
Immunity Depletion, Telomere Imbalance, and Cancer-Associated Metabolism Pathway Aberrations in Intestinal Mucosa upon Short-Term Caloric Restriction



Abstract
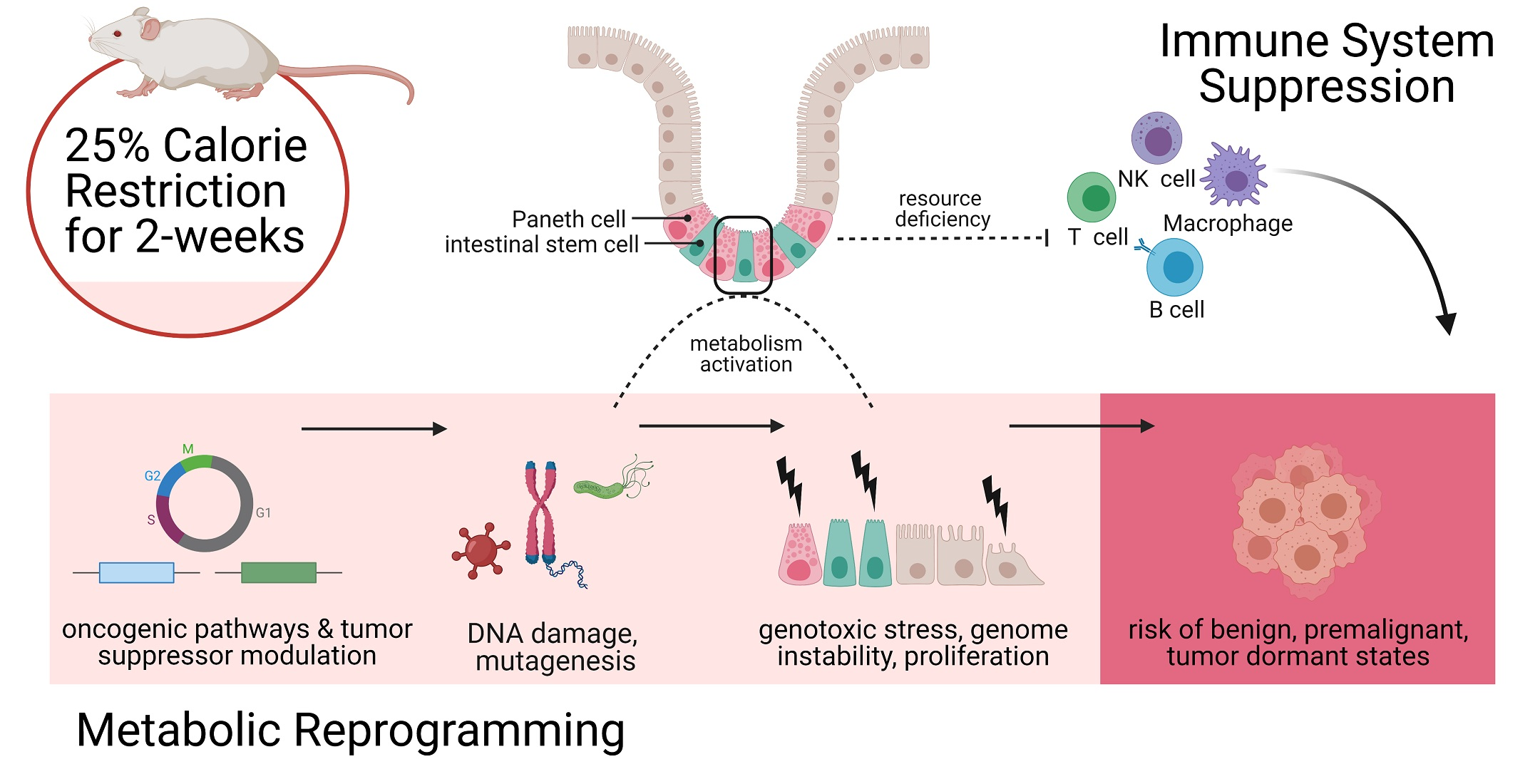
Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Computational Resources
2.2. Immune System and Epithelial Related Gene Curation
2.3. Cancer Related Gene Curation
2.4. qPCR Data Analysis across Gastrointestinal (GI) Tract Tissues
3. Results
3.1. Analysis of Differentially Expressed Genes in Mouse Duodenum Mucosa
3.2. DM Tissue Expressed Genes Are Translatable between Mouse and Human
3.3. 26% of CR Response Genes Are Involved in Mucosa Normal-Adenoma-Carcinoma Differential Gene Expression Patterns

3.4. Telomeric Maintenance Pathways and Cell Cycle Gene Responses in CR Mice
3.5. Mice Mucosa Cellular Immune System Compartments Are Deeply Suppressed by CR and Epithelial Network Interactions Are Activated
3.6. CR Induces Downregulation of Embryonal/Haematopoiesis/Immune CSC Genes, However Upregulation of Epithelial Cell CSC Genes
3.7. Functional Characterization of the Common Subset of DM and Paneth Cells DEGs
3.8. Gene Ontology and Pathway Analysis Revealed That 26% of CR Responded DEGs Are Involved in Malignancy and Cancer–Associated Networks
3.9. Network of the Cancer-Associated, Immune System, Epithelial, and Telomere Gene Subsets
3.10. CR Induces a Network between DEGs of Glutathione, Chemical Carcinogenesis, and Sirtuin Signaling Pathways
3.11. CR Profiles Common between Duodenum and Liver
3.12. DM Microarray-Defined DEG and GI Tract Tissue Samples qPCR Analysis Suggests Correlated Positional Expression Patterns of CR-Induced Response along the GI Tract
3.13. Interferon-Inducible DNA-Binding Gene Family Members on Chr 11 Are CR-Suppressed
3.14. Drug Targets in Energy-Responsive Metabolic Pathways
4. Discussion
Major Findings of This Study
- CR induces tissue homeostasis dysregulation, shifting the transcriptome profile to pro-oncogenomic pathway patterns in DM associated with activation of metabolism and proliferative activity of epithelial cells including Paneth cells, but depletion of intracellular immunity and functions of all immune cell types;
- CR induces transcription of cell cycle genes including a subset of key cancer-associated signaling genes directly involved in reprogramming pathways, premalignancy, malignancy states, and poor outcome in mice and humans;
- In CR response, apoptotic gene expression is reduced or not significantly varied;
- Tissue-specific proliferative epithelial stem DEGs are activated, however, immune-specific stem cell/progenitor genes are suppressed upon CR;
- CR induces transcription activation of key tumor-susceptible and oncogenes gene sets and their networks with tissue-specific risk of carcinogenesis and down-regulates protective mechanisms mediated by key tumor suppressor genes;
- CR induces multiple transcription suppression effects in autophagy, tumor-immune surveillance mechanics, and genes associated with induction and effector stages of NK, T-, B- cells and macrophages immune response;
- Detoxifying exogenous chemicals enzymes and drug metabolism networks with glutathione pathways are activated upon CR but could be involved in anti-cancer and pro-cancer outcomes via mutagenesis and DNA damage/repair mechanisms.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | CR | FC | Annotation | References |
|---|---|---|---|---|
| Fkbp5 | Up | 8.54 | most over-expressed CR-response gene, androgen-responsive gene with high expression in esophageal adenocarcinoma (EAC) tissues and this is associated with decreased patient survival, pro-oncogenic role in EAC | [60,61] |
| Vldlr | Up | 2.81 | pro-oncogene involved in both lipid metabolisms and proliferation, pathogenesis of gastric cancer, breast cancer, and involvement in cancer cell growth | [50,51,59,62] |
| Tlr4 | Down | −1.77 | innate immune system, pathogen recognition, therapeutic target, reduced expression associated with metastatic status of CRC | [63,64,65,66] |
| Arntl | Down | −1.87 | tumor suppressor, circadian rhythms | [67,68,69,70] |
| Plau | Up | 1.78 | CSC, poor pancreatic ductal adenocarcinoma prognosis | [71] |
| Ly6a, Ly6e, Ly6c1 | Down | −2.67, −2.18, −1.63 | immune cell differentiation, cancer stem cell biology | [72,73] |
| Sirt3 | Down | −1.38 | oxidative stress, protection of DNA damage, chromosome maintenance | [48,49,74,75] |
| Rnasel | Down | −1.74 | tumor suppressor, predisposition to prostate cancer, antiviral pathways | [76,77,78,79,80,81,82] |
| Cxcl9, Cxcl10 | Down | −1.79, −2.03 | proinflammatory chemokines, therapeutic target | [56,83] |
| Ndrg1 | Up | 2.65 | tumor suppressor, EMT, therapeutic target | [84,85,86,87] |
| Ugt2b5, Ugt2b35, Ugt2b36 | Up | 2.5, 1.7, 2.56 | detoxifying enzymes, reduce risk of carcinogenesis and toxicities by inactivating aromatic-like metabolites | [88] |
| Cyp2c55 (and family) | Up | 2.70 | metabolizing endogenous compounds, detoxifying exogenous chemicals, drug metabolism | [89] |
| Lamc2 | Up | 1.75 | promotes proliferation, cell migration, and invasion in cancers including colorectal and malignant metastases | [12,47,90] |
| Rrm2 | Up | 4.02 | cell cycle, therapeutic target, oncogene playing a key role in tumorigenesis and cancer progression, poor prognostic factor for colon, breast, and pancreatic cancers, cancer driver | [91,92,93,94,95] |
| Aldh1a1 | Up | 2.45 | EMT-related oncogenic, CSC, upregulated in APCMin/+ mouse model of colorectal cancer | [96] |
| Casp4 | Down | −1.87 | apoptosis, CASP4-deficient mice exhibit a defect in autophagy | [97] |
| Cemip | Up | 4.25 | overexpression correlates with poorer colon cancer patient survival and facilitates colorectal and stomach tumor growth, cancer driver | [24,47] |
5. Conclusions
- CR dramatically reduces immune responses, over-expresses the Paneth cell metabolic reprogramming pro-oncogenic genes forming aberrant networks, dysregulates tissue-specific epithelial proliferation and developmental processes, telomere maintaining processes, and response to chemical carcinogenesis;
