
Genomics of Smoldering Multiple Myeloma: Time for Clinical Translation of Findings?

 , ,
, ,  ,
,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Risk Stratification Models
3. Genomic Landscape and Intrinsic Determinants of SMM Progression
3.1. Genomic Landscape
3.2. Molecular Models of Progression
3.3. Molecular Models of Risk Stratification
3.4. Mutational Signatures and Chromosomal Events
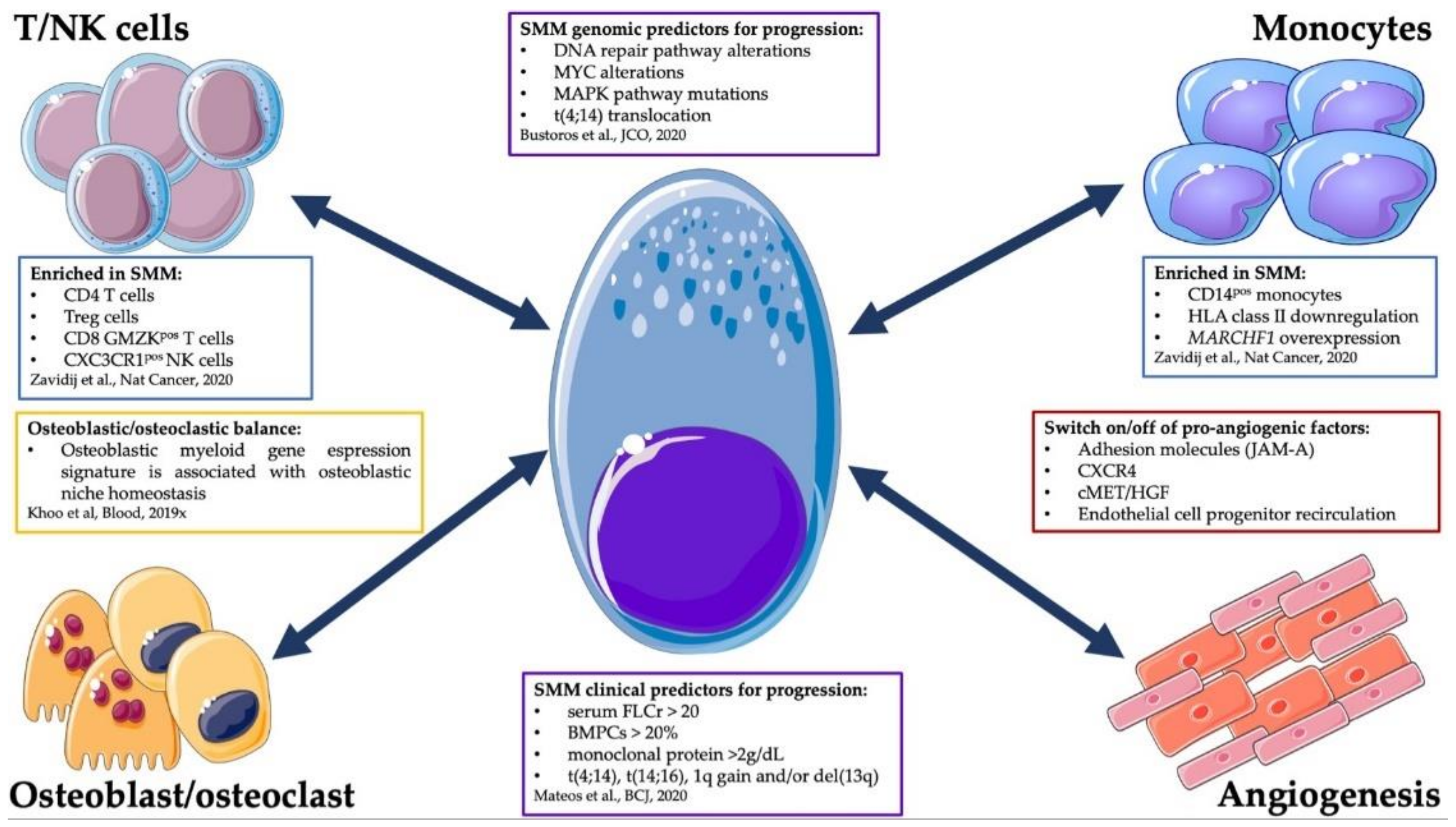
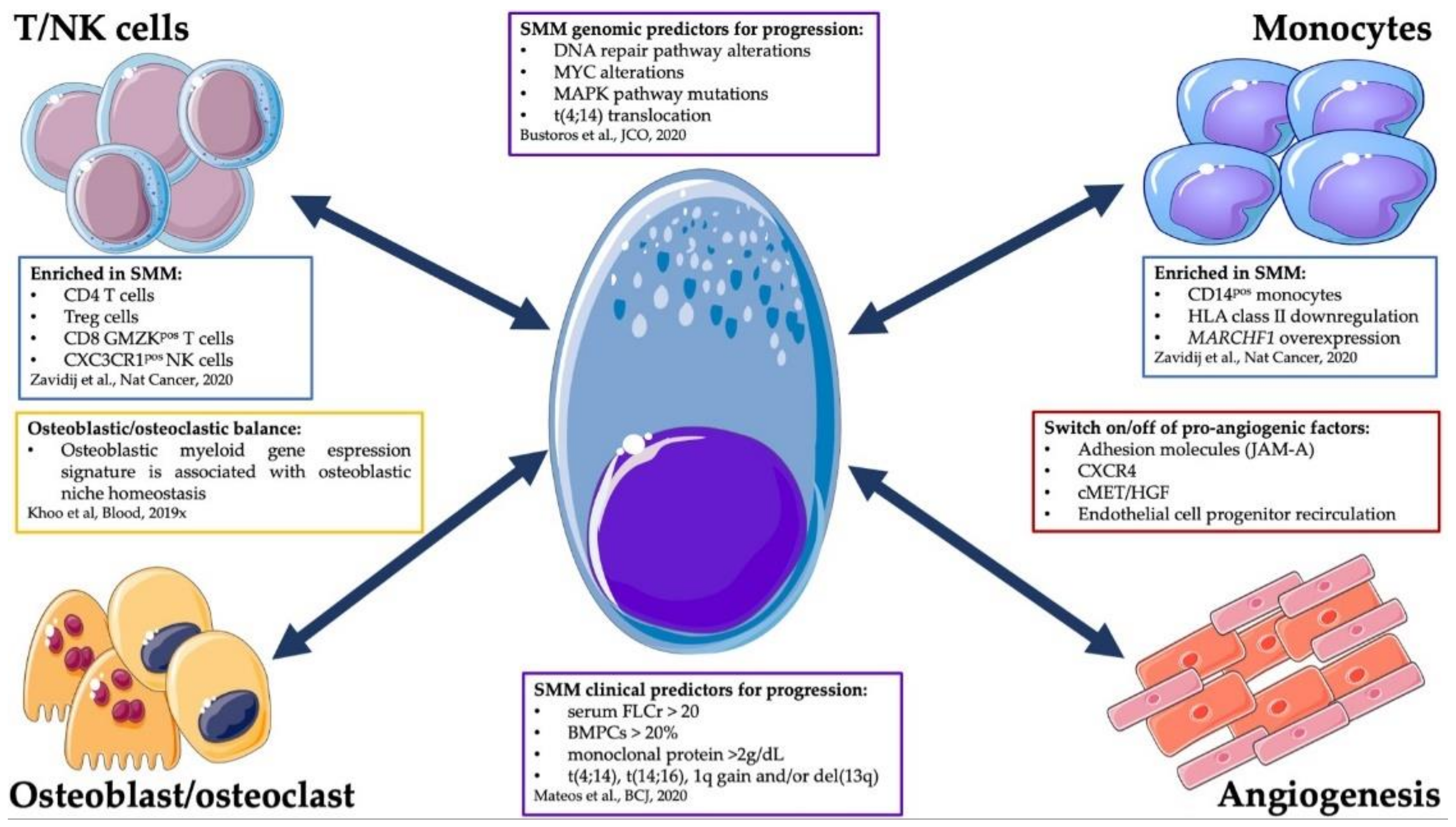
4. Cell-Extrinsic Determinants of SMM Progression
5. Clinical Perspectives
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kristinsson, S.Y.; Holmberg, E.; Blimark, C. Treatment for high-risk smoldering myeloma. N. Engl. J. Med. 2013, 369, 1762–1763. [Google Scholar] [PubMed] [Green Version]
- Ravindran, A.; Bartley, A.C.; Holton, S.J.; Gonsalves, W.I.; Kapoor, P.; Siddiqui, M.A.; Hashmi, S.K.; Marshall, A.L.; Ashrani, A.A.; Dispenzieri, A.; et al. Prevalence, incidence and survival of smoldering multiple myeloma in the united states. Blood Cancer J. 2016, 6, e486. [Google Scholar] [CrossRef] [Green Version]
- Da Via, M.C.; Ziccheddu, B.; Maeda, A.; Bagnoli, F.; Perrone, G.; Bolli, N. A journey through myeloma evolution: From the normal plasma cell to disease complexity. HemaSphere 2020, 4, e502. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Blade, J.; Merlini, G.; Mateos, M.V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International myeloma working group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014, 15, e538–e548. [Google Scholar] [CrossRef]
- Lakshman, A.; Rajkumar, S.V.; Buadi, F.K.; Binder, M.; Gertz, M.A.; Lacy, M.Q.; Dispenzieri, A.; Dingli, D.; Fonder, A.L.; Hayman, S.R.; et al. Risk stratification of smoldering multiple myeloma incorporating revised imwg diagnostic criteria. Blood Cancer J. 2018, 8, 59. [Google Scholar] [CrossRef] [PubMed]
- Mateos, M.V.; Kumar, S.; Dimopoulos, M.A.; Gonzalez-Calle, V.; Kastritis, E.; Hajek, R.; De Larrea, C.F.; Morgan, G.J.; Merlini, G.; Goldschmidt, H.; et al. International myeloma working group risk stratification model for smoldering multiple myeloma (smm). Blood Cancer J. 2020, 10, 102. [Google Scholar] [CrossRef] [PubMed]
- Dhodapkar, M.V.; Sexton, R.; Waheed, S.; Usmani, S.; Papanikolaou, X.; Nair, B.; Petty, N.; Shaughnessy, J.D., Jr.; Hoering, A.; Crowley, J.; et al. Clinical, genomic, and imaging predictors of myeloma progression from asymptomatic monoclonal gammopathies (swog s0120). Blood 2014, 123, 78–85. [Google Scholar] [CrossRef] [Green Version]
