1. Introduction
Kidney cancer is a common malignant tumor in both men and women and accounts for approximately 5% of all cancer incidences [
1]. More than 140,000 people die each year from kidney cancer worldwide, and kidney cancer is the 13th most common cause of cancer death [
2]. The number of patients newly diagnosed with kidney cancer has been increasing for decades [
3]. Advances in imaging technology and the increasing use of health care facilities have led to the early detection of kidney cancer cases [
4]; however, many individuals are still diagnosed with metastatic kidney cancer. The 5-year survival rate of people with kidney cancer is approximately 75%; however, the survival rate depends on a number of factors, including the histologic subtype, tumor size, and stage of cancer upon first diagnosis. Kidney cancer in about two-thirds of patients “diagnosed at early stages is localized to the kidney and these people have a 5-year survival rate of 93%. If kidney cancer has already spread to surrounding tissues or local lymph nodes at the time of diagnosis, then the 5-year survival rate of the patient is 70%. If the cancer is accompanied by distant metastases at the time of diagnosis, the 5-year survival rate is 12% [
5]. Despite the beneficial effects of vascular endothelial growth factor inhibitors on the survival of patients with metastatic kidney cancer, survival may be less than a year [
5]. Therefore, identification of new molecular targets is necessary for the effective treatment of patients with metastatic kidney cancer.
PDLIM2 plays an essential role in cell differentiation and cytoskeleton formation, and is a member of the actinin-associated LIM family associated with tumorigenesis [
6,
7]. PDLIM2, located on chromosome 8p21, is a nuclear protein containing both PDZ and LIM domains, with various cellular functions, such as cell migration regulation, cell polarization, and epithelial–mesenchymal transition (EMT). [
8] It also regulates the activity of transcription factors, such as NF-κB and STAT, and is associated with the development or inhibition of several malignancies [
6,
7,
9]. The role of PDLIM2 in tumor formation and inhibition is controversial. In many cancers, PDLIM2 levels are epigenetically suppressed by promoter hypermethylation, which blocks transcription. In addition, PDLIM2 upregulation has been linked to a pattern of tumorigenesis suppression [
10,
11,
12]; however, in malignant tumors, the chromosomal region containing the PDLIM2 gene is often destroyed, and high expression of PDLIM2 in cancer cell lines derived from metastatic cancer has been associated with the formation of malignant tumors [
13,
14]. Therefore, PDLIM2 can play contradictory roles in various tumors and may be a potential therapeutic target. In this study, we evaluated the expression pattern of PDLIM2 in human renal cancer cells and metastatic kidney cancer and the effect of PDLIM2 inhibition on tumor growth and invasiveness.
3. Discussion
This is the first study to examine the role of PDLIM2 in the tumor growth and metastasis of kidney cancer in a xenograft model. We observed that PDLIM2 is expressed at high levels in metastatic human kidney cancer cell lines comparing non-metastatic kidney cancer cell lines. In particular, the specific inhibition of PDLIM2 was found to result in the reduced proliferation and migration ability of metastatic kidney cancer cells. As metastasis adaptation characteristics were acquired in murine kidney cancer cell line, PDLIM2 expression also was increased. In addition, we provide in vivo evidence that the specific inhibition of PDLIM2 significantly reduces tumor growth and metastasis in a human kidney cancer xenograft model; this is the key finding of our study. Thus, we suggest that PDLIM2 may act as an oncogenic protein in metastatic kidney cancer development and that the selective inhibition of PDLIM2 may be a new therapeutic target for metastatic kidney cancer.
