
Integrative Analysis of Next-Generation Sequencing for Next-Generation Cancer Research toward Artificial Intelligence



{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
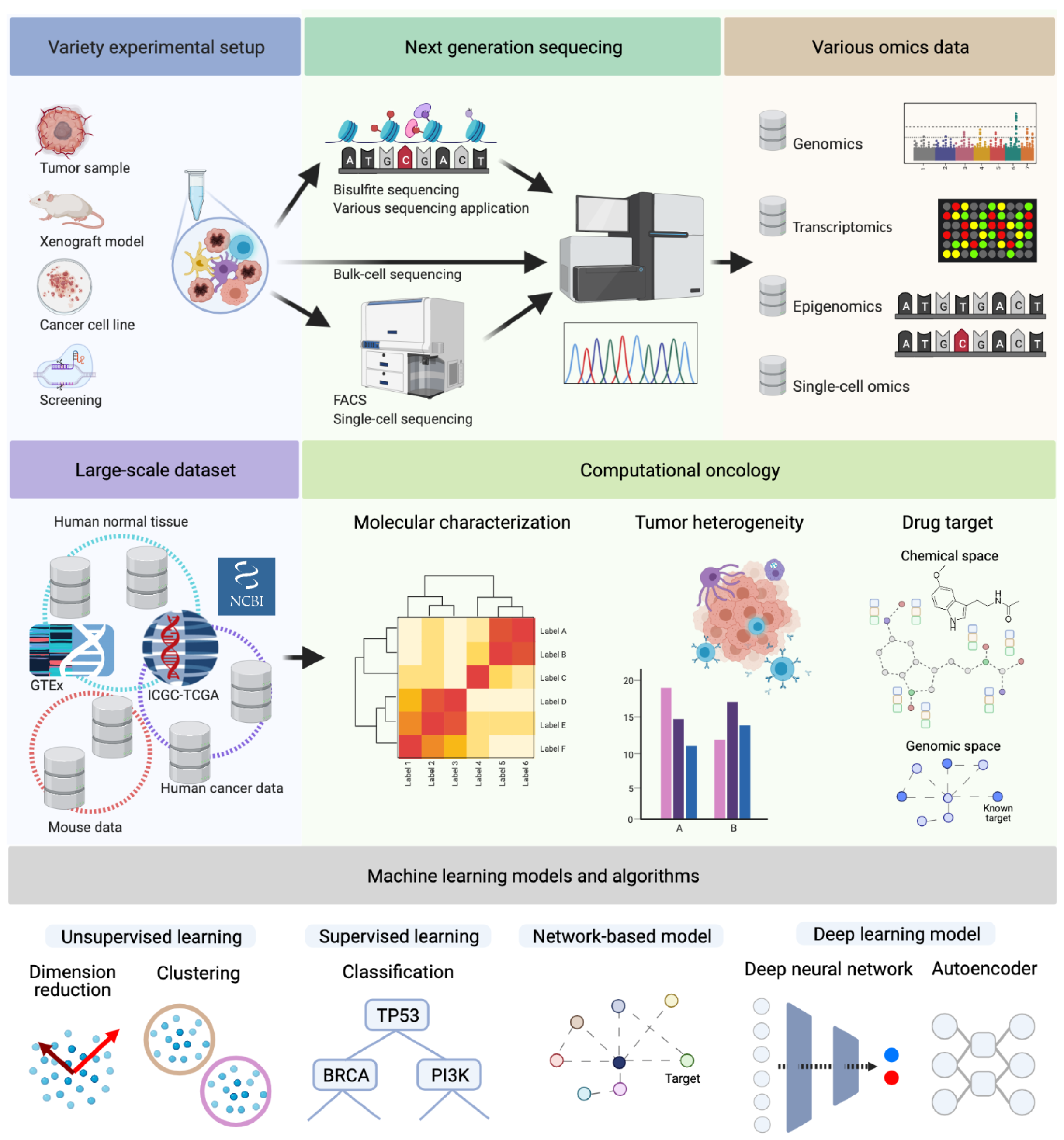
1. Introduction
1.1. Molecular Characterization and Multi-Omics Data
1.2. Tumor Heterogeneity: Cancer Genomics to Translational Medicine
1.3. Drug Target Identification
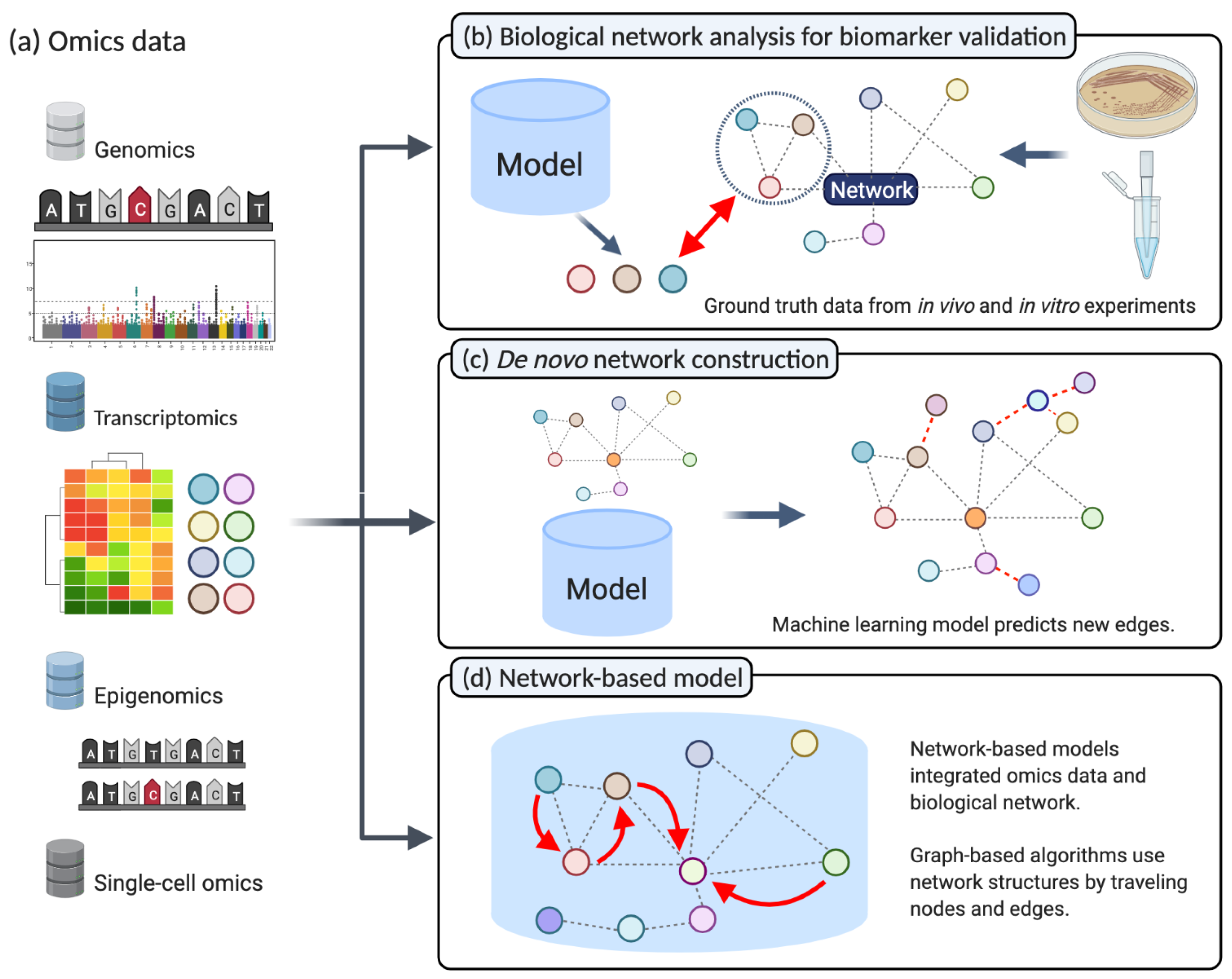
2. Systems Biology in Cancer Research
2.1. Biological Network Analysis for Biomarker Validation
2.2. De Novo Construction of Biological Networks
2.3. Network Based Machine Learning
3. Network-Based Learning in Cancer Research
3.1. Molecular Characterization with Network Information
3.2. Tumor Heterogeneity Study with Network Information
3.3. Drug Target Identification with Network Information
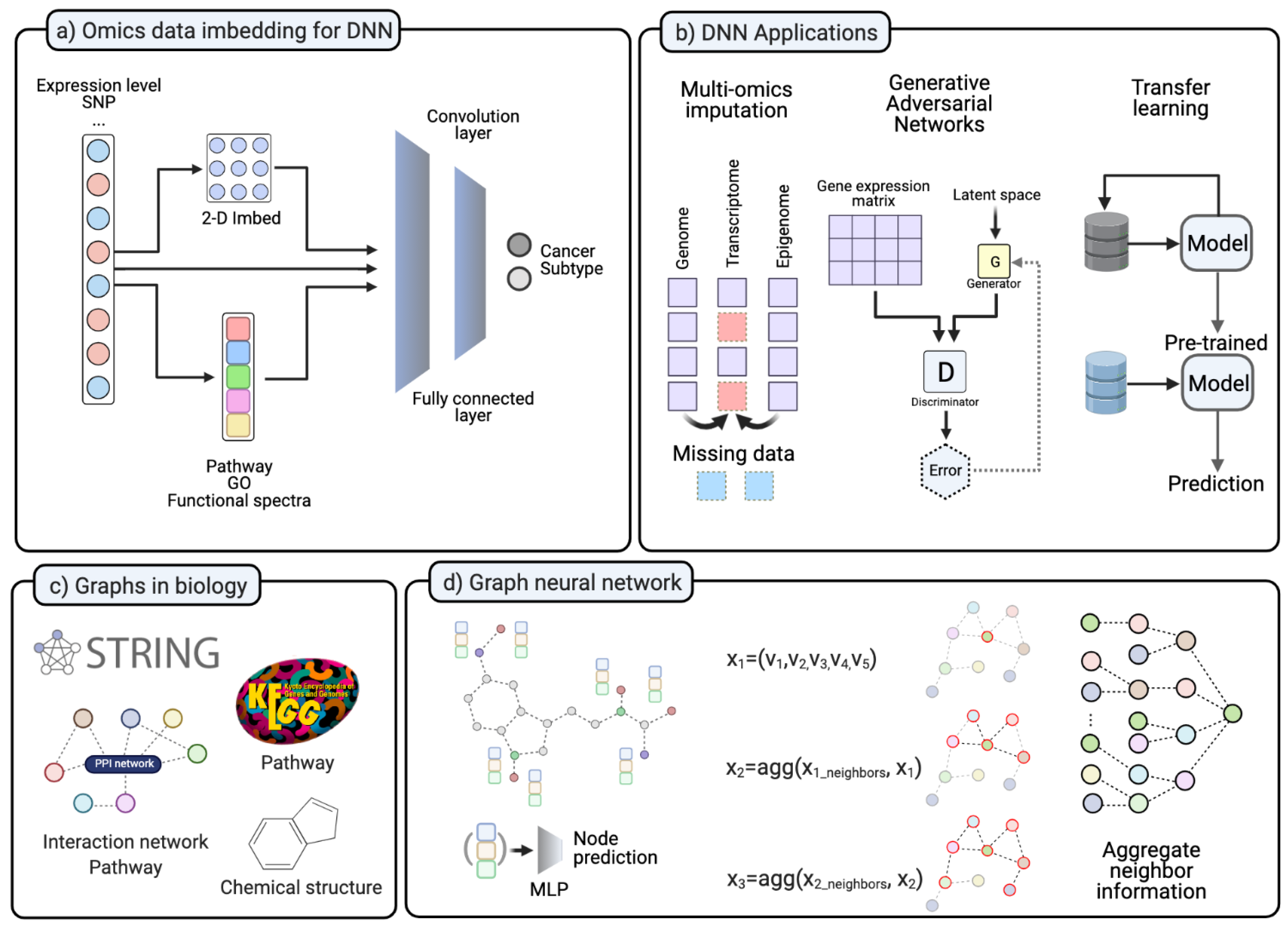
4. Deep Learning in Cancer Research
4.1. Challenges for Deep Learning in Cancer Research
4.2. Molecular Charactization with Network and DNN Model
4.3. Tumor Heterogeneity with Network and DNN Model
4.4. Drug Target Identification with Networks and DNN Models
4.5. Graph Neural Network Model
4.6. Shortcomings in AI and Revisiting Validity of Biological Networks as Prior Knowledge
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Janes, K.A.; Yaffe, M.B. Data-driven modelling of signal-transduction networks. Nat. Rev. Mol. Cell Biol. 2006, 7, 820–828. [Google Scholar] [CrossRef] [PubMed]
- Kreeger, P.K.; Lauffenburger, D.A. Cancer systems biology: A network modeling perspective. Carcinogenesis 2010, 31, 2–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vucic, E.A.; Thu, K.L.; Robison, K.; Rybaczyk, L.A.; Chari, R.; Alvarez, C.E.; Lam, W.L. Translating cancer ‘omics’ to improved outcomes. Genome Res. 2012, 22, 188–195. [Google Scholar] [CrossRef] [Green Version]
- Hoadley, K.A.; Yau, C.; Wolf, D.M.; Cherniack, A.D.; Tamborero, D.; Ng, S.; Leiserson, M.D.; Niu, B.; McLellan, M.D.; Uzunangelov, V.; et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 2014, 158, 929–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutter, C.; Zenklusen, J.C. The cancer genome atlas: Creating lasting value beyond its data. Cell 2018, 173, 283–285. [Google Scholar] [CrossRef]
- Chuang, H.Y.; Lee, E.; Liu, Y.T.; Lee, D.; Ideker, T. Network-based classification of breast cancer metastasis. Mol. Syst. Biol. 2007, 3, 140. [Google Scholar] [CrossRef]
- Zhang, W.; Chien, J.; Yong, J.; Kuang, R. Network-based machine learning and graph theory algorithms for precision oncology. NPJ Precis. Oncol. 2017, 1, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngiam, K.Y.; Khor, W. Big data and machine learning algorithms for health-care delivery. Lancet Oncol. 2019, 20, e262–e273. [Google Scholar] [CrossRef]
- Creixell, P.; Reimand, J.; Haider, S.; Wu, G.; Shibata, T.; Vazquez, M.; Mustonen, V.; Gonzalez-Perez, A.; Pearson, J.; Sander, C.; et al. Pathway and network analysis of cancer genomes. Nat. Methods 2015, 12, 615. [Google Scholar]
- Reyna, M.A.; Haan, D.; Paczkowska, M.; Verbeke, L.P.; Vazquez, M.; Kahraman, A.; Pulido-Tamayo, S.; Barenboim, J.; Wadi, L.; Dhingra, P.; et al. Pathway and network analysis of more than 2500 whole cancer genomes. Nat. Commun. 2020, 11, 729. [Google Scholar] [CrossRef]
- Luo, P.; Ding, Y.; Lei, X.; Wu, F.X. deepDriver: Predicting cancer driver genes based on somatic mutations using deep convolutional neural networks. Front. Genet. 2019, 10, 13. [Google Scholar] [CrossRef] [Green Version]
- Jiao, W.; Atwal, G.; Polak, P.; Karlic, R.; Cuppen, E.; Danyi, A.; De Ridder, J.; van Herpen, C.; Lolkema, M.P.; Steeghs, N.; et al. A deep learning system accurately classifies primary and metastatic cancers using passenger mutation patterns. Nat. Commun. 2020, 11, 728. [Google Scholar] [CrossRef]
- Chaudhary, K.; Poirion, O.B.; Lu, L.; Garmire, L.X. Deep learning–based multi-omics integration robustly predicts survival in liver cancer. Clin. Cancer Res. 2018, 24, 1248–1259. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.; Wang, W.; Tan, M.; Zhu, L.; Zhang, Y.; Fessler, E.; Vermeulen, L.; Wang, X. DeepCC: A novel deep learning-based framework for cancer molecular subtype classification. Oncogenesis 2019, 8, 44. [Google Scholar] [CrossRef]