- Bustoros, M.; Sklavenitis-Pistofidis, R.; Park, J.; Redd, R.; Zhitomirsky, B.; Dunford, A.J.; Salem, K.; Tai, Y.T.; Anand, S.; Mouhieddine, T.H.; et al. Genomic profiling of smoldering multiple myeloma identifies patients at a high risk of disease progression. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 2380–2389. [Google Scholar] [CrossRef] [PubMed]
- Zavidij, O.; Haradhvala, N.J.; Mouhieddine, T.H.; Sklavenitis-Pistofidis, R.; Cai, S.; Reidy, M.; Rahmat, M.; Flaifel, A.; Ferland, B.; Su, N.K.; et al. Single-cell rna sequencing reveals compromised immune microenvironment in precursor stages of multiple myeloma. Nat. Cancer 2020, 1, 493–506. [Google Scholar] [CrossRef]
- Solimando, A.G.; Da Via, M.C.; Leone, P.; Borrelli, P.; Croci, G.A.; Tabares, P.; Brandl, A.; Di Lernia, G.; Bianchi, F.P.; Tafuri, S.; et al. Halting the vicious cycle within the multiple myeloma ecosystem: Blocking jam-a on bone marrow endothelial cells restores the angiogenic homeostasis and suppresses tumor progression. Haematologica 2020. [Google Scholar] [CrossRef]
- Khoo, W.H.; Ledergor, G.; Weiner, A.; Roden, D.L.; Terry, R.L.; McDonald, M.M.; Chai, R.C.; De Veirman, K.; Owen, K.L.; Opperman, K.S.; et al. A niche-dependent myeloid transcriptome signature defines dormant myeloma cells. Blood 2019, 134, 30–43. [Google Scholar] [CrossRef]
- Hill, E.; Dew, A.; Morrison, C.; Yuan, C.; Stetler-Stevenson, M.; Landgren, O.; Kazandjian, D. Assessment of discordance among smoldering multiple myeloma risk models. JAMA Oncol. 2021, 7, 132–134. [Google Scholar] [CrossRef] [PubMed]
- Maura, F.; Bolli, N.; Rustad, E.H.; Hultcrantz, M.; Munshi, N.; Landgren, O. Moving from cancer burden to cancer genomics for smoldering myeloma: A review. JAMA Oncol. 2019, 6, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Merz, M.; Hielscher, T.; Schult, D.; Mai, E.K.; Raab, M.S.; Hillengass, J.; Seckinger, A.; Hose, D.; Granzow, M.; Jauch, A.; et al. Cytogenetic subclone formation and evolution in progressive smoldering multiple myeloma. Leukemia 2020, 34, 1192–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neben, K.; Jauch, A.; Hielscher, T.; Hillengass, J.; Lehners, N.; Seckinger, A.; Granzow, M.; Raab, M.S.; Ho, A.D.; Goldschmidt, H.; et al. Progression in smoldering myeloma is independently determined by the chromosomal abnormalities del(17p), t(4;14), gain 1q, hyperdiploidy, and tumor load. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2013, 31, 4325–4332. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Gupta, V.; Fonseca, R.; Dispenzieri, A.; Gonsalves, W.I.; Larson, D.; Ketterling, R.P.; Lust, J.A.; Kyle, R.A.; Kumar, S.K. Impact of primary molecular cytogenetic abnormalities and risk of progression in smoldering multiple myeloma. Leukemia 2013, 27, 1738–1744. [Google Scholar] [CrossRef] [Green Version]
- Khan, R.; Dhodapkar, M.; Rosenthal, A.; Heuck, C.; Papanikolaou, X.; Qu, P.; van Rhee, F.; Zangari, M.; Jethava, Y.; Epstein, J.; et al. Four genes predict high risk of progression from smoldering to symptomatic multiple myeloma (swog s0120). Haematologica 2015, 100, 1214–1221. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Corral, L.; Gutierrez, N.C.; Vidriales, M.B.; Mateos, M.V.; Rasillo, A.; Garcia-Sanz, R.; Paiva, B.; San Miguel, J.F. The progression from mgus to smoldering myeloma and eventually to multiple myeloma involves a clonal expansion of genetically abnormal plasma cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 1692–1700. [Google Scholar] [CrossRef] [Green Version]
- Schmidt-Hieber, M.; Gutierrez, M.L.; Perez-Andres, M.; Paiva, B.; Rasillo, A.; Tabernero, M.D.; Sayagues, J.M.; Lopez, A.; Barcena, P.; Sanchez, M.L.; et al. Cytogenetic profiles in multiple myeloma and monoclonal gammopathy of undetermined significance: A study in highly purified aberrant plasma cells. Haematologica 2013, 98, 279–287. [Google Scholar] [CrossRef] [Green Version]
- Mikulasova, A.; Smetana, J.; Wayhelova, M.; Janyskova, H.; Sandecka, V.; Kufova, Z.; Almasi, M.; Jarkovsky, J.; Gregora, E.; Kessler, P.; et al. Genomewide profiling of copy-number alteration in monoclonal gammopathy of undetermined significance. Eur. J. Haematol. 2016, 97, 568–575. [Google Scholar] [CrossRef]
- Mikulasova, A.; Wardell, C.P.; Murison, A.; Boyle, E.M.; Jackson, G.H.; Smetana, J.; Kufova, Z.; Pour, L.; Sandecka, V.; Almasi, M.; et al. The spectrum of somatic mutations in monoclonal gammopathy of undetermined significance indicates a less complex genomic landscape than that in multiple myeloma. Haematologica 2017, 102, 1617–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajkumar, S.V.; Hayman, S.R.; Lacy, M.Q.; Dispenzieri, A.; Geyer, S.M.; Kabat, B.; Zeldenrust, S.R.; Kumar, S.; Greipp, P.R.; Fonseca, R.; et al. Combination therapy with lenalidomide plus dexamethasone (rev/dex) for newly diagnosed myeloma. Blood 2005, 106, 4050–4053. [Google Scholar] [CrossRef] [Green Version]