In many previous studies, PDLIM2 has been reported to be a tumor suppressor [
11,
16,
17,
18]. PDLIM2 is known to play a role in inhibiting the activation of the tumorigenic factors NF-κB and STAT3 [
16]. PDLIM2 acts as a tumor suppressor by inhibiting cancer-related genes and increasing the expression of genes involved in antigen presentation and T-cell activation [
16]. Sun et al. reported that PDLIM2 is epigenetically suppressed in lung cancer and is associated with treatment resistance and poor prognosis [
17]. In mice, the inhibition of PDLIM2 resulted in increased lung cancer incidence and was reported to cause resistance against anticancer drugs and immunotherapeutic drugs, such as PD-1 inhibitor [
17]. In breast cancer, PDLIM2 was associated with adhesion signaling and β-catenin activity; the inhibition of PDLIM2 plays a role in inhibiting tumor growth [
18].
However, in our study, higher PDLIM2 expression was observed in the process of in vivo metastasis adaptation of RenCa kidney cancer cells, and we found that PDLIM2 is highly related to tumor growth and metastasis in mouse knockout model, The epigenetic genetic repression of PDLIM2 negatively affects growth and expression of kidney cancer and spontaneous tumorigenesis following aging. These results suggested that it has a role as an oncogenic protein.
In a cancer genome atlas analysis, PDLIM2 was reported to be an unfavorable prognostic factor in kidney cancer. In pathological tissues, high PDLIM2 expression was reported to be linked to poor prognosis [
19]. The principle underlying the role of PDLIM2 in tumor growth and metastasis is not yet clear. In previous studies, we reported that PDLIM2 is associated with the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated protein kinase signaling pathway (ERK) and that an increase in PDLIM2 is associated with the activation of this pathway [
10]. In addition, PDLIM2 was suggested to interact with the MAPK/ERK signaling pathway to regulate MET and various genes, such as cell cycle proteins [
20]. MAPK and mitogen-activated kinases (MKKs) play an essential role in several biological pathways that regulate cell differentiation, proliferation, and survival [
20]. Activated in response to extracellular stimuli, MKK phosphorylates ERK and MAPK [
20]. The MAPK/ERK signaling pathway plays an essential role in cell proliferation and differentiation and in tumor formation and metastasis [
20]. The MAPK/ERK signaling pathway can show both oncogenic and tumor-suppressive effects depending on the tissue-specific tumor microenvironment. In addition, mutations affecting the MAPK/ERK pathway depend on the type of cancer. These differences in tissue and tissue-specific tumor microenvironment may have influenced the role of MAPK/ERK signaling pathway and PDLIM2 gene. The epigenetic genetic repression of PDLIM2 have influenced the MAPK/ERK pathway depending on the tissue-specific tumor microenvironment.
Bowe et al. also reported that PDLIM2 is highly expressed in invasive cancer cells [
21]. In a previous study, we found PDLIM2 to be expressed at higher levels in castration-resistant prostate cancer (CRPC)-like cells in prostate cancer [
10]. Moreover, we confirmed that the specific inhibition of PDLIM2 resulted in significantly reduced tumor growth in human CRPC xenograft models. The inhibition of PDLIM2 resulted in a reduced number and weakened viability, proliferation, and clonal growth of prostate cancer cells; furthermore, PDLIM2 may play a carcinogenic role in human CRPC-like cells [
10]. The expression level of PDLIM2 in androgen-sensitive prostate cancer cells and CRPC-like cells was significantly different, and PDLIM2 was expressed at a high level in CRPC-like cells [
10]. This suggests that PDLIM2 may play several roles in regulating tumor growth and progression in various organs and circumstances. Bowe et al. reported that PDLIM2 is highly expressed in invasive cancer cells [
19]. Additionally, the inhibition of PDLIM2 has been reported to reduce the expression of several oncogenes and increase tumor suppressor gene expression [
19].