- Zeng, X.; Zhu, S.; Liu, X.; Zhou, Y.; Nussinov, R.; Cheng, F. deepDR: A network-based deep learning approach to in silico drug repositioning. Bioinformatics 2019, 35, 5191–5198. [Google Scholar] [CrossRef]
- Issa, N.T.; Stathias, V.; Schürer, S.; Dakshanamurthy, S. Machine and deep learning approaches for cancer drug repurposing. In Seminars in Cancer Biology; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M.; Network, C.G.A.R.; et al. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013, 45, 1113. [Google Scholar] [CrossRef]
- The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, M.C.; Marks, J.H.; Mandell, J.B. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science 2003, 302, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Courtney, K.D.; Corcoran, R.B.; Engelman, J.A. The PI3K pathway as drug target in human cancer. J. Clin. Oncol. 2010, 28, 1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, J.S.; Mullins, M.; Cheang, M.C.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009, 27, 1160. [Google Scholar] [CrossRef]
- Yersal, O.; Barutca, S. Biological subtypes of breast cancer: Prognostic and therapeutic implications. World J. Clin. Oncol. 2014, 5, 412. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Lee, V.H.; Ng, M.K.; Yan, H.; Bijlsma, M.F. Molecular subtyping of cancer: Current status and moving toward clinical applications. Brief. Bioinform. 2019, 20, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Issa, J.P.J.; Baylin, S. Targeting the cancer epigenome for therapy. Nat. Rev. Genet. 2016, 17, 630. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Chaudhary, K.; Garmire, L.X. More is better: Recent progress in multi-omics data integration methods. Front. Genet. 2017, 8, 84. [Google Scholar] [CrossRef] [Green Version]
- Chin, L.; Andersen, J.N.; Futreal, P.A. Cancer genomics: From discovery science to personalized medicine. Nat. Med. 2011, 17, 297. [Google Scholar] [CrossRef] [PubMed]
- Yohe, S.; Thyagarajan, B. Review of clinical next-generation sequencing. Arch. Pathol. Lab. Med. 2017, 141, 1544–1557. [Google Scholar] [CrossRef] [Green Version]
- Prinz, F.; Schlange, T.; Asadullah, K. Believe it or not: How much can we rely on published data on potential drug targets? Nat. Rev. Drug Discov. 2011, 10, 712. [Google Scholar] [CrossRef] [Green Version]
- Allen, M.; Bjerke, M.; Edlund, H.; Nelander, S.; Westermark, B. Origin of the U87MG glioma cell line: Good news and bad news. Sci. Transl. Med. 2016, 8, 354re3. [Google Scholar] [CrossRef]
- Gay, L.; Baker, A.M.; Graham, T.A. Tumour cell heterogeneity. F1000Research 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Dexter, D.L.; Leith, J.T. Tumor heterogeneity and drug resistance. J. Clin. Oncol. 1986, 4, 244–257. [Google Scholar] [CrossRef]
- Kleppe, M.; Levine, R.L. Tumor heterogeneity confounds and illuminates: Assessing the implications. Nat. Med. 2014, 20, 342–344. [Google Scholar] [CrossRef]
- Lawson, D.A.; Kessenbrock, K.; Davis, R.T.; Pervolarakis, N.; Werb, Z. Tumour heterogeneity and metastasis at single-cell resolution. Nat. Cell Biol. 2018, 20, 1349–1360. [Google Scholar] [CrossRef]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef] [Green Version]
- Whiteside, T. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.; Park, Y.; Kim, S. Towards multi-omics characterization of tumor heterogeneity: A comprehensive review of statistical and machine learning approaches. Brief. Bioinform. 2021, 22, bbaa188. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef]
- Kotlov, N.; Bagaev, A.; Revuelta, M.V.; Phillip, J.M.; Cacciapuoti, M.T.; Antysheva, Z.; Svekolkin, V.; Tikhonova, E.; Miheecheva, N.; Kuzkina, N.; et al. Clinical and biological subtypes of B-cell lymphoma revealed by microenvironmental signatures. Cancer Discov. 2021, 11, 1468–1489. [Google Scholar] [CrossRef] [PubMed]
- Roth, A.; Khattra, J.; Yap, D.; Wan, A.; Laks, E.; Biele, J.; Ha, G.; Aparicio, S.; Bouchard-Côté, A.; Shah, S.P. PyClone: Statistical inference of clonal population structure in cancer. Nat. Methods 2014, 11, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lin, F.; Xing, K.; He, X. The reverse evolution from multicellularity to unicellularity during carcinogenesis. Nat. Commun. 2015, 6, 6367. [Google Scholar] [CrossRef] [PubMed]