- Bolli, N.; Maura, F.; Minvielle, S.; Gloznik, D.; Szalat, R.; Fullam, A.; Martincorena, I.; Dawson, K.J.; Samur, M.K.; Zamora, J.; et al. Genomic patterns of progression in smoldering multiple myeloma. Nat. Commun. 2018, 9, 3363. [Google Scholar] [CrossRef]
- Boyle, E.M.; Deshpande, S.; Tytarenko, R.; Ashby, C.; Wang, Y.; Bauer, M.A.; Johnson, S.K.; Wardell, C.P.; Thanendrarajan, S.; Zangari, M.; et al. The molecular make up of smoldering myeloma highlights the evolutionary pathways leading to multiple myeloma. Nat. Commun. 2021, 12, 293. [Google Scholar] [CrossRef]
- Dutta, A.K.; Fink, J.L.; Grady, J.P.; Morgan, G.J.; Mullighan, C.G.; To, L.B.; Hewett, D.R.; Zannettino, A.C.W. Subclonal evolution in disease progression from mgus/smm to multiple myeloma is characterised by clonal stability. Leukemia 2019, 33, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Maura, F.; Bolli, N.; Angelopoulos, N.; Dawson, K.J.; Leongamornlert, D.; Martincorena, I.; Mitchell, T.J.; Fullam, A.; Gonzalez, S.; Szalat, R.; et al. Genomic landscape and chronological reconstruction of driver events in multiple myeloma. Nat. Commun. 2019, 10, 3835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oben, B.; Froyen, G.; Maclachlan, K.H.; Leongamornlert, D.; Abascal, F.; Zheng-Lin, B.; Yellapantula, V.; Derkach, A.; Geerdens, E.; Diamond, B.T.; et al. Whole-genome sequencing reveals progressive versus stable myeloma precursor conditions as two distinct entities. Nat. Commun. 2021, 12, 1861. [Google Scholar] [CrossRef] [PubMed]
- Maura, F.; Rustad, E.H.; Boyle, E.M.; Morgan, G.J. Reconstructing the evolutionary history of multiple myeloma. Best Pract. Res. Clin. Haematol. 2020, 33, 101145. [Google Scholar] [CrossRef] [PubMed]
- Rustad, E.H.; Yellapantula, V.; Leongamornlert, D.; Bolli, N.; Ledergor, G.; Nadeu, F.; Angelopoulos, N.; Dawson, K.J.; Mitchell, T.J.; Osborne, R.J.; et al. Timing the initiation of multiple myeloma. Nat. Commun. 2020, 11, 1917. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.A.; Wardell, C.P.; Melchor, L.; Brioli, A.; Johnson, D.C.; Kaiser, M.F.; Mirabella, F.; Lopez-Corral, L.; Humphray, S.; Murray, L.; et al. Intraclonal heterogeneity is a critical early event in the development of myeloma and precedes the development of clinical symptoms. Leukemia 2014, 28, 384–390. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Choi, M.; Heuck, C.; Mane, S.; Barlogie, B.; Lifton, R.P.; Dhodapkar, M.V. Serial exome analysis of disease progression in premalignant gammopathies. Leukemia 2014, 28, 1548–1552. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Maura, F.; Petljak, M.; Lionetti, M.; Cifola, I.; Liang, W.; Pinatel, E.; Alexandrov, L.B.; Fullam, A.; Martincorena, I.; Dawson, K.J.; et al. Biological and prognostic impact of apobec-induced mutations in the spectrum of plasma cell dyscrasias and multiple myeloma cell lines. Leukemia 2018, 32, 1044–1048. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.A.; Wardell, C.P.; Murison, A.; Boyle, E.M.; Begum, D.B.; Dahir, N.M.; Proszek, P.Z.; Melchor, L.; Pawlyn, C.; Kaiser, M.F.; et al. Apobec family mutational signatures are associated with poor prognosis translocations in multiple myeloma. Nat. Commun. 2015, 6, 6997. [Google Scholar] [CrossRef]
- Maura, F.; Degasperi, A.; Nadeu, F.; Leongamornlert, D.; Davies, H.; Moore, L.; Royo, R.; Ziccheddu, B.; Puente, X.S.; Avet-Loiseau, H.; et al. A practical guide for mutational signature analysis in hematological malignancies. Nat. Commun. 2019, 10, 2969. [Google Scholar] [CrossRef]
- Phan, T.G.; Croucher, P.I. The dormant cancer cell life cycle. Nat. Rev. Cancer 2020, 20, 398–411. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Smyth, M.J.; Martinet, L. Cancer immunoediting and immune dysregulation in multiple myeloma. Blood 2020, 136, 2731–2740. [Google Scholar] [CrossRef]
- Chesi, M.; Robbiani, D.F.; Sebag, M.; Chng, W.J.; Affer, M.; Tiedemann, R.; Valdez, R.; Palmer, S.E.; Haas, S.S.; Stewart, A.K.; et al. Aid-dependent activation of a myc transgene induces multiple myeloma in a conditional mouse model of post-germinal center malignancies. Cancer Cell 2008, 13, 167–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillerey, C.; Ferrari de Andrade, L.; Vuckovic, S.; Miles, K.; Ngiow, S.F.; Yong, M.C.; Teng, M.W.; Colonna, M.; Ritchie, D.S.; Chesi, M.; et al. Immunosurveillance and therapy of multiple myeloma are cd226 dependent. J. Clin. Investig. 2015, 125, 2077–2089. [Google Scholar] [CrossRef] [Green Version]
