Simple Summary
Neurofibromatosis type 1 (NF1) is a genetic disorder caused by pathogenic variants in the NF1 tumor suppressor gene. In 5–10% of NF1 patients, a large heterozygous deletion of the whole NF1 gene is identified, leading to the commonly called “NF1 microdeletion syndrome”. NF1-deleted patients were previously reported to develop a particularly severe form of the disease with frequent cognitive impairment and an increased risk of benign and malignant tumors. Here, we performed a comprehensive clinical assessment of the largest NF1-deleted cohort to date, including 126 NF1 patients with a deletion of the NF1 gene. This work provides new insights into the specific phenotype associated with NF1 deletions and may contribute to improve the follow-up care of NF1 patients.
Abstract
Complete deletion of the NF1 gene is identified in 5–10% of patients with neurofibromatosis type 1 (NF1). Several studies have previously described particularly severe forms of the disease in NF1 patients with deletion of the NF1 locus, but comprehensive descriptions of large cohorts are still missing to fully characterize this contiguous gene syndrome. NF1-deleted patients were enrolled and phenotypically characterized with a standardized questionnaire between 2005 and 2020 from a large French NF1 cohort. Statistical analyses for main NF1-associated symptoms were performed versus an NF1 reference population. A deletion of the NF1 gene was detected in 4% (139/3479) of molecularly confirmed NF1 index cases. The median age of the group at clinical investigations was 21 years old. A comprehensive clinical assessment showed that 93% (116/126) of NF1-deleted patients fulfilled the NIH criteria for NF1. More than half had café-au-lait spots, skinfold freckling, Lisch nodules, neurofibromas, neurological abnormalities, and cognitive impairment or learning disabilities. Comparison with previously described “classic” NF1 cohorts showed a significantly higher proportion of symptomatic spinal neurofibromas, dysmorphism, learning disabilities, malignancies, and skeletal and cardiovascular abnormalities in the NF1-deleted group. We described the largest NF1-deleted cohort to date and clarified the more severe phenotype observed in these patients.
1. Introduction
Neurofibromatosis type 1 (NF1; MIM#162200) is an autosomal dominant disease with a worldwide incidence estimated between 1 in 2500 and 1 in 3000 individuals [1]. In more than 95% of NF1 patients, a loss-of-function pathogenic variant is identified in the NF1 gene (MIM*613113) [2,3]. NF1 is a tumor suppressor located at 17q11.2; it contains 57 constitutive and three alternative exons and spans over ~350 kb. The 8454-nucleotide coding region (NM_00267.3) encodes a 2818-amino-acid protein, neurofibromin, which shows tumor suppressor functions [4] by negatively regulating the RAS-MAPK pathway [5]. About half of NF1 cases result from de novo pathogenic variants.
NF1 diagnosis relies on the National Institutes of Health (NIH) criteria edited in 1988 [6]. These clinical criteria include multiple café-au-lait spots (CALS), axillary or inguinal freckling, Lisch nodules, optic pathway gliomas (OPGs), bone lesions, and neurofibromas (NFs). NFs are benign peripheral nervous tumors and are a pathognomonic manifestation of NF1. They may be cutaneous (cNFs), subcutaneous (scNFs), or plexiform (pNFs) neurofibromas. Plexiform neurofibromas are usually congenital or appear in early childhood in 30% to 50% of NF1 patients; pNFs present as a subtle enlargement of soft tissue with a “wrinkled” texture or a patch of hyperpigmentation with or without hypertrichosis. Although benign, pNFs are often life-threatening by their proximity to internal organs and can undermine quality of life. In ~10% cases, pNFs can transform into malignant peripheral nerve sheath tumors (MPNSTs) [7]. Other less specific clinical features can be observed in NF1 patients, such as musculoskeletal [8] or neurological [9] manifestations, cardiovascular malformations [10,11], endocrine alterations [12], cognitive behavioral disorders [13], Noonan-like features [14], or malignancies [15,16].
Despite a complete penetrance, NF1 displays a highly variable inter- and intrafamilial expressivity in its major features and in the occurrence of complications. Heritability studies have suggested the existence of a genetic component of the variable expressivity [17,18,19]. A large spectrum of pathogenic variations has been reported in the NF1 gene [2,20,21], with 2783 unique variants reported in the Global Variome shared Leiden Open Variation Database (LOVD) on 19 February 2021. However, genetic studies have generally failed to establish clear-cut genotype–phenotype correlations according to the type of NF1 pathogenic variant [2,22]. Recently, a study suggested an association between the presence of NF1 truncating/splicing pathogenic variations or large deletions and the occurrence of “phenotypes requiring medical attention” [23]. International multicenter studies allowed the description of significant genotype-phenotype correlations for a few specific point pathogenic variants of the NF1 gene. The first correlation was identified in 2007 and later confirmed in 2019 for the in-frame deletion of methionine 992 of neurofibromin [24,25]. The Met992 deletions resulted in a milder phenotype characterized by the quasi-absence of neurofibromas and OPGs but with an increased occurrence of Noonan-like features and pulmonic stenosis, when compared to the “classic” NF1 population. Similarly, missense variants affecting the arginine 1809 in neurofibromin were associated with significantly more frequent Noonan-like features, including pulmonic stenosis and short stature, and an increased tendency to develop cognitive impairment and/or learning disorders [26,27,28,29,30]. Arg1809 variants were also associated with an absence of NFs and OPGs. Absence of NFs was also observed in patients with missense variants affecting the methionine 1149, together with the presence of multiple CALS, skinfold freckling, and Noonan-like features [31]. More severe phenotypes were described for individuals with pathogenic missense variants of codons 844 to 848, located in the cysteine-serine rich domain (CSRD), or Arg1276 and Lys1423 in the GAP-related domain (GRD) of neurofibromin [31,32]. These patients suffered from more frequent pNFs (codons 844–848, Lys1423) or spinal NFs (codons 844–848, Arg1276), OPGs (codons 844–848), skeletal abnormalities (codons 844–848, Lys1423), and Noonan-like features including cardiovascular abnormalities (Arg1276, Lys1423).
Interestingly, a particularly severe presentation of NF1 was also evidenced in patients with large deletions encompassing NF1 and several neighboring genes [33,34]. Patients suffered from a high burden of cutaneous NFs (cNFs) with an increased risk of developing MPNSTs. Clinical manifestations of NF1-deleted patients also typically included severe learning disabilities, and dysmorphic and Noonan-like features. NF1-deleted patients did not tend to have a short stature in adulthood, unlike NF1-mutated patients [35].
The emergence of genomic medicine, linked to the development of next generation sequencing (NGS) technologies 10 years ago, has enabled a more precise and rapid molecular characterization of many NF1 patients. These advances have led to the description of a growing number of NF1 pathogenic variants. Cohort analysis has provided the description of a “classical” NF1 phenotype, a necessary basis for the identification of genotype–phenotype correlations. In the present study, we aimed to confirm the distinctive phenotype associated with large deletions of the NF1 locus.
2. Materials and Methods
2.1. Study Cohort
Databases from routine molecular diagnosis at the Cochin Hospital, Paris, France, between 2013 and 2020 were retrospectively analyzed. Between 2013 and 2020, 4091 index cases (IC) with a clinical presentation suggestive of NF1 were molecularly screened for NF1 in the Cochin Hospital (APHP, Paris, France): 2875 index cases (IC) were confirmed with an NF1 pathogenic variant. In addition, 648 NF1 patients were enrolled between 2005 and 2013 in two French clinical research programs (Programme Hospitalier de Recherche Clinique, PHRC) entitled ‘Patients at risk of scalability in neurofibromatosis type 1: phenotypic, genotypic and proteomic comparative study in a cohort’ (PHRC ‘EvoNF’; 2005–2008; inclusion of 219 unrelated adult NF1 patients) and ‘Study of the expressivity of neurofibromatosis 1: identification of modifier genes’ (PHRC GenModif 2010–2013; inclusion of 429 unrelated adult NF1 patients) as part of the ‘NF-France database’ [19]. In these two programs, a pathogenic variant of the NF1 gene was identified in 604 index cases. All patients gave their written informed consent.