- Eirew, P.; Steif, A.; Khattra, J.; Ha, G.; Yap, D.; Farahani, H.; Gelmon, K.; Chia, S.; Mar, C.; Wan, A.; et al. Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature 2015, 518, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Demere, Z.; Nair, K.; Ali, A.; Ferraro, G.B.; Natoli, T.; Deik, A.; Petronio, L.; Tang, A.A.; Zhu, C.; et al. A metastasis map of human cancer cell lines. Nature 2020, 588, 331–336. [Google Scholar] [CrossRef]
- Mills, C.C.; Kolb, E.; Sampson, V.B. Development of chemotherapy with cell-cycle inhibitors for adult and pediatric cancer therapy. Cancer Res. 2018, 78, 320–325. [Google Scholar] [CrossRef] [Green Version]
- Hyman, D.M.; Solit, D.B.; Arcila, M.E.; Cheng, D.T.; Sabbatini, P.; Baselga, J.; Berger, M.F.; Ladanyi, M. Precision medicine at Memorial Sloan Kettering Cancer Center: Clinical next-generation sequencing enabling next-generation targeted therapy trials. Drug Discov. Today 2015, 20, 1422–1428. [Google Scholar] [CrossRef] [Green Version]
- McGranahan, N.; Swanton, C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell 2015, 27, 15–26. [Google Scholar] [CrossRef] [Green Version]
- Ben-David, U.; Ha, G.; Tseng, Y.Y.; Greenwald, N.F.; Oh, C.; Shih, J.; McFarland, J.M.; Wong, B.; Boehm, J.S.; Beroukhim, R.; et al. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat. Genet. 2017, 49, 1567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, M.R.; Wegmann, D.; Ehm, M.G.; Kessner, D.; Jean, P.S.; Verzilli, C.; Shen, J.; Tang, Z.; Bacanu, S.A.; Fraser, D.; et al. An abundance of rare functional variants in 202 drug target genes sequenced in 14,002 people. Science 2012, 337, 100–104. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhodes, D.R.; Barrette, T.R.; Rubin, M.A.; Ghosh, D.; Chinnaiyan, A.M. Meta-analysis of microarrays: Interstudy validation of gene expression profiles reveals pathway dysregulation in prostate cancer. Cancer Res. 2002, 62, 4427–4433. [Google Scholar]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Murohashi, M.; Hinohara, K.; Kuroda, M.; Isagawa, T.; Tsuji, S.; Kobayashi, S.; Umezawa, K.; Tojo, A.; Aburatani, H.; Gotoh, N. Gene set enrichment analysis provides insight into novel signalling pathways in breast cancer stem cells. Br. J. Cancer 2010, 102, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Stolovitzky, G.; Monroe, D.; Califano, A. Dialogue on reverse-engineering assessment and methods: The DREAM of high-throughput pathway inference. Ann. N. Y. Acad. Sci. 2007, 1115, 1–22. [Google Scholar] [CrossRef]
- Mo, Q.; Wang, S.; Seshan, V.E.; Olshen, A.B.; Schultz, N.; Sander, C.; Powers, R.S.; Ladanyi, M.; Shen, R. Pattern discovery and cancer gene identification in integrated cancer genomic data. Proc. Natl. Acad. Sci. USA 2013, 110, 4245–4250. [Google Scholar] [CrossRef] [Green Version]
- Cozzetto, D.; Buchan, D.W.; Bryson, K.; Jones, D.T. Protein function prediction by massive integration of evolutionary analyses and multiple data sources. BMC Bioinform. 2013, 14, S1. [Google Scholar] [CrossRef] [Green Version]
- García-Campos, M.A.; Espinal-Enríquez, J.; Hernández-Lemus, E. Pathway analysis: State of the art. Front. Physiol. 2015, 6, 383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Anda-Jáuregui, G.; Hernández-Lemus, E. Computational Oncology in the Multi-Omics Era: State of the Art. Front. Oncol. 2020, 10, 423. [Google Scholar] [CrossRef]
- Lim, S.; Lee, S.; Jung, I.; Rhee, S.; Kim, S. Comprehensive and critical evaluation of individualized pathway activity measurement tools on pan-cancer data. Brief. Bioinform. 2020, 21, 36–46. [Google Scholar] [CrossRef]
- Du, W.; Elemento, O. Cancer systems biology: Embracing complexity to develop better anticancer therapeutic strategies. Oncogene 2015, 34, 3215–3225. [Google Scholar] [CrossRef]
- de Leeuw, C.A.; Mooij, J.M.; Heskes, T.; Posthuma, D. MAGMA: Generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 2015, 11, e1004219. [Google Scholar] [CrossRef]