- CR induces metabolic reprogramming processes driving pro-oncogenic mechanisms, cell cycle, and EMT pathways, and collectively increases the risk of malignancy;
- CR-induced Rrm2, Lamc2, Fkbp5 and aberrant glutathione gene family activation coupled with Sirtuin3 and RNaseL suppression could play tumorigenic roles in mucosa pathophysiology;
- Interferon-inducible gene family members on Chr11qB1.2 are suppressed by CR.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Colman, R.J.; Anderson, R.M.; Johnson, S.C.; Kastman, E.; Kosmatka, K.J.; Beasley, T.M.; Allison, D.; Cruzen, C.; Simmons, H.A.; Kemnitz, J.W.; et al. Caloric Restriction Delays Disease Onset and Mortality in Rhesus Monkeys. Science 2009, 325, 201–204. [Google Scholar] [CrossRef]
- Hursting, S.D.; Smith, S.M.; Lashinger, L.M.; Harvey, A.E.; Perkins, S.N. Calories and carcinogenesis: Lessons learned from 30 years of calorie restriction research. Carcinogenesis 2009, 31, 83–89. [Google Scholar] [CrossRef]
- Kritchevsky, D. Caloric Restriction and Cancer. J. Nutr. Sci. Vitaminol. 2001, 47, 13–19. [Google Scholar] [CrossRef]
- Hursting, S.D.; Dunlap, S.M.; Ford, N.; Hursting, M.J.; Lashinger, L.M. Calorie restriction and cancer prevention: A mechanistic perspective. Cancer Metab. 2013, 1, 10. [Google Scholar] [CrossRef]
- Klebanov, S. Can Short-Term Dietary Restriction and Fasting Have a Long-Term Anticarcinogenic Effect? Interdiscip. Top. Gerontol. 2006, 35, 176–192. [Google Scholar] [CrossRef]
- Harvie, M.; Howell, A. Energy restriction and the prevention of breast cancer. Proc. Nutr. Soc. 2012, 71, 263–275. [Google Scholar] [CrossRef]
- O’Flanagan, C.H.; Smith, L.A.; McDonell, S.B.; Hursting, S.D. When less may be more: Calorie restriction and response to cancer therapy. BMC Med. 2017, 15, 106. [Google Scholar] [CrossRef]
- Kakuni, M.; Morimura, K.; Wanibuchi, H.; Ogawa, M.; Min, W.; Hayashi, S.; Fukushima, S. Food Restriction Inhibits the Growth of Intestinal Polyps in Multiple Intestinal Neoplasia Mouse. Jpn. J. Cancer Res. 2002, 93, 236–241. [Google Scholar] [CrossRef]
- Tsao, J.-L.; Dudley, S.; Kwok, B.; Nickel, A.E.; Laird, P.W.; Siegmund, K.D.; Liskay, R.M.; Shibata, D. Diet, cancer and aging in DNA mismatch repair deficient mice. Carcinogenesis 2002, 23, 1807–1810. [Google Scholar] [CrossRef]
- Pugh, T.D.; Oberley, T.D.; Weindruch, R. Dietary intervention at middle age: Caloric restriction but not dehydroepiandrosterone sulfate increases lifespan and lifetime cancer incidence in mice. Cancer Res. 1999, 59, 1642–1648. [Google Scholar]
- Mihaylova, M.M.; Sabatini, D.M.; Yilmaz, Ö.H. Dietary and Metabolic Control of Stem Cell Function in Physiology and Cancer. Cell Stem Cell 2014, 14, 292–305. [Google Scholar] [CrossRef] [PubMed]
- Demark-Wahnefried, W.; Rais-Bahrami, S.; Desmond, R.A.; Gordetsky, J.B.; Hunter, G.R.; Yang, E.S.; Azrad, M.; Frugé, A.; Tsuruta, Y.; Norian, L.; et al. Presurgical weight loss affects tumour traits and circulating biomarkers in men with prostate cancer. Br. J. Cancer 2017, 117, 1303–1313. [Google Scholar] [CrossRef] [PubMed]
- Kristal, A.R.; Blount, P.L.; Schenk, J.M.; Sanchez, C.A.; Rabinovitch, P.S.; Odze, R.D.; Standley, J.; Vaughan, T.L.; Reid, B.J. Low-Fat, High Fruit and Vegetable Diets and Weight Loss Do Not Affect Biomarkers of Cellular Proliferation in Barrett Esophagus. Cancer Epidemiol. Biomark. Prev. 2005, 14, 2377–2383. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Demark-Wahnefried, W.; Rogers, L.Q.; Gibson, J.; Harada, S.; Frugé, A.; Oster, R.A.; Grizzle, W.E.; Norian, L.A.; Yang, E.S.; Della Manna, D.; et al. Randomized trial of weight loss in primary breast cancer: Impact on body composition, circulating biomarkers and tumor characteristics. Int. J. Cancer 2019, 146, 2784–2796. [Google Scholar] [CrossRef] [PubMed]
- Duszka, K.; Ellero-Simatos, S.; Ow, G.S.; Defernez, M.; Paramalingam, E.; Tett, A.; Ying, S.; König, J.; Narbad, A.; Kuznetsov, V.A.; et al. Complementary intestinal mucosa and microbiota responses to caloric restriction. Sci. Rep. 2018, 8, 1–15. [Google Scholar] [CrossRef]
- Hammer, S.T.G.; Greenson, J.K. The Clinical Significance of Duodenal Lymphocytosis with Normal Villus Architecture. Arch. Pathol. Lab. Med. 2013, 137, 1216–1219. [Google Scholar] [CrossRef]
- Marsh, M.N.; Rostami, K. What Is A Normal Intestinal Mucosa? Gastroenterology 2016, 151, 784–788. [Google Scholar] [CrossRef]
- Rostami, K.; Marsh, M.N.; Johnson, M.W.; Mohaghegh, H.; Heal, C.; Holmes, G.; Ensari, A.; Aldulaimi, D.; Bancel, B.; Bassotti, G.; et al. ROC-king onwards: Intraepithelial lymphocyte counts, distribution & role in coeliac disease mucosal interpretation. Gut 2017, 66, 2080–2086. [Google Scholar] [CrossRef]
- Yousefi, M.; Nakauka-Ddamba, A.; Berry, C.; Li, N.; Schoenberger, J.; Simeonov, K.P.; Cedeno, R.J.; Yu, Z.; Lengner, C.J. Calorie Restriction Governs Intestinal Epithelial Regeneration through Cell-Autonomous Regulation of mTORC1 in Reserve Stem Cells. Stem Cell Rep. 2018, 10, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Peña-Villalobos, I.; Casanova-Maldonado, I.; Lois, P.; Sabat, P.; Palma, V. Adaptive Physiological and Morphological Adjustments Mediated by Intestinal Stem Cells in Response to Food Availability in Mice. Front. Physiol. 2019, 9, 1821. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, Ö.H.; Katajisto, P.; Lamming, D.; Gültekin, Y.; Bauer-Rowe, K.E.; Sengupta, S.; Birsoy, K.; Dursun, A.; Yilmaz, V.O.; Selig, M.; et al. mTORC1 in the Paneth cell niche couples intestinal stem-cell function to calorie intake. Nature 2012, 486, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Huels, D.J.; Bruens, L.; Hodder, M.C.; Cammareri, P.; Campbell, A.D.; Ridgway, R.A.; Gay, D.M.; Solar-Abboud, M.; Faller, W.J.; Nixon, C.; et al. Wnt ligands influence tumour initiation by controlling the number of intestinal stem cells. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Bultman, S.J. Interplay between diet, gut microbiota, epigenetic events, and colorectal cancer. Mol. Nutr. Food Res. 2017, 61, 61. [Google Scholar] [CrossRef] [PubMed]
