
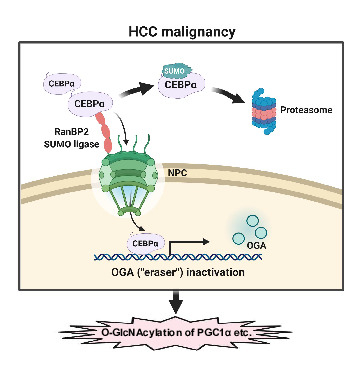
RANBP2 Activates O-GlcNAcylation through Inducing CEBPα-Dependent OGA Downregulation to Promote Hepatocellular Carcinoma Malignant Phenotypes

 , and
, and
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
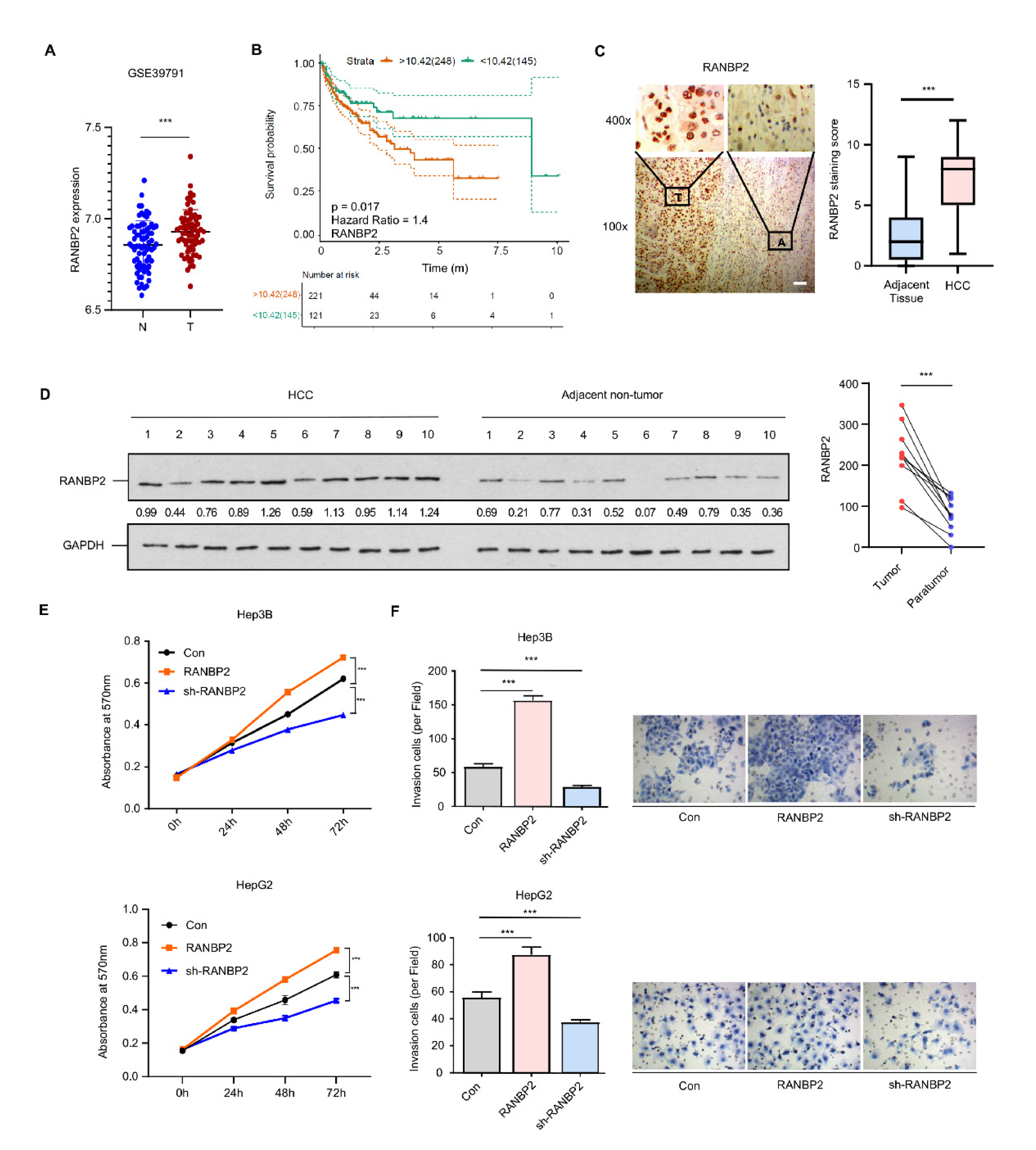
2.1. RANBP2 Is Enriched in HCC and Correlates with Malignant Phenotypes
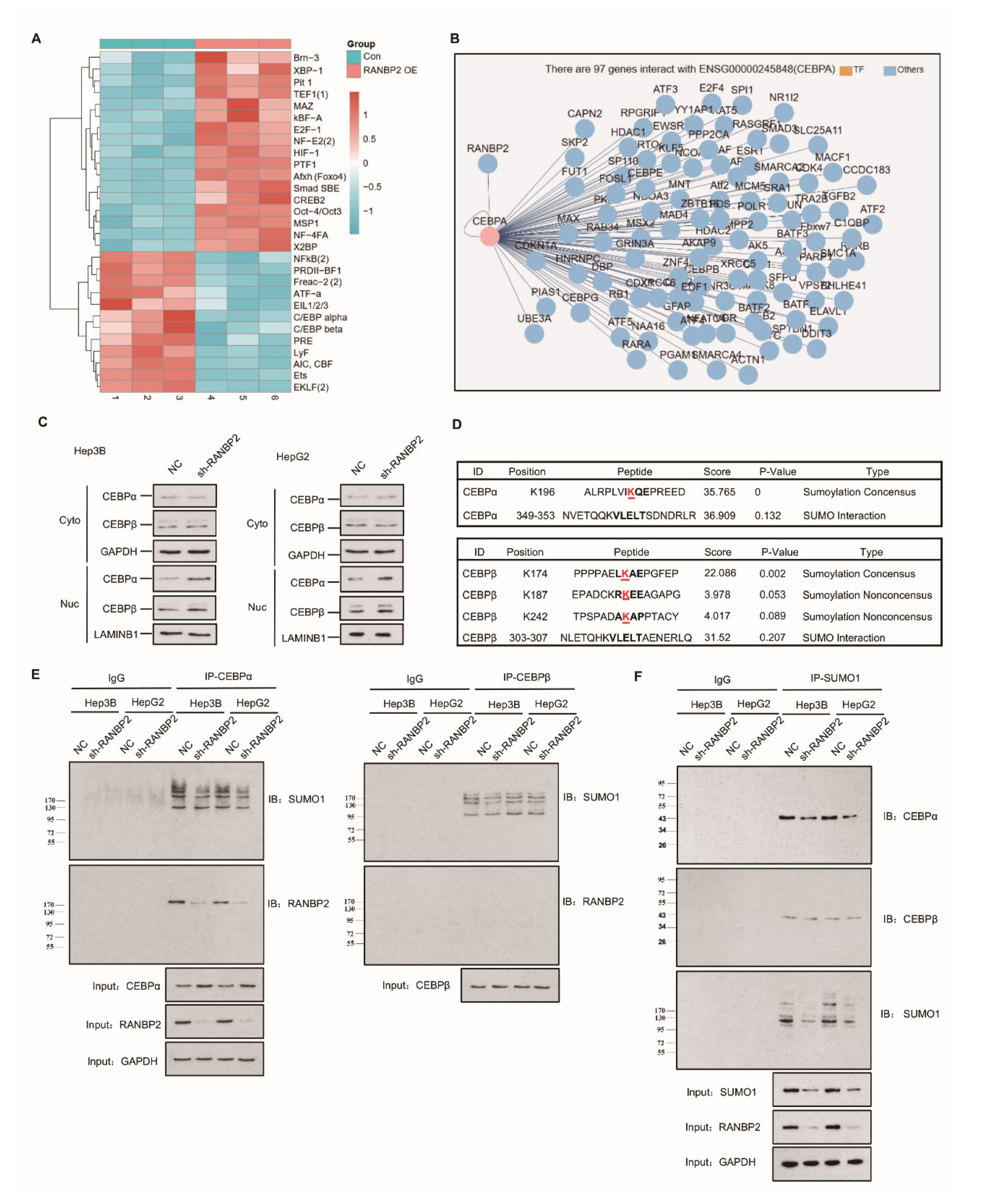
2.2. Delineation of RANBP2 Functions via Interaction with CEBPα and Negatively Regulating Its Expression
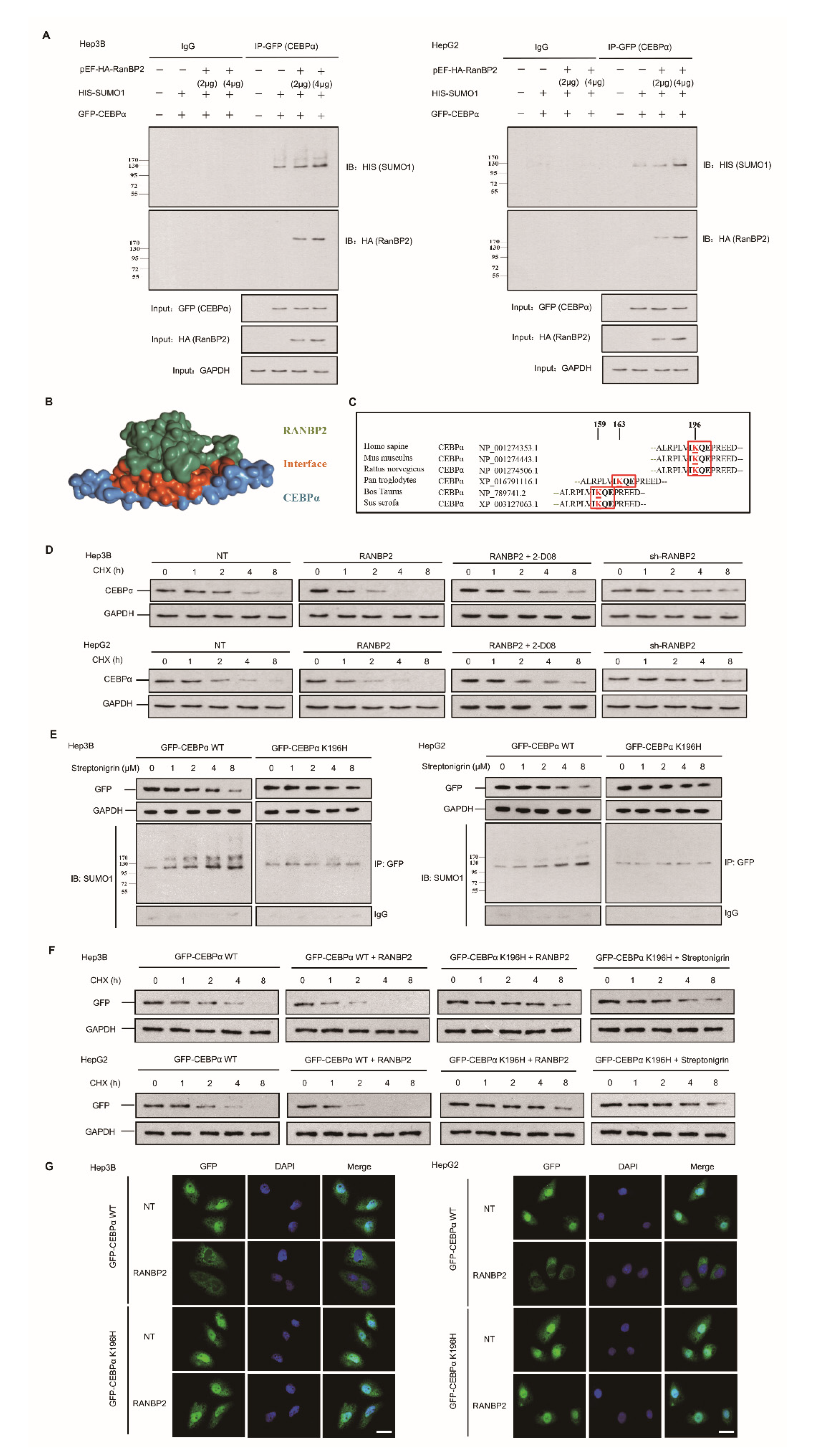
2.3. RANBP2 Overexpression Promotes CEBPα SUMOylation and Its Subsequent Degradation
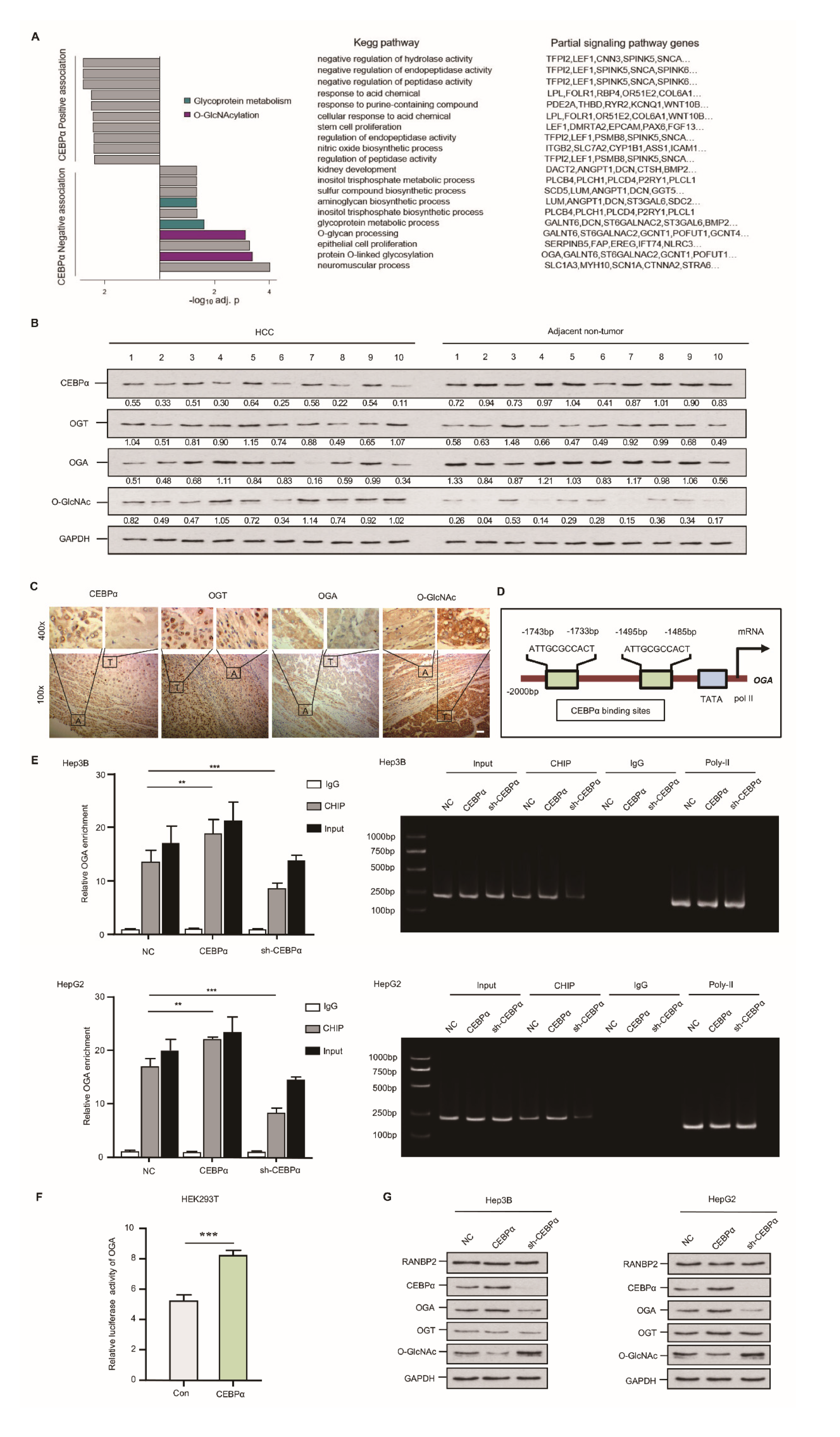
2.4. CEBPα Attenuates O-glycosylation through Triggering OGA Transcriptional Activity
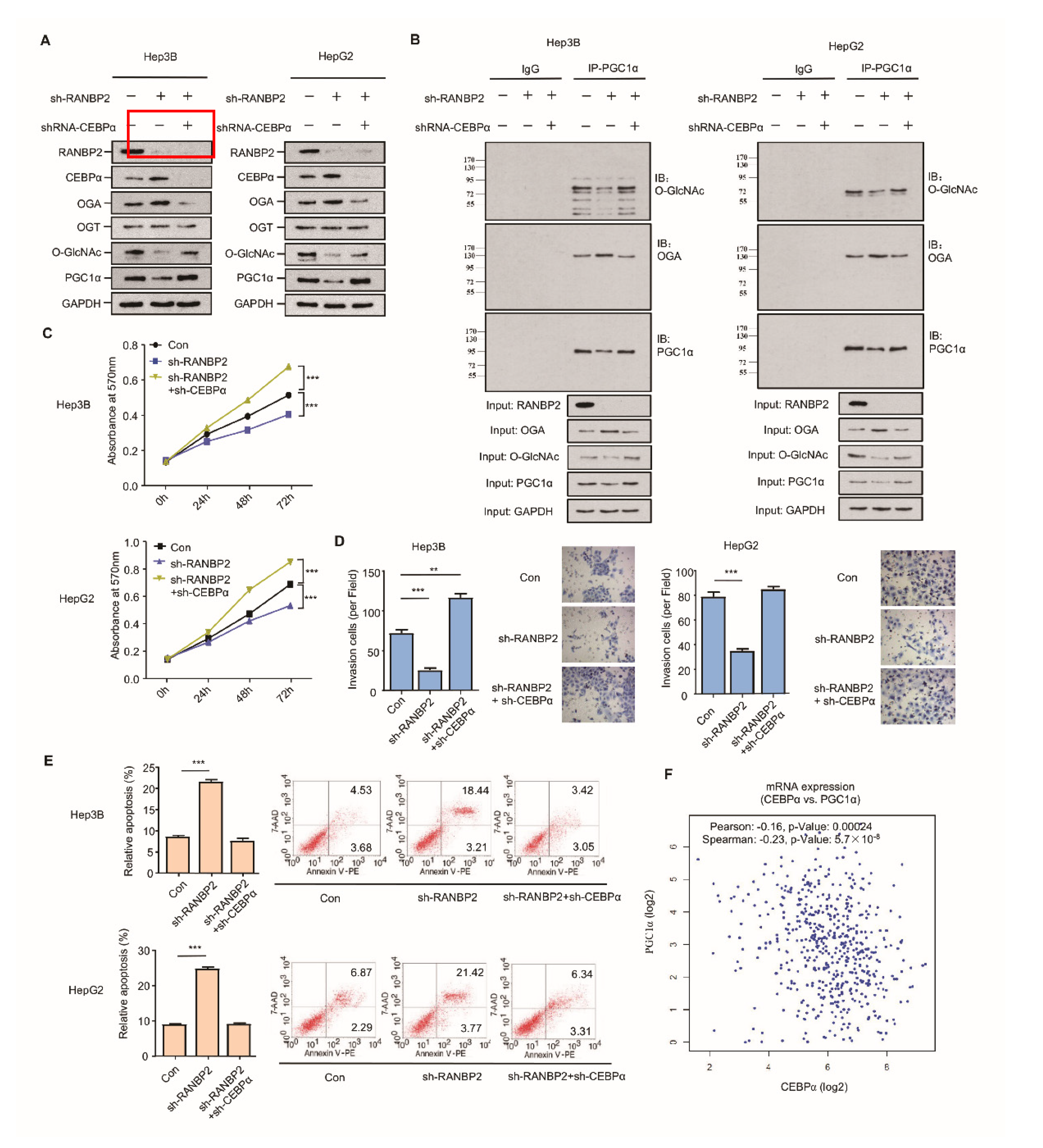
2.5. RANBP2-Associated OGA Modulation Is Transcriptionally Regulated and Independent of SUMOylation
2.6. RANBP2 Promotes CEBPα-Dependent O-glycosylation Imbalance in HCC
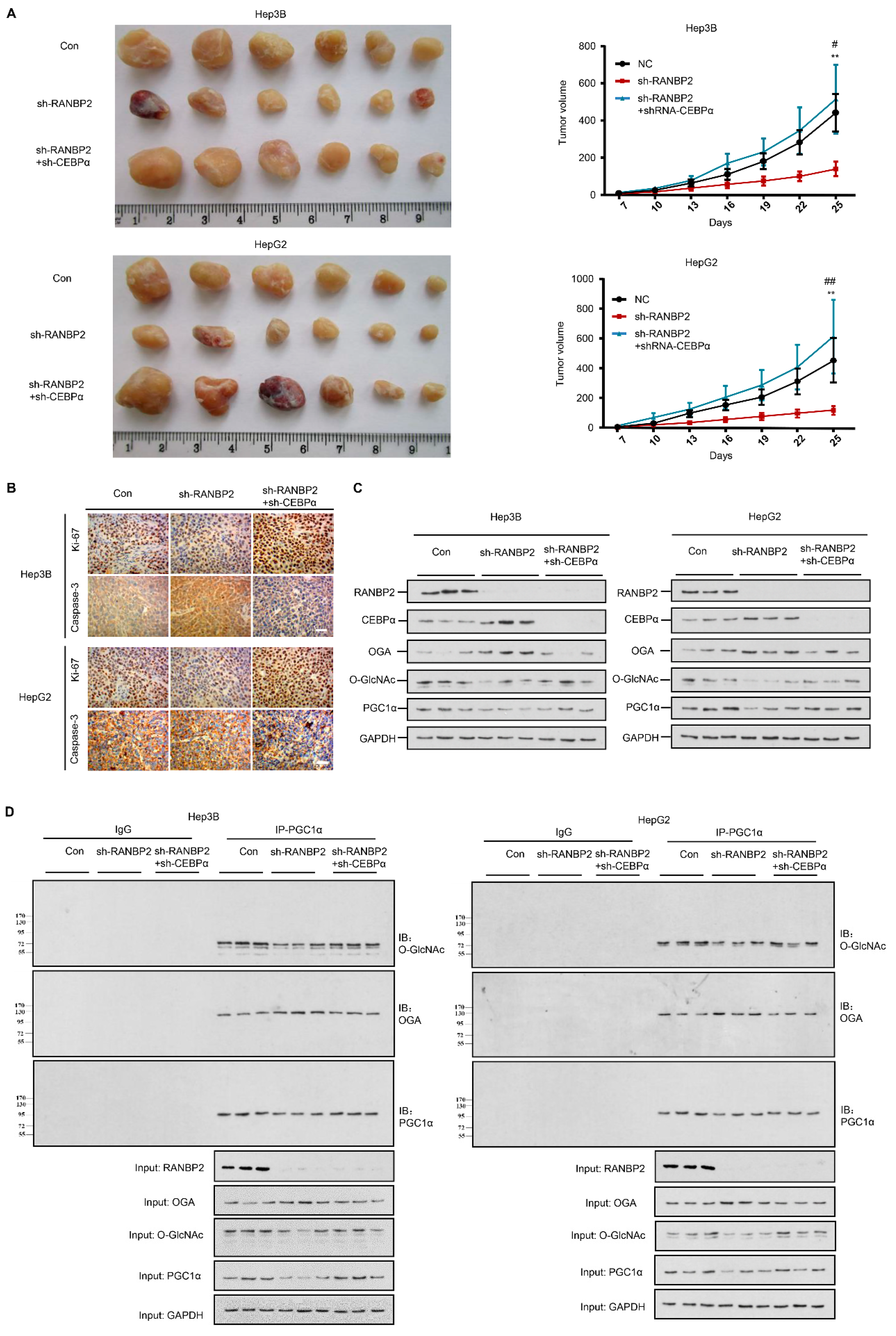
2.7. Silencing of CEBPα Is Essential for RANBP2-Mediated Aberrant O-GlcNAcylation and HCC Tumor Growth In Vivo
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture
4.2. Plasmid Construction, RNA Interference, and Transfection
4.3. Reagents
4.4. Mutagenesis
4.5. Cell Viability, Apoptosis, and Invasion Assay
4.6. Luciferase Assay
4.7. Cycloheximide (CHX)-Based Protein Stability Assay
4.8. mRNA Decay Assay
4.9. Immunohistochemistry
4.10. Immunofluorescence Staining
4.11. Transcriptional Factor/DNA Array
4.12. Quantitative Real-Time RT-PCR and RNA Array
4.13. Chromatin Immunoprecipitation (ChIP) Assay
4.14. Protein Extraction, Immunoprecipitation, and Western Blot Analysis
4.15. Tumor Xenograft Models
4.16. Bioinformatic Analyses
