
Valproic Acid and Breast Cancer: State of the Art in 2021


Abstract
Simple Summary
Abstract
1. Introduction
2. Molecular Subtypes of Breast Cancers and Limitations in the Therapy of Patients Harboring These Subtypes
3. Histone Deacetylase Inhibitors (HDIs)
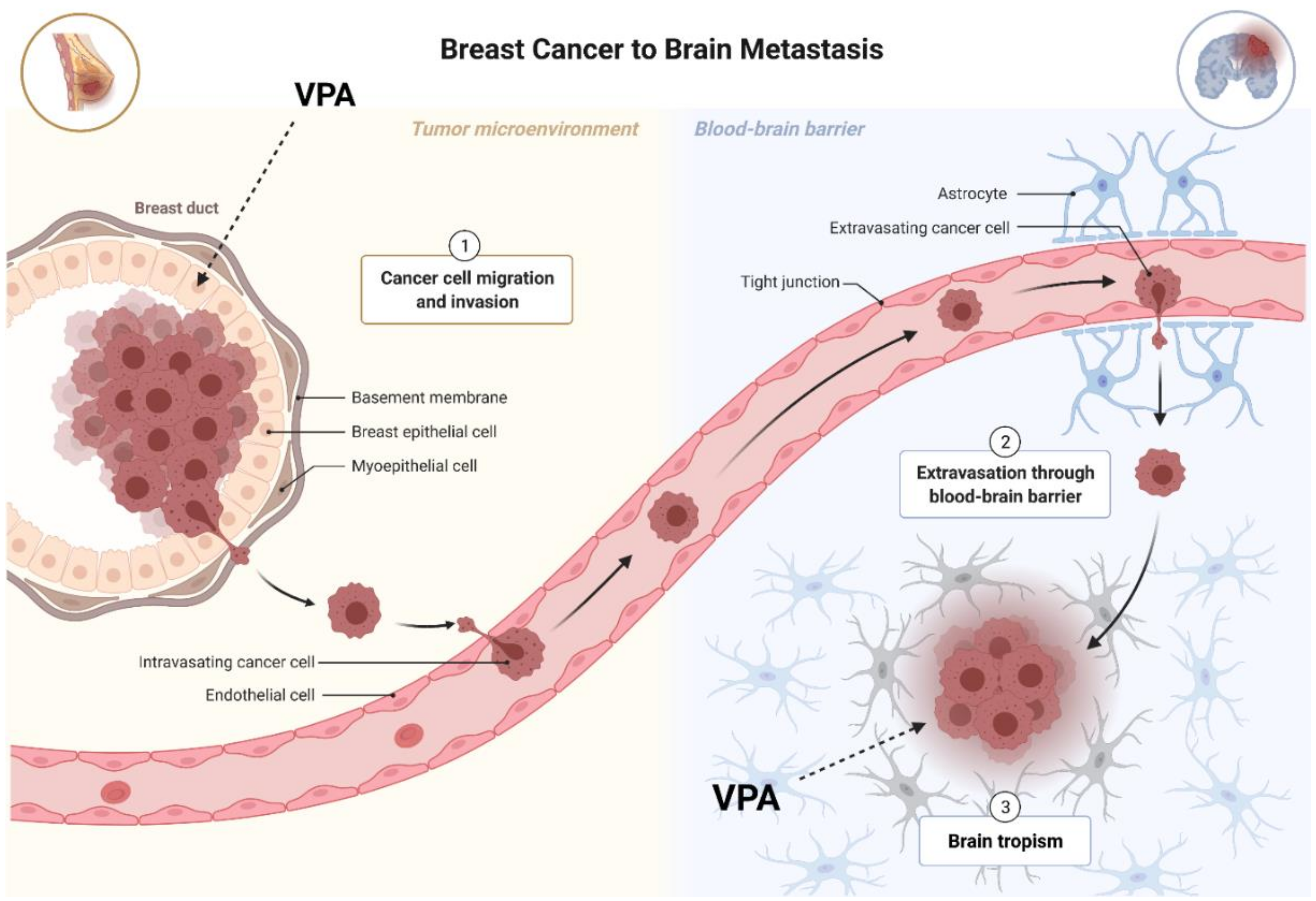
4. Valproic Acid and Breast Cancer
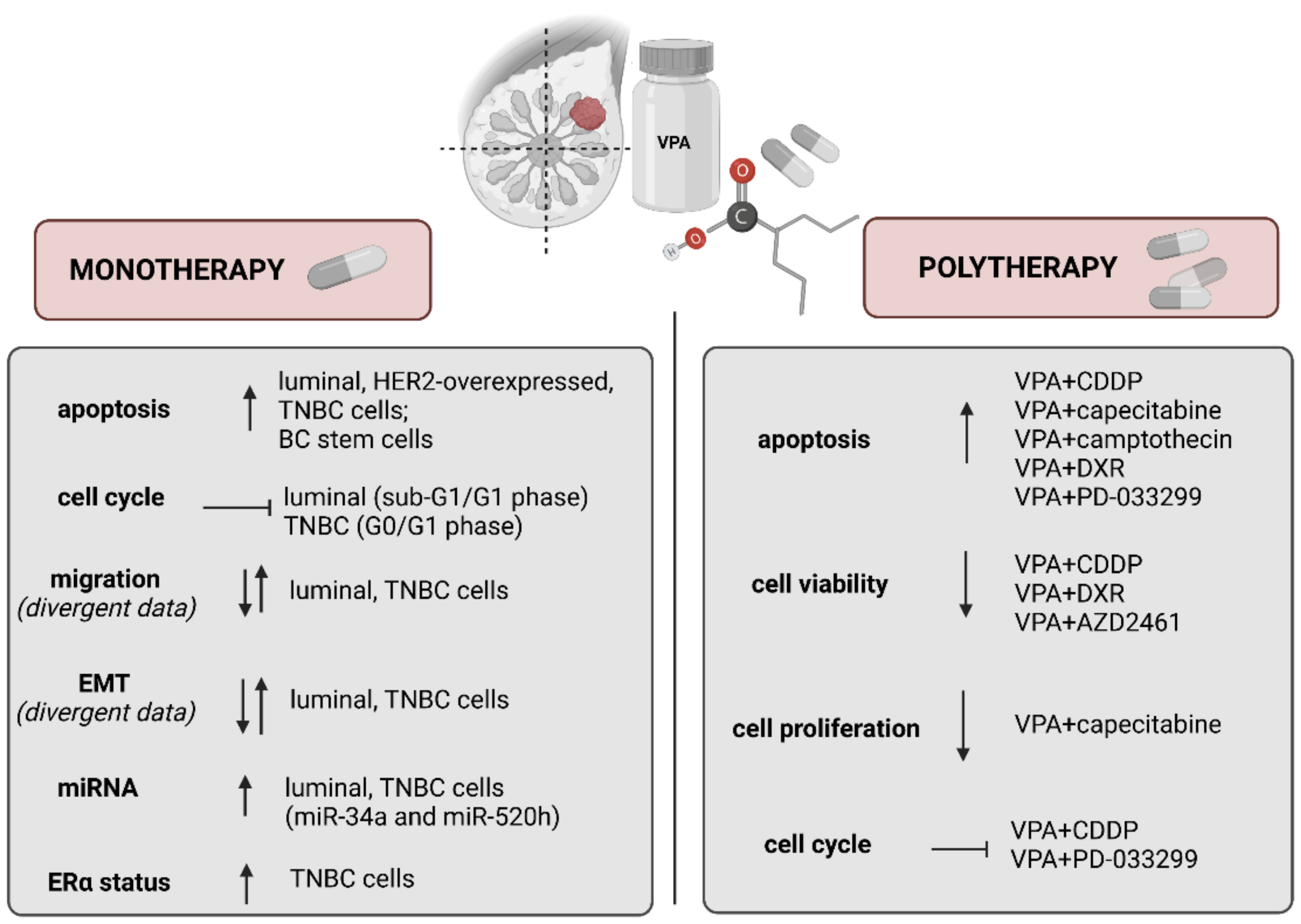
4.1. VPA Induces Apoptosis and Inhibits Cell Cycle
4.2. VPA Regulates Migration and Epithelial-Mesenchymal Transition (EMT)
4.3. VPA Affects microRNAs (miRNAs) Expression
4.4. VPA and Estrogen Receptor Status
4.5. VPA Affects Metabolic Pathways
5. “Valproic Acid et al.” and Breast Cancer
6. Valproic Acid Derivatives and Drug Carriers
7. Clinical Trials
8. Discussion
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Momenimovahed, Z.; Salehiniya, H. Epidemiological characteristics of and risk factors for breast cancer in the world. Breast Cancer Targets Ther. 2019, 11, 151–164. [Google Scholar] [CrossRef] [PubMed]
- The Lancet GLOBOCAN 2018: Counting the toll of cancer. Lancet 2018, 392, 985. [CrossRef]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Janni, W. Targeted Therapy of Breast Cancer. Oncol. Res. Treat. 2016, 39, 100–101. [Google Scholar] [CrossRef]
- Nagini, S. Breast Cancer: Current Molecular Therapeutic Targets and New Players. Anticancer Agents Med. Chem. 2017, 17, 152–163. [Google Scholar] [CrossRef]
- Cai, F.F.; Kohler, C.; Zhang, B.; Wang, M.H.; Chen, W.J.; Zhong, X.Y. Epigenetic therapy for breast cancer. Int. J. Mol. Sci. 2011, 12, 4465–4476. [Google Scholar] [CrossRef]
- Damaskos, C.; Valsami, S.; Kontos, M.; Spartalis, E.; Kalampokas, T.; Kalampokas, E.; Athanasiou, A.; Moris, D.; Daskalopoulou, A.; Davakis, S.; et al. Histone deacetylase inhibitors: An attractive therapeutic strategy against breast cancer. Anticancer Res. 2017, 37, 35–46. [Google Scholar] [CrossRef]
- Gediya, P.; Parikh, P.K.; Vyas, V.K.; Ghate, M.D. Histone deacetylase 2: A potential therapeutic target for cancer and neurodegenerative disorders. Eur. J. Med. Chem. 2021, 216. [Google Scholar] [CrossRef] [PubMed]
- Wawruszak, A.; Kalafut, J.; Okon, E.; Czapinski, J.; Halasa, M.; Przybyszewska, A.; Miziak, P.; Okla, K.; Rivero-Muller, A.; Stepulak, A. Histone deacetylase inhibitors and phenotypical transformation of cancer cells. Cancers 2019, 11, 148. [Google Scholar] [CrossRef]
- Gumbarewicz, E.; Luszczki, J.J.; Wawruszak, A.; Dmoszynska-Graniczka, M.; Grabarska, A.J.; Jarzab, A.M.; Polberg, K.; Stepulak, A. Isobolographic analysis demonstrates additive effect of cisplatin and HDIs combined treatment augmenting their anti-cancer activity in lung cancer cell lines. Am. J. Cancer Res. 2016, 6, 2831–2845. [Google Scholar]
- Dai, X.; Li, T.; Bai, Z.; Yang, Y.; Liu, X.; Zhan, J.; Shi, B. Breast cancer intrinsic subtype classification, clinical use and future trends. Am. J. Cancer Res. 2015, 5, 2929–2943. [Google Scholar] [PubMed]
- Szymiczek, A.; Lone, A.; Akbari, M.R. Molecular intrinsic versus clinical subtyping in breast cancer: A comprehensive review. Clin. Genet. 2021, 99, 613–637. [Google Scholar] [CrossRef]
- McDonald, E.S.; Clark, A.S.; Tchou, J.; Zhang, P.; Freedman, G.M. Clinical diagnosis and management of breast cancer. J. Nucl. Med. 2016, 57, 9S–16S. [Google Scholar] [CrossRef] [PubMed]
- Bonacho, T.; Rodrigues, F.; Liberal, J. Immunohistochemistry for diagnosis and prognosis of breast cancer: A review. Biotech. Histochem. 2020, 95, 71–91. [Google Scholar] [CrossRef]
- Cheang, M.C.U.; Chia, S.K.; Voduc, D.; Gao, D.; Leung, S.; Snider, J.; Watson, M.; Davies, S.; Bernard, P.S.; Parker, J.S.; et al. Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. J. Natl. Cancer Inst. 2009, 101, 736–750. [Google Scholar] [CrossRef]
- Smid, M.; Wang, Y.; Zhang, Y.; Sieuwerts, A.M.; Yu, J.; Klijn, J.G.M.; Foekens, J.A.; Martens, J.W.M. Subtypes of breast cancer show preferential site of relapse. Cancer Res. 2008, 68, 3108–3114. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Liu, Y.R.; Ji, P.; Hu, X.; Shao, Z.M. Impact of molecular subtypes on metastatic breast cancer patients: A SEER population-based study. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Perou, C.M.; Sørile, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; Ress, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef]
- Van Ramshorst, M.S.; van der Voort, A.; van Werkhoven, E.D.; Mandjes, I.A.; Kemper, I.; Dezentjé, V.O.; Oving, I.M.; Honkoop, A.H.; Tick, L.W.; van de Wouw, A.J.; et al. Neoadjuvant chemotherapy with or without anthracyclines in the presence of dual HER2 blockade for HER2-positive breast cancer (TRAIN-2): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1630–1640. [Google Scholar] [CrossRef]
- Harbeck, N.; Gluz, O. Neoadjuvant therapy for triple negative and HER2-positive early breast cancer. Breast 2017, 34, S99–S103. [Google Scholar] [CrossRef]
