
An Algorithm Combining Patient Performance Status, Second Hit Analysis, PROVEAN and Dann Prediction Tools Could Foretell Sensitization to PARP Inhibitors in Digestive, Skin, Ovarian and Breast Cancers

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Approval and Consent to Participate
2.2. DNA Extraction
2.3. Exome Sequencing
2.4. Complex Analyses
2.5. Statistical Analysis
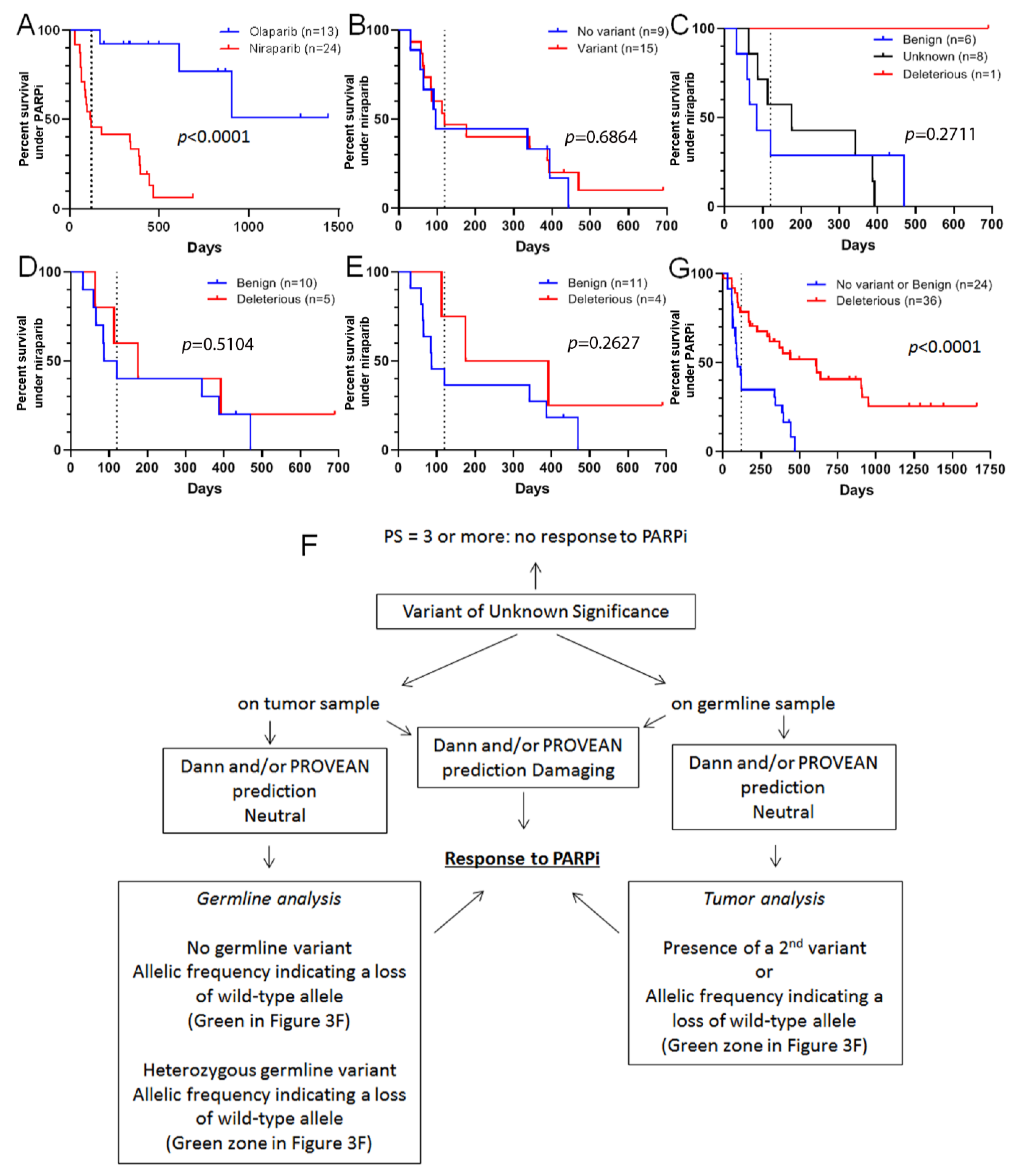
3. Results
3.1. Patient Characteristics
3.2. Some VUS May Respond to Olaparib
3.3. Response of VUS to Olaparib Could Be Predicted before Treatment
3.4. VUS Classification Allows one to Predict Benefit from Particular PARP Inhibitors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
List of Abbreviations
References
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N. Engl. J. Med. 2012, 366, 1382–1392. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive.; Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised; double-blind; placebo-controlled; phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef] [Green Version]
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Goncalves, A.; Lee, K.H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- de Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef]
- Kim, D.S.; Camacho, C.V.; Kraus, W.L. Alternate therapeutic pathways for PARP inhibitors and potential mechanisms of resistance. Exp. Mol. Med. 2021, 53, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Goldgar, D.E.; Easton, D.F.; Byrnes, G.B.; Spurdle, A.B.; Iversen, E.S.; Greenblatt, M.S. IARC Unclassified Genetic Variants Working Group. Genetic evidence and integration of various data sources for classifying uncertain variants into a single model. Hum. Mutat. 2008, 29, 1265–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindor, N.M.; Guidugli, L.; Wang, X.; Vallee, M.P.; Monteiro, A.N.; Tavtigian, S.; Goldgar, D.E.; Couch, F.J. A review of a multifactorial probability-based model for classification of BRCA1 and BRCA2 variants of uncertain significance (VUS). Hum. Mutat. 2012, 33, 8–21. [Google Scholar] [CrossRef] [Green Version]
- Federici, G.; Soddu, S. Variants of uncertain significance in the era of high-throughput genome sequencing: A lesson from breast and ovary cancers. J. Exp. Clin. Cancer Res. 2020, 39, 46. [Google Scholar] [CrossRef] [Green Version]
- Reda, M.; Richard, C.; Bertaut, A.; Niogret, J.; Collot, T.; Fumet, J.D.; Blanc, J.; Truntzer, C.; Desmoulins, I.; Ladoire, S.; et al. Implementation and use of whole exome sequencing for metastatic solid cancer. EBioMedicine 2020, 51, 102624. [Google Scholar] [CrossRef] [PubMed]