- Fink, S.P.; Myeroff, L.L.; Kariv, R.; Platzer, P.; Xin, B.; Mikkola, D.; Lawrence, E.; Morris, N.; Nosrati, A.; Willson, J.K.V.; et al. Induction of KIAA1199/CEMIP is associated with colon cancer phenotype and poor patient survival. Oncotarget 2015, 6, 30500–30515. [Google Scholar] [CrossRef]
- Schulze, A. Cancer metabolism—An update. Mol. Metab. 2020, 33, 1. [Google Scholar] [CrossRef]
- Jones, R.G.; Thompson, C.B. Tumor suppressors and cell metabolism: A recipe for cancer growth. Genes Dev. 2009, 23, 537–548. [Google Scholar] [CrossRef]
- Jia, D.; Li, X.; Bocci, F.; Tripathi, S.; Deng, Y.; Jolly, M.K.; Onuchic, J.N.; Levine, H. Quantifying Cancer Epithelial-Mesenchymal Plasticity and its Association with Stemness and Immune Response. J. Clin. Med. 2019, 8, 725. [Google Scholar] [CrossRef] [PubMed]
- Schulze, A.; Harris, A. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature 2012, 491, 364–373. [Google Scholar] [CrossRef]
- Frezza, C. Metabolism and cancer: The future is now. Br. J. Cancer 2020, 122, 133–135. [Google Scholar] [CrossRef]
- Thiruvengadam, S.S.; O’Malley, M.; LaGuardia, L.; Lopez, R.; Wang, Z.; Shadrach, B.L.; Chen, Y.; Li, C.; Veigl, M.L.; Barnholtz-Sloan, J.S.; et al. Gene Expression Changes Accompanying the Duodenal Adenoma-Carcinoma Sequence in Familial Adenomatous Polyposis. Clin. Transl. Gastroenterol. 2019, 10, e00053. [Google Scholar] [CrossRef]
- Krämer, A.; Green, J.; Pollard, J.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Gregor, A.; Pignitter, M.; Fahrngruber, C.; Bayer, S.; Somoza, V.; König, J.; Duszka, K. Caloric restriction increases levels of taurine in the intestine and stimulates taurine uptake by conjugation to glutathione. J. Nutr. Biochem. 2021, 96, 108781. [Google Scholar] [CrossRef]
- Lyons, Y.A.; Wu, S.Y.; Overwijk, W.W.; Baggerly, K.A.; Sood, A.K. Immune cell profiling in cancer: Molecular approaches to cell-specific identification. Precis. Oncol. 2017, 1, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Paoni, N.F.; Feldman, M.W.; Gutierrez, L.S.; Ploplis, V.A.; Castellino, F.J. Transcriptional profiling of the transition from normal intestinal epithelia to adenomas and carcinomas in the APCMin/+ mouse. Physiol. Genom. 2003, 15, 228–235. [Google Scholar] [CrossRef][Green Version]
- Sansom, O.J.; Reed, K.R.; Hayes, A.J.; Ireland, H.; Brinkmann, H.; Newton, I.P.; Batlle, E.; Simon-Assmann, P.; Clevers, H.; Näthke, I.; et al. Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes Dev. 2004, 18, 1385–1390. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, Y.; Yamamichi, N.; Tomida, S.; Takeuchi, C.; Kageyama-Yahara, N.; Takahashi, Y.; Shiogama, K.; Inada, K.-I.; Ichinose, M.; Fujishiro, M.; et al. Identification of marker genes and pathways specific to precancerous duodenal adenomas and early stage adenocarcinomas. J. Gastroenterol. 2019, 54, 131–140. [Google Scholar] [CrossRef]
- Pesson, M.; Volant, A.; Uguen, A.; Trillet, K.; De La Grange, P.; Aubry, M.; Daoulas, M.; Robaszkiewicz, M.; LE Gac, G.; Morel, A.; et al. A Gene Expression and Pre-mRNA Splicing Signature That Marks the Adenoma-Adenocarcinoma Progression in Colorectal Cancer. PLoS ONE 2014, 9, e87761. [Google Scholar] [CrossRef]
- Sabates-Bellver, J.; Van Der Flier, L.G.; De Palo, M.; Cattaneo, E.; Maake, C.; Rehrauer, H.; Laczko, E.; Kurowski, M.A.; Bujnicki, J.M.; Menigatti, M.; et al. Transcriptome Profile of Human Colorectal Adenomas. Mol. Cancer Res. 2007, 5, 1263–1275. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Yao, Y.; Yang, W.; Zheng, C.-H.; Zhao, M.; Xia, J. dbCPG: A web resource for cancer predisposition genes. Oncotarget 2016, 7, 37803–37811. [Google Scholar] [CrossRef]
- Zhao, M.; Kim, P.; Mitra, R.; Zhao, J.; Zhao, Z. TSGene 2.0: An updated literature-based knowledgebase for tumor suppressor genes. Nucleic Acids Res. 2016, 44, D1023–D1031. [Google Scholar] [CrossRef]
- Zhao, M.; Liu, Y.; Zheng, C.; Qu, H. dbEMT 2.0: An updated database for epithelial-mesenchymal transition genes with experimentally verified information and precalculated regulation information for cancer metastasis. J. Genet. Genom. 2019, 46, 595–597. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Perrotti, N.; Stournaras, C. Colorectal carcinoma cells—Regulation of survival and growth by SGK1. Int. J. Biochem. Cell Biol. 2010, 42, 1571–1575. [Google Scholar] [CrossRef]
- Wang, C.; Shi, M.; Ji, J.; Cai, Q.; Zhao, Q.; Jiang, J.; Liu, J.; Zhang, H.; Zhu, Z.; Zhang, J. Stearoyl-CoA desaturase 1 (SCD1) facilitates the growth and anti-ferroptosis of gastric cancer cells and predicts poor prognosis of gastric cancer. Aging 2020, 12, 15374–15391. [Google Scholar] [CrossRef]
- Hoesel, B.; Schmid, J. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef]
- Shen, Y.; Yao, H.; Li, A.; Wang, M. CSCdb: A cancer stem cells portal for markers, related genes and functional information. Database 2016, 2016. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Uhlén, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef]
- Haigis, M.C.; Deng, C.-X.; Finley, L.W.; Kim, H.-S.; Gius, D. SIRT3 Is a Mitochondrial Tumor Suppressor: A Scientific Tale That Connects Aberrant Cellular ROS, the Warburg Effect, and Carcinogenesis. Cancer Res. 2012, 72, 2468–2472. [Google Scholar] [CrossRef]
- Cheng, Y.; Fu, L.L.; Wen, X.; Wang, X.Y.; Liu, J.; Huang, J. Sirtuin-3 (SIRT3), a therapeutic target with oncogenic and tumor-suppressive function in cancer. Cell Death Dis. 2014, 5, e1047. [Google Scholar] [CrossRef]
- Nakamura, Y.; Yamamoto, M.; Kumamaru, E. Very Low-Density Lipoprotein Receptor in Fetal Intestine and Gastric Adenocarcinoma Cells. Arch. Pathol. Lab. Med. 2000, 124, 119–122. [Google Scholar] [CrossRef]
- Chen, T.; Wu, F.; Chen, F.-M.; Tian, J.; Qu, S. Variations of very low-density lipoprotein receptor subtype expression in gastrointestinal adenocarcinoma cells with various differentiations. World J. Gastroenterol. 2005, 11, 2817–2821. [Google Scholar] [CrossRef]
