
NUAK1 and NUAK2 Fine-Tune TGF-β Signaling


Abstract
Simple Summary
Abstract
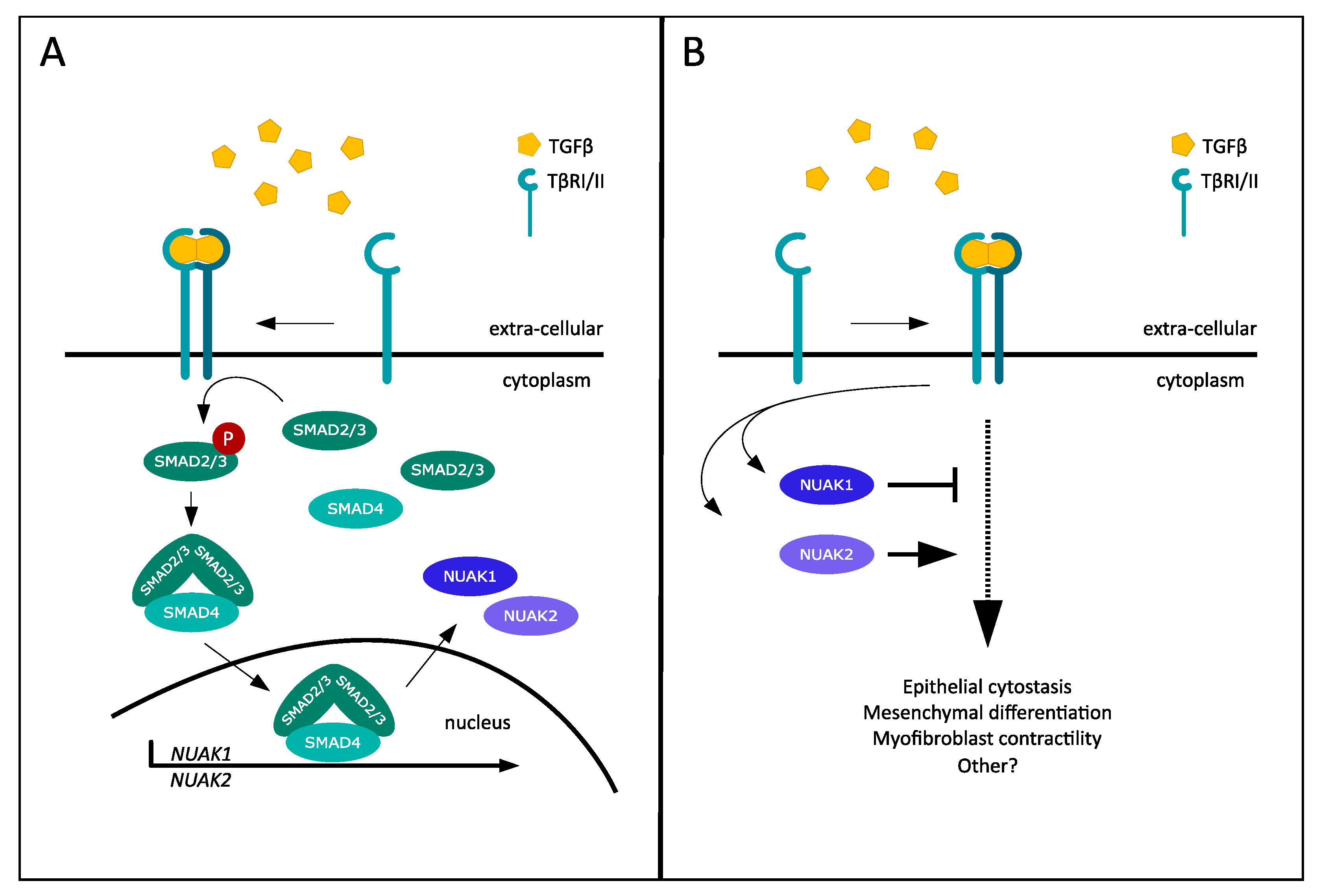
1. TGF-β Signaling, SMADs and Negative Feedback
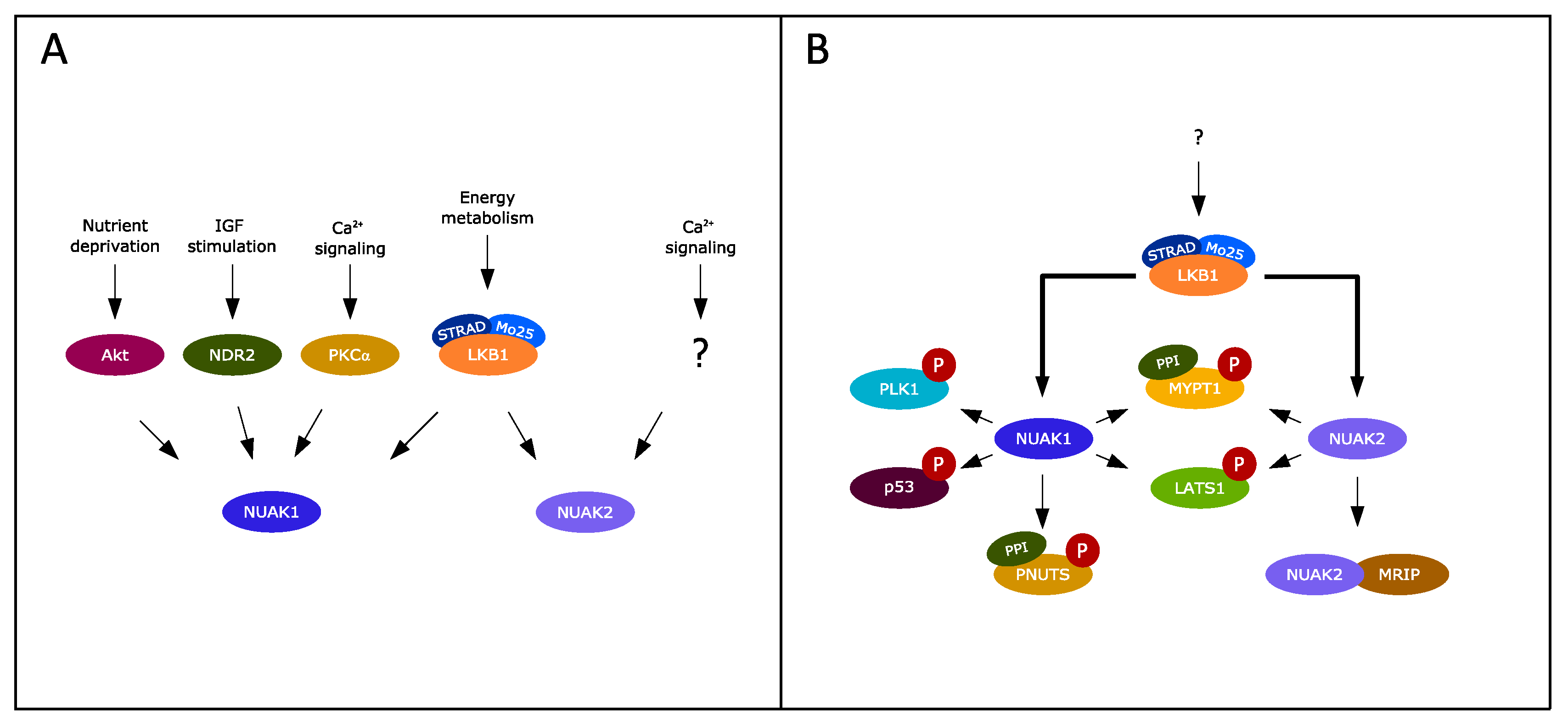
2. Exploring the NUAK Kinase Family
3. Opposing Roles of NUAK1 and NUAK2 in TGF-β Signaling
4. NUAKs at the Crossroads of TGF-β and LKB1 Signaling
5. Additional Dynamics of NUAK Kinase Function
6. Selective NUAK Inhibitors
7. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tzavlaki, K.; Moustakas, A. TGF-β Signaling. Biomolecules 2020, 10, 487. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Akhurst, R.J.; Hata, A. Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.-H.; Moustakas, A. Signaling Receptors for TGF-β Family Members. Cold Spring Harb. Perspect. Biol. 2016, 8, a022053. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Budi, E.H. Specificity, versatility, and control of TGF-β family signaling. Sci. Signal. 2019, 12, eaav5183. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Xiong, X.; Chen, Y.-G. Feedback regulation of TGF-β signaling. Acta Biochim. Biophys. Sin. 2018, 50, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.-M.; Kim, W.; Bae, E.; Gim, J.; Weist, B.M.; Jung, Y.; Hyun, J.-S.; Hernandez, J.B.; Leem, S.-H.; Park, T.; et al. DRAK2 Participates in a Negative Feedback Loop to Control TGF-β/Smads Signaling by Binding to Type I TGF-β Receptor. Cell Rep. 2012, 2, 1286–1299. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kowanetz, M.; Lönn, P.; Vanlandewijck, M.; Kowanetz, K.; Heldin, C.-H.; Moustakas, A. TGFβ induces SIK to negatively regulate type I receptor kinase signaling. J. Cell Biol. 2008, 182, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Lönn, P.; Vanlandewijck, M.; Raja, E.; Kowanetz, M.; Watanabe, Y.; Kowanetz, K.; Vasilaki, E.; Heldin, C.-H.; Moustakas, A. Transcriptional Induction of Salt-inducible Kinase 1 by Transforming Growth Factor β Leads to Negative Regulation of Type I Receptor Signaling in Cooperation with the Smurf2 Ubiquitin Ligase. J. Biol. Chem. 2012, 287, 12867–12878. [Google Scholar] [CrossRef] [PubMed]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The Protein Kinase Complement of the Human Genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef]
- Alessi, D.R.; Sakamoto, K.; Bayascas, J.R. LKB1-Dependent Signaling Pathways. Annu. Rev. Biochem. 2006, 75, 137–163. [Google Scholar] [CrossRef]
