
Activation of DNA Damage Tolerance Pathways May Improve Immunotherapy of Mesothelioma

 , , , , and
, , , , and
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
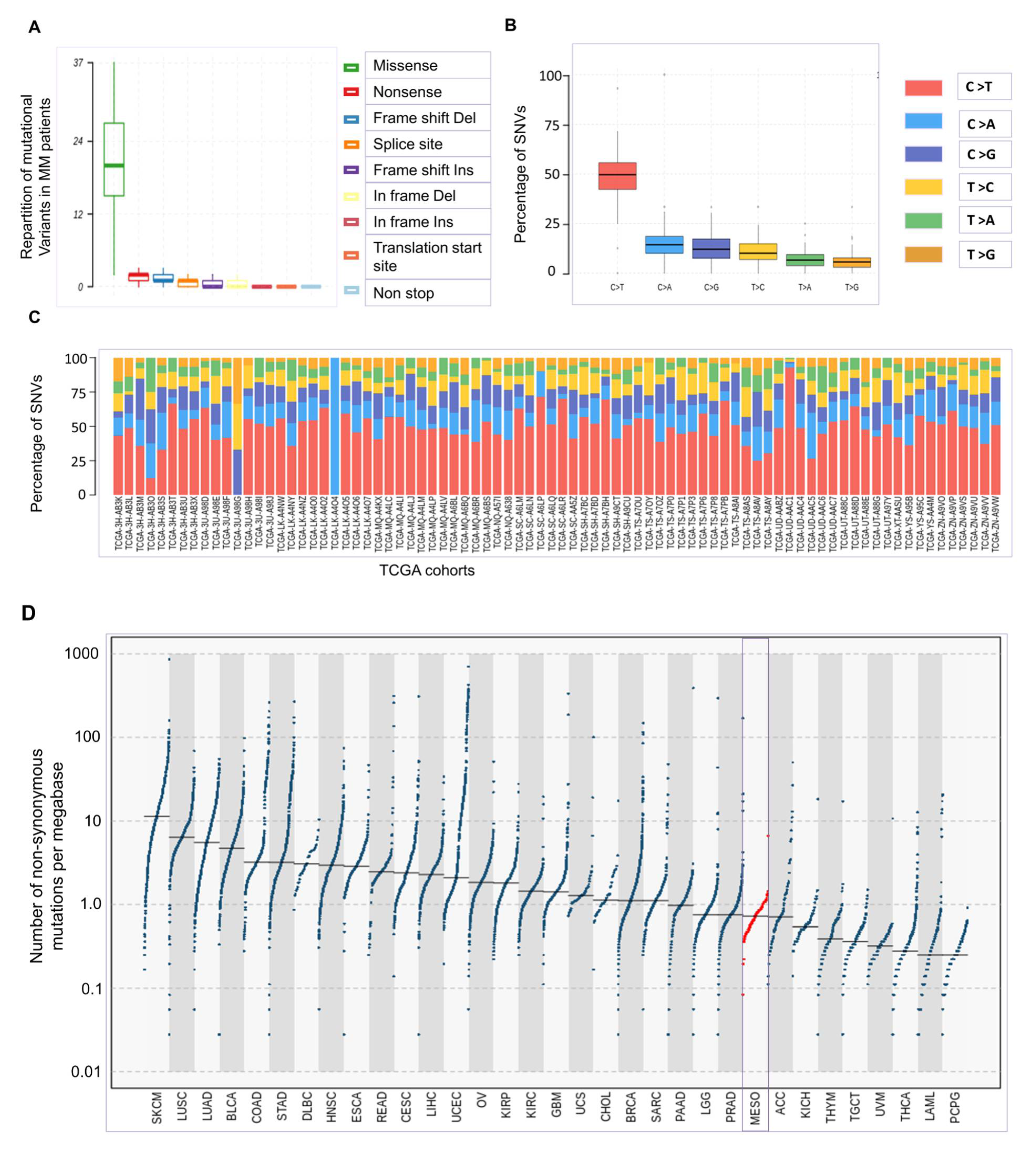
1. Oncogenesis of Mesothelioma Occurs at Low Tumor Mutational Burden
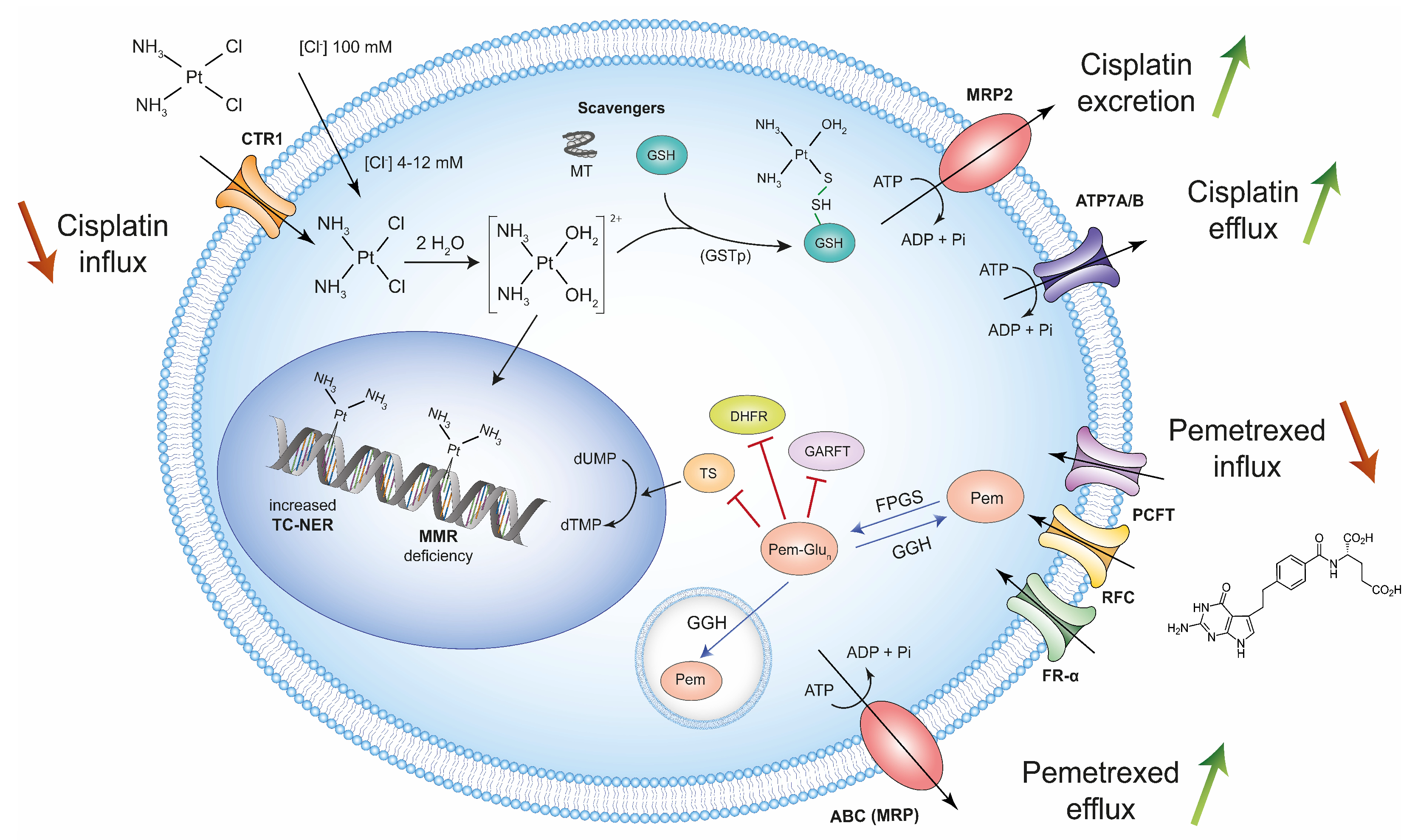
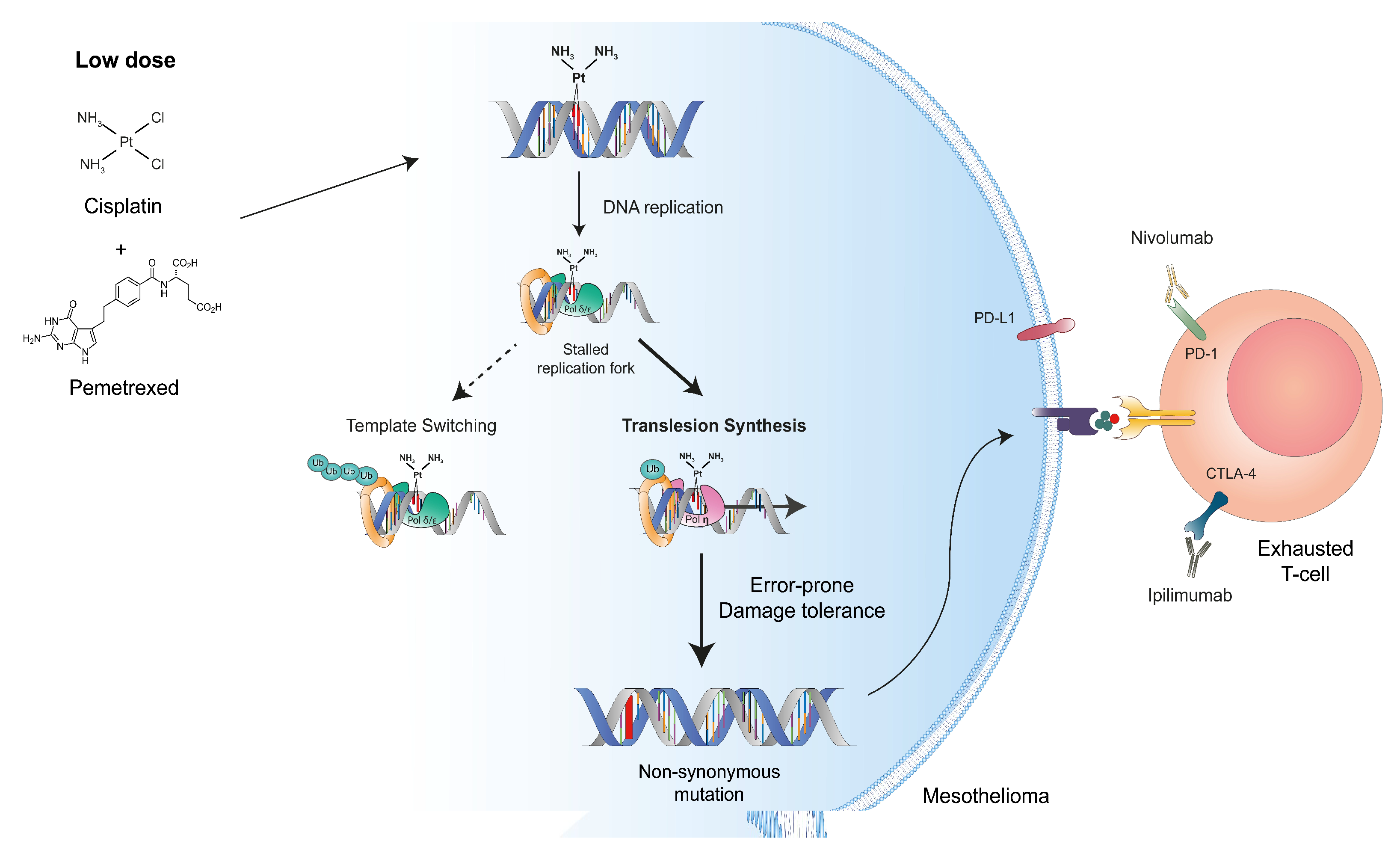
2. High-Dose Treatment with Cisplatin and Pemetrexed Selects Chemoresistant Mesothelioma Cells
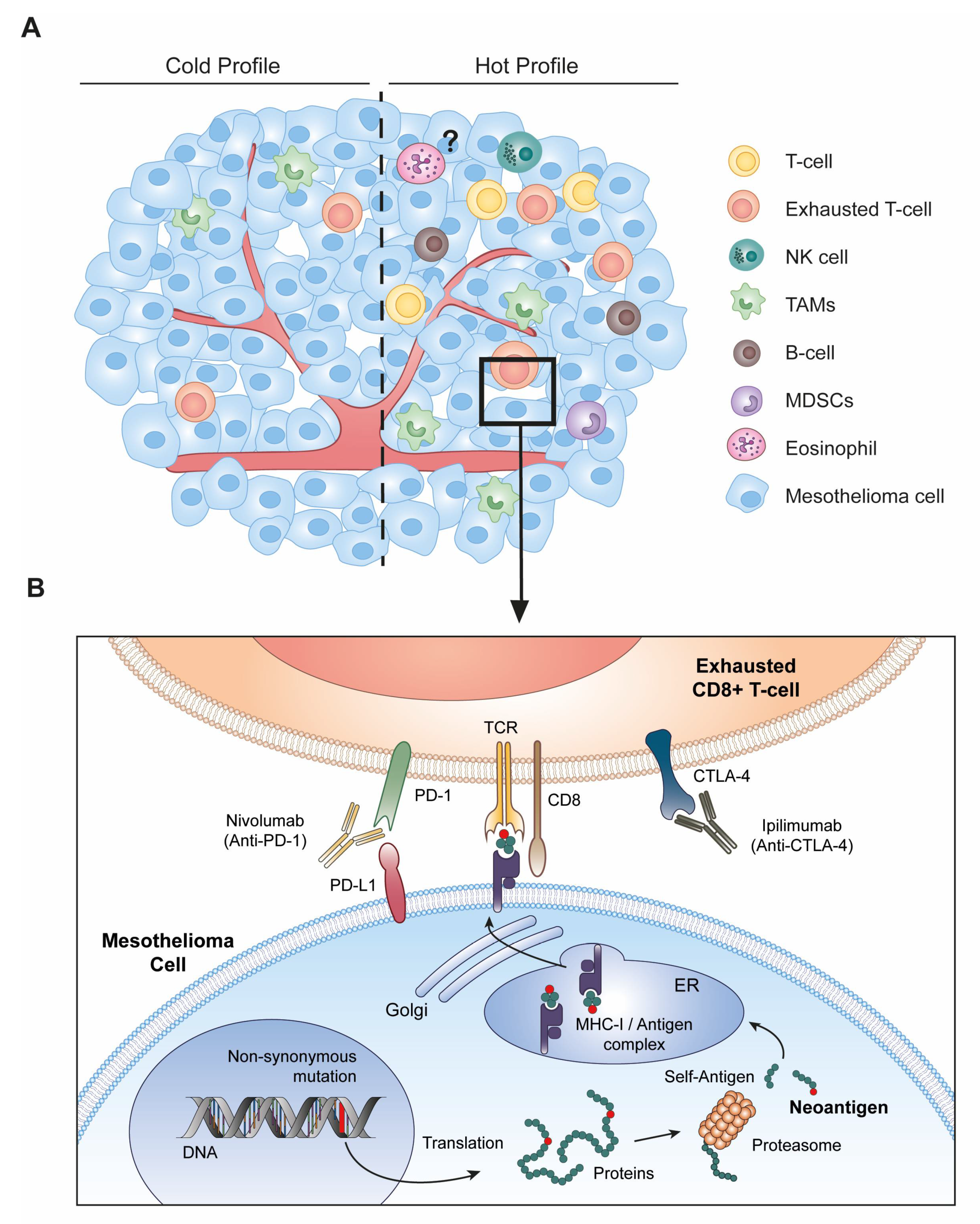
3. Immune Checkpoint Blockade Therapy Requires Altered-Self Antigens
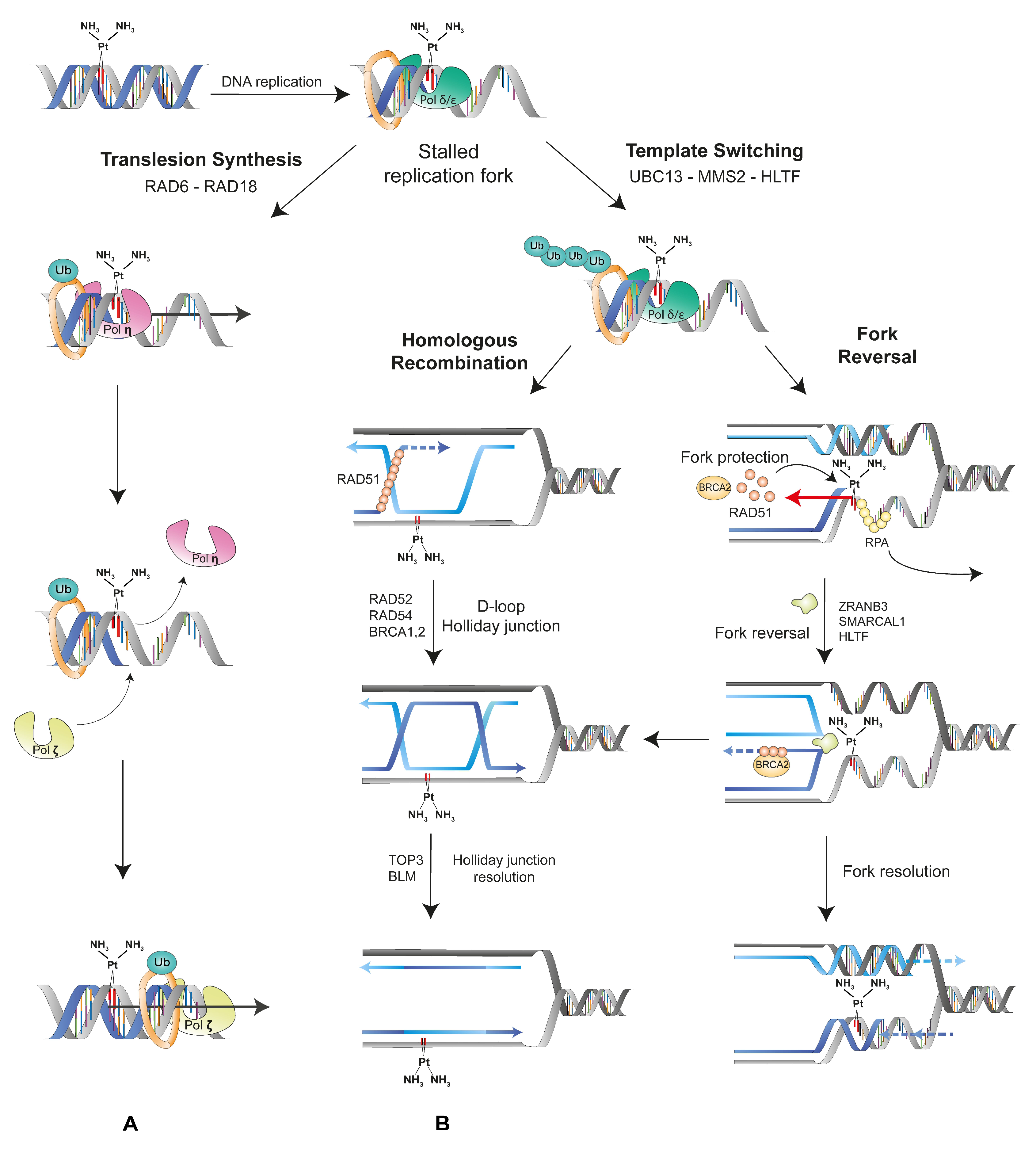
4. Mutations Generated by the DNA Damage Tolerance Pathways May Promote Neo-Antigen Production by MM Cells
5. Fine-Tuning of the DNA Damage Tolerance Pathways May Improve Immunotherapy
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Asciak, R.; George, V.; Rahman, N.M. Update on biology and management of mesothelioma. Eur. Respir. Rev. 2021, 30, 200226. [Google Scholar] [CrossRef]
- Carbone, M.; Harbour, J.W.; Brugarolas, J.; Bononi, A.; Pagano, I.; Dey, A.; Krausz, T.; Pass, H.; Yang, H.; Gaudino, G. Biological Mechanisms and Clinical Significance of BAP1 Mutations in Human Cancer. Cancer Discov. 2020, 10, 1103–1120. [Google Scholar] [CrossRef] [PubMed]
- Berquist, B.R.; Wilson, D.M. Pathways for repairing and tolerating the spectrum of oxidative DNA lesions. Cancer Lett. 2012, 327, 61–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yap, T.A.; Aerts, J.G.; Popat, S.; Fennell, D.A. Novel insights into mesothelioma biology and implications for therapy. Nat. Rev. Cancer 2017, 17, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Fuso Nerini, I.; Roca, E.; Mannarino, L.; Grosso, F.; Frapolli, R.; D’Incalci, M. Is DNA repair a potential target for effective therapies against malignant mesothelioma? Cancer Treat Rev. 2020, 90, 102101. [Google Scholar] [CrossRef]