- Alsadoun, N.; MacGrogan, G.; Truntzer, C.; Lacroix-Triki, M.; Bedgedjian, I.; Koeb, M.H.; Alam, E.E.; Medioni, D.; Parent, M.; Wuithier, P.; et al. Solid papillary carcinoma with reverse polarity of the breast harbors specific morphologic.; immunohistochemical and molecular profile in comparison with other benign or malignant papillary lesions of the breast: A comparative study of 9 additional cases. Mod. Pathol. 2018, 31, 1367–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenthal, R.; McGranahan, N.; Herrero, J.; Taylor, B.S.; Swanton, C. DeconstructSigs: Delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016, 17, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43. [Google Scholar] [CrossRef]
- Hundal, J.; Carreno, B.M.; Petti, A.A.; Linette, G.P.; Griffith, O.L.; Mardis, E.R.; Griffith, M. pVAC-Seq: A genome-guided in silico approach to identifying tumor neoantigens. Genome Med. 2016, 8, 11. [Google Scholar] [CrossRef] [Green Version]
- Ha, G.; Roth, A.; Khattra, J.; Ho, J.; Yap, D.; Prentice, L.M.; Melnyk, N.; McPherson, A.; Bashashati, A.; Laks, E.; et al. TITAN: Inference of copy number architectures in clonal cell populations from tumor whole-genome sequence data. Genome Res. 2014, 24, 1881–1893. [Google Scholar] [CrossRef] [PubMed]
- Sztupinszki, Z.; Diossy, M.; Krzystanek, M.; Reiniger, L.; Csabai, I.; Favero, F.; Birkbak, N.J.; Eklund, A.C.; Syed, A.; Szallasi, Z. Migrating the SNP array-based homologous recombination deficiency measures to next generation sequencing data of breast cancer. NPJ Breast Cancer 2018, 4, 16. [Google Scholar] [CrossRef]
- O’Kane, G.M.; Connor, A.A.; Gallinger, S. Characterization, Detection, and Treatment Approaches for Homologous Recombination Deficiency in Cancer. Trends Mol. Med. 2017, 23, 1121–1137. [Google Scholar] [CrossRef]
- Gulhan, D.C.; Lee, J.J.; Melloni, G.E.M.; Cortes-Ciriano, I.; Park, P.J. Detecting the mutational signature of homologous recombination deficiency in clinical samples. Nat. Genet. 2019, 51, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, D.R.; Dougherty, B.A.; Lai, Z.; Fielding, A.; Grinsted, L.; Spencer, S.; O’Connor, M.J.; Ho, T.W.; Robertson, J.D.; Lanchbury, J.S.; et al. Candidate biomarkers of PARP inhibitor sensitivity in ovarian cancer beyond the BRCA genes. Br. J. Cancer 2018, 119, 1401–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quang, D.; Chen, Y.; Xie, X. DANN: A deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 2015, 31, 761–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Martin, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2391–2402. [Google Scholar] [CrossRef] [Green Version]
- Ghiringhelli, F.; Richard, C.; Chevrier, S.; Vegran, F.; Boidot, R. Efficiency of olaparib in colorectal cancer patients with an alteration of the homologous repair protein. World J. Gastroenterol. 2016, 22, 10680–10686. [Google Scholar] [CrossRef] [PubMed]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Perol, D.; Gonzalez-Martin, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef]