- Kolliopoulos, C.; Raja, E.; Razmara, M.; Heldin, P.; Heldin, C.-H.; Moustakas, A.; van der Heide, L.P. Transforming growth factor β (TGFβ) induces NUAK kinase expression to fine-tune its signaling output. J. Biol. Chem. 2019, 294, 4119–4136. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Kusakai, G.-I.; Kishimoto, A.; Lu, J.; Ogura, T.; Lavin, M.F.; Esumi, H. Identification of a Novel Protein Kinase Mediating Akt Survival Signaling to the ATM Protein. J. Biol. Chem. 2003, 278, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, D.L.; Bai, Y.; Shahmolky, N.; Sharma, M.; Poon, R.; Drucker, D.J.; Rosen, C.F. Identification and characterization of a novel sucrose-non-fermenting protein kinase/AMP-activated protein kinase-related protein kinase, SNARK. Biochem. J. 2001, 355, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Bright, N.J.; Thornton, C.; Carling, D. The regulation and function of mammalian AMPK-related kinases. Acta Physiol. 2009, 196, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Lizcano, J.M.; Göransson, O.; Toth, R.; Deak, M.; Morrice, N.A.; Boudeau, J.; Hawley, S.A.; Udd, L.; Makela, T.P.; Hardie, D.G.; et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004, 23, 833–843. [Google Scholar] [CrossRef] [PubMed]
- Koh, H.-J.; Toyoda, T.; Fujii, N.; Jung, M.M.; Rathod, A.; Middelbeek, R.J.-W.; Lessard, S.J.; Treebak, J.T.; Tsuchihara, K.; Esumi, H.; et al. Sucrose nonfermenting AMPK-related kinase (SNARK) mediates contraction-stimulated glucose transport in mouse skeletal muscle. Proc. Natl. Acad. Sci. USA 2010, 107, 15541–15546. [Google Scholar] [CrossRef]
- Zagorska, A.; Deak, M.; Campbell, D.G.; Banerjee, S.; Hirano, M.; Aizawa, S.; Prescott, A.; Alessi, D. New Roles for the LKB1-NUAK Pathway in Controlling Myosin Phosphatase Complexes and Cell Adhesion. Sci. Signal. 2010, 3, ra25. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Liu, J.-E.; Liu, W.; Liu, C.-Y.; Liu, Z.-Y.; Sun, Z.-Y. A new role of NUAK1: Directly phosphorylating p53 and regulating cell proliferation. Oncogene 2011, 30, 2933–2942. [Google Scholar] [CrossRef] [PubMed]
- Courchet, J.; Lewis, T.L.; Lee, S.; Courchet, V.; Liou, D.-Y.; Aizawa, S.; Polleux, F. Terminal Axon Branching Is Regulated by the LKB1-NUAK1 Kinase Pathway via Presynaptic Mitochondrial Capture. Cell 2013, 153, 1510–1525. [Google Scholar] [CrossRef] [PubMed]
- Monteverde, T.; Tait-Mulder, J.; Hedley, A.; Knight, J.; Sansom, O.J.; Murphy, D.J. Calcium signalling links MYC to NUAK1. Oncogene 2017, 37, 982–992. [Google Scholar] [CrossRef]