- Wuerstlein, R.; Harbeck, N. Neoadjuvant Therapy for HER2-positive Breast Cancer. Rev. Recent Clin. Trials 2017, 12, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Maximiano, S.; Magalhães, P.; Guerreiro, M.P.; Morgado, M. Trastuzumab in the Treatment of Breast Cancer. BioDrugs 2016, 30, 75–86. [Google Scholar] [CrossRef]
- Cameron, D.; Piccart-Gebhart, M.J.; Gelber, R.D.; Procter, M.; Goldhirsch, A.; de Azambuja, E.; Castro, G.; Untch, M.; Smith, I.; Gianni, L.; et al. 11 years’ follow-up of trastuzumab after adjuvant chemotherapy in HER2-positive early breast cancer: Final analysis of the HERceptin Adjuvant (HERA) trial. Lancet 2017, 389, 1195–1205. [Google Scholar] [CrossRef]
- Drooger, J.C.; van Tinteren, H.; de Groot, S.M.; ten Tije, A.J.; de Graaf, H.; Portielje, J.E.A.; Jager, A.; Honkoop, A.; Linn, S.C.; Kroep, J.R.; et al. A randomized phase 2 study exploring the role of bevacizumab and a chemotherapy-free approach in HER2-positive metastatic breast cancer: The HAT study (BOOG 2008–2003), a Dutch Breast Cancer Research Group trial. Cancer 2016, 122, 2961–2970. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nagata, Y.; Lan, K.H.; Zhou, X.; Tan, M.; Esteva, F.J.; Sahin, A.A.; Klos, K.S.; Li, P.; Monia, B.P.; Nguyen, N.T.; et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 2004, 6, 117–127. [Google Scholar] [CrossRef]
- Marotti, J.D.; de Abreu, F.B.; Wells, W.A.; Tsongalis, G.J. Triple-Negative Breast Cancer: Next-Generation Sequencing for Target Identification. Am. J. Pathol. 2017, 187, 2133–2138. [Google Scholar] [CrossRef]
- Jitariu, A.A.; Cîmpean, A.M.; Ribatti, D.; Raica, M. Triple negative breast cancer: The kiss of death. Oncotarget 2017, 8, 46652–46662. [Google Scholar] [CrossRef] [PubMed]
- Jhan, J.R.; Andrechek, E.R. Triple-negative breast cancer and the potential for targeted therapy. Pharmacogenomics 2017, 18, 1595–1609. [Google Scholar] [CrossRef] [PubMed]
- Chun, K.H.; Park, J.H.; Fan, S. Predicting and overcoming chemotherapeutic resistance in breast cancer. In Advances in Experimental Medicine and Biology; Springer LLC: New York, NY, USA, 2017; Volume 1026, pp. 59–104. [Google Scholar]
- Reilly, D.O.; al Sendi, M.; Kelly, C.M. Overview of recent advances in metastatic triple negative breast cancer. World J. Clin. Oncol. 2021, 12, 164–182. [Google Scholar] [CrossRef]
- Mezi, S.; Botticelli, A.; Pomati, G.; Cerbelli, B.; Scagnoli, S.; Amirhassankhani, S.; D’amati, G.; Marchetti, P. Standard of care and promising new agents for the treatment of mesenchymal triple-negative breast cancer. Cancers 2021, 13, 1080. [Google Scholar] [CrossRef] [PubMed]
- Loibl, S.; Poortmans, P.; Morrow, M.; Denkert, C.; Curigliano, G. Breast cancer. Lancet 2021. [Google Scholar] [CrossRef]
- Liu, D.; Vadgama, J.; Wu, Y. Basal-like breast cancer with low TGFβ and high TNFα pathway activity is rich in activated memory CD4 T cells and has a good prognosis. Int. J. Biol. Sci. 2021, 17, 670–682. [Google Scholar] [CrossRef]
- Cejalvo, J.M.; De Dueñas, E.M.; Galván, P.; García-Recio, S.; Gasión, O.B.; Paré, L.; Antolín, S.; Martinello, R.; Blancas, I.; Adamo, B.; et al. Intrinsic subtypes and gene expression profiles in primary and metastatic breast cancer. Cancer Res. 2017, 77, 2213–2221. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, X.S.; Zhang, S. Breast tumor subgroups reveal diverse clinical prognostic power. Sci. Rep. 2014, 4, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Riaz, S.K.; Saeed, M.; Malik, M.F.A. Clinical and therapeutic implications of histone acetylation in breast cancer. West Indian Med. J. 2016, 65, 337–344. [Google Scholar]
- Guo, P.; Chen, W.; Li, H.; Li, M.; Li, L. The Histone Acetylation Modifications of Breast Cancer and their Therapeutic Implications. Pathol. Oncol. Res. 2018, 24, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Ediriweera, M.K.; Tennekoon, K.H.; Samarakoon, S.R. Emerging role of histone deacetylase inhibitors as anti-breast-cancer agents. Drug Discov. Today 2019, 24, 685–702. [Google Scholar] [CrossRef]
- Garmpis, N.; Damaskos, C.; Garmpi, A.; Kalampokas, E.; Kalampokas, T.; Spartalis, E.; Daskalopoulou, A.; Valsami, S.; Kontos, M.; Nonni, A.; et al. Histone deacetylases as new therapeutic targets in triple-negative breast cancer: Progress and promises. Cancer Genom. Proteom. 2017, 14, 299–313. [Google Scholar]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA Approval Summary: Vorinostat for Treatment of Advanced Primary Cutaneous T-Cell Lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Eleutherakis-Papaiakovou, E.; Kanellias, N.; Kastritis, E.; Gavriatopoulou, M.; Terpos, E.; Dimopoulos, M.A. Efficacy of Panobinostat for the Treatment of Multiple Myeloma. J. Oncol. 2020. [Google Scholar] [CrossRef]