- Jiao, X.; Sherman, B.T.; Huang, D.W.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. DAVID-WS: A stateful web service to facilitate gene/protein list analysis. Bioinformatics 2012, 28, 1805–1806. [Google Scholar] [CrossRef] [Green Version]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g: Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [Green Version]
- Vêncio, R.Z.; Koide, T.; Gomes, S.L.; de B Pereira, C.A. BayGO: Bayesian analysis of ontology term enrichment in microarray data. BMC Bioinform. 2006, 7, 86. [Google Scholar] [CrossRef] [Green Version]
- Bauer, S.; Gagneur, J.; Robinson, P.N. GOing Bayesian: Model-based gene set analysis of genome-scale data. Nucleic Acids Res. 2010, 38, 3523–3532. [Google Scholar] [CrossRef] [Green Version]
- Huttenhower, C.; Flamholz, A.I.; Landis, J.N.; Sahi, S.; Myers, C.L.; Olszewski, K.L.; Hibbs, M.A.; Siemers, N.O.; Troyanskaya, O.G.; Coller, H.A. Nearest Neighbor Networks: Clustering expression data based on gene neighborhoods. BMC Bioinform. 2007, 8, 250. [Google Scholar] [CrossRef] [Green Version]
- Ratnakumar, A.; Weinhold, N.; Mar, J.C.; Riaz, N. protein–protein interactions uncover candidate ‘core genes’ within omnigenic disease networks. PLoS Genet. 2020, 16, e1008903. [Google Scholar] [CrossRef]
- List, M.; Alcaraz, N.; Dissing-Hansen, M.; Ditzel, H.J.; Mollenhauer, J.; Baumbach, J. KeyPathwayMinerWeb: Online multi-omics network enrichment. Nucleic Acids Res. 2016, 44, W98–W104. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Gulbahce, N.; Yu, H. Network-based methods for human disease gene prediction. Brief. Funct. Genom. 2011, 10, 280–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, S.P.; Zhu, L.; Huang, D.S. Predicting hub genes associated with cervical cancer through gene co-expression networks. IEEE/ACM Trans. Comput. Biol. Bioinform. 2015, 13, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Shojaie, A.; Michailidis, G. Network-based pathway enrichment analysis with incomplete network information. Bioinformatics 2016, 32, 3165–3174. [Google Scholar] [CrossRef] [Green Version]
- Alexeyenko, A.; Lee, W.; Pernemalm, M.; Guegan, J.; Dessen, P.; Lazar, V.; Lehtiö, J.; Pawitan, Y. Network enrichment analysis: Extension of gene-set enrichment analysis to gene networks. BMC Bioinform. 2012, 13, 226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahmati, S.; Abovsky, M.; Pastrello, C.; Jurisica, I. pathDIP: An annotated resource for known and predicted human gene-pathway associations and pathway enrichment analysis. Nucleic Acids Res. 2017, 45, D419–D426. [Google Scholar] [CrossRef] [Green Version]
- Wadi, L.; Meyer, M.; Weiser, J.; Stein, L.D.; Reimand, J. Impact of outdated gene annotations on pathway enrichment analysis. Nat. Methods 2016, 13, 705–706. [Google Scholar] [CrossRef]
- Röttger, R.; Rückert, U.; Taubert, J.; Baumbach, J. How little do we actually know? On the size of gene regulatory networks. IEEE/ACM Trans. Comput. Biol. Bioinform. 2012, 9, 1293–1300. [Google Scholar] [CrossRef]
- Hawe, J.S.; Theis, F.J.; Heinig, M. Inferring interaction networks from multi-omics data. Front. Genet. 2019, 10, 535. [Google Scholar] [CrossRef]
- Carter, H.; Chen, S.; Isik, L.; Tyekucheva, S.; Velculescu, V.E.; Kinzler, K.W.; Vogelstein, B.; Karchin, R. Cancer-specific high-throughput annotation of somatic mutations: Computational prediction of driver missense mutations. Cancer Res. 2009, 69, 6660–6667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Hu, B. Mathematical modeling and computational prediction of cancer drug resistance. Brief. Bioinform. 2018, 19, 1382–1399. [Google Scholar] [CrossRef]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iancu, O.D.; Kawane, S.; Bottomly, D.; Searles, R.; Hitzemann, R.; McWeeney, S. Utilizing RNA-Seq data for de novo coexpression network inference. Bioinformatics 2012, 28, 1592–1597. [Google Scholar] [CrossRef] [Green Version]
- Ballouz, S.; Verleyen, W.; Gillis, J. Guidance for RNA-seq co-expression network construction and analysis: Safety in numbers. Bioinformatics 2015, 31, 2123–2130. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Han, L.; Yuan, Y.; Li, J.; Hei, N.; Liang, H. Gene co-expression network analysis reveals common system-level properties of prognostic genes across cancer types. Nat. Commun. 2014, 5, 3231. [Google Scholar] [CrossRef] [Green Version]