2.2. Phenotypes
Phenotypical data were recorded with a standardized questionnaire including biometrics and the main clinical features observed in NF1. When clinical assessment was missing or incomplete in the laboratory databases, referent clinicians were contacted to obtain additional clinical data recorded during the follow-up of their patients. Patients with missing data were considered as ‘not specified’ (NS) for the trait and were not included in the statistical analysis for that trait. CALS were counted regardless of their size. Most features were identified by physical examination, with Lisch nodules being diagnosed, or excluded, by slit-lamp examination; individuals not given a slit lamp examination were coded as ‘not documented’ (ND) and excluded from further analysis of the trait. The presence or absence of OPGs was determined by cranial MRI or CT examination, with individuals not given cranial imaging being encoded as ‘ND’. Facial dysmorphism was evaluated on the following aspects: coarse face, flat occiput/brachycephaly, facial asymmetry, prominent forehead, frontal bossing, ptosis, hypertelorism, midface hypoplasia, triangular face, small and down-slanting palpebral fissures, eversion of the lateral eyelid, epicanthic folds, large and low set ears, high and broad nasal bridge, bulbous nasal tip, hooked nose, wide and prominent philtrum, micrognathia, small pointed chin, low posterior hairline, webbed neck. Stature and weight were evaluated according to the 2018 AFPA (Association Française de Pédiatrie Ambulatoire) reference curves. Head circumference was evaluated according to the 2018 AFPA reference curves for children under 5 years old and according to the Nellhaus charts when older. Short and tall stature were defined as a height that was more than 2 standard deviations (sd) respectively below or above the age- and sex-matched population mean. Macrocephaly was defined as a head circumference that was more than 2 sd above the age- and sex-matched population mean. The diagnosis of learning disabilities was performed on specific testing of cognitive abilities and/or history of scholar difficulties leading to repeating at least one level. As the small number of observations of each individual malignancies would have resulted in a lack of power in statistical analyses, we pooled all types of malignant tumors into a single item (namely “malignancies”) and performed analyses on this composed trait.
2.3. NF1 Molecular Analysis
The molecular analysis of the NF1 gene was performed using a variety of screening methodologies including DNA and RNA sequencing, polymorphic microsatellite (MS) marker analysis, and multiplex ligation-dependent probe amplification (MLPA), comparative genomic hybridization array (CGHa), or real-time PCR-based gene-dosage analysis to permit deletion assessment, as previously described [36,37,38]. NGS experiments were performed at the NGS facility of Cochin Hospital, Paris (Assistance Publique-Hôpitaux de Paris AP-HP, France). A custom-made amplification panel (Thermo Fisher Scientific, Courtaboeuf, France) and an Ion S5 XL system (Thermo Fisher Scientific) were used to sequence the coding and IVS flanking (25bp) regions of the NF1 and SPRED1 genes. The sequence analysis was performed according to the Genome Analysis Tool Kit (GATK) guidelines using Polyquery (Paris Descartes University, Paris, France). Assessment of variants implication was performed based on population databases (dbSNP and gnomAD), variant databases (HGMD, LOVD, and COSMIC), and predictions softwares (Alamut and mutation taster). An assessment of variants’ pathogenicity was performed according to the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG-AMP) guidelines [39]. Copy number variation analysis (CNA) was performed using the quantitative values obtained from the number of reads for each amplicon of each sample. For NF1 CNA analysis, SPRED1 was considered as the control gene, and reciprocally for the SPRED1 CNAs. Read number for each NF1 and SPRED1 amplicon was normalized by dividing each amplicon read number by the total of amplicon read numbers of a control gene from the same sample. Copy number ratios of <0.7 and >1.3 were considered deleted and duplicated, respectively.
2.4. Statistical Analyses
Univariate analysis was performed using a two-tailed Fisher’s exact test to compare categorical variables. Resulting p values were adjusted using the Benjamini-Hochberg (B-H) correction for multiple comparisons [40]. Reference cohort for comparison (“classic NF1”) was obtained from Koczkowska et al. [31]. Statistical analyses were performed with the R stats package in RStudio v4.0.3. Radar charts were obtained with the fmsb package in RStudio v4.0.3.
2.5. Characterization of the Deletions
The SALSA MLPA P122 NF1-area (MRC Holland, Amsterdam, Netherlands) was used to determine the NF1 deletion type in patients: type 1, type 2, type 3, or atypical deletions, as previously described [41]. Briefly, 100 ng of patients’ DNA were denatured at 98 °C and hybridized to the kit probes at 60 °C. Ligation was performed at 54 °C, followed by an amplification step with 35 PCR cycles. Fragment separation and detection using GS-500LIZ marker were performed on ABI Prism 3130 automatic DNA sequencer (Thermo Fisher Scientific). The results were analyzed with the GeneMapper® v.4.0 software package (Thermo Fisher Scientific). The kit contains ten control probes on autosomes, one on X, and one on Y chromosomes. Twenty-three probes are distributed along the 17q11.2 region (5 NF1-intragenic, 11 centromeric, and 7 telomeric) and two are located in ASPA (17p13.2) and PMP22 (17p12) genes, respectively. Deletions encompassing SUZ12P1 to LRRC37B probes were classified as type 1, and those including probes in exon 1 of NF1 to LRRC37B were classified as type 3. Type 2 deletions included at least probes from CRLF3 to UTP6, and probes in SUZ12 and/or SUZ12P1 to different degrees. Other cases were classified as atypical deletions.
3. Results
3.1. Description of the Cohort
Between 2013 and 2020, 4091 index cases (IC) with a clinical presentation suggestive of NF1 were molecularly screened for NF1 in the Cochin Hospital. Pathogenic variants for the gene were detected in 2875, among which 114 were deleted. In addition, between 2007 and 2013 in the two French clinical research programs EvoNF and GeneModif, 648 IC were tested for NF1 variant. In 604, a NF1 pathogenic variant was identified, including 25 patients with a whole NF1 gene heterozygous deletion. Altogether, deletions of the NF1 locus represented 4% (139/3479) of positive IC of the cohorts. Patients who refused to be included in research programs or with insufficient clinical data were excluded from the present study. A total of 126 patients were enrolled in this study, with 122 index cases.
Patients ranged between 4 months and 69 years old, with a median at 21 years old (Table 1, Figure S1) and a mean at 22. Forty-five percent (57/126) of the individuals were less than 18 years old. Forty percent (51/126) were males. About 92% (116/126) of all patients fulfilled the clinical NIH criteria, among which the most frequent were: the presence of (i) 6 or more CALS, (ii) axillary or inguinal freckling, and (iii) 2 or more NFs or one pNF (Figure S2). Ninety-one percent (84/92) of patients for whom quantitative data was available had 6 or more CALS. Although about 50% (52/107) of patients had musculoskeletal abnormalities, only 7% (8/107) showed bone lesions such as sphenoid dysplasia, tibial pseudarthrosis or dystrophic scoliosis. Among the 9 patients not fulfilling the NIH criteria for NF1, two were more than 18 years old: a 62-year-old female had a late-onset NF1 and started to notice the presence of neurofibromas around 35 years old; the other adult patient was a 46-year-old woman without any familial history of NF1. She presented with several cutaneous neurofibromas and learning disabilities.

Table 1.
Clinical findings in the NF1-deleted cohort.
3.1.1. Pigmentary Manifestations
Almost all patients with deletion of the NF1 locus presented with CALS (99%; 122/123). The only patient with no detectable CALS had more than 100 cNFs that may have rendered the research for other cutaneous manifestations complex. She also had Lisch nodules evidenced by slit lamp examination. Lisch nodules were predominantly observed in adults, with an increased prevalence with age as previously described (17%, 3/18, before 9 years old; 40%, 6/15, between 9 and 18; 70%, 31/44, in adults) [42]. Other cutaneous findings included xanthogranuloma (n = 1), birthmark (n = 1), verrucous nevus (n = 1), and anemic nevus (n = 1). One 3-year-old patient had hypertrichosis. Blue–red macules were not systematically searched for during the follow-up of the patients, although it constitutes a precocious sign of the disease [43]. They were evidenced in 20% (15/74) of patients.
3.1.2. Neurofibromas
In the present cohort, 69% (84/121) of the patients had at least two cNFs or scNFs or one pNF. Among the remaining 31% (37/121), 5/37 patients were more than 18 years old. Most underage individuals did not receive an MRI examination; thus, the existence of asymptomatic pNFs could not be excluded. Fifteen patients were massively affected and had more than 100 cNFs all over the body, in association with scNFs in 10 cases and with pNFs in 5 cases. pNFs were identified in 23% of patients for whom data was available (17/75) (Table 1). They were never isolated and were most often associated with more than 10 cNFs and/or scNFs. One teenage patient received a treatment with trametinib against multiple deep and spinal NFs resulting in spinal cord compression, walking difficulties and neuropathic pain. Another adult patient underwent multiple excisions of bulky cNFs and of two spinal NFs. Symptomatic spinal NFs were generally associated with back pain or motor deficits.