- Iyer, S.P.; Foss, F.F. Romidepsin for the Treatment of Peripheral T-Cell Lymphoma. Oncologist 2015, 20, 1084–1091. [Google Scholar] [CrossRef]
- Lee, H.Z.; Kwitkowski, V.E.; Del Valle, P.L.; Ricci, M.S.; Saber, H.; Habtemariam, B.A.; Bullock, J.; Bloomquist, E.; Shen, Y.L.; Chen, X.H.; et al. FDA approval: Belinostat for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma. Clin. Cancer Res. 2015, 21, 2666–2670. [Google Scholar] [CrossRef] [PubMed]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Spange, S.; Wagner, T.; Heinzel, T.; Krämer, O.H. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int. J. Biochem. Cell Biol. 2009, 41, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Leng, Y.; Tsai, L.K.; Leeds, P.; Chuang, D.M. Valproic acid attenuates blood-brain barrier disruption in a rat model of transient focal cerebral ischemia: The roles of HDAC and MMP-9 inhibition. J. Cereb. Blood Flow Metab. 2011, 31, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Jang, E.R.; Lim, S.J.; Lee, E.S.; Jeong, G.; Kim, T.Y.; Bang, Y.J.; Lee, J.S. The histone deacetylase inhibitor trichostatin a sensitizes estrogen receptor α-negative breast cancer cells to tamoxifen. Oncogene 2004, 23, 1724–1736. [Google Scholar] [CrossRef]
- Chen, S.Y.; Zheng, X.W.; Cai, J.X.; Zhang, W.P.; You, H.S.; Xing, J.F.; Dong, Y.L. Histone deacetylase inhibitor reverses multidrug resistance by attenuating the nucleophosmin level through PI3K/Akt pathway in breast cancer. Int. J. Oncol. 2016, 49, 294–304. [Google Scholar] [CrossRef][Green Version]
- Lipska, K.; Gumieniczek, A.; Filip, A.A. Anticonvulsant valproic acid and other short-chain fatty acids as novel anticancer therapeutics: Possibilities and challenges. Acta Pharm. 2020, 70, 291–301. [Google Scholar] [CrossRef]
- Romoli, M.; Mazzocchetti, P.; D’Alonzo, R.; Siliquini, S.; Rinaldi, V.E.; Verrotti, A.; Calabresi, P.; Costa, C. Valproic Acid and Epilepsy: From Molecular Mechanisms to Clinical Evidences. Curr. Neuropharmacol. 2018, 17, 926–946. [Google Scholar] [CrossRef] [PubMed]
- Sanaei, M.; Kavoosi, F. Effect of Valproic Acid on the Class I Histone Deacetylase 1, 2 and 3, Tumor Suppressor Genes p21WAF1/CIP1 and p53, and Intrinsic Mitochondrial Apoptotic Pathway, Pro- (Bax, Bak, and Bim) and anti- (Bcl-2, Bcl-xL, and Mcl-1) Apoptotic Genes Expression, Ce. Asian Pacific J. Cancer Prev. 2021, 22, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Pang, B.; Zhang, J.; Zhang, X.; Yuan, J.; Shi, Y.; Qiao, L. Inhibition of lipogenesis and induction of apoptosis by valproic acid in prostate cancer cells via the C/EBPα/SREBP-1 pathway. Acta Biochim. Biophys. Sin. 2021, 53, 354–364. [Google Scholar] [CrossRef]
- Jahani, M.; Khanahmad, H.; Nikpour, P. Evaluation of the Effects of Valproic Acid Treatment on Cell Survival and Epithelial-Mesenchymal Transition-Related Features of Human Gastric Cancer Cells. J. Gastrointest. Cancer 2020. [Google Scholar] [CrossRef]
- Injinari, N.; Amini-Farsani, Z.; Yadollahi-Farsani, M.; Teimori, H. Apoptotic effects of valproic acid on miR-34a, miR-520h and HDAC1 gene in breast cancer. Life Sci. 2021, 269, 119027. [Google Scholar] [CrossRef]
- Ozman, Z.; Ozbek Iptec, B.; Sahin, E.; Guney Eskiler, G.; Deveci Ozkan, A.; Kaleli, S. Regulation of valproic acid induced EMT by AKT/GSK3β/β-catenin signaling pathway in triple negative breast cancer. Mol. Biol. Rep. 2021, 48, 1335–1343. [Google Scholar] [CrossRef] [PubMed]
- Wawruszak, A.; Luszczki, J.J.; Kalafut, J.; Okla, K.; Halasa, M.; Rivero-Muller, A.; Stepulak, A. Additive pharmacological interaction between cisplatin (CDDP) and histone deacetylase inhibitors (HDIs) in MDA-MB-231 triple negative breast cancer (TNBC) cells with altered notch1 activity—an isobolographic analysis. Int. J. Mol. Sci. 2019, 20, 3663. [Google Scholar] [CrossRef]
- Wawruszak, A.; Luszczki, J.J.; Grabarska, A.; Gumbarewicz, E.; Dmoszynska-Graniczka, M.; Polberg, K.; Stepulak, A. Assessment of interactions between cisplatin and two histone deacetylase inhibitors in MCF7, T47D and MDA-MB-231 human breast cancer cell lines—An isobolographic analysis. PLoS ONE 2015, 10, e0143013. [Google Scholar] [CrossRef]
- Fortunati, N.; Bertino, S.; Costantino, L.; Bosco, O.; Vercellinatto, I.; Catalano, M.G.; Boccuzzi, G. Valproic acid is a selective antiproliferative agent in estrogen-sensitive breast cancer cells. Cancer Lett. 2008, 259, 156–164. [Google Scholar] [CrossRef]
- Aztopal, N.; Erkisa, M.; Erturk, E.; Ulukaya, E.; Tokullugil, A.H.; Ari, F. Valproic acid, a histone deacetylase inhibitor, induces apoptosis in breast cancer stem cells. Chem. Biol. Interact. 2018, 280, 51–58. [Google Scholar] [CrossRef]