4.17. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
- HCC: Hepatocellular carcinoma
- RANBP2: RAN-binding protein 2
- SUMO: Small ubiquitin-like modifier
- NPC: Nuclear pore complex
- CEBPα: CCAAT/enhancer-binding protein alpha
- CEBPβ: CCAAT/enhancer-binding protein beta
- O-GlcNAc: O-linked N-acetylglucosamine
- OGT: O-GlcNAc transferase
- OGA: O-GlcNAcase
- PGC1α: Peroxisome proliferative-activated receptor gamma coactivator 1 alpha
- PTM: Post-translational modification
- ChIP: Chromatin immunoprecipitation
- IHC: Immunohistochemistry
- CHX: Cycloheximide
- GSEA: Gene set enrichment analysis
- 2-D08: 2′,3′,4′-trihydroxyflavone
- SN: Streptonigrin
References
- El-Serag, H.B. Hepatocellular carcinoma. N. Engl. J. Med. 2011, 365, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef]
- Kanda, M.; Sugimoto, H.; Kodera, Y. Genetic and epigenetic aspects of initiation and progression of hepatocellular carcinoma. World J. Gastroenterol. 2015, 21, 10584–10597. [Google Scholar] [CrossRef]
- Cui, J.; Huang, W.; Wu, B.; Jin, J.; Jing, L.; Shi, W.P.; Liu, Z.Y.; Yuan, L.; Luo, D.; Li, L.; et al. N-glycosylation by N-acetylglucosaminyltransferase V enhances the interaction of CD147/basigin with integrin beta1 and promotes HCC metastasis. J. Pathol. 2018, 245, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Santos-Laso, A.; Perugorria, M.J.; Banales, J.M. O-GlcNAcylation: Undesired tripmate but an opportunity for treatment in NAFLD-HCC. J. Hepatol. 2017, 67, 218–220. [Google Scholar] [CrossRef] [PubMed]
- Park, S.K.; Zhou, X.; Pendleton, K.E.; Hunter, O.V.; Kohler, J.J.; O’Donnell, K.A.; Conrad, N.K. A Conserved Splicing Silencer Dynamically Regulates O-GlcNAc Transferase Intron Retention and O-GlcNAc Homeostasis. Cell Rep. 2017, 20, 1088–1099. [Google Scholar] [CrossRef]
