
Overexpression of Replication-Dependent Histone Signifies a Subset of Dedifferentiated Liposarcoma with Increased Aggressiveness

 ,
,  , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Subjects and Tumor Samples
2.2. Tumor Cell Indexing
2.3. Whole-Exome Sequencing
2.4. Bulk and Single-Cell RNA-Sequencing
2.5. LPS Cell Lines and Patient-Derived 18DDLPS
2.6. Western Blots for Fresh, Frozen, and FFPE Tissues
2.7. Immunohistochemistry
2.8. Lentivirus Production and Transduction
2.9. Western Blots for HMGA2 Knockdown and Overexpression
2.10. Colony Forming Assay
2.11. Mouse Xenograft Model and Subcutaneous Transplants
2.12. Cell Viability Assays
2.13. Real-Time Quantitative PCR (RT-qPCR)
2.14. Statistical Analyses
3. Results
3.1. Comparison of WD- and DDLPS Expression Profiles
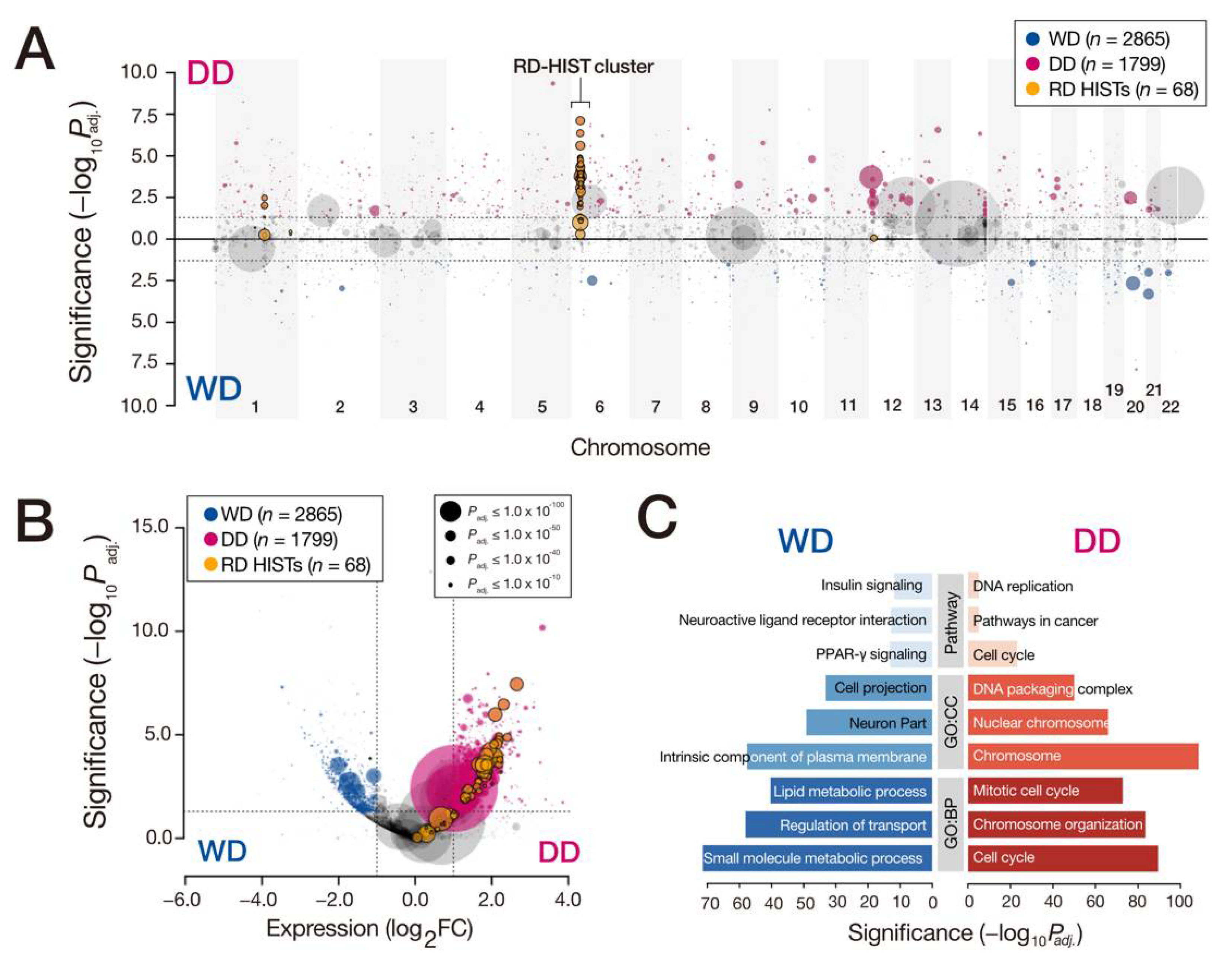
3.2. Overexpression of Replication-Dependent Histone mRNAs in DDLPS
3.3. Overexpression of HMGA2 by the SCNAs
3.4. HMGA2 Regulated RD-HISTs’ Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Crago, A.M.; Singer, S. Clinical and molecular approaches to well differentiated and dedifferentiated liposarcoma. Curr. Opin. Oncol. 2011, 23, 373–378. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.T.J.; Thway, K.; Huang, P.H.; Jones, R.L. Clinical and Molecular Spectrum of Liposarcoma. J. Clin. Oncol. 2018, 36, 151–159. [Google Scholar] [CrossRef]
- Rosai, J.; Akerman, M.; Dal Cin, P.; DeWever, I.; Fletcher, C.D.; Mandahl, N.; Mertens, F.; Mitelman, F.; Rydholm, A.; Sciot, R.; et al. Combined morphologic and karyotypic study of 59 atypical lipomatous tumors. Evaluation of their relationship and differential diagnosis with other adipose tissue tumors (a report of the CHAMP Study Group). Am. J. Surg. Pathol. 1996, 20, 1182–1189. [Google Scholar] [CrossRef]
- Henricks, W.H.; Chu, Y.C.; Goldblum, J.R.; Weiss, S.W. Dedifferentiated liposarcoma: A clinicopathological analysis of 155 cases with a proposal for an expanded definition of dedifferentiation. Am. J. Surg. Pathol. 1997, 21, 271–281. [Google Scholar] [CrossRef]
- Garsed, D.W.; Marshall, O.J.; Corbin, V.D.; Hsu, A.; Di Stefano, L.; Schroder, J.; Li, J.; Feng, Z.P.; Kim, B.W.; Kowarsky, M.; et al. The architecture and evolution of cancer neochromosomes. Cancer Cell 2014, 26, 653–667. [Google Scholar] [CrossRef] [Green Version]