The incidence, metastasis, and angiogenesis of several malignant tumors, including kidney cancer, have been reported to be associated with the activation of the MAPK signaling pathway [
22]. Angiogenesis is essential for kidney cancer. Vascular endothelial growth factor inhibitors are commonly used against metastatic kidney cancer targets, such as angiogenesis-related targets. MAPK/ERK activation is associated with angiogenic growth factor signaling in tumor cells, suggesting that PDLIM2 may act as an oncogenic protein that promotes tumor formation and metastasis by activating MAPK/ERK [
10]. Furthermore, Huang et al. reported that the inhibition of the MAPK and ERK pathways in kidney cancer markedly inhibits the growth of anchorage-independent RCC cells, reduces the extension of tumor cells, and reduces tumor angiogenesis. Therefore, the inhibition of the MAPK signaling pathway leads to the destruction of tumor vasculature [
22].
In addition, PDLIM2 may contribute to the ability of tumor cells to migrate [
10,
19,
23]. In the present study, a decreased migration ability of metastatic kidney cancer cells was observed in the PDLIM2 knockdown group, in which PDLIM2 was selectively inhibited. The PDLIM2 gene encodes a member of the ALP subfamily of proteins in the PDZ-LIM domain [
23]. The encoded protein inhibits anchorage-dependent growth and promotes cell migration and adhesion by interacting with the actin cytoskeleton [
23]. Bowe et al. reported that the inhibition of PDLIM2 inhibited the reversal of the EMT phenotype, loss of orientation, and cytoskeleton polarization [
19].
The preclinical data of this study showed that the regulation of PDLIM2 expression, which regulates the MAPK/ERK signaling pathway, is involved in tumor growth and proliferation in metastatic renal cancer. In addition to the tumor, it was found that the regulation of PDLIM2 expression of the individual was also involved in the growth and proliferation of the tumor. Furthermore, the epigenetic genetic repression of PDLIM2 influenced spontaneous development of tumors. PDLIM2 has an essential regulatory role that may alter the response of cancer cells, and current evidence shows that PDLIM2 is a viable target for cancer therapy. However, further studies are needed to determine the applicability of PDLIM2-targeted treatment in actual clinical practice.
Our study has some limitations. First, we observed an association between PDLIM2 and carcinogenesis. There is only partial in vitro evidence for the oncogenic signaling pathway associated with PDLIM2. Second, to utilize our results in clinical settings, it is necessary to confirm whether PDLIM2 is highly expressed in the kidneys of patients with kidney cancer, whether there is a difference between primary kidney cancer and metastatic kidney cancer in terms of PDLIM2 expression, and whether PDLIM2 is correlated with the prognosis of kidney cancer.
4. Materials and Methods
4.1. Materials
Dulbecco’s modified Eagle’s medium (DMEM), trypsin, antibiotics, TRIzol, and Lipofectamine 2000 were purchased from Invitrogen (Carlsbad, CA, USA). Fetal bovine serum and culture media were obtained from HyClone Laboratories Inc. (South Logan, UT, USA). Anti-PDLIM2 antibodies were purchased from Novus (Littleton, CO, USA) and Abcam (Cambridge, UK). Antibodies against ECAD and NCAD were obtained from Cell Signaling Technology (Danvers, MA, USA). Anti-β-actin antibody (Cat. No. A2066) and other chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA).
4.2. Cell Lines, Plasmids, Virus Production, and Infection
HK-2, ACHN, and 7860 cell lines were purchased from American Type Culture Collection (Manassas, VA, USA). Caki-1, RenCa, and 293T cell lines were purchased from Korean Cell Line Bank (Seoul, Korea). HK-2 cells were cultured in Keratinocyte SFM medium (GIBCO, Carlsbad, CA, USA). Caki-1, ACHN, 786O, and RenCa cells were cultured in MEM (WELGENE, Gyeongsan-si, Korea), and 293T cells for lentiviral packaging were cultured in DMEM (WELGENE, Gyeongsan-si, Korea) supplemented with 10% fetal bovine serum at 37 °C under 5% CO
2. For gene silencing, control and PDLIM2 shRNA-expressing lentivirus infected and stable cell lines were established as previously described [
15]. The oligonucleotide sequences for the PDLIM2 shRNA are listed in
Supplementary Table S1. The FUGW-luc vector was obtained from the Molecular Imaging and Neurovascular Research Laboratory, Dongguk University Ilsan Hospital, Goyang, Republic of Korea; FUGW-luciferase-expressing cells were produced as follows. The FUGW-luc vector was cut using the XhoI enzyme and transfected into the RenCa cell line (RenCa-GFP). The cells incorporated with FUGW-luc were sorted with the GFP channel in BD FACSAria II (BD Biosciences, Franklin Lakes, NJ, USA).