- Renaud, H.J.; Cui, J.Y.; Lu, H.; Klaassen, C.D. Effect of Diet on Expression of Genes Involved in Lipid Metabolism, Oxidative Stress, and Inflammation in Mouse Liver–Insights into Mechanisms of Hepatic Steatosis. PLoS ONE 2014, 9, e88584. [Google Scholar] [CrossRef] [PubMed]
- Mehto, S.; Jena, K.K.; Nath, P.; Chauhan, S.; Kolapalli, S.P.; Das, S.K.; Sahoo, P.K.; Jain, A.; Taylor, G.A.; Chauhan, S. The Crohn’s Disease Risk Factor IRGM Limits NLRP3 Inflammasome Activation by Impeding Its Assembly and by Mediating Its Selective Autophagy. Mol. Cell 2019, 73, 429–445.e7. [Google Scholar] [CrossRef]
- Rozewicki, J.; Li, S.; Amada, K.M.; Standley, D.M.; Katoh, K. MAFFT-DASH: Integrated protein sequence and structural alignment. Nucleic Acids Res. 2019, 47, W5–W10. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Uthaiah, R.; Howard, J.; Herrmann, C.; Wolf, E. Crystal Structure of IIGP1. Mol. Cell 2004, 15, 727–739. [Google Scholar] [CrossRef]
- Liu, M.; Guo, S.; Hibbert, J.M.; Jain, V.; Singh, N.; Wilson, N.O.; Stiles, J.K. CXCL10/IP-10 in infectious diseases pathogenesis and potential therapeutic implications. Cytokine Growth Factor Rev. 2011, 22, 121–130. [Google Scholar] [CrossRef]
- Lee, C.; Lin, Y.; Huang, M.; Lin, C.; Liu, C.; Chow, J.; Liu, H.E. Increased cellular glutathione and protection by bone marrow stromal cells account for the resistance of non-acute promylocytic leukemia acute myeloid leukemia cells to arsenic trioxidein vivo. Leuk. Lymphoma 2006, 47, 521–529. [Google Scholar] [CrossRef]
- Ohshima, K.; Morii, E. Metabolic Reprogramming of Cancer Cells during Tumor Progression and Metastasis. Metabolites 2021, 11, 28. [Google Scholar] [CrossRef] [PubMed]
- García-Miranda, P.; Peral, M.J.; Ilundain, A.A. Rat small intestine expresses the reelin-Disabled-1 signalling pathway. Exp. Physiol. 2010, 95, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Palethorpe, H.M.; Drew, P.A.; Smith, E. Androgen Signaling in Esophageal Adenocarcinoma Cell Lines In Vitro. Dig. Dis. Sci. 2017, 62, 3402–3414. [Google Scholar] [CrossRef]
- Smith, E.; Palethorpe, H.M.; Ruszkiewicz, A.R.; Edwards, S.; Leach, D.A.; Underwood, T.J.; Need, E.F.; Drew, P.A. Androgen Receptor and Androgen-Responsive Gene FKBP5 Are Independent Prognostic Indicators for Esophageal Adenocarcinoma. Dig. Dis. Sci. 2015, 61, 433–443. [Google Scholar] [CrossRef]
- Zhou, H.; Guo, W.; Zhao, Y.; Wang, Y.; Zha, R.; Ding, J.; Liang, L.; Yang, G.; Chen, Z.; Ma, B.; et al. Micro RNA -135a acts as a putative tumor suppressor by directly targeting very low density lipoprotein receptor in human gallbladder cancer. Cancer Sci. 2014, 105, 956–965. [Google Scholar] [CrossRef]
- Santaolalla, R.; Sussman, D.A.; Ruiz, J.R.; Davies, J.M.; Pastorini, C.; España, C.L.; Sotolongo, J.; Burlingame, O.; Bejarano, P.A.; Philip, S.; et al. TLR4 Activates the β-catenin Pathway to Cause Intestinal Neoplasia. PLoS ONE 2013, 8, e63298. [Google Scholar] [CrossRef]
- Fukata, M.; Chen, A.; Vamadevan, A.S.; Cohen, J.; Breglio, K.; Krishnareddy, S.; Hsu, D.; Xu, R.; Harpaz, N.; Dannenberg, A.J.; et al. Toll-Like Receptor-4 Promotes the Development of Colitis-Associated Colorectal Tumors. Gastroenterology 2007, 133, 1869–1869.e14. [Google Scholar] [CrossRef]
- Niedzielska, I.; Niedzielski, Z.; Tkacz, M.; Orawczyk, T.; Ziaja, K.; Starzewski, J.; Mazurek, U.; Markowski, J. Toll-like receptors and the tendency of normal mucous membrane to transform to polyp or colorectal cancer. J. Physiol. Pharmacol. 2009, 60, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Simiantonaki, N.; Kurzik-Dumke, U.; Karyofylli, G.; Jayasinghe, C.; Michel-Schmidt, R.; Kirkpatrick, C.J. Reduced expression of TLR4 is associated with the metastatic status of human colorectal cancer. Int. J. Mol. Med. 2007, 20, 21–29. [Google Scholar] [CrossRef]
- Gwon, D.H.; Lee, W.-Y.; Shin, N.; I Kim, S.; Jeong, K.; Lee, W.-H.; Kim, D.W.; Hong, J.; Lee, S.Y. BMAL1 Suppresses Proliferation, Migration, and Invasion of U87MG Cells by Downregulating Cyclin B1, Phospho-AKT, and Metalloproteinase-9. Int. J. Mol. Sci. 2020, 21, 2352. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Cheng, B.; Xie, M.; Chen, Y.; Zhao, J.; Zhou, X.; Chen, L. Circadian Clock Gene Bmal1 Inhibits Tumorigenesis and Increases Paclitaxel Sensitivity in Tongue Squamous Cell Carcinoma. Cancer Res. 2017, 77, 532–544. [Google Scholar] [CrossRef]
- Fekry, B.; Ribas-Latre, A.; Baumgartner, C.; Deans, J.R.; Kwok, C.; Patel, P.; Fu, L.; Berdeaux, R.; Sun, K.; Kolonin, M.G.; et al. Incompatibility of the circadian protein BMAL1 and HNF4α in hepatocellular carcinoma. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.-M.; Shay, J.; Zeng, T.-C.; Chou, J.-L.; Huang, T.H.-M.; Lai, H.-C.; Chan, M. Epigenetic silencing of ARNTL, a circadian gene and potential tumor suppressor in ovarian cancer. Int. J. Oncol. 2014, 45, 2101–2107. [Google Scholar] [CrossRef]
- Asuthkar, S.; Stepanova, V.; Lebedeva, T.; Holterman, A.L.; Estes, N.; Cines, D.B.; Rao, J.S.; Gondi, C.S. Multifunctional roles of urokinase plasminogen activator (uPA) in cancer stemness and chemoresistance of pancreatic cancer. Mol. Biol. Cell 2013, 24, 2620–2632. [Google Scholar] [CrossRef]
- Al Hossiny, M.; Luo, L.; Frazier, W.R.; Steiner, N.; Gusev, Y.; Kallakury, B.; Glasgow, E.; Creswell, K.; Madhavan, S.; Kumar, R.; et al. Ly6E/K Signaling to TGFβ Promotes Breast Cancer Progression, Immune Escape, and Drug Resistance. Cancer Res. 2016, 76, 3376–3386. [Google Scholar] [CrossRef]
- Park, J.W.; Park, J.M.; Park, D.M.; Kim, D.-Y.; Kim, H.K. Stem Cells Antigen-1 Enriches for a Cancer Stem Cell-Like Subpopulation in Mouse Gastric Cancer. Stem Cells 2016, 34, 1177–1187. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yang, H.; Zhu, R.; Zhao, X.; Liu, L.; Zhou, Z.; Zhao, L.; Liang, B.; Ma, W.; Zhao, J.; Liu, J.; et al. Sirtuin-mediated deacetylation of hnRNP A1 suppresses glycolysis and growth in hepatocellular carcinoma. Oncogene 2019, 38, 4915–4931. [Google Scholar] [CrossRef] [PubMed]
- Amano, H.; Chaudhury, A.; Rodriguez-Aguayo, C.; Lu, L.; Akhanov, V.; Catic, A.; Popov, Y.V.; Verdin, E.; Johnson, H.; Stossi, F.; et al. Telomere Dysfunction Induces Sirtuin Repression that Drives Telomere-Dependent Disease. Cell Metab. 2019, 29, 1274–1290.e9. [Google Scholar] [CrossRef]