- Bouwman, P.; van der Gulden, H.; van der Heijden, I.; Drost, R.; Klijn, C.N.; Prasetyanti, P.; Pieterse, M.; Wientjens, E.; Seibler, J.; Hogervorst, F.B.; et al. A high-throughput functional complementation assay for classification of BRCA1 missense variants. Cancer Discov. 2013, 3, 1142–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirisena, N.; Biswas, K.; Sullivan, T.; Stauffer, S.; Cleveland, L.; Southon, E.; Dissanayake, V.H.W.; Sharan, S.K. Functional evaluation of five BRCA2 unclassified variants identified in a Sri Lankan cohort with inherited cancer syndromes using a mouse embryonic stem cell-based assay. Breast Cancer Res. 2020, 22, 43. [Google Scholar] [CrossRef] [PubMed]
- Boonen, R.; Rodrigue, A.; Stoepker, C.; Wiegant, W.W.; Vroling, B.; Sharma, M.; Rother, M.B.; Celosse, N.; Vreeswijk, M.P.G.; Couch, F.; et al. Functional analysis of genetic variants in the high-risk breast cancer susceptibility gene PALB2. Nat. Commun. 2019, 10, 5296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Campbell, D.; Patnaik, A.; Shapiro, J.D.; Sautois, B.; Vogelzang, N.J.; Voog, E.G.; Bryce, A.H.; McDermott, R.; Ricci, F.; et al. Non-BRCA DNA Damage Repair Gene Alterations and Response to the PARP Inhibitor Rucaparib in Metastatic Castration-Resistant Prostate Cancer: Analysis from the Phase II TRITON2 Study. Clin. Cancer Res. 2020, 26, 2487–2496. [Google Scholar] [CrossRef] [Green Version]
- Ip, L.R.; Poulogiannis, G.; Viciano, F.C.; Sasaki, J.; Kofuji, S.; Spanswick, V.J.; Hochhauser, D.; Hartley, J.A.; Sasaki, T.; Gewinner, C.A. Loss of INPP4B causes a DNA repair defect through loss of BRCA1.; ATM and ATR and can be targeted with PARP inhibitor treatment. Oncotarget 2015, 6, 10548–10562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forster, M.D.; Dedes, K.J.; Sandhu, S.; Frentzas, S.; Kristeleit, R.; Ashworth, A.; Poole, C.J.; Weigelt, B.; Kaye, S.B.; Molife, L.R. Treatment with olaparib in a patient with PTEN-deficient endometrioid endometrial cancer. Nat. Rev. Clin. Oncol. 2011, 8, 302–306. [Google Scholar] [CrossRef]
- Min, A.; Im, S.A.; Yoon, Y.K.; Song, S.H.; Nam, H.J.; Hur, H.S.; Kim, H.P.; Lee, K.H.; Han, S.W.; Oh, D.Y.; et al. RAD51C-deficient cancer cells are highly sensitive to the PARP inhibitor olaparib. Mol. Cancer Ther. 2013, 12, 865–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heeke, A.L.; Pishvaian, M.J.; Lynce, F.; Xiu, J.; Brody, J.R.; Chen, W.J.; Baker, T.M.; Marshall, J.L.; Isaacs, C. Prevalence of Homologous Recombination-Related Gene Mutations Across Multiple Cancer Types. JCO Precis. Oncol. 2018, 2, 1–18. [Google Scholar] [CrossRef]
- Póti, A.; Gyergyak, H.; Nemeth, E.; Rusz, O.; Toth, S.; Kovacshazi, C.; Chen, D.; Szikriszt, B.; Spisák, S.; Takeda, S.; et al. Correlation of homologous recombination deficiency induced mutational signatures with sensitivity to PARP inhibitors and cytotoxic agents. Genome Biol. 2019, 20, 240. [Google Scholar] [CrossRef]