4.3. Colony Formation Assay and Cell Viability Assay
For the colony formation assay, 1000 cells/well were plated in 6-well plates. The cells were cultured for 14 days and then stained with 0.1% crystal violet. The cell colonies were photographed, and the number of colonies comprising more than 50 cells was counted using a SZX7 stereo microscope (Olympus, Tokyo, Japan). For the cell viability assay, cells (2000 to 3000 cells/well) were dispensed in 100-μL culture medium in a 96-well plate and incubated for certain periods. Then, a 10 μL solution of the EZ-Cytox cell viability kit (Daeil-Lab, Seoul, Korea) was mixed with the culture medium in each well of the plate. Samples were incubated for 1 h at 37 °C, and the absorbance of each sample at 450 nm was measured using a microplate reader (PerkinElmer, Waltham, MA, USA).
4.4. RNA Isolation and Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR)
Total cellular RNA was extracted using TRIzol reagent (Ambion, Austin, TX, USA) according to the manufacturer’s instructions. For each reverse-transcription reaction, 1 μg total RNA was used for cDNA synthesis using the MultiScribe Reverse Transcription Kit from Life Technologies (Carlsbad, CA, USA). RT-qPCR was performed using the EvaGreen qPCR Master Mix Kit from Applied Biological Materials Inc. (Richmond, BC, Canada) and the StepOne™ Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). The quantity of 18S ribosomal RNA was measured as an internal control. The sequences of the primers used for reverse transcription and qPCR are listed in
Supplementary Table S1.
4.5. Western Blotting
The cells (5 × 106) were lysed in 1 mL RIPA buffer (150 mM NaCl, 50 mM Tris-HCl [pH 7.2], 0.5% NP-40, 1% Triton X-100, and 1% sodium deoxycholate) containing a protease/phosphatase inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA). The cell lysates were separated on sodium dodecyl sulfate-polyacrylamide gels and transferred onto an Immobilon-P PVDF Membrane (Millipore, Darmstadt, Germany). The membranes were blocked with 5% skim milk in 0.1% Tween-20 for 1 h, followed by overnight incubation at 4 °C with primary antibodies. The membranes were incubated with a horseradish peroxidase-conjugated secondary antibody (1:5000) for 1 h and developed using the ECL-Plus Kit (Thermo Scientific, Rockford, IL, USA).
4.6. Wound Healing and Cell Invasion Assays
The wound healing assay was performed on 100% confluent cells plated in 6-well culture plates. The cells were scratched using a pipette tip and washed twice to remove debris; fresh medium was then added. The cells were incubated in a humidified atmosphere containing 5% CO2 at 37 °C and observed using a SZX7 stereo microscope at certain time points. After the indicated time period, the scratched areas were measured using the ImageJ program (ver. 1.43u; www.rsb.info.nih.gov/ij, accessed on 10 December 2020). For the invasion assay, cells (5 × 104/well) were plated in the upper chambers of transwell culture plates without serum using Matrigel-coated polycarbonate membranes (Corning, Big Flats, NY, USA). Basal medium containing 10% fetal bovine serum was added to the lower chambers as a chemoattractant for cell migration. After 48 h, the cells that did not migrate (non-migrating cells) were removed from the upper chambers, whereas the cells that migrated through the chambers were fixed using 10% ethanol (Sigma-Aldrich, St. Louis, MO, USA). Afterwards, the cells were stained with 0.01% crystal violet solution (Sigma-Aldrich), and the cells that migrated were randomly counted across five different microscopic fields at 20× magnification.