3.1.3. OPGs and Neurological Findings
OPGs were identified in 11% (9/80) of patients and were mainly diagnosed in underage patients. They resulted in loss of visual acuity in four patients, with one presenting bilateral optic atrophy.
The other main neurological finding in the population of individuals with a deletion of the NF1 gene was the presence of unidentified bright objects (UBOs) on cranial MRI (56%, 35/62). These UBOs were virtually detected in every region of the brain, with a predominance in cerebellum and cerebellar peduncles (n = 14), basal ganglia (n = 7), and thalamus (n = 6).
Two individuals suffered from left hearing loss. One was congenital and due to ear canal stenosis with a deformation of the squama.
Learning disabilities or cognitive impairments were present in 73% (74/102) of patients and were found in association with language delay (n = 12), global developmental delay (n = 9), attention deficit disorders (n = 7), psychomotor delay (n = 5), hyperactivity (n = 4), behavior disorders and self-aggressiveness (n = 3), and dysgraphia (n = 2).
3.1.4. Malignancies
In the present cohort, 12% (10/84) of patients developed a cancer, including a 12-year-old girl and three adults who developed MPNSTs. A 35-year-old woman without malignancy had a family history of a sister who died from an MPNST and could not be included in the present study. Three patients developed a breast cancer, of which one was less than 50 years old.
3.1.5. Cardiovascular Abnormalities
Cardiovascular abnormalities concerned 23% (15/64) of NF1-deleted patients and included hypertension (n = 7), pulmonic stenosis (n = 2), aortic stenosis (n = 1), extra-dural hematoma (n = 1), patent ductus arteriosus (n = 1), septal hypertrophy (n = 1), aberrant vessel in ascending aorta (n = 1), and occlusion of the left internal carotid artery (n = 1). The presence of pulmonic stenosis was not ascertained in the 25 patients from the two PHRC.
3.1.6. Other Findings
As short stature is frequently reported in NF1 patients, we investigated that in our cohort. We observed that 1% (1/77) of patients was more than 2sd taller than the mean age- and sex-matched general population, 19% (15/77) at +1sd, 65% (50/77) at mean, 11% (8/77) at −1sd, 3% (2/77) at −2sd, and 1% (1/77) at −3sd. Co-occurrence of tall stature and OPGs was not observed in our population [44]. Deep palmar and plantar creases were identified in two patients, with excess of skin in one of them. Ten patients showed hyperflexibility of joints.
Two patients were diagnosed with hypothyroidism. Additionally, one had a goiter, and one had a surgical removal of a neurofibroma of the thyroid gland. One patient had a keratoconus.
Co-occurrence of NF1 and other genetic diseases within the same family was noticed in three cases: one 15-year-old patient had concomitant familial polycystic kidney (this condition was already described and genetically confirmed in [45]); another patient inherited Charcot-Marie-Tooth disease type 1A from her mother, a condition that was already suggested or observed in the past [46,47,48,49]; also, one patient had a daughter diagnosed with Costello syndrome but no NF1.
3.2. Comparison of the Clinical Findings in the French NF1-Deleted Cohort with “Classic NF1” Phenotype
Statistical comparisons of the main clinical findings in NF1 versus the reference “classic” NF1 population are summarized in Table 2 and Figure 1; p values were adjusted using the Benjamini-Hochberg (B-H) correction for multiple comparisons.
Table 2.
Comparison of the main clinical findings in the French deleted cohort with “classic” NF1.
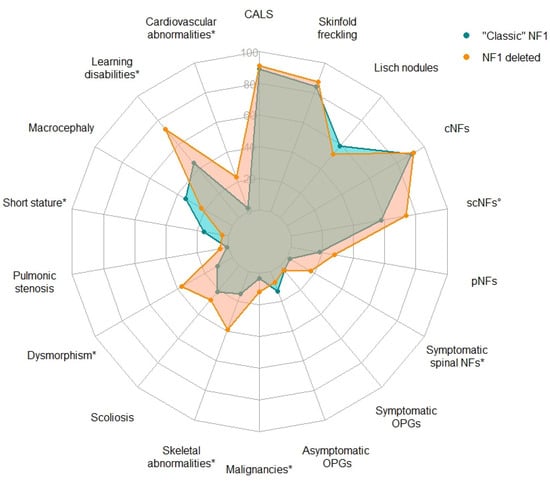
Figure 1.
Radar chart of the frequency of the main clinical findings in the French NF1-deleted cohort and in “classic” NF1. Frequency of features with a white circle (°) or an asterisk (*) significantly differ between the two cohorts, respectively only before or after correction for multiple testing with the Benjamini-Hochberg (B-H) procedure. CALS: café-au-lait spots; cNFs: cutaneous neurofibromas; OPGs: optic pathway gliomas; pNFs: plexiform neurofibromas; scNFs: subcutaneous neurofibromas.
For clinical features that usually develop during adolescence or adulthood (cNFs, scNFs, scoliosis), only patients >18 years old were considered for statistical analyses. Benign pigmentary lesions were not significantly more frequent in the NF1-deleted group than in the “classic” NF1 population. We observed a significantly higher proportion of individuals with scNFs and spinal NFs in the NF1-deleted patients. The presence of scNFs, particularly when they are in great number, have previously been associated with the development of internal neurofibromas at risk for malignant transformation in NF1 patients [50]. Related to this observation, malignant tumors were significantly over-represented in the NF1-deleted group (p value = 0.0072 after correction for multiple testing). As pNFs mostly grow in young children [51], we focused on individuals >8 years-old and considered all types of pNFs. We observed a tendency towards a higher number of pNFs in the NF1-deleted patients, though not significant (p value = 0.089 and 0.13 respectively before and after correction).
Skeletal abnormalities were twice as frequent in NF1-deleted patients as in the general NF1 population (42/107 vs. 144/948). However, it must be taken into consideration that we included more clinical features in that category than previous studies that mainly focused on scoliosis, pseudarthrosis, or long bone dysplasia. As previously described in [34], we confirmed the higher prevalence of dysmorphism and learning disabilities in NF1-deleted patients (adjusted p values 6.3 × 10−7 and 2.2 × 10−6, respectively).
An important difference was observed for the cardiovascular abnormalities which were identified in >23% of NF1-deleted patients versus less than 3% for the general NF1 group (15/64 vs. 54/2322, adjusted p value = 1.7 × 10−9). Pulmonic stenosis was found in 5% (2/39) of NF1-deleted patients, but the group for which cardiovascular issues were assessed was too small to achieve significance against the reference cohort (adjusted p value = 0.13).
3.3. Characterization of the Deletions of the NF1 Locus
MLPA analysis of the NF1 locus was performed in the DNA samples from 108 NF1-deleted patients. Among them, 66% (71/108) carried type-1, 19% (21/108) carried type-2, and 5% (5/108) carried type-3 deletions (Table S1). Atypical profile was observed in 11 samples (10%). A significantly higher proportion of scNFs, pNFs, skeletal abnormalities, dysmorphism, cognitive impairment, learning disabilities, and cardiovascular abnormalities were observed in type-1 deleted patients when compared to the “classic” NF1 group. The occurrence of malignancies did not achieve significance anymore in any category, as DNA samples from patients with MPNST (n = 2), breast cancer (n = 2) and intestinal tumor (n = 1) were either insufficient or of low quality and could not allow the classification of these patients. Non-Hodgkin lymphoma was observed in a type-2 deleted patient, but no causality of the NF1 deletion was established.
4. Discussion
Phenotypical variability in NF1 is a challenge for health professionals and may complicate genetic counselling. Recent advances in the NF1 field and large international multicenter studies have broadened our knowledge on the NF1 clinical expressivity. Publications in the last ten years have led to the identification of rare but specific genotype–phenotype correlations: variations of three codons of neurofibromin (Met992, Met1149, Arg1809) were shown to be associated with milder forms of NF1, while pathogenic variants in seven other codons (codons 844–848, Arg1276, Lys1423) located in functional domains of the protein were associated with more severe symptoms (Table S2) [24,25,26,27,28,29,30,31,32]. A more severe presentation of patients with complete deletions of the NF1 gene was also suggested in past studies [33], but only two investigations in large well characterized NF1 cohorts were previously conducted [34,52].