- de Queiroz, R.M.; Madan, R.; Chien, J.; Dias, W.B.; Slawson, C. Changes in O-Linked N-Acetylglucosamine (O-GlcNAc) Homeostasis Activate the p53 Pathway in Ovarian Cancer Cells. J. Biol. Chem. 2016, 291, 18897–18914. [Google Scholar] [CrossRef]
- Qian, K.; Wang, S.; Fu, M.; Zhou, J.; Singh, J.P.; Li, M.D.; Yang, Y.; Zhang, K.; Wu, J.; Nie, Y.; et al. Transcriptional regulation of O-GlcNAc homeostasis is disrupted in pancreatic cancer. J. Biol. Chem. 2018, 293, 13989–14000. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.B.; Han, X.; Li, M.D.; Singh, J.P.; Qian, K.; Azarhoush, S.; Zhao, L.; Bennett, A.M.; Samuel, V.T.; Wu, J.; et al. O-GlcNAc transferase/host cell factor C1 complex regulates gluconeogenesis by modulating PGC-1alpha stability. Cell Metab. 2012, 16, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.Y.; Liu, H.; Tin, K.X.; Huang, Y.; Yeh, K.H.; Peng, H.W.; Chen, H.D.; He, J.Y.; Chiang, Y.J.; Liu, C.S.; et al. Identification of UAP1L1 as a critical factor for protein O-GlcNAcylation and cell proliferation in human hepatoma cells. Oncogene 2019, 38, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Duan, F.; Wu, H.; Jia, D.; Wu, W.; Ren, S.; Wang, L.; Song, S.; Guo, X.; Liu, F.; Ruan, Y.; et al. O-GlcNAcylation of RACK1 promotes hepatocellular carcinogenesis. J. Hepatol. 2018, 68, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Zhang, X.; Wu, J.L.; Fu, L.; Liu, K.; Liu, D.; Chen, G.G.; Lai, P.B.; Wong, N.; Yu, J. O-GlcNAc transferase promotes fatty liver-associated liver cancer through inducing palmitic acid and activating endoplasmic reticulum stress. J. Hepatol. 2017, 67, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.J.; Feng, Y.H.; Gu, B.H.; Li, Y.M.; Chen, H. The post-translational modification, SUMOylation, and cancer (Review). Int. J. Oncol. 