- Petralia, F.; Wang, P.; Yang, J.; Tu, Z. Integrative random forest for gene regulatory network inference. Bioinformatics 2015, 31, i197–i205. [Google Scholar] [CrossRef] [Green Version]
- Omranian, N.; Eloundou-Mbebi, J.M.; Mueller-Roeber, B.; Nikoloski, Z. Gene regulatory network inference using fused LASSO on multiple data sets. Sci. Rep. 2016, 6, 20533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, J.; Braun, C.J.; Saur, D.; Rad, R. In vivo functional screening for systems-level integrative cancer genomics. Nat. Rev. Cancer 2020, 20, 573–593. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, L.; Xie, N.; Nice, E.C.; Zhang, T.; Cui, Y.; Huang, C. Overcoming cancer therapeutic bottleneck by drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 1–25. [Google Scholar] [CrossRef]
- Dimitrakopoulos, C.; Hindupur, S.K.; Häfliger, L.; Behr, J.; Montazeri, H.; Hall, M.N.; Beerenwinkel, N. Network-based integration of multi-omics data for prioritizing cancer genes. Bioinformatics 2018, 34, 2441–2448. [Google Scholar] [CrossRef]
- Yan, J.; Risacher, S.L.; Shen, L.; Saykin, A.J. Network approaches to systems biology analysis of complex disease: Integrative methods for multi-omics data. Brief. Bioinform. 2018, 19, 1370–1381. [Google Scholar] [CrossRef] [Green Version]
- Bersanelli, M.; Mosca, E.; Remondini, D.; Giampieri, E.; Sala, C.; Castellani, G.; Milanesi, L. Methods for the integration of multi-omics data: Mathematical aspects. BMC Bioinform. 2016, 17, 167–177. [Google Scholar] [CrossRef] [Green Version]
- Leiserson, M.D.; Vandin, F.; Wu, H.T.; Dobson, J.R.; Eldridge, J.V.; Thomas, J.L.; Papoutsaki, A.; Kim, Y.; Niu, B.; McLellan, M.; et al. Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat. Genet. 2015, 47, 106–114. [Google Scholar] [CrossRef]
- Hofree, M.; Shen, J.P.; Carter, H.; Gross, A.; Ideker, T. Network-based stratification of tumor mutations. Nat. Methods 2013, 10, 1108–1115. [Google Scholar] [CrossRef]
- O’Neil, N.J.; Bailey, M.L.; Hieter, P. Synthetic lethality and cancer. Nat. Rev. Genet. 2017, 18, 613–623. [Google Scholar] [CrossRef]
- Ku, A.A.; Hu, H.M.; Zhao, X.; Shah, K.N.; Kongara, S.; Wu, D.; McCormick, F.; Balmain, A.; Bandyopadhyay, S. Integration of multiple biological contexts reveals principles of synthetic lethality that affect reproducibility. Nat. Commun. 2020, 11, 2375. [Google Scholar] [CrossRef]
- Barbie, D.A.; Tamayo, P.; Boehm, J.S.; Kim, S.Y.; Moody, S.E.; Dunn, I.F.; Schinzel, A.C.; Sandy, P.; Meylan, E.; Scholl, C.; et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009, 462, 108–112. [Google Scholar] [CrossRef]
- Vaske, C.J.; Benz, S.C.; Sanborn, J.Z.; Earl, D.; Szeto, C.; Zhu, J.; Haussler, D.; Stuart, J.M. Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics 2010, 26, i237–i245. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Swanton, C. Clonal heterogeneity and tumor evolution: Past, present, and the future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciriello, G.; Cerami, E.; Sander, C.; Schultz, N. Mutual exclusivity analysis identifies oncogenic network modules. Genome Res. 2012, 22, 398–406. [Google Scholar] [CrossRef] [Green Version]
- Leiserson, M.D.; Wu, H.T.; Vandin, F.; Raphael, B.J. CoMEt: A statistical approach to identify combinations of mutually exclusive alterations in cancer. Genome Biol. 2015, 16, 160. [Google Scholar] [CrossRef] [PubMed]
- Ooi, S.L.; Shoemaker, D.D.; Boeke, J.D. DNA helicase gene interaction network defined using synthetic lethality analyzed by microarray. Nat. Genet. 2003, 35, 277–286. [Google Scholar] [CrossRef]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The Tumor Microenvironment at a Glance; The Company of Biologists Ltd.: Cambridge, UK, 2012. [Google Scholar]
- McGrail, D.J.; Federico, L.; Li, Y.; Dai, H.; Lu, Y.; Mills, G.B.; Yi, S.; Lin, S.Y.; Sahni, N. Multi-omics analysis reveals neoantigen-independent immune cell infiltration in copy-number driven cancers. Nat. Commun. 2018, 9, 1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darzi, M.; Gorgin, S.; Majidzadeh-A, K.; Esmaeili, R. Gene co-expression network analysis reveals immune cell infiltration as a favorable prognostic marker in non-uterine leiomyosarcoma. Sci. Rep. 2021, 11, 2339. [Google Scholar] [CrossRef]