- Carbone, E.; Neri, P.; Mesuraca, M.; Fulciniti, M.T.; Otsuki, T.; Pende, D.; Groh, V.; Spies, T.; Pollio, G.; Cosman, D.; et al. Hla class i, nkg2d, and natural cytotoxicity receptors regulate multiple myeloma cell recognition by natural killer cells. Blood 2005, 105, 251–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, R.; Strowig, T.; Verma, R.; Koduru, S.; Hafemann, A.; Hopf, S.; Kocoglu, M.H.; Borsotti, C.; Zhang, L.; Branagan, A.; et al. Microenvironment-dependent growth of preneoplastic and malignant plasma cells in humanized mice. Nat. Med. 2016, 22, 1351–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailur, J.K.; McCachren, S.S.; Doxie, D.B.; Shrestha, M.; Pendleton, K.; Nooka, A.K.; Neparidze, N.; Parker, T.L.; Bar, N.; Kaufman, J.L.; et al. Early alterations in stem-like/resident t cells, innate and myeloid cells in the bone marrow in preneoplastic gammopathy. JCI Insight 2019, 5, e127807. [Google Scholar] [CrossRef]
- Hashimoto, M.; Kamphorst, A.O.; Im, S.J.; Kissick, H.T.; Pillai, R.N.; Ramalingam, S.S.; Araki, K.; Ahmed, R. Cd8 t cell exhaustion in chronic infection and cancer: Opportunities for interventions. Annu. Rev. Med. 2018, 69, 301–318. [Google Scholar] [CrossRef] [PubMed]
- Calcinotto, A.; Brevi, A.; Chesi, M.; Ferrarese, R.; Garcia Perez, L.; Grioni, M.; Kumar, S.; Garbitt, V.M.; Sharik, M.E.; Henderson, K.J.; et al. Microbiota-driven interleukin-17-producing cells and eosinophils synergize to accelerate multiple myeloma progression. Nat. Commun. 2018, 9, 4832. [Google Scholar] [CrossRef]
- Leone, P.; Solimando, A.G.; Malerba, E.; Fasano, R.; Buonavoglia, A.; Pappagallo, F.; De Re, V.; Argentiero, A.; Silvestris, N.; Vacca, A.; et al. Actors on the scene: Immune cells in the myeloma niche. Front. Oncol. 2020, 10, 599098. [Google Scholar] [CrossRef]
- Giuliani, N.; Storti, P.; Bolzoni, M.; Palma, B.D.; Bonomini, S. Angiogenesis and multiple myeloma. Cancer Microenviron. Off. J. Int. Cancer Microenviron. Soc. 2011, 4, 325–337. [Google Scholar] [CrossRef] [Green Version]
- Moschetta, M.; Mishima, Y.; Kawano, Y.; Manier, S.; Paiva, B.; Palomera, L.; Aljawai, Y.; Calcinotto, A.; Unitt, C.; Sahin, I.; et al. Targeting vasculogenesis to prevent progression in multiple myeloma. Leukemia 2016, 30, 1103–1115. [Google Scholar] [CrossRef]
- Bianchi, G.; Czarnecki, P.G.; Ho, M.; Roccaro, A.M.; Sacco, A.; Kawano, Y.; Gulla, A.; Aktas Samur, A.; Chen, T.; Wen, K.; et al. Robo1 promotes homing, dissemination, and survival of multiple myeloma within the bone marrow microenvironment. Blood Cancer Discov. 2021. [Google Scholar] [CrossRef]