2018, 52, 1081–1094. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, P.A.; Nakayama, T.A.; Pak, W.L.; Travis, G.H. Cyclophilin-related protein RanBP2 acts as chaperone for red/green opsin. Nature 1996, 383, 637–640. [Google Scholar] [CrossRef]
- Singh, B.B.; Patel, H.H.; Roepman, R.; Schick, D.; Ferreira, P.A. The zinc finger cluster domain of RanBP2 is a specific docking site for the nuclear export factor, exportin-1. J. Biol. Chem. 1999, 274, 37370–37378. [Google Scholar] [CrossRef] [PubMed]
- Aslanukov, A.; Bhowmick, R.; Guruju, M.; Oswald, J.; Raz, D.; Bush, R.A.; Sieving, P.A.; Lu, X.; Bock, C.B.; Ferreira, P.A. RanBP2 modulates Cox11 and hexokinase I activities and haploinsufficiency of RanBP2 causes deficits in glucose metabolism. PLoS Genet. 2006, 2, e177. [Google Scholar] [CrossRef]
- Ben-Efraim, I.; Gerace, L. Gradient of increasing affinity of importin beta for nucleoporins along the pathway of nuclear import. J. Cell Biol. 2001, 152, 411–417. [Google Scholar] [CrossRef]
- Kaminsky, R.; Denison, C.; Bening-Abu-Shach, U.; Chisholm, A.D.; Gygi, S.P.; Broday, L. SUMO regulates the assembly and function of a cytoplasmic intermediate filament protein in C. elegans. Dev. Cell 2009, 17, 724–735. [Google Scholar] [CrossRef]
- Liu, X.; Liu, J.; Xiao, W.; Zeng, Q.; Bo, H.; Zhu, Y.; Gong, L.; He, D.; Xing, X.; Li, R.; et al. SIRT1 Regulates N(6)-Methyladenosine RNA Modification in Hepatocarcinogenesis by Inducing RANBP2-Dependent FTO SUMOylation. Hepatology 2020, 72, 2029–2050. [Google Scholar] [CrossRef]
- Mahadevan, K.; Zhang, H.; Akef, A.; Cui, X.A.; Gueroussov, S.; Cenik, C.; Roth, F.P.; Palazzo, A.F. RanBP2/Nup358 potentiates the translation of a subset of mRNAs encoding secretory proteins. PLoS Biol. 2013, 11, e1001545. [Google Scholar] [CrossRef]