- Mawatari, T.; Ninomiya, I.; Inokuchi, M.; Harada, S.; Hayashi, H.; Oyama, K.; Makino, I.; Nakagawara, H.; Miyashita, T.; Tajima, H.; et al. Valproic acid inhibits proliferation of HER2-expressing breast cancer cells by inducing cell cycle arrest and apoptosis through Hsp70 acetylation. Int. J. Oncol. 2015, 47, 2073–2081. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Tang, Z.; Qing, B.; Tang, R.; Duan, Q.; Ding, S.; Deng, D. Valproic acid promotes the epithelial-to-mesenchymal transition of breast cancer cells through stabilization of Snail and transcriptional upregulation of Zeb1. Eur. J. Pharmacol. 2019, 865. [Google Scholar] [CrossRef]
- Wawruszak, A.; Gumbarewicz, E.; Okon, E.; Jeleniewicz, W.; Czapinski, J.; Halasa, M.; Okla, K.; Smok-Kalwat, J.; Bocian, A.; Rivero-Muller, A.; et al. Histone deacetylase inhibitors reinforce the phenotypical markers of breast epithelial or mesenchymal cancer cells but inhibit their migratory properties. Cancer Manag. Res. 2019, 11, 8345–8358. [Google Scholar] [CrossRef]
- Li, G.F.; Qian, T.L.; Li, G.S.; Yang, C.X.; Qin, M.; Huang, J.; Sun, M.; Han, Y.Q. Sodium valproate inhibits MDA-MB-231 breast cancer cell migration by upregulating NM23H1 expression. Genet. Mol. Res. 2012, 11, 77–86. [Google Scholar] [CrossRef]
- Fortunati, N.; Bertino, S.; Costantino, L.; De Bortoli, M.; Compagnone, A.; Bandino, A.; Catalano, M.G.; Boccuzzi, G. Valproic acid restores ERα and antiestrogen sensitivity to ERα-negative breast cancer cells. Mol. Cell. Endocrinol. 2010, 314, 17–22. [Google Scholar] [CrossRef]
- Zhou, X.; Li, Z.; Wang, X.; Jiang, G.; Shan, C.; Liu, S. Metabolomics reveals the effect of valproic acid on MCF-7 and MDA-MB-231 cells. Xenobiotica 2020, 50, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Zhang, C.; Cao, F.-Y.; Cao, F.; Jiang, L.; Ding, H.Z. Prognostic and clinicopathological value of NM23 expression in patients with breast cancer: A systematic review and meta-analysis. Curr. Probl. Cancer 2017, 41, 80–93. [Google Scholar] [CrossRef]
- Mátyási, B.; Farkas, Z.; Kopper, L.; Sebestyén, A.; Boissan, M.; Mehta, A.; Takács-Vellai, K. The Function of NM23-H1/NME1 and Its Homologs in Major Processes Linked to Metastasis. Pathol. Oncol. Res. 2020, 26, 49–61. [Google Scholar] [CrossRef]
- Das, V.; Bhattacharya, S.; Chikkaputtaiah, C.; Hazra, S.; Pal, M. The basics of epithelial–mesenchymal transition (EMT): A study from a structure, dynamics, and functional perspective. J. Cell. Physiol. 2019, 234, 14535–14555. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; San Juan, B.P.; Lim, E.; Weinberg, R.A. EMT, cell plasticity and metastasis. Cancer Metastasis Rev. 2016, 35, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Kotiyal, S.; Bhattacharya, S. Breast cancer stem cells, EMT and therapeutic targets. Biochem. Biophys. Res. Commun. 2014, 453, 112–116. [Google Scholar] [CrossRef]
- Song, K.; Farzaneh, M. Signaling pathways governing breast cancer stem cells behavior. Stem Cell Res. Ther. 2021, 12, 245. [Google Scholar] [CrossRef]
- Kaszak, I.; Witkowska-Piłaszewicz, O.; Niewiadomska, Z.; Dworecka-Kaszak, B.; Toka, F.N.; Jurka, P. Role of cadherins in cancer—A review. Int. J. Mol. Sci. 2020, 21, 7624. [Google Scholar] [CrossRef] [PubMed]
- Kar, R.; Jha, N.K.; Jha, S.K.; Sharma, A.; Dholpuria, S.; Asthana, N.; Chaurasiya, K.; Singh, V.K.; Burgee, S.; Nand, P. A “NOTCH” deeper into the epithelial-to-mesenchymal transition (EMT) program in breast cancer. Genes 2019, 10, 961. [Google Scholar] [CrossRef] [PubMed]
- Nawshad, A.; Lagamba, D.; Hay, E.D. Transforming growth factor β (TGFβ) signalling in palatal growth, apoptosis and epithelial mesenchymal transformation (EMT). Arch. Oral Biol. 2004, 49, 675–689. [Google Scholar] [CrossRef] [PubMed]
- Santibanez, J.F.; Obradović, H.; Kukolj, T.; Krstić, J. Transforming growth factor-β, matrix metalloproteinases, and urokinase-type plasminogen activator interaction in the cancer epithelial to mesenchymal transition. Dev. Dyn. 2018, 247, 382–395. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, R.K.; Varshney, A.K.; Yadava, P.K. Diversity and functional evolution of the plasminogen activator system. Biomed. Pharmacother. 2018, 98, 886–898. [Google Scholar] [CrossRef]
- Makena, M.R.; Gatla, H.; Verlekar, D.; Sukhavasi, S.; Pandey, M.K.; Pramanik, K.C. Wnt/β-catenin signaling: The culprit in pancreatic carcinogenesis and therapeutic resistance. Int. J. Mol. Sci. 2019, 20, 4242. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Chen, L.; Zhang, G.; Shan, A.; Ye, C.; Liang, B.; Sun, J.; Liao, X.; Zhu, C.; Chen, Y.; et al. MicroRNAs target the Wnt/β-catenin signaling pathway to regulate epithelial-mesenchymal transition in cancer. Oncol. Rep. 2020, 44, 1299–1313. [Google Scholar] [CrossRef]
- Su, C.M.; Wang, M.Y.; Hong, C.C.; Chen, H.A.; Su, Y.H.; Wu, C.H.; Huang, M.T.; Chang, Y.W.; Jiang, S.S.; Sung, S.Y.; et al. MiR-520h is crucial for DAPK2 regulation and breast cancer progression. Oncogene 2016, 35, 1134–1142. [Google Scholar] [CrossRef]