- Plummer, R.; Dua, D.; Cresti, N.; Drew, Y.; Stephens, P.; Foegh, M.; Knudsen, S.; Sachdev, P.; Mistry, B.M.; Dixit, V.; et al. First-in-human study of the PARP/tankyrase inhibitor E7449 in patients with advanced solid tumours and evaluation of a novel drug-response predictor. Br. J. Cancer 2020, 123, 525–533. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical and Pathologic Characteristics | n (%) | Median Age (Min–Max), Years |
|---|---|---|
| Age at olaparib treatment | 41 | 63 (31–84) |
| Organs | ||
| Ovary | 23 (56.2%) | 63 (43–84) |
| Breast | 8 (19.5%) | 62 (31–83) |
| Digestive tract | 8 (19.5%) | 62 (50–76) |
| (pancreas, colon, rectum) | (5, 2, 1) | |
| Endometrium | 1 (2.4%) | 56 |
| Skin | 1 (2.4%) | 73 |
| Histology | ||
| Adenocarcinoma (breast, digestive tract) | 16 (39%) | |
| High-grade serous adenocarcinoma | 22 (53.8%) | |
| Clear cell adenocarcinoma | 1 (2.4%) | |
| Basal Cell carcinoma | 1 (2.4%) | |
| Unknown | 1 (2.4%) | |
| Response to platinum salts | Days (min–max) | |
| Number of patients treated | 39 (92.9%) | |
| Progression Free Survival | 126 (30–637) | |
| PFS < 90 days | 7 (18%) | |
| PFS > 90 days | 32 (82%) |
| Cancer Type, Patient No. | Gene(s) | Nucleotide Variant | Protein Variant | Impact | PFS (Days) |
|---|---|---|---|---|---|
| Ovarian #1 | BRCA1 | c.798_799delTT | Ser267LysfsTer19 | Pathogenic | 1218 (still under olaparib) |
| Ovarian #2 | BRCA1 | c.53T > C | p.Met18Thr | Unknown | 240 |
| Ovarian #3 | BRCA1 | c.2477_2478delCA | p.Thr826ArgfsTer4 | Pathogenic | 953 |
| Ovarian #4 | BRCA2 | c.7617 + 1G > T | Pathogenic | 441 | |
| Ovarian #5 | BRCA1 | c.181T > G | p.Cys61Gly | Pathogenic | 59 |
| Ovarian #6 | PALB2 | c.656A > G | p.Asp219Gly | Unknown | 224 |
| Ovarian #7 | BRCA2 | c.3847_3848delGT | p.Val1283LysfsTer3 | Pathogenic | 1659 (still under olaparib) |
| Ovarian #8 | BRCA1 | c.3708T > G | p.Asn1236Lys | Benign | 81 |
| Ovarian #9 | BRCA1 | c.2066_2069delGTAA | p.Ser689LysfsTer11 | Pathogenic | 910 |
| Ovarian #10 | BRCA1 | c.3839_3843delinsAGGC | p.Ser1280_Gln1281delinsTer | Pathogenic | 94 |
| Ovarian #11 | BRCA2 | c.8504C > G | p.Ser2835Ter | Pathogenic | 1359 (still under olaparib) |
| Ovarian #12 | BRCA1 | c.4956G > A | p.Met1652Ile | Benign | 58 |
| BRCA2 | c.9976A > T | p.Lys3326Ter | Benign | ||
| Ovarian #13 | BRCA1 | c.349C > T | p.His117Tyr | Unknown | 120 |
| BRCA2 | c.8494G > T | p.Glu2832Ter | Pathogenic | ||
| Ovarian #14 * | BRCA1 | c.4204C > T | p.Gln1402Ter | Pathogenic | 101 |
| Ovarian #15 * | BRCA1 | c.4204C > T | p.Gln1402Ter | Pathogenic | 168 |
| Ovarian #16 | BRCA1 | c.68_69delAG | p.Glu23ValfsTer17 | Pathogenic | 79 |
| Ovarian #17 | BRCA2 | c.3267_3268delGA | p.Gln1089HisfsTer9 | Pathogenic | 614 |
| Ovarian #18 | BRCA1 | c.191G > A | p.Cys64Tyr | Pathogenic | 636 |
| Ovarian #19 | BRCA2 | c.5350_5351delAA | p.Asn1784HisfsTer2 | Pathogenic | 1 (allergic reaction) |
| Ovarian #20 | BRCA1 | c.2744C > T | p.Ser915Phe | Unknown | 368 |
| Ovarian #21 | BRCA2 | c.2539A > T | p. Arg847Ter | Pathogenic | 288 |