- Zhang, M.; Luo, J.-L.; Sun, Q.; Harber, J.; Dawson, A.G.; Nakas, A.; Busacca, S.; Sharkey, A.J.; Waller, D.; Sheaff, M.T.; et al. Clonal architecture in mesothelioma is prognostic and shapes the tumour microenvironment. Nat. Commun. 2021, 12, 1751. [Google Scholar] [CrossRef]
- Schutsky, E.K.; Nabel, C.S.; Davis, A.K.F.; DeNizio, J.E.; Kohli, R.M. APOBEC3A efficiently deaminates methylated, but not TET-oxidized, cytosine bases in DNA. Nucleic Acids Res. 2017, 45, 7655–7665. [Google Scholar] [CrossRef]
- Willems, L.; Gillet, N.A. APOBEC3 Interference during Replication of Viral Genomes. Viruses 2015, 7, 2999–3018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hmeljak, J.; Sanchez-Vega, F.; Hoadley, K.A.; Shih, J.; Stewart, C.; Heiman, D.; Tarpey, P.; Danilova, L.; Drill, E.; Gibb, E.A.; et al. Integrative Molecular Characterization of Malignant Pleural Mesothelioma. Cancer Discov. 2018, 8, 1548–1565. [Google Scholar] [CrossRef] [Green Version]
- Bueno, R.; Stawiski, E.W.; Goldstein, L.D.; Durinck, S.; De Rienzo, A.; Modrusan, Z.; Gnad, F.; Nguyen, T.; Jaiswal, B.S.; Chirieac, L.R.; et al. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat. Genet. 2016, 48, 407–416. [Google Scholar] [CrossRef]
- Cedrés, S.; Ponce-Aix, S.; Iranzo, P.; Callejo, A.; Pardo, N.; Navarro, A.; Martinez-Marti, A.; Gómez-Abecia, S.; Zucchiatti, A.C.; Sansano, I.; et al. Analysis of mismatch repair (MMR) proteins expression in a series of malignant pleural mesothelioma (MPM) patients. Clin. Transl. Oncol. 2020, 22, 1390–1398. [Google Scholar] [CrossRef]
- Badhai, J.; Pandey, G.K.; Song, J.-Y.; Krijgsman, O.; Bhaskaran, R.; Chandrasekaran, G.; Kwon, M.-C.; Bombardelli, L.; Monkhorst, K.; Grasso, C.; et al. Combined deletion of Bap1, Nf2, and Cdkn2ab causes rapid onset of malignant mesothelioma in mice. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koppens, M.; Van Lohuizen, M. Context-dependent actions of Polycomb repressors in cancer. Oncogene 2016, 35, 1341–1352. [Google Scholar] [CrossRef] [PubMed]