4.7. Animal Studies
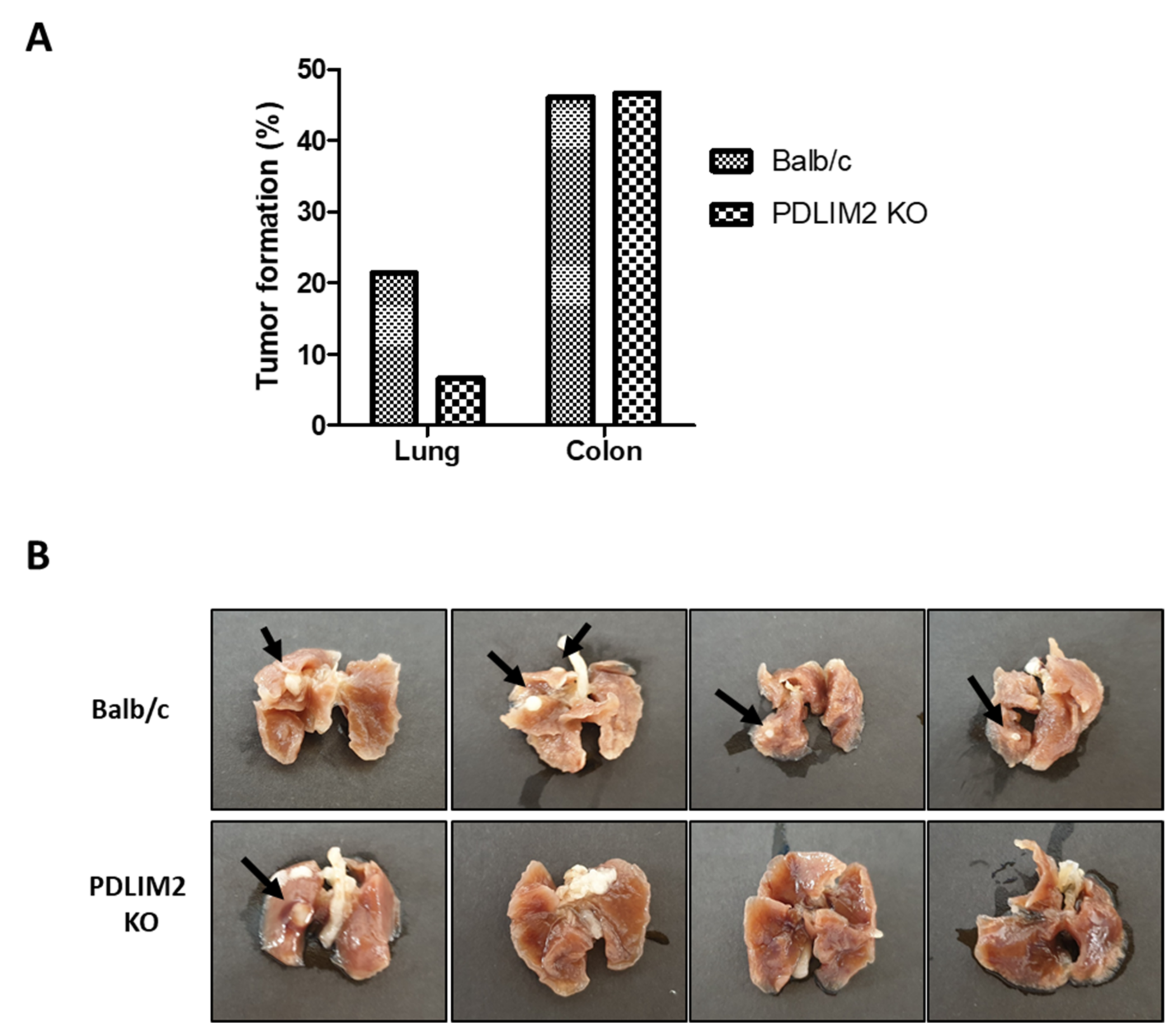
All animal experiments were performed in accordance with the Seoul National University Hospital institutional guidelines under IACUC protocol No.19-0058-S1A0. PDLIM2 knockout mice were kindly provided by Dr. Takashi Tanaka (RIKEN Laboratory, Kanagawa, Japan). To establish a blood-borne metastatic model, 1 × 106 RenCa-GFP cells in 100 μLPBS were injected into the tail vein of six-week-old BALB/c or PDLIM2 knockout mice (n = 8 for each group). Bioluminescent signals were measured every week for 4 weeks; the lungs with tumors were excised, and bioluminescent signals were measured. The lungs with tumors were photographed and fixed with paraformaldehyde (PFA). The tumor specimens were stained with H&E. To establish a metastatic model with orthotopic tumors, 1 × 105 RenCa-GFP cells expressing a luciferase expression vector were injected into the kidneys of six-week-old male BALB/c mice or PDLIM2 knockout mice (n = 5 for each group). For the injection step, cells were suspended in 100 µL of 50% Matrigel (BD Biosciences) in complete medium. Bioluminescent signals were measured every week for 3 weeks until the nephrectomy of tumor-bearing kidney was performed. After nephrectomy, metastatic signals were measured for another 3 weeks. The mice were euthanized, and organs were extracted, measured for bioluminescent signals, fixed in 4% PFA at 4 °C, and embedded in paraffin. Then, the tumor specimens were stained with H&E.