- Suzuki, A.; Ogura, T.; Esumi, H. NDR2 Acts as the Upstream Kinase of ARK5 during Insulin-like Growth Factor-1 Signaling. J. Biol. Chem. 2006, 281, 13915–13921. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Zagórska, A.; Deak, M.; Campbell, D.G.; Prescott, A.R.; Alessi, D.R. Interplay between Polo kinase, LKB1-activated NUAK1 kinase, PP1βMYPT1 phosphatase complex and the SCFβTrCP E3 ubiquitin ligase. Biochem. J. 2014, 461, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Humbert, N.; Navaratnam, N.; Augert, A.; Da Costa, M.; Martien, S.; Wang, J.; Martinez, D.; Abbadie, C.; Carling, D.; De Launoit, Y.; et al. Regulation of ploidy and senescence by the AMPK-related kinase NUAK1. EMBO J. 2009, 29, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, D.L.; Rosen, C.F. Regulation of SNARK activity in response to cellular stresses. Biochim. Biophys. Acta (BBA) Gen. Subj. 2005, 1724, 71–85. [Google Scholar] [CrossRef]
- Lessard, S.J.; Rivas, D.; So, K.; Koh, H.-J.; Queiroz, A.L.; Hirshman, M.F.; Fielding, R.A.; Goodyear, L.J. The AMPK-related kinase SNARK regulates muscle mass and myocyte survival. J. Clin. Investig. 2015, 126, 560–570. [Google Scholar] [CrossRef]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the Human Tissue-specific Expression by Genome-wide Integration of Transcriptomics and Antibody-based Proteomics. Mol. Cell. Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Lu, J.; Kusakai, G.-I.; Kishimoto, A.; Ogura, T.; Esumi, H. ARK5 Is a Tumor Invasion-Associated Factor Downstream of Akt Signaling. Mol. Cell. Biol. 2004, 24, 3526–3535. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Kusakai, G.-I.; Kishimoto, A.; Shimojo, Y.; Miyamoto, S.; Ogura, T.; Ochiai, A.; Esumi, H. Regulation of caspase-6 and FLIP by the AMPK family member ARK5. Oncogene 2004, 23, 7067–7075. [Google Scholar] [CrossRef]
- Legembre, P.; Schickel, R.; Barnhart, B.C.; Peter, M.E. Identification of SNF1/AMP Kinase-related Kinase as an NF-κB-regulated Anti-apoptotic Kinase Involved in CD95-induced Motility and Invasiveness. J. Biol. Chem. 2004, 279, 46742–46747. [Google Scholar] [CrossRef]
- Namiki, T.; Yaguchi, T.; Nakamura, K.; Valencia, J.C.; Coelho, S.G.; Yin, L.; Kawaguchi, M.; Vieira, W.D.; Kaneko, Y.; Tanemura, A.; et al. NUAK2 Amplification Coupled with PTEN Deficiency Promotes Melanoma Development via CDK Activation. Cancer Res. 2015, 75, 2708–2715. [Google Scholar] [CrossRef] [PubMed]
- Namiki, T.; Tanemura, A.; Valencia, J.C.; Coelho, S.G.; Passeron, T.; Kawaguchi, M.; Vieira, W.D.; Ishikawa, M.; Nishijima, W.; Izumo, T.; et al. AMP kinase-related kinase NUAK2 affects tumor growth, migration, and clinical outcome of human melanoma. Proc. Natl. Acad. Sci. USA 2011, 108, 6597–6602. [Google Scholar] [CrossRef]