| Ovarian #22 | ATM | c.103C > T | p.Arg35Ter | Pathogenic | 306 |
| Ovarian #23 | BRCA2 | c.1690T > C | p.Met990Lys | Unknown | 93 |
| Breast #1 | BRCA2 | c.1981_1984dup | p.Ser662Ter | Pathogenic | 231 |
| Breast #2 | BRIP1 | c.2002delG | p.Glu668LysfsTer20 | Pathogenic | 98 |
| Breast #3 | BRCA1 | c.5341G > T | p.Glu1781Ter | Pathogenic | 190 |
| Breast #4 | BRCA2 | c.7654dupA | p.Ile2552AsnfsTer2 | Pathogenic | 223 |
| BRCA2 | c.7645_7668delTGCATAAAAATTAACAGCAAAAAT | p.Cys2549_Asn2556del | Unknown | ||
| Breast #5 | BRCA1 | c.4251_4252delG > T | p.Leu1418ArgfsTer9 | Pathogenic | 1212 (still under olaparib) |
| Breast #6 | BRCA1 | c.2783G > T | p.Gly928Val | Unknown | 136 |
| Breast #7 | BRCA1 | c.3485delA | Asp1162ValfsTer48 | Pathogenic | 116 |
| Breast #8 | BRCA2 | c.4860A > T | p.Leu1620Phe | Unknown | 144 |
| RAD51D | c.328G > A | p.Asp110Asn | Unknown | ||
| Digestive tract #1 (colon) | PALB2 | c.2719G > A | p.Glu907Lys | Unknown | 36 |
| Digestive tract #2 (pancreas) | BRCA1 | c.2521C > T | p.Arg841Trp | Unknown | 12 |
| UIMC1 | c.1690T > C | p.Tyr564His | Unknown | ||
| Digestive tract #3 (pancreas) | CHEK2 | c.349A > G | p.Arg160Gly | Pathogenic | 64 |
| Digestive tract #4 (rectum) | BRCA1 | c.2521C > T | p. Arg841Trp | Probably Benign | 62 |
| Digestive tract #5 (pancreas) | BRCA1 | c.5128A > C | p.Met1710Leu | Unknown | 54 |
| Digestive tract #6 (pancreas) | BRCA1 | c.5295A > C | p.Glu1786Asp | Unknown | 12 |
| Digestive tract #7 (pancreas) | BRIP1 | c.3G > A | p.Met1? | Unknown | 27 |
| Digestive tract #8 (pancreas) | ATM | c.598C > T | p.Gln200Ter | Pathogenic | 20 |
| Endometrium #1 | PTEN | c.867dupA | p.Val290SerfsTer8 | Pathogenic | 190 |
| Skin #1 | PALB2 | c.2431C > T | p.Pro811Ser | Unknown | 210 |
| RAD50 | c.3041A > G | p.Gln1014Arg | Unknown | ||
| RAD51C | c.584C > T | p.Ala195Val | Unknown |
| Cancer Type, Patient No. | Gene(s) | Nucleotide Variant | Protein Variant | Second Hit Prediction * | Dann Prediction | PROVEAN Prediction | P S | Final Prediction | PFS (Days) |
|---|---|---|---|---|---|---|---|---|---|
| Ovarian #2 | BRCA1 | c.53T > C | p.Met18Thr | B (0.19, Red) | D | D | 1 | S | 240 |
| Ovarian #6 | PALB2 | c.656A > G | p.Asp219Gly | D (1.12, Green) | B | B | 1 | S | 224 |
| Ovarian #20 | BRCA1 | c.2744C > T | p.Ser915Phe | B (0.14, Red) | D | D | 2 | S | 368 |
| Ovarian #23 | BRCA2 | c.1690T > C | p.Met990Lys | B (0.26, Red) | B | D | 1 | S or R | 93 |
| Breast #6 | BRCA1 | c.2783G > T | p.Gly928Val | D (1.04, Green) | U | D | 1 | S | 136 |
| Breast #8 | BRCA2 | c.4860A > T | p.Leu1620Phe | B (0.44, Red) | B | D | 2 | S | 144 |
| RAD51D | c.328G > A | p.Asp110Asn | B (0.16, Red) | D | D | ||||
| Digestive tract #1 (colon) | PALB2 | c.2719G > A | p.Glu907Lys | B (0.66, Red) | B | B | 1 | R | 36 |
| Digestive tract #2 (pancreas) | BRCA1 | c.2521C > T | p.Arg841Trp | D (1.48, Green) | B | D | 3 | R | 12 |
| UIMC1 | c.1690T > C | p.Tyr564His | D (0.91, Green) | D | D | ||||