In the present study, we clinically described the largest NF1-deleted cohort including 126 patients with molecularly confirmed heterozygous deletion of the NF1 gene. Pigmentary manifestations of NF1 mainly include CALS, skinfold freckling and Lisch nodules. All three are benign and do not cause any functional disturbance. They originate from the deregulation of melanogenesis in melanocytes [53] as a result of activation of the cAMP-mediated PKA and ERK1/2 signaling pathways [54]. We observed that more than half of the NF1-deleted patients had café-au-lait spots, skinfold freckling, Lisch nodules, neurofibromas, neurological abnormalities, and cognitive impairment or learning disabilities. Musculoskeletal abnormalities were diagnosed in 49% (52/107) of patients, mainly implying scoliosis (25/107), hyperflexibility of joints (10/82), and pectus abnormalities (9/82). Though the two latest seem to be more specific to deleted patients [52], scoliosis is a frequent outcome in the “classic” NF1 population [55]. However, scoliosis seems to be more frequent in underage NF1-deleted patients, though not significantly (8/48 vs. 2/39 [56], p value = 0.183). UBOs were frequent in our study cohort but they were also reported in up to 94% of a “classic” NF1 population [57]. UBOs appear as hyperintense lesions on T2-weighted images, but their clinical significance is uncertain and their association with neurocognitive functions remained elusive [58]. Their number and size tend to increase until the age of 10 years old and then to spontaneously regress over time [59]. UBOs are a non-specific but early sign of the disease, like CALS and skinfold freckling which can be found in Legius syndrome or other syndromes [60] but also in the general population [61]. UBOs could be considered as a good candidate criterion for the diagnosis of NF1 in children.
Neurofibromas constitute the pathognomonic symptom of NF1. However, as for the case of Lisch nodules, cNFs and scNFs mainly appear after the first decade of life [62]; pNFs can be earlier and even appear during the neonatal period. In our study, cNFs and scNFs were more frequent in adults (59/64 and 39/53, respectively) than in children under 8 years old (2/31 and 3/27) or in patients between 9 and 18 years old (10/19 and 11/14). Although most studies did not provide detailed information about underage NF1 patients, two of them were of interest to explore the prevalence of cNFs and scNFs [56,63]. In Huson et al., the proportion of cNFs was estimated at 10% before 11 years old, and at 57% between 11 and 20 years old. In McGaughran et al., the estimation was at 23% before 10, and at 47% between 10 and 19 years old for cNFs, and at 13% before 10 and 40% between 10 and 19 years old for scNFs. Although the age ranges were slightly different, these data are comparable to what we observed in our cohort for cNFs: 2/31 (6%) before 9, and 10/19 (53%) between 9 and 18 years old. For scNFs, they were also in line with what we observed in patients before 9 (3/27: 11%) and confirm the higher prevalence of scNFs in our NF1-deleted cohort between 9 and 18 years old (11/14: 79%). We observed that 28% (17/61) of patients over 8 years old for which the trait was evaluated presented with pNFs, either externally visible or assessed by MRI. Previously described cohorts used as a reference for pNFs (“classic” NF1) only focused on externally visible pNFs (18%, 120/648), which could argue for the discrepancies observed in the two groups. However, it might be of interest to explore further for a possible higher risk or an earlier appearance of pNFs in NF1-deleted patients. In our cohort, symptomatic spinal neurofibromas were significantly more frequent in the NF1-deleted patients (6/35 vs. 36/2,058; p value = 1.7 × 10−4 after correction for multiple testing). Altogether, NF1-deleted patients had a higher tumor burden than non-deleted NF1 patients [64].
The most frequent central nervous system (CNS) tumors observed in NF1 are gliomas which concern about 20% of patients [65]. They are mainly low-grade gliomas of the optic pathways and are most often classified as pilocytic astrocytoma [66,67,68]. In our cohort, OPGs were present in 11% (9/80) of patients and were responsible for loss of visual acuity in three patients, with one presenting bilateral optic atrophy. NF1 also predisposes to a wide spectrum of malignancies, of which MPNSTs and brain tumors are the most represented [55]. NF1-deleted patients would be particularly at risk of developing malignancies, especially MPNSTs [33] which occurred in 5% (4/84) of patients in the present study. This prevalence may be underestimated in our cohort showing a median age of 21, with 45% (57/126) of patients being minor at the time of clinical investigation. Only long-term follow-up in prospective studies could give a definitive confirmation of an increased MPNST risk in NF1-deleted patients. In our cohort, breast cancer was diagnosed in three patients, of whom one was less than fifty years old. A previous work suggested that NF1 deletions would not be responsible for an increased risk for breast cancer as no cases were observed in patients with NF1 deletions in this report [69]. These results are not corroborated by our study. Reported malignancies also included a non-Hodgkin lymphoma (n = 1), an intestinal tumor (n = 1), and a thoracic synovial sarcoma (n = 1). Reported malignancies in the “classic” NF1 group included MPNSTs (n = 15), chronic myeloid leukemia (n = 1), rhabdomyosarcoma (n = 1), and breast cancers (n = 2), with some patients developing several malignant or non-malignant tumors.
Cardiovascular malformations have been reported in many patients with NF1 [10,11], but overall frequency of these findings in NF1-deleted patients had never been evaluated before. Pulmonic stenosis is the most frequently reported congenital heart disease in NF1 and is almost always found in association with Noonan-like features [20], as it is the case in our NF1-deleted group (n = 2/2 patients with pulmonic stenosis and dysmorphic features). Altogether, cardiovascular abnormalities were frequent in our group (23%, 15/64) and included hypertension (n = 7), pulmonic stenosis (n = 2), aortic stenosis (n = 1), extra-dural hematoma (n = 1), patent ductus arteriosus (n = 1), septal hypertrophy (n = 1), aberrant vessel in ascending aorta (n = 1), and occlusion of the left internal carotid artery (n = 1). Previous studies also identified ventricular septal defect, atrial septal defect, aortic stenosis, mitral valve prolapse or insufficiency, and hypertrophic cardiomyopathy [33].
While NF1 patients are usually described with a short stature [70,71], NF1-deleted populations are depicted to be taller [35], with large hands and feet. The proportion of short stature patients was significantly lower in our deleted cohort when compared to the “classic” NF1 population (3/77 vs. 109/684, p value = 0.0086 after correction for multiple testing). In comparison to previously reported NF1 populations, we observed a significantly higher proportion of symptomatic spinal neurofibromas, dysmorphism, learning disabilities, malignancies, and skeletal and cardiovascular abnormalities in the present NF1-deleted cohort. These results are in line with previous observations [33,34] and confirm the overall more severe presentation of the disease in NF1-deleted patients.
The majority of NF1-associated symptoms (including pigmentary lesions, pseudarthrosis, neurofibromas, and other tumors) are due to the complete loss-of-function of the NF1 gene (resulting from the somatic inactivation of the NF1 wild-type allele) [54,72,73,74]. Thus, constitutional loss of the NF1 gene alone may not explain the vast spectrum of variable expressivity observed in NF1-deleted patients. In NF1-deleted patients, NF1 locus deletion generally encompasses not only the gene itself, but also its flanking genes [75]. Three different types of recurrent deletions of the NF1 locus were previously described; they implied low-copy repeats (LCRs) located at 17q11.2 region: NF1-REP-c and NF1-REP-a or NF1-REP-b (for type 1 and type 3 deletions, respectively) or SUZ12 and its pseudogene SUZ12P for type 2 deletions [33,34,76]. These deletions vary in size (approximately 1.4 Mb for type 1, 1.2 Mb for type 2, and 1 Mb for type 3 deletions) and in the number of deleted genes. These recurrent deletions of the NF1 locus were suggested to be responsible for a contiguous gene syndrome. Loss of SUZ12 in type 1 and type 2 deletions may promote MPNST development in NF1-deleted patients through its role in the polycomb repressive complex 2 (PRC2): recent advances in genomic studies of MPNSTs identified critical involvement of PRC2 core components SUZ12 and EED in transition to malignancy [77,78,79]. Further functional studies are needed to provide confirmation of the other phenotypes suggested to be more often associated with large deletions of the NF1 locus. Interestingly, haploinsufficiency of the ADAP2 gene has been suggested as causal for the increased cardiovascular abnormalities found in NF1-deleted patients. Morpholino-mediated knockdown of adap2 in zebrafish embryos resulted in variable circulatory defects at two days postfertilization, caused by abnormal heart development and function [80]. Even less frequent, atypical NF1 microdeletions with non-recurring breakpoints have also been reported [33]. Molecular characterization of large prospective cohorts would be needed to delineate the specific implications of each subtype of NF1 locus deletions and to specify the role of the different deleted genes on the clinical outcomes and phenotypes of deleted patients.