- Sakaue-Sawano, A.; Kurokawa, H.; Morimura, T.; Hanyu, A.; Hama, H.; Osawa, H.; Kashiwagi, S.; Fukami, K.; Miyata, T.; Miyoshi, H.; et al. Visualizing Spatiotemporal Dynamics of Multicellular Cell-Cycle Progression. Cell 2008, 132, 487–498. [Google Scholar] [CrossRef]
- Lodyga, M.; Hinz, B. TGF-β1—A truly transforming growth factor in fibrosis and immunity. Semin. Cell Dev. Biol. 2020, 101, 123–139. [Google Scholar] [CrossRef]
- Caja, L.; Dituri, F.; Mancarella, S.; Caballero-Diaz, D.; Moustakas, A.; Giannelli, G.; Fabregat, I. TGF-β and the Tissue Microenvironment: Relevance in Fibrosis and Cancer. Int. J. Mol. Sci. 2018, 19, 1294. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Black, B.L.; Derynck, R. TGF-beta inhibits muscle differentiation through functional repression of myogenic transcription factors by Smad3. Genes Dev. 2001, 15, 2950–2966. [Google Scholar] [CrossRef]
- Morén, A.; Raja, E.; Heldin, C.-H.; Moustakas, A. Negative Regulation of TGFβ Signaling by the Kinase LKB1 and the Scaffolding Protein LIP1. J. Biol. Chem. 2011, 286, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Katajisto, P.; Vaahtomeri, K.; Ekman, N.; Ventelä, E.; Ristimäki, A.; Bardeesy, N.; Feil, R.; A DePinho, R.; Makela, T. LKB1 signaling in mesenchymal cells required for suppression of gastrointestinal polyposis. Nat. Genet. 2008, 40, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Vaahtomeri, K.; Ventelä, E.; Laajanen, K.; Katajisto, P.; Wipff, P.-J.; Hinz, B.; Vallenius, T.; Tiainen, M.; Makela, T. Lkb1 is required for TGFβ-mediated myofibroblast differentiation. J. Cell Sci. 2008, 121, 3531–3540. [Google Scholar] [CrossRef] [PubMed]
- Vallenius, T.; Vaahtomeri, K.; Kovac, B.; Osiceanu, A.-M.; Viljanen, M.; Makela, T. An association between NUAK2 and MRIP reveals a novel mechanism for regulation of actin stress fibers. J. Cell Sci. 2011, 124, 384–393. [Google Scholar] [CrossRef]
- Port, J.; Muthalagu, N.; Raja, M.; Ceteci, F.; Monteverde, T.; Kruspig, B.; Hedley, A.; Kalna, G.; Lilla, S.; Neilson, L.; et al. Colorectal Tumors Require NUAK1 for Protection from Oxidative Stress. Cancer Discov. 2018, 8, 632–647. [Google Scholar] [CrossRef]
- Cossa, G.; Roeschert, I.; Prinz, F.; Baluapuri, A.; Vidal, R.S.; Schülein-Völk, C.; Chang, Y.-C.; Ade, C.P.; Mastrobuoni, G.; Girard, C.; et al. Localized Inhibition of Protein Phosphatase 1 by NUAK1 Promotes Spliceosome Activity and Reveals a MYC-Sensitive Feedback Control of Transcription. Mol. Cell 2020, 77, 1322–1339.e11. [Google Scholar] [CrossRef]