- Zauderer, M.G.; Szlosarek, P.; Le Moulec, S.; Popat, S.; Taylor, P.; Planchard, D.; Scherpereel, A.; Jahan, T.; Koczywas, M.; Forster, M.; et al. Phase 2, multicenter study of the EZH2 inhibitor tazemetostat as monotherapy in adults with relapsed or refractory (R/R) malignant mesothelioma (MM) with BAP1 inactivation. J. Clin. Oncol. 2018, 36, 8515. [Google Scholar] [CrossRef]
- Cantini, L.; Hassan, R.; Sterman, D.H.; Aerts, J.G.J.V. Emerging Treatments for Malignant Pleural Mesothelioma: Where Are We Heading? Front. Oncol. 2020, 10, 343. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Sekido, Y. NF2/Merlin Inactivation and Potential Therapeutic Targets in Mesothelioma. Int. J. Mol. Sci. 2018, 19, 988. [Google Scholar] [CrossRef] [Green Version]
- Vogelzang, N.J.; Rusthoven, J.J.; Symanowski, J.; Denham, C.; Kaukel, E.; Ruffie, P.; Gatzemeier, U.; Boyer, M.; Emri, S.; Manegold, C.; et al. Phase III Study of Pemetrexed in Combination With Cisplatin Versus Cisplatin Alone in Patients With Malignant Pleural Mesothelioma. J. Clin. Oncol. 2003, 21, 2636–2644. [Google Scholar] [CrossRef]
- Chen, S.-H.; Chang, J.-Y. New Insights into Mechanisms of Cisplatin Resistance: From Tumor Cell to Microenvironment. Int. J. Mol. Sci. 2019, 20, 4136. [Google Scholar] [CrossRef] [Green Version]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2011, 31, 1869–1883. [Google Scholar] [CrossRef] [Green Version]