We noticed a high prevalence of sporadic cases in our cohort (103/119, 87%), consistent with our previous descriptions [34]. In the cohort, three patients showed NF1 mosaic deletions with 30–40% allelic frequencies (variant allele frequency, VAF) estimated by NGS and confirmed by MLPA. The first patient was a 46-year-old woman. MLPA on extracted blood DNA gave an estimate of 30% VAF type-1 deletion of the NF1 locus. Clinically, she had few CALS, unilateral Lisch nodules, multiple cNFs limited to the arm and the head, two deep neurofibromas, cervical scoliosis, large hands and feet, and had personal history of psychotic episodes and ductal carcinoma in situ of the breast. The second patient with mosaic NF1 was a 23-year-old woman with type-2 deletion. She presented with a few CALS and freckling, bilateral Lisch nodules, symptomatic OPG, UBOs on MRI, headaches, learning disabilities, and a precocious puberty. She also had one scNF with no histological confirmation. The third mosaic patient was a type-2 NF1-deleted 2-year-old girl with only pigmentary manifestations (CALS, freckling, and Lisch nodules). MLPA analysis of the NF1 locus suggested a fourth case of mosaicism in a 24-year-old man. Allelic frequency of the type-2 deletion was estimated around 40% (ratios 0.58 to 0.63 between the CRLF3 and UTP6 deleted probes). The patient presented CALS, freckling and Lisch nodules. Altogether, at least 3 of the 21 type-2 NF1 deletions (14%) were post-zygotic alterations. The frequent occurrence of de novo NF1 deletions recommends that potential somatic mosaicism should be considered when performing phenotypical assessment.
5. Conclusions
In conclusion, the retrospective study of a large cohort of patients with complete deletion of the NF1 locus (the most frequent genetic alteration found in NF1 patients) allowed us to confirm the association between NF1 deletion and a particularly severe form of the disease. These results may help in adapting medical care and genetic counselling for these patients. Resorting to imaging techniques should be encouraged in this at-risk population when confronted with an unusual symptomatology. Our study also suggests the need to implement molecular techniques allowing the detection and sizing of the NF1 locus deletions, which will make it possible to refine the genotype–phenotype correlations related to the large deletions in NF1 patients.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/cancers13122963/s1, Figure S1: Violin plot showing the distribution of ages in the deleted cohort, Figure S2: Radar plot for the fulfilling of the 6 clinical NIH criteria in the deleted cohort, Table S1: Main clinical findings in patients with type-1, 2 and 3 NF1 deletions in the French NF1 cohort and comparison with “classic” NF1, Table S2: Summary of clinical findings and statistical analysis versus “classic NF1” population from previously published and current genotype–phenotype correlations in NF1.
Author Contributions
Conceptualization, L.P., D.V. and E.P.; Data curation, L.P.; Formal analysis, L.P., D.V. and B.P.; Funding acquisition, P.W. and E.P.; Investigation, A.S., I.L., A.C., T.M., C.B., F.M.-P., S.S., O.O.G., L.D., V.L., C.Q., B.G.-D., F.A., H.D., A.-M.G., J.L., S.J., M.-C.V., M.D., M.L., B.L., Y.A., A.L., F.P., A.D.C., E.M., A.T., M.F., Y.C., H.N., L.R., B.D., S.B., D.G., S.A.-B., M.N., R.S., G.H., G.N., J.M.-H., A.J., S.F. and P.W.; Methodology, L.P., D.V. and E.P.; Project administration, M.V. and E.P.; Resources, A.S., M.V. and P.W.; Software, A.B.-S. and P.N.; Supervision, D.V. and E.P.; Validation, E.P.; Writing—original draft, L.P.; Writing—review & editing, D.V. and E.P. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants by Association Neurofibromatoses et Recklinghausen Fondation CAP NF; Ligue Française Contre les Neurofibromatoses; INSERM (Nf1GeneModif project), and Ministère de l’Enseignement Supérieur et de la Recherche.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki and was approved by the local ethics committee CERAPHP (IRB registration #00011928—Ref. 3 March 2021).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Data are contained within the article or Supplementary Materials and the crude data are available on request.
Acknowledgments
We would like to give our special thanks to all the members of the NF-France Network for their precious collaboration: H. Adamski, C. Baumann-Morel, S. Bastuji-Garin, C. Bellanne, E. Bieth, P. Bousquet, C. Brandt, X. Balguerie, L. Boudali, P. Berbis, S. Buche, P. Castelnau, Y. Chaix, J. Chevrant-Breton, E. Collet, J.F. Cuny, P. Chastagner, M.L. Chandeclerc, E. Cheuret, P. Cintas, H. Dollfus, C. Derancourt, V. Drouin-Garraud, M. D’Incan, H. De Leersnyder, O. Dereure, D. Doumar, N. Fabre, V. Ferraro, C. Francannet, L. Faivre, F. Fellmann, N. Feugier, D. Gaillard, A. Goldenberg, L. Guyant-Maréchal, J.S. Guillamo, S. Hadj-Rabia, D. Hamel Teillac, I. Kemlin, J.P. Lacour, V. Laithier, J.C. Leonard, N. Lesavre, S. Lyonnet, K. Maincent, S. Maradeix, L. Machet, E. Mansat, N. Meyer, L. Mortier, M. Mozelle, J.C. Moreno, O. Montagne, C. Moret, E. Puzenat, S. Pinson, D. Rodriguez, E. Schweitzer, S. Sportich, J.F. Stalder, D. Staumont-Sallé, C. Thalamas, C. Thauvin, L. Taillandier, A. Verloes, and J. Zeller.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Williams, V.C.; Lucas, J.; Babcock, M.A.; Gutmann, D.H.; Korf, B.; Maria, B.L. Neurofibromatosis Type 1 Revisited. Pediatrics 2009, 123, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, A.; Pasmant, E.; Imbard, A.; Luscan, A.; Soares, M.; Blanché, H.; Laurendeau, I.; Ferkal, S.; Vidaud, M.; Pinson, S.; et al. NF1 Molecular Characterization and Neurofibromatosis Type I Genotype-Phenotype Correlation: The French Experience. Hum. Mutat. 2013, 34, 1510–1518. [Google Scholar] [CrossRef] [PubMed]
- Messiaen, L.M.; Callens, T.; Mortier, G.; Beysen, D.; Vandenbroucke, I.; Van Roy, N.; Speleman, F.; Paepe, A.D. Exhaustive Mutation Analysis of the NF1 Gene Allows Identification of 95% of Mutations and Reveals a High Frequency of Unusual Splicing Defects. Hum. Mutat. 2000, 15, 541–555. [Google Scholar] [CrossRef]
- von Deimling, A.; Krone, W.; Menon, A.G. Neurofibromatosis Type 1: Pathology, Clinical Features and Molecular Genetics. Brain Pathol. 1995, 5, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.F.; O’Connell, P.; Viskochil, D.; Cawthon, R.; Robertson, M.; Culver, M.; Dunn, D.; Stevens, J.; Gesteland, R.; White, R. The Neurofibromatosis Type 1 Gene Encodes a Protein Related to GAP. Cell 1990, 62, 599–608. [Google Scholar] [CrossRef]
- National Institutes of Health Consensus Development Conference Statement: Neurofibromatosis. Bethesda, MD, USA, July 13–15, 1987. Neurofibromatosis 1988, 1, 172–178.