- Long, T.M.; Chakrabarti, A.; Ezelle, H.J.; Brennan-Laun, S.E.; Raufman, J.-P.; Polyakova, I.; Silverman, R.H.; Hassel, B.A. RNase-L deficiency exacerbates experimental colitis and colitis-associated cancer. Inflamm. Bowel Dis. 2013, 19, 1295–1305. [Google Scholar] [CrossRef]
- Burke, J.M.; Moon, S.; Matheny, T.; Parker, R. RNase L Reprograms Translation by Widespread mRNA Turnover Escaped by Antiviral mRNAs. Mol. Cell 2019, 75, 1203–1217.e5. [Google Scholar] [CrossRef] [PubMed]
- Mullan, P.B.; Hosey, A.M.; Buckley, N.E.; Quinn, J.E.; Kennedy, R.D.; Johnston, P.G.; Harkin, D.P. The 2,5 oligoadenylate synthetase/RNaseL pathway is a novel effector of BRCA1- and interferon-γ-mediated apoptosis. Oncogene 2005, 24, 5492–5501. [Google Scholar] [CrossRef]
- Krüger, S.; Silber, A.-S.; Engel, C.; Görgens, H.; Mangold, E.; Pagenstecher, C.; Holinski-Feder, E.; Doeberitz, M.V.K.; Moeslein, G.; Dietmaier, W.; et al. Arg462Gln sequence variation in the prostate-cancer-susceptibility gene RNASEL and age of onset of hereditary non-polyposis colorectal cancer: A case-control study. Lancet Oncol. 2005, 6, 566–572. [Google Scholar] [CrossRef]
- Krüger, S.; Engel, C.; Bier, A.; Silber, A.-S.; Görgens, H.; Mangold, E.; Pagenstecher, C.; Holinski-Feder, E.; Doeberitz, M.V.K.; Royer-Pokora, B.; et al. The additive effect of p53 Arg72Pro and RNASEL Arg462Gln genotypes on age of disease onset in Lynch syndrome patients with pathogenic germline mutations in MSH2 or MLH1. Cancer Lett. 2007, 252, 55–64. [Google Scholar] [CrossRef]
- Batman, G.; Oliver, A.W.; Zehbe, I.; Richard, C.; Hampson, L.; Hampson, I.N. Lopinavir up-regulates expression of the antiviral protein ribonuclease L in human papillomavirus-positive cervical carcinoma cells. Antivir. Ther. 2011, 16, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Li, G.; Li, Y.; Gaughan, C.; Baskar, D.; Parker, Y.; Lindner, D.J.; Weiss, S.R.; Silverman, R.H. RNase L is a negative regulator of cell migration. Oncotarget 2015, 6, 44360–44372. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, N.; Ye, J.; Hirakawa, J.; Kawashima, H. Forced Expression of CXCL10 Prevents Liver Metastasis of Colon Carcinoma Cells by the Recruitment of Natural Killer Cells. Biol. Pharm. Bull. 2019, 42, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Wangpu, X.; Yang, X.; Zhao, J.; Lu, J.; Guan, S.; Lu, J.; Kovacevic, Z.; Liu, W.; Mi, L.; Jin, R.; et al. The metastasis suppressor, NDRG1, inhibits “stemness” of colorectal cancer via down-regulation of nuclear beta-catenin and CD44. Oncotarget 2015, 6, 33893–33911. [Google Scholar] [CrossRef]
- Sahni, S.; Bae, D.H.; Lane, D.J.; Kovacevic, Z.; Kalinowski, D.S.; Jansson, P.J.; Richardson, D.R. The Metastasis Suppressor, N-myc Downstream-Regulated Gene 1 (NDRG1), Inhibits Stress-Induced Autophagy in Cancer Cells. J. Biol. Chem. 2014, 289, 9692–9709. [Google Scholar] [CrossRef] [PubMed]
- Strzelczyk, B.; Szulc, A.; Rzepko, R.; Kitowska, A.; Skokowski, J.; Szutowicz, A.; Pawelczyk, T. Identification of High-Risk Stage II Colorectal Tumors by Combined Analysis of the NDRG1 Gene Expression and the Depth of Tumor Invasion. Ann. Surg. Oncol. 2009, 16, 1287–1294. [Google Scholar] [CrossRef] [PubMed]
- Park, K.C.; Paluncic, J.; Kovacevic, Z.; Richardson, D.R. Pharmacological targeting and the diverse functions of the metastasis suppressor, NDRG1, in cancer. Free Radic. Biol. Med. 2020, 157, 154–175. [Google Scholar] [CrossRef]
- Sundararaghavan, V.L.; Sindhwani, P.; Hinds, T.D. Glucuronidation and UGT isozymes in bladder: New targets for the treatment of uroepithelial carcinomas? Oncotarget 2017, 8, 3640–3648. [Google Scholar] [CrossRef]
- Guengerich, F.P. Cytochrome P450 research and The Journal of Biological Chemistry. J. Biol. Chem. 2019, 294, 1671–1680. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Du, C.; Ji, D.; Xi, J.; Gu, J. Overexpression of LAMC2 predicts poor prognosis in colorectal cancer patients and promotes cancer cell proliferation, migration, and invasion. Tumour Biol. 2017, 39, 1010428317705849. [Google Scholar] [CrossRef]
- Furuta, E.; Okuda, H.; Kobayashi, A.; Watabe, K. Metabolic genes in cancer: Their roles in tumor progression and clinical implications. Biochim. Biophys. Acta 2010, 1805, 141–152. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, H.; Lai, L.; Wang, X.; Loera, S.; Xue, L.; He, H.; Zhang, K.; Hu, S.; Huang, Y.; et al. Ribonucleotide reductase small subunit M2 serves as a prognostic biomarker and predicts poor survival of colorectal cancers. Clin. Sci. 2013, 124, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Fujita, H.; Ohuchida, K.; Mizumoto, K.; Itaba, S.; Ito, T.; Nakata, K.; Yu, J.; Kayashima, T.; Souzaki, R.; Tajiri, T.; et al. Gene Expression Levels as Predictive Markers of Outcome in Pancreatic Cancer after Gemcitabine-Based Adjuvant Chemotherapy. Neoplasia 2010, 12, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.Y.; Chou, C.J.; Lo, H.L.; Yang, P.M. Repositioning of a cyclin-dependent kinase inhibitor GW8510 as a ribonucleotide reductase M2 inhibitor to treat human colorectal cancer. Cell Death Discov. 2016, 2, 16027. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Aldrees, M.; Arif, M.; Li, X.; Mardinoglu, A.; Aziz, M.A. Elucidating the Reprograming of Colorectal Cancer Metabolism Using Genome-Scale Metabolic Modeling. Front. Oncol. 2019, 9, 681. [Google Scholar] [CrossRef]
- Tomita, H.; Tanaka, K.; Tanaka, T.; Hara, A. Aldehyde dehydrogenase 1A1 in stem cells and cancer. Oncotarget 2016, 7, 11018–11032. [Google Scholar] [CrossRef]
- Krause, K.; Caution, K.; Badr, A.; Hamilton, K.; Saleh, A.; Patel, K.; Seveau, S.; Hall-Stoodley, L.; Hegazi, R.; Zhang, X.; et al. CASP4/caspase-11 promotes autophagosome formation in response to bacterial infection. Autophagy 2018, 14, 1928–1942. [Google Scholar] [CrossRef]
- Glickman, J.N. The Nonneoplastic Small Intestine. Abdominal Key. 2018. Available online: https://abdominalkey.com/the-nonneoplastic-small-intestine (accessed on 1 January 2021).