- Cast, A.; Valanejad, L.; Wright, M.; Nguyen, P.; Gupta, A.; Zhu, L.; Shin, S.; Timchenko, N. C/EBPalpha-dependent preneoplastic tumor foci are the origin of hepatocellular carcinoma and aggressive pediatric liver cancer. Hepatology 2018, 67, 1857–1871. [Google Scholar] [CrossRef]
- Tian, Y.; Yang, B.; Qiu, W.; Hao, Y.; Zhang, Z.; Yang, B.; Li, N.; Cheng, S.; Lin, Z.; Rui, Y.C.; et al. ER-residential Nogo-B accelerates NAFLD-associated HCC mediated by metabolic reprogramming of oxLDL lipophagy. Nat. Commun. 2019, 10, 3391. [Google Scholar] [CrossRef]
- Seipel, K.; Marques, M.T.; Bozzini, M.A.; Meinken, C.; Mueller, B.U.; Pabst, T. Inactivation of the p53-KLF4-CEBPA Axis in Acute Myeloid Leukemia. Clin. Cancer Res. 2016, 22, 746–756. [Google Scholar] [CrossRef]
- Zhao, X.; Reebye, V.; Hitchen, P.; Fan, J.; Jiang, H.; Saetrom, P.; Rossi, J.; Habib, N.A.; Huang, K.W. Mechanisms involved in the activation of C/EBPalpha by small activating RNA in hepatocellular carcinoma. Oncogene 2019, 38, 3446–3457. [Google Scholar] [CrossRef]
- Huang, Q.; Li, J.; Xing, J.; Li, W.; Li, H.; Ke, X.; Zhang, J.; Ren, T.; Shang, Y.; Yang, H.; et al. CD147 promotes reprogramming of glucose metabolism and cell proliferation in HCC cells by inhibiting the p53-dependent signaling pathway. J. Hepatol. 2014, 61, 859–866. [Google Scholar] [CrossRef]
- Tohme, S.; Yazdani, H.O.; Liu, Y.; Loughran, P.; Van der Windt, D.J.; Huang, H.; Simmons, R.L.; Shiva, S.; Tai, S.; Tsung, A. Hypoxia mediates mitochondrial biogenesis in hepatocellular carcinoma to promote tumor growth through HMGB1 and TLR9 interaction. Hepatology 2017, 66, 182–197. [Google Scholar] [CrossRef] [PubMed]