- Tu, J.J.; Ou-Yang, L.; Yan, H.; Zhang, X.F.; Qin, H. Joint reconstruction of multiple gene networks by simultaneously capturing inter-tumor and intra-tumor heterogeneity. Bioinformatics 2020, 36, 2755–2762. [Google Scholar] [CrossRef]
- Hopkins, A.L. Network pharmacology: The next paradigm in drug discovery. Nat. Chem. Biol. 2008, 4, 682–690. [Google Scholar] [CrossRef]
- Yamanishi, Y.; Araki, M.; Gutteridge, A.; Honda, W.; Kanehisa, M. Prediction of drug–target interaction networks from the integration of chemical and genomic spaces. Bioinformatics 2008, 24, i232–i240. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, L. A survey and systematic assessment of computational methods for drug response prediction. Brief. Bioinform. 2021, 22, 232–246. [Google Scholar] [CrossRef]
- Hernandez-Lemus, E.; Martínez-García, M. Pathway-based drug-repurposing schemes in cancer: The role of translational bioinformatics. Front. Oncol. 2020, 10, 605680. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Das, A.; Jerby-Arnon, L.; Arafeh, R.; Auslander, N.; Davidson, M.; McGarry, L.; James, D.; Amzallag, A.; Park, S.G.; et al. Harnessing synthetic lethality to predict the response to cancer treatment. Nat. Commun. 2018, 9, 2546. [Google Scholar] [CrossRef] [PubMed]
- Angermueller, C.; Pärnamaa, T.; Parts, L.; Stegle, O. Deep learning for computational biology. Mol. Syst. Biol. 2016, 12, 878. [Google Scholar] [CrossRef]
- Samiei, M.; Würfl, T.; Deleu, T.; Weiss, M.; Dutil, F.; Fevens, T.; Boucher, G.; Lemieux, S.; Cohen, J.P. The TCGA Meta-Dataset Clinical Benchmark. arXiv 2019, arXiv:1910.08636. [Google Scholar]
- Webb, S. Deep learning for biology. Nature 2018, 554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, S.; Zeng, X.; Xia, F.; Huang, W.; Liu, X. Application of deep learning methods in biological networks. Brief. Bioinform. 2021, 22, 1902–1917. [Google Scholar] [CrossRef]
- GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 2020, 369, 1318–1330. [Google Scholar] [CrossRef]
- 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 2015, 526, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmud, M.; Kaiser, M.S.; McGinnity, T.M.; Hussain, A. Deep learning in mining biological data. Cogn. Comput. 2021, 13, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Huss, M.; Abid, A.; Mohammadi, P.; Torkamani, A.; Telenti, A. A primer on deep learning in genomics. Nat. Genet. 2019, 51, 12–18. [Google Scholar] [CrossRef]
- Troyanskaya, O.; Cantor, M.; Sherlock, G.; Brown, P.; Hastie, T.; Tibshirani, R.; Botstein, D.; Altman, R.B. Missing value estimation methods for DNA microarrays. Bioinformatics 2001, 17, 520–525. [Google Scholar] [CrossRef] [Green Version]
- Song, M.; Greenbaum, J.; Luttrell, J., IV; Zhou, W.; Wu, C.; Shen, H.; Gong, P.; Zhang, C.; Deng, H.W. A Review of Integrative Imputation for Multi-Omics Datasets. Front. Genet. 2020, 11, 570255. [Google Scholar] [CrossRef]
- Voillet, V.; Besse, P.; Liaubet, L.; San Cristobal, M.; González, I. Handling missing rows in multi-omics data integration: Multiple imputation in multiple factor analysis framework. BMC Bioinform. 2016, 17, 402. [Google Scholar] [CrossRef] [Green Version]
- Fang, Z.; Ma, T.; Tang, G.; Zhu, L.; Yan, Q.; Wang, T.; Celedón, J.C.; Chen, W.; Tseng, G.C. Bayesian integrative model for multi-omics data with missingness. Bioinformatics 2018, 34, 3801–3808. [Google Scholar] [CrossRef]
- Zhou, X.; Chai, H.; Zhao, H.; Luo, C.H.; Yang, Y. Imputing missing RNA-sequencing data from DNA methylation by using a transfer learning–based neural network. GigaScience 2020, 9, giaa076. [Google Scholar] [CrossRef] [PubMed]
- Arisdakessian, C.; Poirion, O.; Yunits, B.; Zhu, X.; Garmire, L.X. DeepImpute: An accurate, fast, and scalable deep neural network method to impute single-cell RNA-seq data. Genome Biol. 2019, 20, 211. [Google Scholar] [CrossRef] [Green Version]
- Goodfellow, I.J.; Pouget-Abadie, J.; Mirza, M.; Xu, B.; Warde-Farley, D.; Ozair, S.; Courville, A.; Bengio, Y. Generative adversarial networks. arXiv 2014, arXiv:1406.2661. [Google Scholar]
- Lopez, R.; Regier, J.; Cole, M.B.; Jordan, M.I.; Yosef, N. Deep generative modeling for single-cell transcriptomics. Nat. Methods 2018, 15, 1053–1058. [Google Scholar] [CrossRef]