- Hussen, B.M.; Shoorei, H.; Mohaqiq, M.; Dinger, M.E.; Hidayat, H.J.; Taheri, M.; Ghafouri-Fard, S. The Impact of Non-coding RNAs in the Epithelial to Mesenchymal Transition. Front. Mol. Biosci. 2021, 8. [Google Scholar] [CrossRef]
- Pan, G.; Liu, Y.; Shang, L.; Zhou, F.; Yang, S. EMT-associated microRNAs and their roles in cancer stemness and drug resistance. Cancer Commun. 2021, 41, 199–217. [Google Scholar] [CrossRef] [PubMed]
- Dong, B.; Li, S.; Zhu, S.; Yi, M.; Luo, S.; Wu, K. MiRNA-mediated EMT and CSCs in cancer chemoresistance. Exp. Hematol. Oncol. 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.; Cho, J.G.; Yun, E.; Lee, A.; Ryu, H.Y.; Lee, Y.J.; Yoon, S.; Chang, W.; Lee, M.S.; Kwon, B.S.; et al. MicroRNA 34A–AXL axis regulates vasculogenic mimicry formation in breast cancer cells. Genes 2021, 12, 9. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Ren, Q.; Zuo, W.; Jia, R.; Xie, L.; Lin, R.; Zhao, H.; Chen, J.; Lei, Y.; Wang, P.; et al. Valproic acid exhibits anti-tumor activity selectively against EGFR/ErbB2/ErbB3-coexpressing pancreatic cancer via induction of ErbB family members-targeting microRNAs. J. Exp. Clin. Cancer Res. 2019, 38. [Google Scholar] [CrossRef]
- Hu, S.; Liu, L.; Chang, E.B.; Wang, J.Y.; Raufman, J.P. Butyrate inhibits pro-proliferative miR-92a by diminishing c-Myc-induced miR-17-92a cluster transcription in human colon cancer cells. Mol. Cancer 2015, 14. [Google Scholar] [CrossRef]
- Bellissimo, T.; Ganci, F.; Gallo, E.; Sacconi, A.; Tito, C.; De Angelis, L.; Pulito, C.; Masciarelli, S.; Diso, D.; Anile, M.; et al. Thymic Epithelial Tumors phenotype relies on miR-145-5p epigenetic regulation. Mol. Cancer 2017, 16. [Google Scholar] [CrossRef]
- Rücker, F.G.; Lang, K.M.; Fütterer, M.; Komarica, V.; Schmid, M.; Döhner, H.; Schlenk, R.F.; Döhner, K.; Knudsen, S.; Bullinger, L. Molecular dissection of valproic acid effects in acute myeloid leukemia identifies predictive networks. Epigenetics 2016, 11, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Wang, J.; Zheng, R.; Song, B.; Huang, L.; Liu, Y.; Hao, Y.; Bai, X. MiR-133 Targets YES1 and Inhibits the Growth of Triple-Negative Breast Cancer Cells. Technol. Cancer Res. Treat. 2020, 19. [Google Scholar] [CrossRef]
- Shen, Y.; Xu, Y.; Huang, L.; Chi, Y.; Meng, L. MiR-205 suppressed the malignant behaviors of breast cancer cells by targeting CLDN11 via modulation of the epithelial-to-mesenchymal transition. Aging 2021, 13. [Google Scholar] [CrossRef]
- Chernyi, V.S.; Tarasova, P.V.; Kozlov, V.V.; Saik, O.V.; Kushlinskii, N.E.; Gulyaeva, L.F. Search of MicroRNAs Regulating the Receptor Status of Breast Cancer In Silico and Experimental Confirmation of Their Expression in Tumors. Bull. Exp. Biol. Med. 2017, 163, 655–659. [Google Scholar] [CrossRef] [PubMed]
- Cun, J.; Yang, Q. Bioinformatics-based interaction analysis of miR-92a-3p and key genes in tamoxifen-resistant breast cancer cells. Biomed. Pharmacother. 2018, 107, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Zhang, X.; Tan, W.; Gao, J.; Pan, L.; Ye, X.; Chen, L.; Zheng, W. miR-145-5p Suppresses Breast Cancer Progression by Inhibiting SOX2. J. Surg. Res. 2019, 236, 278–287. [Google Scholar] [CrossRef]
- Chaudhary, S.; Islam, Z.; Mishra, V.; Rawat, S.; Ashraf, G.M.; Kolatkar, P.R. Sox2: A Regulatory Factor in Tumorigenesis and Metastasis. Curr. Protein Pept. Sci. 2019, 20, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.; Garikapati, K.R.; Ramaiah, M.J.; Polavarapu, K.K.; Bhadra, U.; Bhadra, M.P. miR-15a/miR-16 induces mitochondrial dependent apoptosis in breast cancer cells by suppressing oncogene BMI1. Life Sci. 2016, 164, 60–70. [Google Scholar] [CrossRef]
- Heers, H.; Stanislaw, J.; Harrelson, J.; Lee, M.W. Valproic acid as an adjunctive therapeutic agent for the treatment of breast cancer. Eur. J. Pharmacol. 2018, 835, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Olsen, C.M.; Meussen-Elholm, E.T.M.; Røste, L.S.; Taubøll, E. Antiepileptic drugs inhibit cell growth in the human breast cancer cell line MCF7. Mol. Cell. Endocrinol. 2004, 213, 173–179. [Google Scholar] [CrossRef]
- Tayanloo-Beik, A.; Sarvari, M.; Payab, M.; Gilany, K.; Alavi-Moghadam, S.; Gholami, M.; Goodarzi, P.; Larijani, B.; Arjmand, B. OMICS insights into cancer histology; Metabolomics and proteomics approach. Clin. Biochem. 2020, 84, 13–20. [Google Scholar] [CrossRef]
- Sarin, N.; Engel, F.; Rothweiler, F.; Cinatl, J.; Michaelis, M.; Frötschl, R.; Fröhlich, H.; Kalayda, G. Key Players of Cisplatin Resistance: Towards a Systems Pharmacology Approach. Int. J. Mol. Sci. 2018, 19, 767. [Google Scholar] [CrossRef] [PubMed]
- Al-malky, H.S.; Al Harthi, S.E.; Osman, A.M.M. Major obstacles to doxorubicin therapy: Cardiotoxicity and drug resistance. J. Oncol. Pharm. Pract. 2020, 26, 434–444. [Google Scholar] [CrossRef]