- Jozwiak, P.; Forma, E.; Brys, M.; Krzeslak, A. O-GlcNAcylation and Metabolic Reprograming in Cancer. Front. Endocrinol. 2014, 5, 145. [Google Scholar]
- Ma, Z.; Vosseller, K. Cancer metabolism and elevated O-GlcNAc in oncogenic signaling. J. Biol. Chem. 2014, 289, 34457–34465. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.L.; Cheng, Y.Y.; Ding, D.; Mei, S.; Xu, J.W.; Yu, J.; Ou-Yang, Q.; Deng, L.; Chen, Q.; Li, Q.Q.; et al. C/EBP-alpha ameliorates CCl(4)-induced liver fibrosis in mice through promoting apoptosis of hepatic stellate cells with little apoptotic effect on hepatocytes in vitro and in vivo. Apoptosis 2012, 17, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.D.; Leung, C.H.; Yan, B.; Tan, C.M.; Low, S.Y.; Aung, M.O.; Salto-Tellez, M.; Lim, S.G.; Hooi, S.C. C/EBPalpha is up-regulated in a subset of hepatocellular carcinomas and plays a role in cell growth and proliferation. Gastroenterology 2010, 139, 632–643. [Google Scholar] [CrossRef]
- Shi, Y.C.; Zhao, H.; Yin, C.; Zeng, X.; Zhang, Q.; Xu, W.P.; Wei, J.; Chen, F.; Xie, W.F. C/EBPalpha inhibits hepatocellular carcinoma by reducing Notch3/Hes1/p27 cascades. Dig. Liver Dis. 2013, 45, 844–851. [Google Scholar] [CrossRef]
- Reebye, V.; Saetrom, P.; Mintz, P.J.; Huang, K.W.; Swiderski, P.; Peng, L.; Liu, C.; Liu, X.; Lindkaer-Jensen, S.; Zacharoulis, D.; et al. Novel RNA oligonucleotide improves liver function and inhibits liver carcinogenesis in vivo. Hepatology 2014, 59, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Tomizawa, M.; Watanabe, K.; Saisho, H.; Nakagawara, A.; Tagawa, M. Down-regulated expression of the CCAAT/enhancer binding protein alpha and beta genes in human hepatocellular carcinoma: A possible prognostic marker. Anticancer. Res. 2003, 23, 351–354. [Google Scholar]