4.8. In Vivo Bioluminescence Imaging
The mice from each group were intraperitoneally injected with 150 mg/kg VivoGloTM D-luciferin (Promega, Madison, WI, USA) and imaged after 15 min. After anesthetizing the mice using 1–3% isoflurane, the photons emitted from the tumor were detected using Xenogen IVIS Imaging System 200 (Alameda, CA, USA). Depending on the experimental conditions, the settings for the target max count (3000 or 30,000 photon counts) and exposure time (10 s. or auto-exposure with a maximum exposure time of 1 min) were applied for imaging. Regions of interest (ROI) of the same size and shape were used for all acquired images to measure the total flux (photons per sec) in the ROI. Living Image (Version 2.20, Xenogen, Alameda, CA, USA) was used to quantify signals emitted from the ROI.
4.9. Generation of the Metastasis-Prone Adapted RenCa Cells
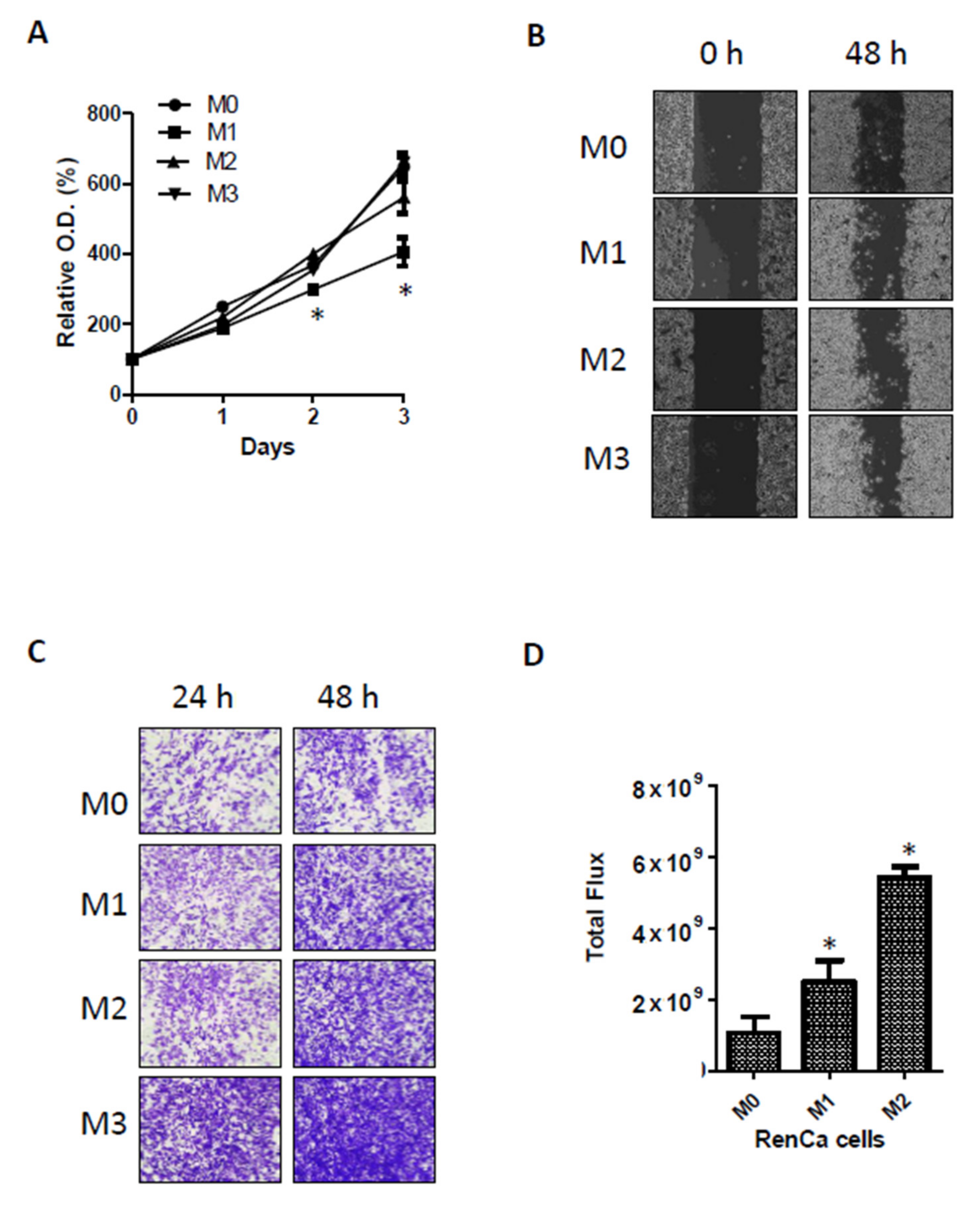
Metastasis-prone adapted RenCa cells were established as follows. RenCa-GFP cells were injected into 6-week-old male BALB/c mouse kidneys. Primary tumors were monitored weekly using bioluminescence signals, and 3 weeks after injection, the tumor-bearing kidneys from each mouse were removed; metastatic signals were monitored for another 3 weeks. On the day of euthanasia, the tumor masses in the lungs were surgically dissected under sterile conditions and incubated with trypsin for 2 h at 37 °C. The dissociated cells were selected in vitro by adding zeocin under normal cell culture conditions for 1 week. These metastatic cells were considered as metastatic round 1 (RenCa-M1), and the expanded tumor cells were re-implanted into the mouse kidney. This in vivo metastatic cycle was performed two more times until highly metastatic RenCa cells were established (M2,M3).
4.10. Immunohistochemistry
Kidney tumors arising from orthotopically injected original (M0) and metastatic RenCa cells (M1,M2) were fixed using 4% formaldehyde solution overnight. After dehydration, the tissues were embedded in paraffin. Serial sections were sliced, mounted on silanized glass slides, and stained with H&E. The tumor slides were immune-stained with PDLIM2 antibody.
4.11. Statistical Analyses
All data were analyzed using Microsoft Excel 2010 software, unless otherwise stated. Normally distributed continuous variables were analyzed using Student’s t-test. All the statistical tests were two-tailed. p < 0.05 was considered statistically significant.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