- Alfarouk, K.O.; Stock, C.M.; Taylor, S.; Walsh, M.; Muddathir, A.K.; Verduzco, D.; Bashir, A.H.H.; Mohammed, O.Y.; Elhassan, G.O.; Harguindey, S.; et al. Resistance to cancer chemotherapy: Failure in drug response from ADME to P-gp. Cancer Cell Int. 2015, 15, 71. [Google Scholar] [CrossRef] [PubMed]
- Okon, E.; Luszczki, J.J.; Kukula-Koch, W.; Halasa, M.; Jarzab, A.; Khurelbat, D.; Stepulak, A.; Wawruszak, A. Synergistic or Additive Pharmacological Interactions between Magnoflorine and Cisplatin in Human Cancer Cells of Different Histological Origin. Int. J. Mol. Sci. 2020, 21, 2848. [Google Scholar] [CrossRef] [PubMed]
- Kukula-Koch, W.; Grabarska, A.; Łuszczki, J.; Czernicka, L.; Nowosadzka, E.; Gumbarewicz, E.; Jarząb, A.; Audo, G.; Upadhyay, S.; Głowniak, K.; et al. Superior anticancer activity is demonstrated by total extract of Curcuma longa L. as opposed to individual curcuminoids separated by centrifugal partition chromatography. Phyther. Res. 2018, 32, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Wróblewska-łuczka, P.; Grabarska, A.; Łuszczki, J.J.; Florek-łuszczki, M.; Plewa, Z. Synergy, additivity, and antagonism between cisplatin and selected coumarins in human melanoma cells. Int. J. Mol. Sci. 2021, 22, 537. [Google Scholar] [CrossRef] [PubMed]
- Jarzab, A.; Luszczki, J.; Guz, M.; Skalicka-Wozniak, K.; Halasa, M.; Smok-Kalwat, J.; Polberg, K.; Stepulak, A. Combination of osthole and cisplatin against rhabdomyosarcoma TE671 cells yielded additive pharmacologic interaction by means of isobolographic analysis. Anticancer Res. 2018, 38, 205–210. [Google Scholar] [PubMed]
- Dasari, S.; Bernard Tchounwou, P. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Minegaki, T.; Suzuki, A.; Mori, M.; Tsuji, S.; Yamamoto, S.; Watanabe, A.; Tsuzuki, T.; Tsunoda, T.; Yamamoto, A.; Tsujimoto, M.; et al. Histone deacetylase inhibitors sensitize 5-fluorouracil-resistant MDA-MB-468 breast cancer cells to 5-fluorouracil. Oncol. Lett. 2018, 16, 6202–6208. [Google Scholar] [CrossRef]
- Terranova-Barberio, M.; Roca, M.S.; Zotti, A.I.; Leone, A.; Bruzzese, F.; Vitagliano, C.; Scogliamiglio, G.; Russo, D.; D’Angelo, G.; Franco, R.; et al. Valproic acid potentiates the anticancer activity of capecitabine in vitro and in vivo in breast cancer models via induction of thymidine phosphorylase expression. Oncotarget 2016, 7, 7715–7731. [Google Scholar] [CrossRef]
- Arakawa, Y.; Saito, S.; Yamada, H.; Aiba, K. Simultaneous treatment with camptothecin and valproic acid suppresses induction of Bcl-XL and promotes apoptosis of MCF-7 breast cancer cells. Apoptosis 2009, 14, 1076–1085. [Google Scholar] [CrossRef]
- Phillips, S.L.; Williams, C.B.; Zambrano, J.N.; Williams, C.J.; Yeh, E.S. Connexin 43 in the development and progression of breast cancer: What’s the connection? Int. J. Oncol. 2017, 51, 1005–1013. [Google Scholar] [CrossRef]
- Sargazi, S.; Kooshkaki, O.; Zavar Reza, J.; Saravani, R.; Zarei Jaliani, H.; Mirinejad, S.; Meshkini, F. Mild antagonistic effect of Valproic acid in combination with AZD2461 in MCF-7 breast cancer cells. Med. J. Islamic Repub. Iran 2019, 33, 29. [Google Scholar] [CrossRef]
- Soldi, R.; Cohen, A.L.; Cheng, L.; Sun, Y.; Moos, P.J.; Bild, A.H. A genomic approach to predict synergistic combinations for breast cancer treatment. Pharm. J. 2013, 13, 94–104. [Google Scholar] [CrossRef]
- Ponce-Cusi, R.; Calaf, G.M. Apoptotic activity of 5-fluorouracil in breast cancer cells transformed by low doses of ionizing α-particle radiation. Int. J. Oncol. 2016, 48, 774–782. [Google Scholar] [CrossRef] [PubMed]
- Varshavsky-Yanovsky, A.N.; Goldstein, L.J. Role of Capecitabine in Early Breast Cancer. J. Clin. Oncol. 2020, 38, 179–182. [Google Scholar] [CrossRef]
- Zunino, F.; Pratesi, G. Camptothecins in clinical development. Expert Opin. Investig. Drugs 2004, 13, 269–284. [Google Scholar] [CrossRef]
- Ulukan, H.; Swaan, P.W. Camptothecins: A review of their chemotherapeutic potential. Drugs 2002, 62, 2039–2057. [Google Scholar] [CrossRef]
- Lovitt, C.J.; Shelper, T.B.; Avery, V.M. Doxorubicin resistance in breast cancer cells is mediated by extracellular matrix proteins. BMC Cancer 2018, 18. [Google Scholar] [CrossRef] [PubMed]
- Christowitz, C.; Davis, T.; Isaacs, A.; Van Niekerk, G.; Hattingh, S.; Engelbrecht, A.M. Mechanisms of doxorubicin-induced drug resistance and drug resistant tumour growth in a murine breast tumour model. BMC Cancer 2019, 19, 757. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.H.; Zheng, C.; Jiang, G.J.; Dong, S.Y. Sodium valproate enhances doxorubicin cytotoxicity in breast cancer cells in vitro. Nan Fang Yi Ke Da Xue Xue Bao 2015, 35, 62–65. Available online: https://pubmed.ncbi.nlm.nih.gov/25613611/ (accessed on 21 April 2021).