- Martin, L.P.; Hamilton, T.C.; Schilder, R.J. Platinum Resistance: The Role of DNA Repair Pathways. Clin. Cancer Res. 2008, 14, 1291–1295. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Lu, T.; Chen, Z.; Zhan, C.; Wang, Q. Mechanisms of resistance to pemetrexed in non-small cell lung cancer. Transl. Lung Cancer Res. 2019, 8, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Hotta, K.; Fujimoto, N. Current evidence and future perspectives of immune-checkpoint inhibitors in unresectable malignant pleural mesothelioma. J. Immunother. Cancer 2019, 8, e000461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baas, P.; Scherpereel, A.; Nowak, A.K.; Fujimoto, N.; Peters, S.; Tsao, A.S.; Mansfield, A.S.; Popat, S.; Jahan, T.; Antonia, S.; et al. First-line nivolumab plus ipilimumab in unresectable malignant pleural mesothelioma (CheckMate 743): A multicentre, randomised, open-label, phase 3 trial. Lancet 2021, 397, 375–386. [Google Scholar] [CrossRef]
- Zhuo, M.; Gorgun, F.M.; Tyler, D.S.; Englander, E.W. Transient activation of tumoral DNA damage tolerance pathway coupled with immune checkpoint blockade exerts durable tumor regression in mouse melanoma. Pigment. Cell Melanoma Res. 2021, 34, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, G.J.; Van Zandwijk, N.; Rasko, J.E.J. The Immune Microenvironment in Mesothelioma: Mechanisms of Resistance to Immunotherapy. Front. Oncol. 2019, 9, 1366. [Google Scholar] [CrossRef] [PubMed]
- Mutsaers, S.E.; Pixley, F.J.; Prêle, C.M.; Hoyne, G.F. Mesothelial cells regulate immune responses in health and disease: Role for immunotherapy in malignant mesothelioma. Curr. Opin. Immunol. 2020, 64, 88–109. [Google Scholar] [CrossRef] [PubMed]
- Hamaidia, M.; Gazon, H.; Hoyos, C.; Hoffmann, G.B.; Louis, R.; Duysinx, B.; Willems, L. Inhibition of EZH2 methyltransferase decreases immunoediting of mesothelioma cells by autologous macrophages through a PD-1-dependent mechanism. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Minnema-Luiting, J.; Vroman, H.; Aerts, J.; Cornelissen, R. Heterogeneity in Immune Cell Content in Malignant Pleural Mesothelioma. Int. J. Mol. Sci. 2018, 19, 1041. [Google Scholar] [CrossRef] [Green Version]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- Pauken, K.; Wherry, J. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015, 36, 265–276. [Google Scholar] [CrossRef] [Green Version]
- Šmahel, M. PD-1/PD-L1 Blockade Therapy for Tumors with Downregulated MHC Class I Expression. Int. J. Mol. Sci. 2017, 18, 1331. [Google Scholar] [CrossRef] [Green Version]
- Blum, Y.; Meiller, C.; Quetel, L.; Elarouci, N.; Ayadi, M.; Tashtanbaeva, D.; Armenoult, L.; Montagne, F.; Tranchant, R.; Renier, A.; et al. Dissecting heterogeneity in malignant pleural mesothelioma through histo-molecular gradients for clinical applications. Nat. Commun. 2019, 10, 1333. [Google Scholar] [CrossRef] [PubMed]
- Alcala, N.; Mangiante, L.; Le-Stang, N.; Gustafson, C.E.; Boyault, S.; Damiola, F.; Alcala, K.; Brevet, M.; Thivolet-Bejui, F.; Blanc-Fournier, C.; et al. Redefining malignant pleural mesothelioma types as a continuum uncovers immune-vascular interactions. EBioMedicine 2019, 48, 191–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, S.G.; Mutti, L. Immunotherapy for mesothelioma: A critical review of current clinical trials and future perspectives. Transl. Lung Cancer Res. 2020, 9, S100–S119. [Google Scholar] [CrossRef] [PubMed]
- Toumpanakis, D.; Theocharis, S.E. DNA repair systems in malignant mesothelioma. Cancer Lett. 2011, 312, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Stojic, L.; Brun, R.; Jiricny, J. Mismatch repair and DNA damage signalling. DNA Repair 2004, 3, 1091–1101. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef] [Green Version]
- Neelsen, K.J.; Lopes, M. Replication fork reversal in eukaryotes: From dead end to dynamic response. Nat. Rev. Mol. Cell Biol. 2015, 16, 207–220. [Google Scholar] [CrossRef]