- Wang, Y.; Yao, Q.; Kwok, J.T.; Ni, L.M. Generalizing from a few examples: A survey on few-shot learning. ACM Comput. Surv. 2020, 53. [Google Scholar] [CrossRef]
- Wang, X.; Ghasedi Dizaji, K.; Huang, H. Conditional generative adversarial network for gene expression inference. Bioinformatics 2018, 34, i603–i611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Agarwal, D.; Huang, M.; Hu, G.; Zhou, Z.; Ye, C.; Zhang, N.R. Data denoising with transfer learning in single-cell transcriptomics. Nat. Methods 2019, 16, 875–878. [Google Scholar] [CrossRef] [PubMed]
- Snell, J.; Swersky, K.; Zemel, R.S. Prototypical networks for few-shot learning. arXiv 2017, arXiv:1703.05175. [Google Scholar]
- Yang, Z.; Shu, J.; Liang, Y.; Meng, D.; Xu, Z. Select-ProtoNet: Learning to Select for Few-Shot Disease Subtype Prediction. arXiv 2020, arXiv:2009.00792. [Google Scholar]
- Ma, T.; Zhang, A. AffinityNet: Semi-supervised few-shot learning for disease type prediction. In Proceedings of the AAAI Conference on Artificial Intelligence, Honolulu, HI, USA, 27 January–1 February 2019; Volume 33, pp. 1069–1076. [Google Scholar]
- Yuan, Y.; Shi, Y.; Li, C.; Kim, J.; Cai, W.; Han, Z.; Feng, D.D. DeepGene: An advanced cancer type classifier based on deep learning and somatic point mutations. BMC Bioinform. 2016, 17, 243–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyu, B.; Haque, A. Deep learning based tumor type classification using gene expression data. In Proceedings of the 2018 ACM International Conference on Bioinformatics, Computational Biology, and Health Informatics, Washington, DC, USA, 29 August–1 September 2018; pp. 89–96. [Google Scholar]
- Joseph, M.; Devaraj, M.; Leung, C.K. DeepGx: Deep learning using gene expression for cancer classification. In Proceedings of the 2019 IEEE/ACM International Conference on Advances in Social Networks Analysis and Mining (ASONAM), Vancouver, BC, Canada, 27–30 August 2019; pp. 913–920. [Google Scholar]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Menden, K.; Marouf, M.; Oller, S.; Dalmia, A.; Magruder, D.S.; Kloiber, K.; Heutink, P.; Bonn, S. Deep learning–based cell composition analysis from tissue expression profiles. Sci. Adv. 2020, 6, eaba2619. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Preissl, S.; Ren, B. Single-cell multimodal omics: The power of many. Nat. Methods 2020, 17, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Wan, J.; Song, Q.; Wei, Z. Clustering single-cell RNA-seq data with a model-based deep learning approach. Nat. Mach. Intell. 2019, 1, 191–198. [Google Scholar] [CrossRef]
- Tran, D.; Nguyen, H.; Tran, B.; La Vecchia, C.; Luu, H.N.; Nguyen, T. Fast and precise single-cell data analysis using a hierarchical autoencoder. Nat. Commun. 2021, 12, 1029. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xu, J.; Li, S.C. DeepMF: Deciphering the latent patterns in omics profiles with a deep learning method. BMC Bioinform. 2019, 20, 648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Way, G.P.; Zietz, M.; Rubinetti, V.; Himmelstein, D.S.; Greene, C.S. Compressing gene expression data using multiple latent space dimensionalities learns complementary biological representations. Genome Biol. 2020, 21, 109. [Google Scholar] [CrossRef]
- Chang, Y.; Park, H.; Yang, H.J.; Lee, S.; Lee, K.Y.; Kim, T.S.; Jung, J.; Shin, J.M. Cancer drug response profile scan (CDRscan): A deep learning model that predicts drug effectiveness from cancer genomic signature. Sci. Rep. 2018, 8, 8857. [Google Scholar] [CrossRef] [PubMed]
- Sharifi-Noghabi, H.; Zolotareva, O.; Collins, C.C.; Ester, M. MOLI: Multi-omics late integration with deep neural networks for drug response prediction. Bioinformatics 2019, 35, i501–i509. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Pan, S.; Chen, F.; Long, G.; Zhang, C.; Philip, S.Y. A comprehensive survey on graph neural networks. IEEE Trans. Neural Netw. Learn. Syst. 2020. [Google Scholar] [CrossRef] [Green Version]
- Dutil, F.; Cohen, J.P.; Weiss, M.; Derevyanko, G.; Bengio, Y. Towards gene expression convolutions using gene interaction graphs. arXiv 2018, arXiv:1806.06975. [Google Scholar]