- Muñoz, A.M.; Fragoso-Vázquez, M.J.; Martel, B.P.; Chávez-Blanco, A.; Dueñas-González, A.; García-Sánchez, J.R.; Bello, M.; Romero-Castro, A.; Correa-Basurto, J. Targeting Breast Cancer Cells with G4 PAMAM Dendrimers and Valproic Acid Derivative Complexes. Anticancer Agents Med. Chem. 2020, 20, 1857–1872. [Google Scholar] [CrossRef]
- Ribociclib&Belinostat in Patients W Metastatic Triple Neg Breast Cancer & Recurrent Ovarian Cancer W Response Prediction by Genomics. Available online: https://clinicaltrials.gov/ct2/show/NCT04315233?term=Histone+deacetylase+inhibitor&cond=Breast+Cancer&draw=3&rank=5 (accessed on 17 June 2021).
- Pembrolizumab and Tamoxifen with or without Vorinostat for the Treatment of Estrogen Receptor Positive Breast Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT04190056?term=Histone+deacetylase+inhibitor&cond=Breast+Cancer&draw=3 (accessed on 17 June 2021).
- A Pilot Study of the Combination of Entinostat With Capecitabine in High Risk Breast Cancer After Neo-adjuvant Therapy. Available online: https://clinicaltrials.gov/ct2/show/NCT03473639?term=Histone+deacetylase+inhibitor&cond=Breast+Cancer&draw=3&rank=36 (accessed on 17 June 2021).
- Talazoparib in Combination With Belinostat for Metastatic Breast Cancer, Metastatic Castration Resistant Prostate Cancer, and Metastatic Ovarian Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT04703920?term=Histone+deacetylase+inhibitor&cond=Breast+Cancer&draw=3&rank=32 (accessed on 17 June 2021).
- BN-Brachyury, Entinostat, Adotrastuzumab Emtansine and M7824 in Advanced Stage Breast Cancer (BrEAsT). Available online: https://clinicaltrials.gov/ct2/show/NCT04296942?term=Histone+deacetylase+inhibitor&cond=Breast+Cancer&draw=3&rank=17 (accessed on 17 June 2021).
- Olaparib in Combination With Vorinostat in Patients With Relapsed/Refractory and/or Metastatic Breast Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT03742245?term=Histone+deacetylase+inhibitor&cond=Breast+Cancer&draw=3&rank=11 (accessed on 17 June 2021).
- Münster, P.; Marchion, D.; Bicaku, E.; Schmitt, M.; Ji, H.L.; DeConti, R.; Simon, G.; Fishman, M.; Minton, S.; Garrett, C.; et al. Phase I trial of histone deacetylase inhibition by valproic acid followed by the topoisomerase II inhibitor epirubicin in advanced solid tumors: A clinical and translational study. J. Clin. Oncol. 2007, 25, 1979–1985. [Google Scholar] [CrossRef]
- Munster, P.; Marchion, D.; Bicaku, E.; Lacevic, M.; Kim, J.; Centeno, B.; Daud, A.; Neuger, A.; Minton, S.; Sullivan, D. Clinical and biological effects of valproic acid as a histone deacetylase inhibitor on tumor and surrogate tissues: Phase i/ii trial of valproic acid and epirubicin/FEC. Clin. Cancer Res. 2009, 15, 2488–2496. [Google Scholar] [CrossRef]
- Arce, C.; Pérez, C.; González-Fierro, A.; de la Cruz-Hernńdez, E.; Revilla-Vázquez, A.; Chávez-Blanco, A.; Trejo-Becerril, C.; Pérez-Cárdenas, E.; Taja-Chayeb, L.; Bargallo, E.; et al. A proof-of-principle study of epigenetics therapy added to neoadjuvant doxorubicin cyclophosphamide for locally advanced breast cancer. PLoS ONE 2006, 1, e98. [Google Scholar] [CrossRef] [PubMed]
- Bevacizumab and Temsirolimus Alone or in Combination With Valproic Acid or Cetuximab in Treating Patients With Advanced or Metastatic Malignancy or Other Benign Disease. Available online: https://clinicaltrials.gov/ct2/show/study/NCT01552434?term=valproic+acid+and+breast+cancer&draw=2&rank=7 (accessed on 17 June 2021).
- Fardi, M.; Solali, S.; Farshdousti Hagh, M. Epigenetic mechanisms as a new approach in cancer treatment: An updated review. Genes Dis. 2018, 5, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Souza, C.; Chatterji, B. HDAC Inhibitors as Novel Anti-Cancer Therapeutics. Recent Pat. Anticancer Drug Discov. 2015, 10, 145–162. [Google Scholar] [CrossRef]
- Diederich, M.; Chateauvieux, S.; Morceau, F.; Dicato, M. Molecular and therapeutic potential and toxicity of valproic acid. J. Biomed. Biotechnol. 2010, 2010. [Google Scholar] [CrossRef]
- Činčárová, L.; Zdráhal, Z.; Fajkus, J. New perspectives of valproic acid in clinical practice. Expert Opin. Investig. Drugs 2013, 22, 1535–1547. [Google Scholar] [CrossRef]
- Goyal, J.; Rodriguez, R. Evidence from clinical trials for the use of valproic acid in solid tumors: Focus on prostate cancer. Clin. Investig. 2013, 3, 467–478. [Google Scholar] [CrossRef]
- Duenas-Gonzalez, A.; Candelaria, M.; Perez-Plascencia, C.; Perez-Cardenas, E.; de la Cruz-Hernandez, E.; Herrera, L.A. Valproic acid as epigenetic cancer drug: Preclinical, clinical and transcriptional effects on solid tumors. Cancer Treat. Rev. 2008, 34, 206–222. [Google Scholar] [CrossRef]
- Ponzano, A.; Tiboni, G.M. Teratology of valproic acid: An updated review of the possible mediating mechanisms. Minerva Ginecol. 2018, 70, 303–322. [Google Scholar]
- Meisel, J.L.; Venur, V.A.; Gnant, M.; Carey, L. Evolution of Targeted Therapy in Breast Cancer: Where Precision Medicine Began. Am. Soc. Clin. Oncol. Educ. B 2018, 38, 78–86. [Google Scholar] [CrossRef]
- Dhritlahre, R.K.; Saneja, A. Recent advances in HER2-targeted delivery for cancer therapy. Drug Discov. Today 2020. [Google Scholar] [CrossRef]
- Mitsogianni, M.; Trontzas, I.P.; Gomatou, G.; Ioannou, S.; Syrigos, N.K.; Kotteas, E.A. The changing treatment of metastatic her2-positive breast cancer. Oncol. Lett. 2021, 21. [Google Scholar] [CrossRef] [PubMed]
- Catalano, M.G.; Fortunati, N.; Pugliese, M.; Poli, R.; Bosco, O.; Mastrocola, R.; Aragno, M.; Boccuzzi, G. Valproic acid, a histone deacetylase inhibitor, enhances sensitivity to doxorubicin in anaplastic thyroid cancer cells. J. Endocrinol. 2006, 191, 465–472. [Google Scholar] [CrossRef] [PubMed]



 —stop).