- Chen, Z.; Orlowski, R.Z.; Wang, M.; Kwak, L.; McCarty, N. Osteoblastic niche supports the growth of quiescent multiple myeloma cells. Blood 2014, 123, 2204–2208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terpos, E.; Zamagni, E.; Lentzsch, S.; Drake, M.T.; Garcia-Sanz, R.; Abildgaard, N.; Ntanasis-Stathopoulos, I.; Schjesvold, F.; de la Rubia, J.; Kyriakou, C.; et al. Treatment of multiple myeloma-related bone disease: Recommendations from the bone working group of the international myeloma working group. Lancet Oncol. 2021, 22, e119–e130. [Google Scholar] [CrossRef]
- Mateos, M.V.; Hernandez, M.T.; Giraldo, P.; de la Rubia, J.; de Arriba, F.; Lopez Corral, L.; Rosinol, L.; Paiva, B.; Palomera, L.; Bargay, J.; et al. Lenalidomide plus dexamethasone for high-risk smoldering multiple myeloma. N. Engl. J. Med. 2013, 369, 438–447. [Google Scholar] [CrossRef] [Green Version]
- Mateos, M.V.; Hernandez, M.T.; Giraldo, P.; de la Rubia, J.; de Arriba, F.; Corral, L.L.; Rosinol, L.; Paiva, B.; Palomera, L.; Bargay, J.; et al. Lenalidomide plus dexamethasone versus observation in patients with high-risk smouldering multiple myeloma (quiredex): Long-term follow-up of a randomised, controlled, phase 3 trial. Lancet Oncol. 2016, 17, 1127–1136. [Google Scholar] [CrossRef]
- Lonial, S.; Jacobus, S.; Fonseca, R.; Weiss, M.; Kumar, S.; Orlowski, R.Z.; Kaufman, J.L.; Yacoub, A.M.; Buadi, F.K.; O’Brien, T.; et al. Randomized trial of lenalidomide versus observation in smoldering multiple myeloma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 1126–1137. [Google Scholar] [CrossRef] [PubMed]
- Bolli, N.; Sgherza, N.; Curci, P.; Rizzi, R.; Strafella, V.; Delia, M.; Gagliardi, V.P.; Neri, A.; Baldini, L.; Albano, F.; et al. What is new in the treatment of smoldering multiple myeloma? J. Clin. Med. 2021, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Bolli, N.; Maura, F.; Minvielle, S.; Gloznik, D.; Szalat, R.; Fullam, A.; Samur, M.K.; Tarpey, P.; Davies, H.; Martincorena, I.; et al. Whole genome sequencing of unique paired smm/mgus progressing to mm samples reveals a genomic landscape with diverse evolutionary pattern. Blood 2016, 128, 2088. [Google Scholar] [CrossRef]
- Manzoni, M.; Marchica, V.; Storti, P.; Ziccheddu, B.; Sammarelli, G.; Todaro, G.; Pelizzoni, F.; Salerio, S.; Notarfranchi, L.; Pompa, A.; et al. Application of next-generation sequencing for the genomic characterization of patients with smoldering myeloma. Cancers 2020, 12, 1332. [Google Scholar] [CrossRef]