- Kennedy, L.; Sandhu, J.K.; Harper, M.E.; Cuperlovic-Culf, M. Role of Glutathione in Cancer: From Mechanisms to Therapies. Biomolecules 2020, 10, 1429. [Google Scholar] [CrossRef]
- Yang, E.; Shen, J. The roles and functions of Paneth cells in Crohn’s disease: A critical review. Cell Prolif. 2021, 54, e12958. [Google Scholar] [CrossRef]
- Nakanishi, Y.; Reina-Campos, M.; Nakanishi, N.; Llado, V.; Elmen, L.; Peterson, S.; Campos, A.; De, S.K.; Leitges, M.; Ikeuchi, H.; et al. Control of Paneth Cell Fate, Intestinal Inflammation, and Tumorigenesis by PKClambda/iota. Cell Rep. 2016, 16, 3297–3310. [Google Scholar] [CrossRef]
- Wang, D.; Peregrina, K.; Dhima, E.; Lin, E.Y.; Mariadason, J.M.; Augenlicht, L.H. Paneth cell marker expression in intestinal villi and colon crypts characterizes dietary induced risk for mouse sporadic intestinal cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 10272–10277. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Suzuki, K.; Sifuentes-Dominguez, L.; Miyata, N.; Song, J.; Lopez, A.; Starokadomskyy, P.; Gopal, P.; Dozmorov, I.; Tan, S.; et al. Paneth cell-derived growth factors support tumorigenesis in the small intestine. Life Sci. Alliance 2021, 4. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, D.; Pham, D.N.; Levesque, N.; Truongcao, M.; Foulkes, W.D.; Sapienza, C.; Rozen, R. Oncogenic role of PDK4 in human colon cancer cells. Br. J. Cancer 2017, 116, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yan, L.H.; Manoj, S.; Li, Y.; Lu, L. Central Role of CEMIP in Tumorigenesis and Its Potential as Therapeutic Target. J. Cancer 2017, 8, 2238–2246. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Fang, X.; Yang, Y.; Song, Y. Silencing of CEMIP suppresses Wnt/beta-catenin/Snail signaling transduction and inhibits EMT program of colorectal cancer cells. Acta Histochem. 2018, 120, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Xing, P.Y.; Pettersson, S.; Kundu, P. Microbial Metabolites and Intestinal Stem Cells Tune Intestinal Homeostasis. Proteomics 2020, 20, e1800419. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Sun, T.; Yu, Y.; Gao, Y.; Wang, X.; Chen, Z. MicroRNA-370 inhibits the proliferation, invasion and EMT of gastric cancer cells by directly targeting PAQR4. J. Pharmacol. Sci. 2018, 138, 96–106. [Google Scholar] [CrossRef]
- Gu, C.; Cai, J.; Xu, Z.; Zhou, S.; Ye, L.; Yan, Q.; Zhang, Y.; Fang, Y.; Liu, Y.; Tu, C.; et al. MiR-532-3p suppresses colorectal cancer progression by disrupting the ETS1/TGM2 axis-mediated Wnt/beta-catenin signaling. Cell Death Dis. 2019, 10, 739. [Google Scholar] [CrossRef]
- Yousefi, M.; Li, L.; Lengner, C.J. Hierarchy and Plasticity in the Intestinal Stem Cell Compartment. Trends Cell Biol. 2017, 27, 753–764. [Google Scholar] [CrossRef]
- Tang, X.; Kuhlenschmidt, T.B.; Li, Q.; Ali, S.; Lezmi, S.; Chen, H.; Pires-Alves, M.; Laegreid, W.W.; Saif, T.A.; Kuhlenschmidt, M.S. A mechanically-induced colon cancer cell population shows increased metastatic potential. Mol. Cancer 2014, 13, 131. [Google Scholar] [CrossRef]
- Vidyasekar, P.; Shyamsunder, P.; Arun, R.; Santhakumar, R.; Kapadia, N.K.; Kumar, R.; Verma, R.S. Genome Wide Expression Profiling of Cancer Cell Lines Cultured in Microgravity Reveals Significant Dysregulation of Cell Cycle and MicroRNA Gene Networks. PLoS ONE 2015, 10, e0135958. [Google Scholar] [CrossRef] [PubMed]
- Regnier, M.; Polizzi, A.; Lippi, Y.; Fouche, E.; Michel, G.; Lukowicz, C.; Smati, S.; Marrot, A.; Lasserre, F.; Naylies, C.; et al. Insights into the role of hepatocyte PPARalpha activity in response to fasting. Mol. Cell. Endocrinol. 2018, 471, 75–88. [Google Scholar] [CrossRef]
- Kim, A.D.; Zhang, R.; Han, X.; Kang, K.A.; Piao, M.J.; Maeng, Y.H.; Chang, W.Y.; Hyun, J.W. Involvement of glutathione and glutathione metabolizing enzymes in human colorectal cancer cell lines and tissues. Mol. Med. Rep. 2015, 12, 4314–4319. [Google Scholar] [CrossRef]
- Bansal, A.; Simon, M.C. Glutathione metabolism in cancer progression and treatment resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar] [CrossRef] [PubMed]
- Narayanankutty, A.; Job, J.T.; Narayanankutty, V. Glutathione, an Antioxidant Tripeptide: Dual Roles in Carcinogenesis and Chemoprevention. Curr. Protein Pept. Sci. 2019, 20, 907–917. [Google Scholar] [CrossRef]
- Brautigam, L.; Zhang, J.; Dreij, K.; Spahiu, L.; Holmgren, A.; Abe, H.; Tew, K.D.; Townsend, D.M.; Kelner, M.J.; Morgenstern, R.; et al. MGST1, a GSH transferase/peroxidase essential for development and hematopoietic stem cell differentiation. Redox Biol. 2018, 17, 171–179. [Google Scholar] [CrossRef]
- Zhang, H.; Liao, L.H.; Liu, S.M.; Lau, K.W.; Lai, A.K.; Zhang, J.H.; Wang, Q.; Chen, X.Q.; Wei, W.; Liu, H.; et al. Microsomal glutathione S-transferase gene polymorphisms and colorectal cancer risk in a Han Chinese population. Int. J. Color. Dis. 2007, 22, 1185–1194. [Google Scholar] [CrossRef]
- Peng, L.; Linghu, R.; Chen, D.; Yang, J.; Kou, X.; Wang, X.Z.; Hu, Y.; Jiang, Y.Z.; Yang, J. Inhibition of glutathione metabolism attenuates esophageal cancer progression. Exp. Mol. Med. 2017, 49, e318. [Google Scholar] [CrossRef]
- Grasmann, G.; Smolle, E.; Olschewski, H.; Leithner, K. Gluconeogenesis in cancer cells—Repurposing of a starvation-induced metabolic pathway? Biochim. Biophys. Acta Rev. Cancer 2019, 1872, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Mears, H.V.; Emmott, E.; Chaudhry, Y.; Hosmillo, M.; Goodfellow, I.G.; Sweeney, T.R. Ifit1 regulates norovirus infection and enhances the interferon response in murine macrophage-like cells. Wellcome Open Res. 2019, 4, 82. [Google Scholar] [CrossRef] [PubMed]