- Kroonen, J.S.; Vertegaal, A.C.O. Targeting SUMO Signaling to Wrestle Cancer. Trends Cancer 2021, 7, 496–510. [Google Scholar] [CrossRef]
- Ong, J.R.; Bamodu, O.A.; Khang, N.V.; Lin, Y.K.; Yeh, C.T.; Lee, W.H.; Cherng, Y.G. SUMO-Activating Enzyme Subunit 1 (SAE1) Is a Promising Diagnostic Cancer Metabolism Biomarker of Hepatocellular Carcinoma. Cells 2021, 10, 178. [Google Scholar] [CrossRef]
- Liu, J.; Wu, Z.; Han, D.; Wei, C.; Liang, Y.; Jiang, T.; Chen, L.; Sha, M.; Cao, Y.; Huang, F.; et al. Mesencephalic Astrocyte-Derived Neurotrophic Factor Inhibits Liver Cancer Through Small Ubiquitin-Related Modifier (SUMO)ylation-Related Suppression of NF-kappaB/Snail Signaling Pathway and Epithelial-Mesenchymal Transition. Hepatology 2020, 71, 1262–1278. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Ge, C.; Li, H.; Zhao, F.; Hou, H.; Chen, T.; Jiang, G.; Xie, H.; Cui, Y.; Yao, M.; et al. Ribonucleotide reductase M2B inhibits cell migration and spreading by early growth response protein 1-mediated phosphatase and tensin homolog/Akt1 pathway in hepatocellular carcinoma. Hepatology 2014, 59, 1459–1470. [Google Scholar] [CrossRef]
- Widagdo, J.; Chai, Y.J.; Ridder, M.C.; Chau, Y.Q.; Johnson, R.C.; Sah, P.; Huganir, R.L.; Anggono, V. Activity-Dependent Ubiquitination of GluA1 and GluA2 Regulates AMPA Receptor Intracellular Sorting and Degradation. Cell Rep. 2015, 10, 783–795. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Weng, H.; Sun, W.; Qin, X.; Shi, H.; Wu, H.; Zhao, B.S.; Mesquita, A.; Liu, C.; Yuan, C.L.; et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol. 2018, 20, 285–295. [Google Scholar] [CrossRef]