- Kanojia, D.; Nagata, Y.; Garg, M.; Lee, D.H.; Sato, A.; Yoshida, K.; Sato, Y.; Sanada, M.; Mayakonda, A.; Bartenhagen, C.; et al. Genomic landscape of liposarcoma. Oncotarget 2015, 6, 42429–42444. [Google Scholar] [CrossRef] [Green Version]
- Thway, K. Well-differentiated liposarcoma and dedifferentiated liposarcoma: An updated review. Semin. Diagn. Pathol. 2019, 36, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirata, M.; Asano, N.; Katayama, K.; Yoshida, A.; Tsuda, Y.; Sekimizu, M.; Mitani, S.; Kobayashi, E.; Komiyama, M.; Fujimoto, H.; et al. Integrated exome and RNA sequencing of dedifferentiated liposarcoma. Nat. Commun. 2019, 10, 5683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome. Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas. Cell 2017, 171, 950–965.e928. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.K.; Liang, D.; Gajjalaiahvari, U.R.; Kabbaj, M.H.; Paik, J.; Gunjan, A. Excess histone levels mediate cytotoxicity via multiple mechanisms. Cell Cycle 2010, 9, 4236–4244. [Google Scholar] [CrossRef] [Green Version]
- Maya Miles, D.; Penate, X.; Sanmartin Olmo, T.; Jourquin, F.; Munoz Centeno, M.C.; Mendoza, M.; Simon, M.N.; Chavez, S.; Geli, V. High levels of histones promote whole-genome-duplications and trigger a Swe1(WEE1)-dependent phosphorylation of Cdc28(CDK1). Elife 2018, 7, e35337. [Google Scholar] [CrossRef]
- Groth, A.; Rocha, W.; Verreault, A.; Almouzni, G. Chromatin challenges during DNA replication and repair. Cell 2007, 128, 721–733. [Google Scholar] [CrossRef] [Green Version]
- Marzluff, W.F.; Wagner, E.J.; Duronio, R.J. Metabolism and regulation of canonical histone mRNAs: Life without a poly(A) tail. Nat. Rev. Genet. 2008, 9, 843–854. [Google Scholar] [CrossRef] [Green Version]
- Brocato, J.; Fang, L.; Chervona, Y.; Chen, D.; Kiok, K.; Sun, H.; Tseng, H.C.; Xu, D.; Shamy, M.; Jin, C.; et al. Arsenic induces polyadenylation of canonical histone mRNA by down-regulating stem-loop-binding protein gene expression. J. Biol. Chem. 2014, 289, 31751–31764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyons, S.M.; Cunningham, C.H.; Welch, J.D.; Groh, B.; Guo, A.Y.; Wei, B.; Whitfield, M.L.; Xiong, Y.; Marzluff, W.F. A subset of replication-dependent histone mRNAs are expressed as polyadenylated RNAs in terminally differentiated tissues. Nucleic Acids Res. 2016, 44, 9190–9205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravasi, T.; Suzuki, H.; Cannistraci, C.V.; Katayama, S.; Bajic, V.B.; Tan, K.; Akalin, A.; Schmeier, S.; Kanamori-Katayama, M.; Bertin, N.; et al. An atlas of combinatorial transcriptional regulation in mouse and man. Cell 2010, 140, 744–752. [Google Scholar] [CrossRef] [Green Version]