| Digestive tract #5 (pancreas) | BRCA1 | c.5128A > C | p.Met1710Leu | B (0.34, Red) | B | B | 0 | R | 54 |
| Digestive tract #6 (pancreas) | BRCA1 | c.5295A > C | p.Glu1786Asp | B (0.27, Red) | U | B | 1 | R | 12 |
| Digestive tract #7 (pancreas) | BRIP1 | c.3G > A | p.Met1? | D (0.96, Green) | U | U | 3 | R | 27 |
| Skin #1 | PALB2 | c.2431C > T | p.Pro811Ser | B (0.53, Red) | B | B | 1 | S | 210 |
| RAD50 | c.3041A > G | p.Gln1014Arg | B (0.54, Red) | B | B | ||||
| RAD51C | c.584C > T | p.Ala195Val | D (0.72, Green) | D | B |
| Characteristics | Olaparib n (%) | Niraparib n (%) |
|---|---|---|
| Total | 14 | 24 |
| Histology | ||
| High grade serous adenocarninoma | 12 (86%) | 24 (100%) |
| Endometrioid carcinoma | 2 (14%) | 0 |
| Response to platin | ||
| Complete response | 5 (36%) | 8 (33%) |
| Partial response | 7 (50%) | 14 (58%) |
| Stable disease | 2 (14%) | 2 (9%) |
| Genes | Nucleotide Variant | Protein Variant | Second Hit Prediction | Dann Prediction | Final Prediction with PROVEAN | PS | Treatment | Final Prediction | PFS (Days) |
|---|---|---|---|---|---|---|---|---|---|
| INPP4B | c.1381T > C | p.Phe461Leu | D | B | N | 1 | Nira | S | 392 |
| ATM | c.2578G > C | p.Asp860His | B | D | D | 1 | Nira | S | 176 |
| FANCF | c.373G > A | p.Asp125Asn | B | B | B | 1 | Nira | S | 113 |
| PALB2 | c.1273G > A | p.Val425Met | D | B | B | ||||
| RAD51B | c.902G > A | p.Ser301Asn | B | D | B | ||||
| ATM | c.4079G > A | p.Ser1360Asn | B | B | B | 1 | Nira | R | 342 |
| BRCA2 | c.9109C > G | p.Gln3037Glu | U | B | B | 2 | Nira | R | 84 |
| BRCA2 | c.1181A > C | p.Glu394Ala | U | B | B | 1 | Nira | R | 87 |
| BRCA1 | c.1692T > A | p.Asn564Lys | B | B | B | 1 | Nira | R | 469 |
| RAD50 | c.527C > T | p.Thr176Ile | U | D | B | 1 | Nira | U | 63 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chevrier, S.; Richard, C.; Collot, T.; Mananet, H.; Arnould, L.; Boidot, R. An Algorithm Combining Patient Performance Status, Second Hit Analysis, PROVEAN and Dann Prediction Tools Could Foretell Sensitization to PARP Inhibitors in Digestive, Skin, Ovarian and Breast Cancers. Cancers 2021, 13, 3113. https://doi.org/10.3390/cancers13133113
Chevrier S, Richard C, Collot T, Mananet H, Arnould L, Boidot R. An Algorithm Combining Patient Performance Status, Second Hit Analysis, PROVEAN and Dann Prediction Tools Could Foretell Sensitization to PARP Inhibitors in Digestive, Skin, Ovarian and Breast Cancers. Cancers. 2021; 13(13):3113. https://doi.org/10.3390/cancers13133113
Chicago/Turabian StyleChevrier, Sandy, Corentin Richard, Thomas Collot, Hugo Mananet, Laurent Arnould, and Romain Boidot. 2021. "An Algorithm Combining Patient Performance Status, Second Hit Analysis, PROVEAN and Dann Prediction Tools Could Foretell Sensitization to PARP Inhibitors in Digestive, Skin, Ovarian and Breast Cancers" Cancers 13, no. 13: 3113. https://doi.org/10.3390/cancers13133113