- Zeng, P.Y.; Vakoc, C.R.; Chen, Z.C.; Blobel, G.A.; Berger, S.L. In vivo dual cross-linking for identification of indirect DNA-associated proteins by chromatin immunoprecipitation. Biotechniques 2006, 41, 694–698. [Google Scholar] [CrossRef]
- Zeng, D.; Li, M.; Zhou, R.; Zhang, J.; Sun, H.; Shi, M.; Bin, J.; Liao, Y.; Rao, J.; Liao, W. Tumor Microenvironment Characterization in Gastric Cancer Identifies Prognostic and Immunotherapeutically Relevant Gene Signatures. Cancer Immunol. Res. 2019, 7, 737–750. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Chen, X.; Xiao, M.; Zhu, Y.; Gong, R.; Liu, J.; Zeng, Q.; Xu, C.; Chen, X.; Wang, F.; et al. RANBP2 Activates O-GlcNAcylation through Inducing CEBPα-Dependent OGA Downregulation to Promote Hepatocellular Carcinoma Malignant Phenotypes. Cancers 2021, 13, 3475. https://doi.org/10.3390/cancers13143475
Liu X, Chen X, Xiao M, Zhu Y, Gong R, Liu J, Zeng Q, Xu C, Chen X, Wang F, et al. RANBP2 Activates O-GlcNAcylation through Inducing CEBPα-Dependent OGA Downregulation to Promote Hepatocellular Carcinoma Malignant Phenotypes. Cancers. 2021; 13(14):3475. https://doi.org/10.3390/cancers13143475
Chicago/Turabian StyleLiu, Xiaoming, Xingyu Chen, Mengqing Xiao, Yuxing Zhu, Renjie Gong, Jianye Liu, Qinghai Zeng, Canxia Xu, Xiong Chen, Fen Wang, and et al. 2021. "RANBP2 Activates O-GlcNAcylation through Inducing CEBPα-Dependent OGA Downregulation to Promote Hepatocellular Carcinoma Malignant Phenotypes" Cancers 13, no. 14: 3475. https://doi.org/10.3390/cancers13143475
APA StyleLiu, X., Chen, X., Xiao, M., Zhu, Y., Gong, R., Liu, J., Zeng, Q., Xu, C., Chen, X., Wang, F., & Cao, K. (2021). RANBP2 Activates O-GlcNAcylation through Inducing CEBPα-Dependent OGA Downregulation to Promote Hepatocellular Carcinoma Malignant Phenotypes. Cancers, 13(14), 3475. https://doi.org/10.3390/cancers13143475