—stop).
—stop).
—stop).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class of HDAC | HDAC | Zinc/Nicotinamide (NAD)-Dependent |
|---|---|---|
| I | HDAC1, 2, 3, 8 | zinc-dependent |
| II | IIa: HDAC4, 5, 7, 9 IIb: HDAC6, 10 | zinc-dependent |
| III | SIRT1-SIRT7 | NAD-dependent |
| IV | HDAC 11 | zinc-dependent |
| Class of HDIs | HDI | Abrreviation | FDA Approval for Cancer Treatment |
|---|---|---|---|
| Hydroxamic acids | Tichostatin A | TSA | Approved for CTCL treatment Approved for PTCLs treatment Approved for multiple myeloma treatment |
| Vorinostat | SAHA | ||
| Belinostat | PXD-101 | ||
| Panobinostat | LBH-589 | ||
| Resminostat | 4SC-201 | ||
| Short chain fatty acids | Sodium butyrate | NaB | |
| Phenylbutyrate | PBA | ||
| Valproic acid | VPA | ||
| Cyclic peptides | Apicidin | CAS183506-66-3 | Approved for PTCLs treatment |
| Romidepsin | FK228 | ||
| Benzamides | Mocetinostat | MGCD103 | |
| Entinostat | MS-275 | ||
| Domatinostat | 4SC-202 |
| Cellular Process | Sub-Type of BC | Cell Line | Mechanism of Action | References |
|---|---|---|---|---|
| Apoptosis | Luminal | MCF7 | ↑apoptosis (↑p21, ↑Bak, ↑Bax/Bcl-2 ratio, ↓Bcl-2 proteins expression, ↓telomerase activity) | [60] |
| MCF7 tem cells | ↑apoptosis (↑M30 protein expression, ↑caspase 3 and 7 activation, ↑nuclear pycnosis) | [61] | ||
| HER-2-overexpressed | SKBR3 | ↑apoptosis (↑cleaves caspase 3, ↑Hsp70 acetyaltion) | [62] | |
| TNBC | MDA-MB-231 | ↑apoptosis | [57] | |
| Cell cycle | Luminal | MCF7 | cell cycle arrest in sub-G1 phase | [60] |
| ZR-75-1 | cell cycle arrest in G1 phase | [60] | ||
| HER-2-overexpressed | SKBR3 | ↑p21WAF1 protein expression | [62] | |
| TNBC | MDA-MB-231 | cell cycle arrest in G0/G1 phase | [57] | |
| Migration | Luminal | MCF7 | ↑migration | [63] |
| MCF7 T47D | ↓migration | [64] | ||
| TNBC | MDA-MB-231 | ↓migration (↑nm23H1 gene expression) | [65] | |
| MDA-MB-231 MDA-MB-468 | ↓migration | [64] | ||
| MDA-MB-231 | ↑migration | [57,63] | ||
| EMT | Luminal | MCF7 | ↑EMT (↑Snail, ↑Zeb-2 genes expression) | [63] |
| MCF7 T47D | ↓EMT (↑E-cadherin gene and protein expresssion) | [64] | ||
| TNBC | MDA-MB-231 | ↑EMT (↑Snail, ↑Zeb-2 genes expression) | [63] | |
| ↑EMT (↑Snail, ↓E-cadherin, ↓GKS3β genes expression) | [57] | |||
| MDA-MB-468 | ↑EMT (↑N-cadherin gene and protein expression) | [64] | ||
| miRNA | Luminal | MCF7 | ↑miR-34a, ↑miR-520h expression | [56] |
| TNBC | MDA-MB-231 | ↑miR-34a, ↑miR-520h expression | [56] | |
| ER receptor status | TNBC | MDA-MB-231 | ↑ERα, ↑FoxA1 genes and proteins expression | [66] |
| Metabolic pathways | Luminal | MCF7 | ↑furfural expression, alteration in alanine, taurine and hypotaurine metabolism | [67] |
| Drug-Drug Combination | BC-Subtype | Cell Line | Mechanism of Action | Type of Pramacological Interaction | References |
|---|---|---|---|---|---|
| VPA+CDDP | Luminal | MCF7 | ↑apoptosis, ↓cell viability, cell cycle arrest | additivity | [59] |
| T47D | additivity with tendency towards synergism | [59] | |||
| TNBC | MDA-MB-231 | ↑apoptosis, ↓cell viability, cell cycle arrest | antagonism | [59] | |
| MDA-MB-231 with decreased and increased Notch1 activity | ↓cell viability | additivity | [58] | ||
| VPA+5-FU | TNBC | MDA-MB-468 | sensitization of BC cells insensitive to 5-FU | N/A | [108] |
| VPA+capecitabine | Luminal, HER2-overexpressed, TNBC | MCF7, SKBR3, MDA-MB-231, MDA-MB-468 | ↓proliferation, ↑apoptosis, ↑thymidine phosphorylase gene and protein expression | synergism, additivity | [109] |
| VPA+camptothecin | Luminal | MCF7 | ↑apoptosis (↓BcL-xl protein espression) | synergism (more than additivity) | [110] |
| VPA+DXR | TNBC | Hs578T | ↓viability, ↑cytotoxicity, ↑apoptosis, ↑Cx43 protein expression | N/A | [111] |
| VPA+AZD2461 (PARP1 inhibitor) | Luminal | MCF7 | ↓viability | mild antagonism | [112] |
| VPA+PD-033299 (CDK inhibitor) | Luminal, HER2-overexpressed, TNBC | Panel of BC cells (MCF7, T47D, BT474, MDA-MB-361, SKBR3, HCC1143, HCC38, HCC1806, BT483, BT549, MDA-MB-435, MDA-MB-453,) and 3D cultures from pleural effusion of patients | ↑apoptosis, cell cycle arrest, overexpression of CDKN1C gene | synergism | [113] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wawruszak, A.; Halasa, M.; Okon, E.; Kukula-Koch, W.; Stepulak, A. Valproic Acid and Breast Cancer: State of the Art in 2021. Cancers 2021, 13, 3409. https://doi.org/10.3390/cancers13143409
Wawruszak A, Halasa M, Okon E, Kukula-Koch W, Stepulak A. Valproic Acid and Breast Cancer: State of the Art in 2021. Cancers. 2021; 13(14):3409. https://doi.org/10.3390/cancers13143409
Chicago/Turabian StyleWawruszak, Anna, Marta Halasa, Estera Okon, Wirginia Kukula-Koch, and Andrzej Stepulak. 2021. "Valproic Acid and Breast Cancer: State of the Art in 2021" Cancers 13, no. 14: 3409. https://doi.org/10.3390/cancers13143409
APA StyleWawruszak, A., Halasa, M., Okon, E., Kukula-Koch, W., & Stepulak, A. (2021). Valproic Acid and Breast Cancer: State of the Art in 2021. Cancers, 13(14), 3409. https://doi.org/10.3390/cancers13143409