- Schulte-Sasse, R.; Budach, S.; Hnisz, D.; Marsico, A. Graph Convolutional Networks Improve the Prediction of Cancer Driver Genes. In Proceedings of the International Conference on Artificial Neural Networks, Munich, Germany, 17–19 September 2019; Springer: Berlin/Heidelberg, Germany, 2019; pp. 658–668. [Google Scholar]
- Schulte-Sasse, R.; Budach, S.; Hnisz, D.; Marsico, A. Integration of multiomics data with graph convolutional networks to identify new cancer genes and their associated molecular mechanisms. Nat. Mach. Intell. 2021, 3, 513–526. [Google Scholar] [CrossRef]
- Cai, R.; Chen, X.; Fang, Y.; Wu, M.; Hao, Y. Dual-Dropout Graph Convolutional Network for Predicting Synthetic Lethality in Human Cancers. Bioinformatics 2020, 36, 4458–4465. [Google Scholar] [CrossRef]
- Lee, S.; Lim, S.; Lee, T.; Sung, I.; Kim, S. Cancer subtype classification and modeling by pathway attention and propagation. Bioinformatics 2020, 36, 3818–3824. [Google Scholar] [CrossRef]
- Sun, M.; Zhao, S.; Gilvary, C.; Elemento, O.; Zhou, J.; Wang, F. Graph convolutional networks for computational drug development and discovery. Brief. Bioinform. 2020, 21, 919–935. [Google Scholar] [CrossRef]
- Wainberg, M.; Merico, D.; Delong, A.; Frey, B.J. Deep learning in biomedicine. Nat. Biotechnol. 2018, 36, 829–838. [Google Scholar] [CrossRef]
- Ching, T.; Himmelstein, D.S.; Beaulieu-Jones, B.K.; Kalinin, A.A.; Do, B.T.; Way, G.P.; Ferrero, E.; Agapow, P.M.; Zietz, M.; Hoffman, M.M.; et al. Opportunities and obstacles for deep learning in biology and medicine. J. R. Soc. Interface 2018, 15, 20170387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [Green Version]
- Bansal, M.; Belcastro, V.; Ambesi-Impiombato, A.; Di Bernardo, D. How to infer gene networks from expression profiles. Mol. Syst. Biol. 2007, 3, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Li, Y.; Narayan, R.; Subramanian, A.; Xie, X. Gene expression inference with deep learning. Bioinformatics 2016, 32, 1832–1839. [Google Scholar] [CrossRef] [Green Version]
- Bertin, P.; Hashir, M.; Weiss, M.; Frappier, V.; Perkins, T.J.; Boucher, G.; Cohen, J.P. Analysis of Gene Interaction Graphs as Prior Knowledge for Machine Learning Models. arXiv 2019, arXiv:1905.02295. [Google Scholar]
- Samek, W.; Wiegand, T.; Müller, K.R. Explainable artificial intelligence: Understanding, visualizing and interpreting deep learning models. arXiv 2017, arXiv:1708.08296. [Google Scholar]
- Holzinger, A. Explainable ai and multi-modal causability in medicine. i-com 2020, 19, 171–179. [Google Scholar] [CrossRef]
- Anguita-Ruiz, A.; Segura-Delgado, A.; Alcalá, R.; Aguilera, C.M.; Alcalá-Fdez, J. eXplainable Artificial Intelligence (XAI) for the identification of biologically relevant gene expression patterns in longitudinal human studies, insights from obesity research. PLoS Comput. Biol. 2020, 16, e1007792. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.; Heider, D. GUESS: Projecting machine learning scores to well-calibrated probability estimates for clinical decision-making. Bioinformatics 2019, 35, 2458–2465. [Google Scholar] [CrossRef] [PubMed]
- Holzinger, A.; Malle, B.; Saranti, A.; Pfeifer, B. Towards multi-modal causability with graph neural networks enabling information fusion for explainable ai. Inf. Fusion 2021, 71, 28–37. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, Y.; Heider, D.; Hauschild, A.-C. Integrative Analysis of Next-Generation Sequencing for Next-Generation Cancer Research toward Artificial Intelligence. Cancers 2021, 13, 3148. https://doi.org/10.3390/cancers13133148
Park Y, Heider D, Hauschild A-C. Integrative Analysis of Next-Generation Sequencing for Next-Generation Cancer Research toward Artificial Intelligence. Cancers. 2021; 13(13):3148. https://doi.org/10.3390/cancers13133148
Chicago/Turabian StylePark, Youngjun, Dominik Heider, and Anne-Christin Hauschild. 2021. "Integrative Analysis of Next-Generation Sequencing for Next-Generation Cancer Research toward Artificial Intelligence" Cancers 13, no. 13: 3148. https://doi.org/10.3390/cancers13133148
APA StylePark, Y., Heider, D., & Hauschild, A.-C. (2021). Integrative Analysis of Next-Generation Sequencing for Next-Generation Cancer Research toward Artificial Intelligence. Cancers, 13(13), 3148. https://doi.org/10.3390/cancers13133148