- Branzei, D.; Szakal, B. DNA damage tolerance by recombination: Molecular pathways and DNA structures. DNA Repair 2016, 44, 68–75. [Google Scholar] [CrossRef]
- Ghosal, G.; Chen, J. DNA damage tolerance: A double-edged sword gurading the genome. Transl. Cancer Res. 2013, 2, 107–129. [Google Scholar] [PubMed]
- Carbone, M.; Yang, H.; Pass, H.; Krausz, T.; Testa, J.R.; Gaudino, G. BAP1 and cancer. Nat. Rev. Cancer 2013, 13, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Goodman, M.F.; Woodgate, R. Translesion DNA Polymerases. Cold Spring Harb. Perspect. Biol. 2013, 5, a010363. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Gao, Y. Translesion and Repair DNA Polymerases: Diverse Structure and Mechanism. Annu. Rev. Biochem. 2018, 87, 239–261. [Google Scholar] [CrossRef]
- Seelinger, O. Helicase-Like Transcription Factor HLTF and E3 Ubiquitin Ligase SHPRH Confer DNA Damage Tolerance through Direct Interactions with Proliferating Cell Nuclear Antigen (PCNA). Int. J. Mol. Sci. 2020, 21, 693. [Google Scholar] [CrossRef] [Green Version]
- Ripley, B.M.; Gildenberg, M.; Washington, M.T. Control of DNA Damage Bypass by Ubiquitylation of PCNA. Genes 2020, 11, 138. [Google Scholar] [CrossRef] [Green Version]
- Szikriszt, B.; Póti, Á.; Pipek, O.; Krzystanek, M.; Kanu, N.; Molnár, J.; Ribli, D.; Szeltner, Z.; Tusnády, G.E.; Csabai, I.; et al. A comprehensive survey of the mutagenic impact of common cancer cytotoxics. Genome Biol. 2016, 17, 99. [Google Scholar] [CrossRef] [Green Version]
- Szikriszt, B.; Póti, Á.; Németh, E.; Kanu, N.; Swanton, C.; Szüts, D. A comparative analysis of the mutagenicity of platinum-containing chemotherapeutic agents reveals direct and indirect mutagenic mechanisms. Mutagenesis 2021, 36, 75–86. [Google Scholar] [CrossRef]