- Tucker, T.; Wolkenstein, P.; Revuz, J.; Zeller, J.; Friedman, J.M. Association between Benign and Malignant Peripheral Nerve Sheath Tumors in NF1. Neurology 2005, 65, 205–211. [Google Scholar] [CrossRef]
- Patel, N.B.; Stacy, G.S. Musculoskeletal Manifestations of Neurofibromatosis Type 1. AJR Am. J. Roentgenol. 2012, 199, W99–W106. [Google Scholar] [CrossRef]
- Bayat, M.; Bayat, A. Neurological Manifestations of Neurofibromatosis: A Review. Neurol. Sci. 2020, 41, 2685–2690. [Google Scholar] [CrossRef]
- Lin, A.E.; Birch, P.H.; Korf, B.R.; Tenconi, R.; Niimura, M.; Poyhonen, M.; Armfield Uhas, K.; Sigorini, M.; Virdis, R.; Romano, C.; et al. Cardiovascular Malformations and Other Cardiovascular Abnormalities in Neurofibromatosis 1. Am. J. Med. Genet. 2000, 95, 108–117. [Google Scholar] [CrossRef]
- Cutruzzolà, A.; Irace, C.; Frazzetto, M.; Sabatino, J.; Gullace, R.; De Rosa, S.; Spaccarotella, C.; Concolino, D.; Indolfi, C.; Gnasso, A. Functional and Morphological Cardiovascular Alterations Associated with Neurofibromatosis 1. Sci. Rep. 2020, 10, 12070. [Google Scholar] [CrossRef]
- Bizzarri, C.; Bottaro, G. Endocrine Implications of Neurofibromatosis 1 in Childhood. Horm. Res. Paediatr. 2015, 83, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Torres Nupan, M.M.; Velez Van Meerbeke, A.; López Cabra, C.A.; Herrera Gomez, P.M. Cognitive and Behavioral Disorders in Children with Neurofibromatosis Type 1. Front. Pediatrics 2017, 5, 227. [Google Scholar] [CrossRef]
- Carey, J.C. Neurofibromatosis-Noonan Syndrome. Am. J. Med. Genet. 1998, 75, 263–264. [Google Scholar] [CrossRef]
- Costa, A.D.A.; Gutmann, D.H. Brain Tumors in Neurofibromatosis Type 1. Neurooncol. Adv. 2019, 1, vdz040. [Google Scholar] [CrossRef]
- Seminog, O.O.; Goldacre, M.J. Risk of Benign Tumours of Nervous System, and of Malignant Neoplasms, in People with Neurofibromatosis: Population-Based Record-Linkage Study. Br. J. Cancer 2013, 108, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Easton, D.F.; Ponder, M.A.; Huson, S.M.; Ponder, B.A. An Analysis of Variation in Expression of Neurofibromatosis (NF) Type 1 (NF1): Evidence for Modifying Genes. Am. J. Hum. Genet. 1993, 53, 305–313. [Google Scholar] [PubMed]
- Szudek, J.; Joe, H.; Friedman, J.M. Analysis of Intrafamilial Phenotypic Variation in Neurofibromatosis 1 (NF1). Genet. Epidemiol. 2002, 23, 150–164. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, A.; Pasmant, E.; Laurendeau, I.; Parfait, B.; Barbarot, S.; Guillot, B.; Combemale, P.; Ferkal, S.; Vidaud, M.; Aubourg, P.; et al. Unravelling the Genetic Basis of Variable Clinical Expression in Neurofibromatosis 1. Hum. Mol. Genet. 2009, 18, 2768–2778. [Google Scholar] [CrossRef] [PubMed]
- Pinna, V.; Daniele, P.; Calcagni, G.; Mariniello, L.; Criscione, R.; Giardina, C.; Lepri, F.R.; Hozhabri, H.; Alberico, A.; Cavone, S.; et al. Prevalence, Type, and Molecular Spectrum of NF1 Mutations in Patients with Neurofibromatosis Type 1 and Congenital Heart Disease. Genes 2019, 10, 675. [Google Scholar] [CrossRef]
- Melloni, G.; Eoli, M.; Cesaretti, C.; Bianchessi, D.; Ibba, M.C.; Esposito, S.; Scuvera, G.; Morcaldi, G.; Micheli, R.; Piozzi, E.; et al. Risk of Optic Pathway Glioma in Neurofibromatosis Type 1: No Evidence of Genotype-Phenotype Correlations in A Large Independent Cohort. Cancers 2019, 11, 1838. [Google Scholar] [CrossRef] [PubMed]
- Castle, B.; Baser, M.E.; Huson, S.M.; Cooper, D.N.; Upadhyaya, M. Evaluation of Genotype-Phenotype Correlations in Neurofibromatosis Type 1. J. Med. Genet. 2003, 40, e109. [Google Scholar] [CrossRef] [PubMed]
- Kang, E.; Kim, Y.-M.; Seo, G.H.; Oh, A.; Yoon, H.M.; Ra, Y.-S.; Kim, E.K.; Kim, H.; Heo, S.-H.; Kim, G.-H.; et al. Phenotype Categorization of Neurofibromatosis Type I and Correlation to NF1 Mutation Types. J. Hum. Genet. 2020, 65, 79–89. [Google Scholar] [CrossRef]
- Upadhyaya, M.; Huson, S.M.; Davies, M.; Thomas, N.; Chuzhanova, N.; Giovannini, S.; Evans, D.G.; Howard, E.; Kerr, B.; Griffiths, S.; et al. An Absence of Cutaneous Neurofibromas Associated with a 3-Bp Inframe Deletion in Exon 17 of the NF1 Gene (c.2970-2972 DelAAT): Evidence of a Clinically Significant NF1 Genotype-Phenotype Correlation. Am. J. Hum. Genet. 2007, 80, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Koczkowska, M.; Callens, T.; Gomes, A.; Sharp, A.; Chen, Y.; Hicks, A.D.; Aylsworth, A.S.; Azizi, A.A.; Basel, D.G.; Bellus, G.; et al. Expanding the Clinical Phenotype of Individuals with a 3-Bp in-Frame Deletion of the NF1 Gene (c.2970_2972del): An Update of Genotype-Phenotype Correlation. Genet. Med. 2019, 21, 867–876. [Google Scholar] [CrossRef]
- Pinna, V.; Lanari, V.; Daniele, P.; Consoli, F.; Agolini, E.; Margiotti, K.; Bottillo, I.; Torrente, I.; Bruselles, A.; Fusilli, C.; et al. P.Arg1809Cys Substitution in Neurofibromin Is Associated with a Distinctive NF1 Phenotype without Neurofibromas. Eur. J. Hum. Genet. 2015, 23, 1068–1071. [Google Scholar] [CrossRef]
- Rojnueangnit, K.; Xie, J.; Gomes, A.; Sharp, A.; Callens, T.; Chen, Y.; Liu, Y.; Cochran, M.; Abbott, M.-A.; Atkin, J.; et al. High Incidence of Noonan Syndrome Features Including Short Stature and Pulmonic Stenosis in Patients Carrying NF1 Missense Mutations Affecting p.Arg1809: Genotype-Phenotype Correlation. Hum. Mutat. 2015, 36, 1052–1063. [Google Scholar] [CrossRef]
- Nyström, A.M.; Ekvall, S.; Allanson, J.; Edeby, C.; Elinder, M.; Holmström, G.; Bondeson, M.L.; Annerén, G. Noonan Syndrome and Neurofibromatosis Type I in a Family with a Novel Mutation in NF1. Clin. Genet. 2009, 76, 524–534. [Google Scholar] [CrossRef]
- Ekvall, S.; Sjörs, K.; Jonzon, A.; Vihinen, M.; Annerén, G.; Bondeson, M.-L. Novel Association of Neurofibromatosis Type 1-Causing Mutations in Families with Neurofibromatosis-Noonan Syndrome. Am. J. Med. Genet. A 2014, 164A, 579–587. [Google Scholar] [CrossRef]
- Santoro, C.; Maietta, A.; Giugliano, T.; Melis, D.; Perrotta, S.; Nigro, V.; Piluso, G. Arg(1809) Substitution in Neurofibromin: Further Evidence of a Genotype-Phenotype Correlation in Neurofibromatosis Type 1. Eur. J. Hum. Genet. 2015, 23, 1460–1461. [Google Scholar] [CrossRef]