- Ma, S.; Meng, Z.; Chen, R.; Guan, K.-L. The Hippo Pathway: Biology and Pathophysiology. Annu. Rev. Biochem. 2019, 88, 577–604. [Google Scholar] [CrossRef]
- Gill, M.K.; Christova, T.; Zhang, Y.Y.; Gregorieff, A.; Zhang, L.; Narimatsu, M.; Song, S.; Xiong, S.; Couzens, A.L.; Tong, J.; et al. A feed forward loop enforces YAP/TAZ signaling during tumorigenesis. Nat. Commun. 2018, 9, 3510. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.-C.; Pepe-Mooney, B.; Galli, G.; Dill, M.T.; Huang, H.-T.; Hao, M.; Wang, Y.; Liang, H.; Calogero, R.; Camargo, F.D. NUAK2 is a critical YAP target in liver cancer. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Al-Busani, H.; Al-Sobaihi, S.; Nojima, K.; Tanemura, A.; Yaguchi, T.; Kawakami, Y.; Matsumura, H.; Nishimura, E.K.; Yokozeki, H.; Namiki, T. NUAK2 localization in normal skin and its expression in a variety of skin tumors with YAP. J. Dermatol. Sci. 2020, 97, 143–151. [Google Scholar] [CrossRef]
- Bonnard, C.; Navaratnam, N.; Ghosh, K.; Chan, P.W.; Tan, T.T.; Pomp, O.; Ng, A.Y.J.; Tohari, S.; Changede, R.; Carling, D.; et al. A loss-of-function NUAK2 mutation in humans causes anencephaly due to impaired Hippo-YAP signaling. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Varelas, X.; Samavarchi-Tehrani, P.; Narimatsu, M.; Weiss, A.; Cockburn, K.; Larsen, B.G.; Rossant, J.; Wrana, J.L. The Crumbs Complex Couples Cell Density Sensing to Hippo-Dependent Control of the TGF-β-SMAD Pathway. Dev. Cell 2010, 19, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Narimatsu, M.; Samavarchi-Tehrani, P.; Varelas, X.; Wrana, J.L. Distinct Polarity Cues Direct Taz/Yap and TGFβ Receptor Localization to Differentially Control TGFβ-Induced Smad Signaling. Dev. Cell 2015, 32, 652–656. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Buhrlage, S.J.; Huang, H.-T.; Deng, X.; Zhou, W.; Wang, J.; Traynor, R.; Prescott, A.; Alessi, D.R.; Gray, N.S. Characterization of WZ4003 and HTH-01-015 as selective inhibitors of the LKB1-tumour-suppressor-activated NUAK kinases. Biochem. J. 2013, 457, 215–225. [Google Scholar] [CrossRef]
- Faisal, M.; Kim, J.H.; Yoo, K.H.; Roh, E.J.; Hong, S.S.; Lee, S.H. Development and Therapeutic Potential of NUAKs Inhibitors. J. Med. Chem. 2021, 64, 2–25. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| NUAK1 | |||||
|---|---|---|---|---|---|
| Model | Upstream Signaling | Direct Activator | Target Substrate | Cellular Phenotype | Ref |