- Ceppi, P.; Novello, S.; Cambieri, A.; Longo, M.; Monica, V.; Iacono, M.L.; Giaj-Levra, M.; Saviozzi, S.; Volante, M.; Papotti, M.; et al. Polymerase η mRNA Expression Predicts Survival of Non–Small Cell Lung Cancer Patients Treated with Platinum-Based Chemotherapy. Clin. Cancer Res. 2009, 15, 1039–1045. [Google Scholar] [CrossRef] [Green Version]
- Teng, K.Y.; Qiu, M.Z.; Li, Z.H.; Luo, H.Y.; Zeng, Z.L.; Luo, R.Z.; Zhang, H.Z.; Wang, Z.Q.; Li, Y.H.; Xu, R.H. DNA polymeraseη protein expression predicts treatment response and survival of metastatic gastric adenocarcinoma patients treated with oxaliplatin-based chemotherapy. J. Transl. Med. 2010, 8, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehmood, R.K.; Parker, J.; Ahmed, S.; Qasem, E.A.; Mohammed, A.; Zeeshan, M.; Jehangir, E. Review of Cisplatin and Oxaliplatin in Current Immunogenic and Monoclonal Antibodies Perspective. World J. Oncol. 2014, 5, 97–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hato, S.V.; Khong, A.; De Vries, I.J.M.; Lesterhuis, W.J. Molecular pathways: The immunogenic effects of platinum-based chemotherapeutics. Clin. Cancer Res. 2014, 20, 2831–2837. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; de Perrot, M. Radio-immunotherapy and chemo-immunotherapy as a novel treatment paradigm in malignant pleural mesothelioma. Transl. Lung Cancer Res. 2017, 6, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Ngwa, W.; Irabor, O.C.; Schoenfeld, J.D.; Hesser, J.; Demaria, S.; Formenti, S.C. Using immunotherapy to boost the abscopal effect. Nat. Rev. Cancer 2018, 18, 313–322. [Google Scholar] [CrossRef]
- Patel, S.A.; Minn, A.J. Combination Cancer Therapy with Immune Checkpoint Blockade: Mechanisms and Strategies. Immunity 2018, 48, 417–433. [Google Scholar] [CrossRef] [Green Version]