- Koczkowska, M.; Callens, T.; Chen, Y.; Gomes, A.; Hicks, A.D.; Sharp, A.; Johns, E.; Uhas, K.A.; Armstrong, L.; Bosanko, K.A.; et al. Clinical Spectrum of Individuals with Pathogenic NF1 Missense Variants Affecting p.Met1149, p.Arg1276, and p.Lys1423: Genotype-Phenotype Study in Neurofibromatosis Type 1. Hum. Mutat. 2020, 41, 299–315. [Google Scholar] [CrossRef] [PubMed]
- Koczkowska, M.; Chen, Y.; Callens, T.; Gomes, A.; Sharp, A.; Johnson, S.; Hsiao, M.-C.; Chen, Z.; Balasubramanian, M.; Barnett, C.P.; et al. Genotype-Phenotype Correlation in NF1: Evidence for a More Severe Phenotype Associated with Missense Mutations Affecting NF1 Codons 844–848. Am. J. Hum. Genet. 2018, 102, 69–87. [Google Scholar] [CrossRef]
- Kehrer-Sawatzki, H.; Mautner, V.-F.; Cooper, D.N. Emerging Genotype-Phenotype Relationships in Patients with Large NF1 Deletions. Hum. Genet. 2017, 136, 349–376. [Google Scholar] [CrossRef]
- Pasmant, E.; Sabbagh, A.; Spurlock, G.; Laurendeau, I.; Grillo, E.; Hamel, M.-J.; Martin, L.; Barbarot, S.; Leheup, B.; Rodriguez, D.; et al. NF1 Microdeletions in Neurofibromatosis Type 1: From Genotype to Phenotype. Hum. Mutat. 2010, 31, E1506–E1518. [Google Scholar] [CrossRef]
- Ning, X.; Farschtschi, S.; Jones, A.; Kehrer-Sawatzki, H.; Mautner, V.-F.; Friedman, J.M. Growth in Neurofibromatosis 1 Microdeletion Patients. Clin. Genet. 2016, 89, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Pasmant, E.; de Saint-Trivier, A.; Laurendeau, I.; Dieux-Coeslier, A.; Parfait, B.; Vidaud, M.; Vidaud, D.; Bièche, I. Characterization of a 7.6-Mb Germline Deletion Encompassing the NF1 Locus and about a Hundred Genes in an NF1 Contiguous Gene Syndrome Patient. Eur. J. Hum. Genet. 2008, 16, 1459–1466. [Google Scholar] [CrossRef]
- Upadhyaya, M.; Spurlock, G.; Monem, B.; Thomas, N.; Friedrich, R.E.; Kluwe, L.; Mautner, V. Germline and Somatic NF1 Gene Mutations in Plexiform Neurofibromas. Hum. Mutat. 2008, 29, E103–E111. [Google Scholar] [CrossRef]
- Pasmant, E.; Parfait, B.; Luscan, A.; Goussard, P.; Briand-Suleau, A.; Laurendeau, I.; Fouveaut, C.; Leroy, C.; Montadert, A.; Wolkenstein, P.; et al. Neurofibromatosis Type 1 Molecular Diagnosis: What Can NGS Do for You When You Have a Large Gene with Loss of Function Mutations? Eur. J. Hum. Genet. 2015, 23, 596–601. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Imbard, A.; Pasmant, E.; Sabbagh, A.; Luscan, A.; Soares, M.; Goussard, P.; Blanché, H.; Laurendeau, I.; Ferkal, S.; Vidaud, M.; et al. NF1 Single and Multi-Exons Copy Number Variations in Neurofibromatosis Type 1. J. Hum. Genet. 2015, 60, 221–224. [Google Scholar] [CrossRef]
- Kinori, M.; Hodgson, N.; Zeid, J.L. Ophthalmic Manifestations in Neurofibromatosis Type 1. Surv. Ophthalmol. 2018, 63, 518–533. [Google Scholar] [CrossRef] [PubMed]
- Boyd, K.P.; Korf, B.R.; Theos, A. Neurofibromatosis Type 1. J. Am. Acad. Dermatol. 2009, 61, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Cambiaso, P.; Galassi, S.; Palmiero, M.; Mastronuzzi, A.; Del Bufalo, F.; Capolino, R.; Cacchione, A.; Buonuomo, P.S.; Gonfiantini, M.V.; Bartuli, A.; et al. Growth Hormone Excess in Children with Neurofibromatosis Type-1 and Optic Glioma. Am. J. Med. Genet. A 2017, 173, 2353–2358. [Google Scholar] [CrossRef] [PubMed]
- Peces, R.; Mena, R.; Martín, Y.; Hernández, C.; Peces, C.; Tellería, D.; Cuesta, E.; Selgas, R.; Lapunzina, P.; Nevado, J. Co-Occurrence of Neurofibromatosis Type 1 and Optic Nerve Gliomas with Autosomal Dominant Polycystic Kidney Disease Type 2. Mol. Genet. Genomic Med. 2020, 8, e1321. [Google Scholar] [CrossRef] [PubMed]
- Koc, F.; Guzel, A.I. Neurofibromatosis Type 1 Associated with Charcot-Marie-Tooth Type 1A. J. Dermatol. 2009, 36, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Lupski, J.R.; Pentao, L.; Williams, L.L.; Patel, P.I. Stable Inheritance of the CMT1A DNA Duplication in Two Patients with CMT1 and NF1. Am. J. Med. Genet. 1993, 45, 92–96. [Google Scholar] [CrossRef]
- Onu, D.O.; Hunn, A.W.; Peters-Willke, J. Charcot-Marie-Tooth Syndrome and Neurofibromatosis Type 1 with Multiple Neurofibromas of the Entire Spinal Nerve Roots. BMJ Case Rep. 2013, 2013. [Google Scholar] [CrossRef]
- Roos, K.L.; Pascuzzi, R.M.; Dunn, D.W. Neurofibromatosis, Charcot-Marie-Tooth Disease, or Both? Neurofibromatosis 1989, 2, 238–243. [Google Scholar]
- Sbidian, E.; Bastuji-Garin, S.; Valeyrie-Allanore, L.; Ferkal, S.; Lefaucheur, J.P.; Drouet, A.; Brugière, P.; Vialette, C.; Combemale, P.; Barbarot, S.; et al. At-Risk Phenotype of Neurofibromatose-1 Patients: A Multicentre Case-Control Study. Orphanet J. Rare Dis. 2011, 6, 51. [Google Scholar] [CrossRef]
- Akshintala, S.; Baldwin, A.; Liewehr, D.J.; Goodwin, A.; Blakeley, J.O.; Gross, A.M.; Steinberg, S.M.; Dombi, E.; Widemann, B.C. Longitudinal Evaluation of Peripheral Nerve Sheath Tumors in Neurofibromatosis Type 1: Growth Analysis of Plexiform Neurofibromas and Distinct Nodular Lesions. Neuro Oncol. 2020, 22, 1368–1378. [Google Scholar] [CrossRef] [PubMed]
- Mautner, V.-F.; Kluwe, L.; Friedrich, R.E.; Roehl, A.C.; Bammert, S.; Högel, J.; Spöri, H.; Cooper, D.N.; Kehrer-Sawatzki, H. Clinical Characterisation of 29 Neurofibromatosis Type-1 Patients with Molecularly Ascertained 1.4 Mb Type-1 NF1 Deletions. J. Med. Genet. 2010, 47, 623–630. [Google Scholar] [CrossRef]
- De Schepper, S.; Boucneau, J.; Lambert, J.; Messiaen, L.; Naeyaert, J.-M. Pigment Cell-Related Manifestations in Neurofibromatosis Type 1: An Overview. Pigment Cell Res. 2005, 18, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Allouche, J.; Bellon, N.; Saidani, M.; Stanchina-Chatrousse, L.; Masson, Y.; Patwardhan, A.; Gilles-Marsens, F.; Delevoye, C.; Domingues, S.; Nissan, X.; et al. In Vitro Modeling of Hyperpigmentation Associated to Neurofibromatosis Type 1 Using Melanocytes Derived from Human Embryonic Stem Cells. Proc. Natl. Acad. Sci. USA 2015, 112, 9034–9039. [Google Scholar] [CrossRef] [PubMed]
- Gutmann, D.H.; Ferner, R.E.; Listernick, R.H.; Korf, B.R.; Wolters, P.L.; Johnson, K.J. Neurofibromatosis Type 1. Nat. Rev. Dis. Primers 2017, 3, 17004. [Google Scholar] [CrossRef]