- Leon-Cabrera, S.; Vazquez-Sandoval, A.; Molina-Guzman, E.; Delgado-Ramirez, Y.; Delgado-Buenrostro, N.L.; Callejas, B.E.; Chirino, Y.I.; Perez-Plasencia, C.; Rodriguez-Sosa, M.; Olguin, J.E.; et al. Deficiency in STAT1 Signaling Predisposes Gut Inflammation and Prompts Colorectal Cancer Development. Cancers 2018, 10, 341. [Google Scholar] [CrossRef] [PubMed]
- Topfer, K.; Kempe, S.; Muller, N.; Schmitz, M.; Bachmann, M.; Cartellieri, M.; Schackert, G.; Temme, A. Tumor evasion from T cell surveillance. J. Biomed. Biotechnol. 2011, 2011, 918471. [Google Scholar] [CrossRef]
- Kaplan, D.H.; Shankaran, V.; Dighe, A.S.; Stockert, E.; Aguet, M.; Old, L.J.; Schreiber, R.D. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc. Natl. Acad. Sci. USA 1998, 95, 7556–7561. [Google Scholar] [CrossRef]
- Shankaran, V.; Ikeda, H.; Bruce, A.T.; White, J.M.; Swanson, P.E.; Old, L.J.; Schreiber, R.D. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 2001, 410, 1107–1111. [Google Scholar] [CrossRef] [PubMed]
- Street, S.E.; Trapani, J.A.; MacGregor, D.; Smyth, M.J. Suppression of lymphoma and epithelial malignancies effected by interferon gamma. J. Exp. Med. 2002, 196, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E.; et al. Loss of IFN-gamma Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 2016, 167, 397–404.e399. [Google Scholar] [CrossRef]
- Meunier, E.; Broz, P. Interferon-inducible GTPases in cell autonomous and innate immunity. Cell Microbiol. 2016, 18, 168–180. [Google Scholar] [CrossRef]
- Liu, B.; Gulati, A.S.; Cantillana, V.; Henry, S.C.; Schmidt, E.A.; Daniell, X.; Grossniklaus, E.; Schoenborn, A.A.; Sartor, R.B.; Taylor, G.A. Irgm1-deficient mice exhibit Paneth cell abnormalities and increased susceptibility to acute intestinal inflammation. Am. J. Physiol. Liver Physiol. 2013, 305, G573–G584. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, Y.; Christian, S.L.; Ho, N.; Pongnopparat, T.; Licursi, M.; Hirasawa, K. Oncogenic Ras inhibits IRF1 to promote viral oncolysis. Oncogene 2015, 34, 3985–3993. [Google Scholar] [CrossRef]
- Hong, M.; Zhang, Z.; Chen, Q.; Lu, Y.; Zhang, J.; Lin, C.; Zhang, F.; Zhang, W.; Li, X.; Zhang, W.; et al. IRF1 inhibits the proliferation and metastasis of colorectal cancer by suppressing the RAS-RAC1 pathway. Cancer Manag. Res. 2019, 11, 369–378. [Google Scholar] [CrossRef]
- Ohsugi, T.; Yamaguchi, K.; Zhu, C.; Ikenoue, T.; Takane, K.; Shinozaki, M.; Tsurita, G.; Yano, H.; Furukawa, Y. Anti-apoptotic effect by the suppression of IRF1 as a downstream of Wnt/beta-catenin signaling in colorectal cancer cells. Oncogene 2019, 38, 6051–6064. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Hou, W.; Scharping, N.E.; Vendetti, F.P.; Srivastava, R.; Roy, C.N.; Menk, A.V.; Wang, Y.; Chauvin, J.M.; Karukonda, P.; et al. IRF1 Inhibits Antitumor Immunity through the Upregulation of PD-L1 in the Tumor Cell. Cancer Immunol. Res. 2019, 7, 1258–1266. [Google Scholar] [CrossRef]
- Kuznetsov, V.A.; Makalkin, I.A.; Taylor, M.A.; Perelson, A.S. Nonlinear dynamics of immunogenic tumors: Parameter estimation and global bifurcation analysis. Bull. Math. Biol. 1994, 56, 295–321. [Google Scholar] [CrossRef]
- Xiang, J.; Zhang, Y.; Tuo, L.; Liu, R.; Gou, D.; Liang, L.; Chen, C.; Xia, J.; Tang, N.; Wang, K. Transcriptomic changes associated with PCK1 overexpression in hepatocellular carcinoma cells detected by RNA-seq. Genes Dis. 2019. [Google Scholar] [CrossRef]
- Kim, S.; Kim, N.; Kang, K.; Kim, W.; Won, J.; Cho, J. Whole Transcriptome Analysis Identifies TNS4 as a Key Effector of Cetuximab and a Regulator of the Oncogenic Activity of KRAS Mutant Colorectal Cancer Cell Lines. Cells 2019, 8, 878. [Google Scholar] [CrossRef]
- Sawazaki, S.; Oshima, T.; Sakamaki, K.; Aoyama, T.; Sato, T.; Shiozawa, M.; Yoshikawa, T.; Rino, Y.; Imada, T.; Masuda, M. Clinical Significance of Tensin 4 Gene Expression in Patients with Gastric Cancer. Vivo 2017, 31, 1065–1071. [Google Scholar] [CrossRef][Green Version]
- Aguirre-Portoles, C.; Feliu, J.; Reglero, G.; Ramirez de Molina, A. ABCA1 overexpression worsens colorectal cancer prognosis by facilitating tumour growth and caveolin-1-dependent invasiveness, and these effects can be ameliorated using the BET inhibitor apabetalone. Mol. Oncol. 2018, 12, 1735–1752. [Google Scholar] [CrossRef]
- Xiao, Y.; Meierhofer, D. Glutathione Metabolism in Renal Cell Carcinoma Progression and Implications for Therapies. Int. J. Mol. Sci. 2019, 20, 3672. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Paffrath, V.; Clima, R.; Busch, J.F.; Rabien, A.; Kilic, E.; Villegas, S.; Timmermann, B.; Attimonelli, M.; Jung, K.; et al. Papillary Renal Cell Carcinomas Rewire Glutathione Metabolism and Are Deficient in Both Anabolic Glucose Synthesis and Oxidative Phosphorylation. Cancers 2019, 11, 1298. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.M.; Lester, E.T.; Tauber, D.; Parker, R. RNase L promotes the formation of unique ribonucleoprotein granules distinct from stress granules. J. Biol. Chem. 2020, 295, 1426–1438. [Google Scholar] [CrossRef]
- Banerjee, S.; Gusho, E.; Gaughan, C.; Dong, B.; Gu, X.; Holvey-Bates, E.; Talukdar, M.; Li, Y.; Weiss, S.R.; Sicheri, F.; et al. OAS-RNase L innate immune pathway mediates the cytotoxicity of a DNA-demethylating drug. Proc. Natl. Acad. Sci. USA 2019, 116, 5071–5076. [Google Scholar] [CrossRef]
- Zenke, K.; Muroi, M.; Tanamoto, K.I. IRF1 supports DNA binding of STAT1 by promoting its phosphorylation. Immunol. Cell Biol. 2018, 96, 1095–1103. [Google Scholar] [CrossRef]