| HepG2 cells | Glucose starvation | Akt | ATM | Stimulated p53-mediated cell survival during glucose starvation | [13] |
| PANC-1 and DLD-1 cell lines | IGF-1 signaling | Akt | Increased MT1-MMP production and subsequent MMP-2 and MMP-9 activation, stimulated tumor invasion and metastasis | [28] | |
| SW480, DLD-1, HCT-15, HCT-116 and WiDr cell lines | IGF-1 signaling | Akt | Caspase-6 | Promoted resistance to the extrinsic Fas/FasL—mediated cell death in colorectal cancer cells | [29] |
| HCT-166, DLD-1 and SW480 cells | IGF-1 signaling | NDR2 | Promoted tumor cell survival and invasion | [22] | |
| WI-38 cells, HEK293 | LKB1 | LATS1 | Increased aneuploidy and induced senescence | [24] | |
| HEK293, Lkb1−/− and Nuak1−/−MEFs | Cell detachment | LKB1 | PP1 subunit MYPT1 | Promoted myosin II-mediated cell detachment | [18] |
| A549 and Hep3B cells | Glucose starvation | LKB1 | p53 | Promoted p21/WAF1 induced cell cycle arrest | [19] |
| Mouse cortical neurons (Lkb1 KO or Nuak1 KO) | LKB1 | Stimulated presynaptic mitochondria immobilization and cortical axon branching | [20] | ||
| U2OS cells | CDK and subsequent PLK | E3 ligase SCFβTrCP binding | SCFβTrCP-mediated degradation of Nuak1. Controlled proliferation by stimulating S-phase and PLK1-mediated mitosis | [23] | |
| HeLa cells | Ca2+ signaling | PKC⍺ | Raptor | Inhibited MTORC1-regulated cell growth | [21] |
| U2OS, SW480 and HCT116 cell lines | Oxidative stress | PP1 subunit MYPT1 | Promoted colorectal cancer formation by suppressing GSK3β-dependent inhibition of NRF2 nuclear mobilization | [41] | |
| NMuMG, HaCaT, HEK293T, AG1523 cell lines | Expression induced by TGF-β | Inhibited TGF-β-mediated epithelial cytostasis, mesenchymal differentiation and myofibroblast contractility | [12] | ||
| U2OS cells | PP1 subunit PNUTS | Promoted spliceosome activity | [42] | ||
| NUAK2 | |||||
|---|---|---|---|---|---|
| Model | Upstream Signaling | Direct Activator | Target Substrate | Cellular Phenotype | Ref |
| BHK fibroblasts, NRKC cells | Elevated AMP concentration (AICAR), glucose deprivation | Observed auto-phosphorylation | SAMS peptide | [14] | |
| MCF7(-FB), HEK293T, ACHN cell lines | Expression induced by Fas receptor activation, requiring NF-κB signaling | Protected tumor cells against Fas-induced apoptosis and promoted motility and invasion | [30] | ||
| BHK, HEK293, INS-1, H4IIE, NRKC cell lines | Diverse cellular stresses | Cell type-specific kinase activity regulated upon nutrient starvation, cellular ATP decrease and/or AMP increase, ER stress, osmotic stress, oxidative stress or UV-B radiation | [25] | ||