- Huson, S.M.; Compston, D.A.; Harper, P.S. A Genetic Study of von Recklinghausen Neurofibromatosis in South East Wales. II. Guidelines for Genetic Counselling. J. Med. Genet. 1989, 26, 712–721. [Google Scholar] [CrossRef]
- Griffith, J.L.; Morris, S.M.; Mahdi, J.; Goyal, M.S.; Hershey, T.; Gutmann, D.H. Increased Prevalence of Brain Tumors Classified as T2 Hyperintensities in Neurofibromatosis 1. Neurol. Clin. Pract. 2018, 8, 283–291. [Google Scholar] [CrossRef]
- Baudou, E.; Nemmi, F.; Biotteau, M.; Maziero, S.; Assaiante, C.; Cignetti, F.; Vaugoyeau, M.; Audic, F.; Peran, P.; Chaix, Y. Are Morphological and Structural MRI Characteristics Related to Specific Cognitive Impairments in Neurofibromatosis Type 1 (NF1) Children? Eur. J. Paediatr. Neurol. 2020, 28, 89–100. [Google Scholar] [CrossRef]
- Jacques, C.; Dietemann, J.L. [Imaging features of neurofibromatosis type 1]. J. Neuroradiol. 2005, 32, 180–197. [Google Scholar] [CrossRef]
- Shah, K.N. The Diagnostic and Clinical Significance of Café-Au-Lait Macules. Pediatric Clin. N. Am. 2010, 57, 1131–1153. [Google Scholar] [CrossRef]
- Bernier, A.; Larbrisseau, A.; Perreault, S. Café-Au-Lait Macules and Neurofibromatosis Type 1: A Review of the Literature. Pediatric Neurol. 2016, 60, 24–29.e1. [Google Scholar] [CrossRef]
- Cannon, A.; Chen, M.-J.; Li, P.; Boyd, K.P.; Theos, A.; Redden, D.T.; Korf, B. Cutaneous Neurofibromas in Neurofibromatosis Type I: A Quantitative Natural History Study. Orphanet J. Rare Dis. 2018, 13, 31. [Google Scholar] [CrossRef] [PubMed]
- McGaughran, J.M.; Harris, D.I.; Donnai, D.; Teare, D.; MacLeod, R.; Westerbeek, R.; Kingston, H.; Super, M.; Harris, R.; Evans, D.G. A Clinical Study of Type 1 Neurofibromatosis in North West England. J. Med. Genet. 1999, 36, 197–203. [Google Scholar]
- Well, L.; Döbel, K.; Kluwe, L.; Bannas, P.; Farschtschi, S.; Adam, G.; Mautner, V.-F.; Salamon, J. Genotype-Phenotype Correlation in Neurofibromatosis Type-1: NF1 Whole Gene Deletions Lead to High Tumor-Burden and Increased Tumor-Growth. PLoS Genet. 2021, 17, e1009517. [Google Scholar] [CrossRef]
- Lobbous, M.; Bernstock, J.D.; Coffee, E.; Friedman, G.K.; Metrock, L.K.; Chagoya, G.; Elsayed, G.; Nakano, I.; Hackney, J.R.; Korf, B.R.; et al. An Update on Neurofibromatosis Type 1-Associated Gliomas. Cancers 2020, 12, 114. [Google Scholar] [CrossRef] [PubMed]
- Hernáiz Driever, P.; von Hornstein, S.; Pietsch, T.; Kortmann, R.; Warmuth-Metz, M.; Emser, A.; Gnekow, A.K. Natural History and Management of Low-Grade Glioma in NF-1 Children. J. Neurooncol. 2010, 100, 199–207. [Google Scholar] [CrossRef]
- Campen, C.J.; Gutmann, D.H. Optic Pathway Gliomas in Neurofibromatosis Type 1. J. Child Neurol. 2018, 33, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Freret, M.E.; Gutmann, D.H. Insights into Optic Pathway Glioma Vision Loss from Mouse Models of Neurofibromatosis Type 1. J. Neurosci. Res. 2019, 97, 45–56. [Google Scholar] [CrossRef]
- Frayling, I.M.; Mautner, V.-F.; van Minkelen, R.; Kallionpaa, R.A.; Aktaş, S.; Baralle, D.; Ben-Shachar, S.; Callaway, A.; Cox, H.; Eccles, D.M.; et al. Breast Cancer Risk in Neurofibromatosis Type 1 Is a Function of the Type of NF1 Gene Mutation: A New Genotype-Phenotype Correlation. J. Med. Genet. 2019, 56, 209–219. [Google Scholar] [CrossRef]
- Clementi, M.; Milani, S.; Mammi, I.; Boni, S.; Monciotti, C.; Tenconi, R. Neurofibromatosis Type 1 Growth Charts. Am. J. Med. Genet. 1999, 87, 317–323. [Google Scholar] [CrossRef]
- Szudek, J.; Birch, P.; Friedman, J.M. Growth Charts for Young Children with Neurofibromatosis 1 (NF1). Am. J. Med. Genet. 2000, 92, 224–228. [Google Scholar] [CrossRef]
- Sant, D.W.; Margraf, R.L.; Stevenson, D.A.; Grossmann, A.H.; Viskochil, D.H.; Hanson, H.; Everitt, M.D.; Rios, J.J.; Elefteriou, F.; Hennessey, T.; et al. Evaluation of Somatic Mutations in Tibial Pseudarthrosis Samples in Neurofibromatosis Type 1. J. Med. Genet. 2015, 52, 256–261. [Google Scholar] [CrossRef]
- Laycock-van Spyk, S.; Thomas, N.; Cooper, D.N.; Upadhyaya, M. Neurofibromatosis Type 1-Associated Tumours: Their Somatic Mutational Spectrum and Pathogenesis. Hum. Genom. 2011, 5, 623–690. [Google Scholar] [CrossRef]
- Serra, E.; Puig, S.; Otero, D.; Gaona, A.; Kruyer, H.; Ars, E.; Estivill, X.; Lázaro, C. Confirmation of a Double-Hit Model for the NF1 Gene in Benign Neurofibromas. Am. J. Hum. Genet. 1997, 61, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Kluwe, L.; Siebert, R.; Gesk, S.; Friedrich, R.E.; Tinschert, S.; Kehrer-Sawatzki, H.; Mautner, V.-F. Screening 500 Unselected Neurofibromatosis 1 Patients for Deletions of the NF1 Gene. Hum. Mutat. 2004, 23, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Pasmant, E.; Sabbagh, A.; Masliah-Planchon, J.; Haddad, V.; Hamel, M.-J.; Laurendeau, I.; Soulier, J.; Parfait, B.; Wolkenstein, P.; Bièche, I.; et al. Detection and Characterization of NF1 Microdeletions by Custom High Resolution Array CGH. J. Mol. Diagn. 2009, 11, 524–529. [Google Scholar] [CrossRef][Green Version]
- De Raedt, T.; Beert, E.; Pasmant, E.; Luscan, A.; Brems, H.; Ortonne, N.; Helin, K.; Hornick, J.L.; Mautner, V.; Kehrer-Sawatzki, H.; et al. PRC2 Loss Amplifies Ras-Driven Transcription and Confers Sensitivity to BRD4-Based Therapies. Nature 2014, 514, 247–251. [Google Scholar] [CrossRef]
- Oppel, F.; Ki, D.H.; Zimmerman, M.W.; Ross, K.N.; Tao, T.; Shi, H.; He, S.; Aster, J.C.; Look, A.T. Suz12 Inactivation in P53- and Nf1-Deficient Zebrafish Accelerates the Onset of Malignant Peripheral Nerve Sheath Tumors and Expands the Spectrum of Tumor Types. Dis. Models Mech. 2020, 13. [Google Scholar] [CrossRef]
- Sohier, P.; Luscan, A.; Lloyd, A.; Ashelford, K.; Laurendeau, I.; Briand-Suleau, A.; Vidaud, D.; Ortonne, N.; Pasmant, E.; Upadhyaya, M. Confirmation of Mutation Landscape of NF1-Associated Malignant Peripheral Nerve Sheath Tumors. Genes Chromosomes Cancer 2017, 56, 421–426. [Google Scholar] [CrossRef]
- Ferrari, L.; Scuvera, G.; Tucci, A.; Bianchessi, D.; Rusconi, F.; Menni, F.; Battaglioli, E.; Milani, D.; Riva, P. Identification of an Atypical Microdeletion Generating the RNF135-SUZ12 Chimeric Gene and Causing a Position Effect in an NF1 Patient with Overgrowth. Hum. Genet. 2017, 136, 1329–1339. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).