- Fusco, A.; Fedele, M. Roles of HMGA proteins in cancer. Nat. Rev. Cancer 2007, 7, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Barretina, J.; Taylor, B.S.; Banerji, S.; Ramos, A.H.; Lagos-Quintana, M.; Decarolis, P.L.; Shah, K.; Socci, N.D.; Weir, B.A.; Ho, A.; et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat. Genet. 2010, 42, 715–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coughlan, A.M.; Harmon, C.; Whelan, S.; O’Brien, E.C.; O’Reilly, V.P.; Crotty, P.; Kelly, P.; Ryan, M.; Hickey, F.B.; O’Farrelly, C.; et al. Myeloid Engraftment in Humanized Mice: Impact of Granulocyte-Colony Stimulating Factor Treatment and Transgenic Mouse Strain. Stem Cells Dev. 2016, 25, 530–541. [Google Scholar] [CrossRef]
- Singh, R.K.; Paik, J.; Gunjan, A. Generation and management of excess histones during the cell cycle. Front Biosci. (Landmark Ed) 2009, 14, 3145–3158. [Google Scholar] [CrossRef] [Green Version]
- Silvestre-Roig, C.; Braster, Q.; Wichapong, K.; Lee, E.Y.; Teulon, J.M.; Berrebeh, N.; Winter, J.; Adrover, J.M.; Santos, G.S.; Froese, A.; et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature 2019, 569, 236–240. [Google Scholar] [CrossRef]
- Fan, Y.; Nikitina, T.; Morin-Kensicki, E.M.; Zhao, J.; Magnuson, T.R.; Woodcock, C.L.; Skoultchi, A.I. H1 linker histones are essential for mouse development and affect nucleosome spacing in vivo. Mol. Cell Biol. 2003, 23, 4559–4572. [Google Scholar] [CrossRef] [Green Version]
- Gould, T.J.; Lysov, Z.; Liaw, P.C. Extracellular DNA and histones: Double-edged swords in immunothrombosis. J. Thromb. Haemost. 2015, 13 (Suppl. 1), S82–S91. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, X.; Pelayo, R.; Monestier, M.; Ammollo, C.T.; Semeraro, F.; Taylor, F.B.; Esmon, N.L.; Lupu, F.; Esmon, C.T. Extracellular histones are major mediators of death in sepsis. Nat. Med. 2009, 15, 1318–1321. [Google Scholar] [CrossRef] [Green Version]
- Sgarra, R.; Pegoraro, S.; Ros, G.; Penzo, C.; Chiefari, E.; Foti, D.; Brunetti, A.; Manfioletti, G. High Mobility Group A (HMGA) proteins: Molecular instigators of breast cancer onset and progression. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 216–229. [Google Scholar] [CrossRef] [PubMed]