- Wang, L.; Fouts, D.E.; Starkel, P.; Hartmann, P.; Chen, P.; Llorente, C.; DePew, J.; Moncera, K.; Ho, S.B.; Brenner, D.A.; et al. Intestinal REG3 Lectins Protect against Alcoholic Steatohepatitis by Reducing Mucosa-Associated Microbiota and Preventing Bacterial Translocation. Cell Host Microbe 2016, 19, 227–239. [Google Scholar] [CrossRef]
- Ducheix, S.; Peres, C.; Hardfeldt, J.; Frau, C.; Mocciaro, G.; Piccinin, E.; Lobaccaro, J.M.; De Santis, S.; Chieppa, M.; Bertrand-Michel, J.; et al. Deletion of Stearoyl-CoA Desaturase-1 From the Intestinal Epithelium Promotes Inflammation and Tumorigenesis, Reversed by Dietary Oleate. Gastroenterology 2018, 155, 1524–1538.e1529. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.; Cheng, F.; Zhao, J.; Zhao, Z. ccmGDB: A database for cancer cell metabolism genes. Nucleic Acids Res. 2016, 44, D959–D968. [Google Scholar] [CrossRef]
- Shinmura, K.; Kato, H.; Kawanishi, Y.; Nagura, K.; Kamo, T.; Okubo, Y.; Inoue, Y.; Kurabe, N.; Du, C.; Iwaizumi, M.; et al. SASS6 overexpression is associated with mitotic chromosomal abnormalities and a poor prognosis in patients with colorectal cancer. Oncol. Rep. 2015, 34, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, N.; Wang, X.; Shen, J.; Hong, X.; Yu, H.; Zhang, Y.; Wan, T.; Zhang, L.; Wang, J.; et al. Membrane protein hMYADM preferentially expressed in myeloid cells is up-regulated during differentiation of stem cells and myeloid leukemia cells. Life Sci. 2007, 80, 420–429. [Google Scholar] [CrossRef]
- Cusanelli, E.; Chartrand, P. Telomeric repeat-containing RNA TERRA: A noncoding RNA connecting telomere biology to genome integrity. Front. Genet. 2015, 6, 143. [Google Scholar] [CrossRef]
- Avogaro, L.; Querido, E.; Dalachi, M.; Jantsch, M.F.; Chartrand, P.; Cusanelli, E. Live-cell imaging reveals the dynamics and function of single-telomere TERRA molecules in cancer cells. RNA Biol. 2018, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Roake, C.M.; Artandi, S.E. Approaching TERRA Firma: Genomic Functions of Telomeric Noncoding RNA. Cell 2017, 170, 8–9. [Google Scholar] [CrossRef] [PubMed]
- Bryan, T.M.; Englezou, A.; Gupta, J.; Bacchetti, S.; Reddel, R.R. Telomere elongation in immortal human cells without detectable telomerase activity. EMBO J. 1995, 14, 4240–4248. [Google Scholar] [CrossRef]
- Dilley, R.L.; Greenberg, R.A. ALTernative Telomere Maintenance and Cancer. Trends Cancer 2015, 1, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Sille, F.C.; Thomas, R.; Smith, M.T.; Conde, L.; Skibola, C.F. Post-GWAS functional characterization of susceptibility variants for chronic lymphocytic leukemia. PLoS ONE 2012, 7, e29632. [Google Scholar] [CrossRef]
- Ozden, O.; Park, S.H.; Kim, H.S.; Jiang, H.; Coleman, M.C.; Spitz, D.R.; Gius, D. Acetylation of MnSOD directs enzymatic activity responding to cellular nutrient status or oxidative stress. Aging 2011, 3, 102–107. [Google Scholar] [CrossRef]
- Makino, N.; Oyama, J.; Maeda, T.; Koyanagi, M.; Higuchi, Y.; Shimokawa, I.; Mori, N.; Furuyama, T. FoxO1 signaling plays a pivotal role in the cardiac telomere biology responses to calorie restriction. Mol. Cell. Biochem. 2016, 412, 119–130. [Google Scholar] [CrossRef]
- Ozer, O.; Hickson, I.D. Pathways for maintenance of telomeres and common fragile sites during DNA replication stress. Open Biol. 2018, 8. [Google Scholar] [CrossRef]
- Hamada, T.; Kurachi, S.; Kurachi, K. Heterogeneous nuclear ribonucleoprotein A3 is the liver nuclear protein binding to age related increase element RNA of the factor IX gene. PLoS ONE 2010, 5, e12971. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.R.; Hung, S.C.; Wang, T.C. Telomeric DNA-binding activities of heterogeneous nuclear ribonucleoprotein A3 in vitro and in vivo. Biochim. Biophys. Acta 2010, 1803, 1164–1174. [Google Scholar] [CrossRef] [PubMed]
- Skaar, J.R.; Richard, D.J.; Saraf, A.; Toschi, A.; Bolderson, E.; Florens, L.; Washburn, M.P.; Khanna, K.K.; Pagano, M. INTS3 controls the hSSB1-mediated DNA damage response. J. Cell Biol. 2009, 187, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Braun, D.M.; Chung, I.; Kepper, N.; Deeg, K.I.; Rippe, K. TelNet—A database for human and yeast genes involved in telomere maintenance. BMC Genet. 2018, 19, 32. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.P.; Cifuentes-Rojas, C.; Kesner, B.; Aeby, E.; Lee, H.G.; Wei, C.; Oh, H.J.; Boukhali, M.; Haas, W.; Lee, J.T. TERRA RNA Antagonizes ATRX and Protects Telomeres. Cell 2017, 170, 86–101.e116. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maestri, E.; Duszka, K.; Kuznetsov, V.A. Immunity Depletion, Telomere Imbalance, and Cancer-Associated Metabolism Pathway Aberrations in Intestinal Mucosa upon Short-Term Caloric Restriction. Cancers 2021, 13, 3180. https://doi.org/10.3390/cancers13133180
Maestri E, Duszka K, Kuznetsov VA. Immunity Depletion, Telomere Imbalance, and Cancer-Associated Metabolism Pathway Aberrations in Intestinal Mucosa upon Short-Term Caloric Restriction. Cancers. 2021; 13(13):3180. https://doi.org/10.3390/cancers13133180
Chicago/Turabian StyleMaestri, Evan, Kalina Duszka, and Vladimir A. Kuznetsov. 2021. "Immunity Depletion, Telomere Imbalance, and Cancer-Associated Metabolism Pathway Aberrations in Intestinal Mucosa upon Short-Term Caloric Restriction" Cancers 13, no. 13: 3180. https://doi.org/10.3390/cancers13133180
APA StyleMaestri, E., Duszka, K., & Kuznetsov, V. A. (2021). Immunity Depletion, Telomere Imbalance, and Cancer-Associated Metabolism Pathway Aberrations in Intestinal Mucosa upon Short-Term Caloric Restriction. Cancers, 13(13), 3180. https://doi.org/10.3390/cancers13133180