
- Kuhn, B.; Tichý, M.; Wang, L.; Robinson, S.; Martin, R.E.; Kuglstatter, A.; Benz, J.R.; Giroud, M.; Schirmeister, T.; Abel, R. Prospective evaluation of free energy calculations for the prioritization of cathepsin L inhibitors. J. Med. Chem. 2017, 60, 2485–2497. [Google Scholar] [CrossRef]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug solubility: Importance and enhancement techniques. Int. Sch. Res. Not. 2012, 2012, 195727. [Google Scholar] [CrossRef] [Green Version]
- Hornedo, J.; Van Echo, D.A. Amsacrine (m-AMSA): A new antineoplastic agent. Pharmacology, clinical activity and toxicity. Pharmacotherapy 1985, 5, 78–90. [Google Scholar] [CrossRef]
- Tran, C.; Ouk, S.; Clegg, N.J.; Chen, Y.; Watson, P.A.; Arora, V.; Wongvipat, J.; Smith-Jones, P.M.; Yoo, D.; Kwon, A.; et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 2009, 324, 787–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brewer, A.D.; Minatelli, J.A.; Plowman, J.; Paull, K.D.; Narayanan, V.L. 5-(N-phenylcarboxamido)-2-thiobarbituric acid (NSC 336628), a novel potential antitumor agent. Biochem. Pharmacol. 1985, 34, 2047–2050. [Google Scholar] [CrossRef]
- Fortune, J.M.; Osheroff, N. Merbarone inhibits the catalytic activity of human topoisomerase IIalpha by blocking DNA cleavage. J. Biol. Chem. 1998, 273, 17643–17650. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, N.; Guchhait, S.K.; Bharatam, P.V. Pharmacoinformatics analysis of merbarone binding site in human topoisomerase IIalpha. J. Mol. Graph. Model. 2019, 86, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Di, L.; Kerns, E.H.; Carter, G.T. Drug-like property concepts in pharmaceutical design. Curr. Pharm. Des. 2009, 15, 2184–2194. [Google Scholar] [CrossRef]
- Wang, L.; Eastmond, D.A. Catalytic inhibitors of topoisomerase II are DNA-damaging agents: Induction of chromosomal damage by merbarone and ICRF-187. Environ. Mol. Mutagen. 2002, 39, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Pastor, N.; Dominguez, I.; Orta, M.L.; Campanella, C.; Mateos, S.; Cortes, F. The DNA topoisomerase II catalytic inhibitor merbarone is genotoxic and induces endoreduplication. Mutat. Res. Mol. Mech. Mutagen. 2012, 738–739, 45–51. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matias-Barrios, V.M.; Radaeva, M.; Ho, C.-H.; Lee, J.; Adomat, H.; Lallous, N.; Cherkasov, A.; Dong, X. Optimization of New Catalytic Topoisomerase II Inhibitors as an Anti-Cancer Therapy. Cancers 2021, 13, 3675. https://doi.org/10.3390/cancers13153675
Matias-Barrios VM, Radaeva M, Ho C-H, Lee J, Adomat H, Lallous N, Cherkasov A, Dong X. Optimization of New Catalytic Topoisomerase II Inhibitors as an Anti-Cancer Therapy. Cancers. 2021; 13(15):3675. https://doi.org/10.3390/cancers13153675
Chicago/Turabian StyleMatias-Barrios, Victor M., Mariia Radaeva, Chia-Hao Ho, Joseph Lee, Hans Adomat, Nada Lallous, Artem Cherkasov, and Xuesen Dong. 2021. "Optimization of New Catalytic Topoisomerase II Inhibitors as an Anti-Cancer Therapy" Cancers 13, no. 15: 3675. https://doi.org/10.3390/cancers13153675
APA StyleMatias-Barrios, V. M., Radaeva, M., Ho, C.-H., Lee, J., Adomat, H., Lallous, N., Cherkasov, A., & Dong, X. (2021). Optimization of New Catalytic Topoisomerase II Inhibitors as an Anti-Cancer Therapy. Cancers, 13(15), 3675. https://doi.org/10.3390/cancers13153675