- Beird, H.C.; Wu, C.C.; Ingram, D.R.; Wang, W.L.; Alimohamed, A.; Gumbs, C.; Little, L.; Song, X.; Feig, B.W.; Roland, C.L.; et al. Genomic profiling of dedifferentiated liposarcoma compared to matched well-differentiated liposarcoma reveals higher genomic complexity and a common origin. Cold Spring Harb Mol. Case Stud. 2018, 4, a002386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saada-Bouzid, E.; Burel-Vandenbos, F.; Ranchere-Vince, D.; Birtwisle-Peyrottes, I.; Chetaille, B.; Bouvier, C.; Chateau, M.C.; Peoc’h, M.; Battistella, M.; Bazin, A.; et al. Prognostic value of HMGA2, CDK4, and JUN amplification in well-differentiated and dedifferentiated liposarcomas. Mod. Pathol. 2015, 28, 1404–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreux, N.; Marty, M.; Chibon, F.; Velasco, V.; Hostein, I.; Ranchere-Vince, D.; Terrier, P.; Coindre, J.M. Value and limitation of immunohistochemical expression of HMGA2 in mesenchymal tumors: About a series of 1052 cases. Mod. Pathol. 2010, 23, 1657–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoo, Y.; Park, S.-Y.; Jo, E.B.; Choi, M.; Lee, K.W.; Hong, D.; Lee, S.; Lee, C.-R.; Lee, Y.; Um, J.-Y.; et al. Overexpression of Replication-Dependent Histone Signifies a Subset of Dedifferentiated Liposarcoma with Increased Aggressiveness. Cancers 2021, 13, 3122. https://doi.org/10.3390/cancers13133122
Yoo Y, Park S-Y, Jo EB, Choi M, Lee KW, Hong D, Lee S, Lee C-R, Lee Y, Um J-Y, et al. Overexpression of Replication-Dependent Histone Signifies a Subset of Dedifferentiated Liposarcoma with Increased Aggressiveness. Cancers. 2021; 13(13):3122. https://doi.org/10.3390/cancers13133122
Chicago/Turabian StyleYoo, Yongjin, Sang-Yoon Park, Eun Byeol Jo, Minji Choi, Kyo Won Lee, Doopyo Hong, Sangmoon Lee, Cho-Rong Lee, Youngha Lee, Jae-Young Um, and et al. 2021. "Overexpression of Replication-Dependent Histone Signifies a Subset of Dedifferentiated Liposarcoma with Increased Aggressiveness" Cancers 13, no. 13: 3122. https://doi.org/10.3390/cancers13133122
APA StyleYoo, Y., Park, S.-Y., Jo, E. B., Choi, M., Lee, K. W., Hong, D., Lee, S., Lee, C.-R., Lee, Y., Um, J.-Y., Park, J. B., Seo, S. W., Choi, Y.-L., Kim, S., Lee, S.-G., & Choi, M. (2021). Overexpression of Replication-Dependent Histone Signifies a Subset of Dedifferentiated Liposarcoma with Increased Aggressiveness. Cancers, 13(13), 3122. https://doi.org/10.3390/cancers13133122