- Manzoni, M.; Pompa, A.; Fabris, S.; Pelizzoni, F.; Ciceri, G.; Seia, M.; Ziccheddu, B.; Bolli, N.; Corradini, P.; Baldini, L.; et al. Limits and applications of genomic analysis of circulating tumor DNA as a liquid biopsy in asymptomatic forms of multiple myeloma. HemaSphere 2020, 4, e402. [Google Scholar] [CrossRef]

{kind=link}
| Feature | Required Extent |
|---|---|
| (1) Serum monoclonal protein (IgG or IgA) | ≥30 g/L |
| or | |
| urinary monoclonal protein | ≥500 mg per 24 h |
| and/or | |
| clonal bone marrow plasma cells | 10–60% |
| (2) myeloma defining events (MDEs) 1 or amyloidosis | absent |
| Model | Risk Factors | Risk Group | Two-Year PD Rate (%) |
|---|---|---|---|
| SWOG 2014 [7] | MC > 3 g/dl, sFLC > 25 mg/dl, GEP-70 > 0.26 | Low (0 factors) | 3.4 |
| Intermediate (1 factor) | 21.9 | ||
| High (> 2 factors) | 66.7 | ||
| IMWG 2020 [6] | MC > 2 g/dL, sFLCr > 20, BMPC > 20% + high risk cytogenetics: [t(4;14), t(14;16), +1q, del(13q)] | Low (0 factors) | 6 |
| Low-intermediate (1 factor) | 22 | ||
| Intermediate (2 factors) | 45.5 | ||
| High (3–4 factors) | 63.1 | ||
| Dana Farber 2020 [8] | DNA repair pathway gene alterations [mutations in TP53 and ATM, del(17p)], MAPK pathway gene mutations (KRAS, NRAS), MYC aberrations (translocations or copy-number variations) | 0 factors | 14.4 |
| At least 1 factor | 86.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lionetti, M.; Da Vià, M.C.; Albano, F.; Neri, A.; Bolli, N.; Musto, P. Genomics of Smoldering Multiple Myeloma: Time for Clinical Translation of Findings? Cancers 2021, 13, 3319. https://doi.org/10.3390/cancers13133319
Lionetti M, Da Vià MC, Albano F, Neri A, Bolli N, Musto P. Genomics of Smoldering Multiple Myeloma: Time for Clinical Translation of Findings? Cancers. 2021; 13(13):3319. https://doi.org/10.3390/cancers13133319
Chicago/Turabian StyleLionetti, Marta, Matteo C. Da Vià, Francesco Albano, Antonino Neri, Niccolò Bolli, and Pellegrino Musto. 2021. "Genomics of Smoldering Multiple Myeloma: Time for Clinical Translation of Findings?" Cancers 13, no. 13: 3319. https://doi.org/10.3390/cancers13133319
APA StyleLionetti, M., Da Vià, M. C., Albano, F., Neri, A., Bolli, N., & Musto, P. (2021). Genomics of Smoldering Multiple Myeloma: Time for Clinical Translation of Findings? Cancers, 13(13), 3319. https://doi.org/10.3390/cancers13133319