| HEK293 cells | LATS1 | [24] | |||
| Mouse skeletal muscle | In situ and in vitro muscle contraction, treadmill activity | LKB1 | Suggested AS160 and TBC1D1 | Stimulated contraction, stimulated glucose transport in muscle | [17] |
| U2OS and HeLa cell lines | Induced expression upon growth signals and actin stress fiber alterations | MRIP (kinase-independent association) | MLCP inhibition and promoted actin stress fibers | [40] | |
| C32, SM2-1 and mel18 melanoma cell lines | PTEN-deficiency | CDK2 activation promoted melanoma tumor growth | [31] | ||
| C2C12 myoblasts and mouse skeletal muscle | Increased expression with muscle differentiation and metabolic stress | MYPT1 | Promoted Rho kinase signaling-mediated myocyte survival during stress | [26] | |
| MDA-MB231 cells, HEK293T | Induced expression by serum-activated YAP/TAZ signaling | LATS1/2 | Positive feedforward reinforcement of YAP/TAZ signaling, increased cell proliferation | [44] | |
| HeLa cells | Ca2+ signaling | [21] | |||
| HuCCT-1, H69 and SNU475 cell lines | YAP-mediated induction | MYPT1 | Actomyosin-regulated YAP amplification, promoted YAP driven liver cancer cell proliferation | [45] | |
| NMuMG, HaCaT, HEK293T, AG1523 | Expression induced by TGF-β | Stimulated TGF-β-mediated epithelial cytostasis, mesenchymal differentiation and myofibroblast contractility | [12] | ||
| Various types of skin tumors | Close association between NUAK2 and YAP expression in squamous cell carcinoma and Bowen’s disease | [46] | |||
| Patient derived NPCs | Loss-of-function NUAK2 germline mutation | LATS2 | Hippo signaling regulated neural tube closure through the apical actomyosin network | [47] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van de Vis, R.A.J.; Moustakas, A.; van der Heide, L.P. NUAK1 and NUAK2 Fine-Tune TGF-β Signaling. Cancers 2021, 13, 3377. https://doi.org/10.3390/cancers13133377
van de Vis RAJ, Moustakas A, van der Heide LP. NUAK1 and NUAK2 Fine-Tune TGF-β Signaling. Cancers. 2021; 13(13):3377. https://doi.org/10.3390/cancers13133377
Chicago/Turabian Stylevan de Vis, Reinofke A. J., Aristidis Moustakas, and Lars P. van der Heide. 2021. "NUAK1 and NUAK2 Fine-Tune TGF-β Signaling" Cancers 13, no. 13: 3377. https://doi.org/10.3390/cancers13133377
APA Stylevan de Vis, R. A. J., Moustakas, A., & van der Heide, L. P. (2021). NUAK1 and NUAK2 Fine-Tune TGF-β Signaling. Cancers, 13(13), 3377. https://doi.org/10.3390/cancers13133377