- Hamaidia, M.; Staumont, B.; Duysinx, B.; Louis, R.; Willems, L. Improvement of malignant pleural mesothelioma immunotherapy by epigenetic modulators. Curr. Top. Med. Chem. 2015, 16, 777–787. [Google Scholar] [CrossRef]
- Sen, T.; Rodriguez, B.L.; Chen, L.; Della Corte, C.M.; Morikawa, N.; Fujimoto, J.; Cristea, S.; Nguyen, T.; Diao, L.; Li, L.; et al. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer Discov. 2019, 9, 646–661. [Google Scholar] [CrossRef] [Green Version]
- Wahi, D.; Soni, D.; Grover, A. A Double-Edged Sword: The Anti-Cancer Effects of Emodin by Inhibiting the Redox-Protective Protein MTH1 and Augmenting ROS in NSCLC. J. Cancer 2021, 12, 652–681. [Google Scholar] [CrossRef] [PubMed]
- Elserafy, M.; Abugable, A.; Atteya, R.; El-Khamisy, S.F. Rad5, HLTF, and SHPRH: A Fresh View of an Old Story. Trends Genet. 2018, 34, 574–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brossel, H.; Fontaine, A.; Hoyos, C.; Jamakhani, M.; Willems, M.; Hamaidia, M.; Willems, L. Activation of DNA Damage Tolerance Pathways May Improve Immunotherapy of Mesothelioma. Cancers 2021, 13, 3211. https://doi.org/10.3390/cancers13133211
Brossel H, Fontaine A, Hoyos C, Jamakhani M, Willems M, Hamaidia M, Willems L. Activation of DNA Damage Tolerance Pathways May Improve Immunotherapy of Mesothelioma. Cancers. 2021; 13(13):3211. https://doi.org/10.3390/cancers13133211
Chicago/Turabian StyleBrossel, Hélène, Alexis Fontaine, Clotilde Hoyos, Majeed Jamakhani, Mégane Willems, Malik Hamaidia, and Luc Willems. 2021. "Activation of DNA Damage Tolerance Pathways May Improve Immunotherapy of Mesothelioma" Cancers 13, no. 13: 3211. https://doi.org/10.3390/cancers13133211
APA StyleBrossel, H., Fontaine, A., Hoyos, C., Jamakhani, M., Willems, M., Hamaidia, M., & Willems, L. (2021). Activation of DNA Damage Tolerance Pathways May Improve Immunotherapy of Mesothelioma. Cancers, 13(13), 3211. https://doi.org/10.3390/cancers13133211