
Epigenomic and Metabolomic Integration Reveals Dynamic Metabolic Regulation in Bladder Cancer


Abstract
Simple Summary
Abstract

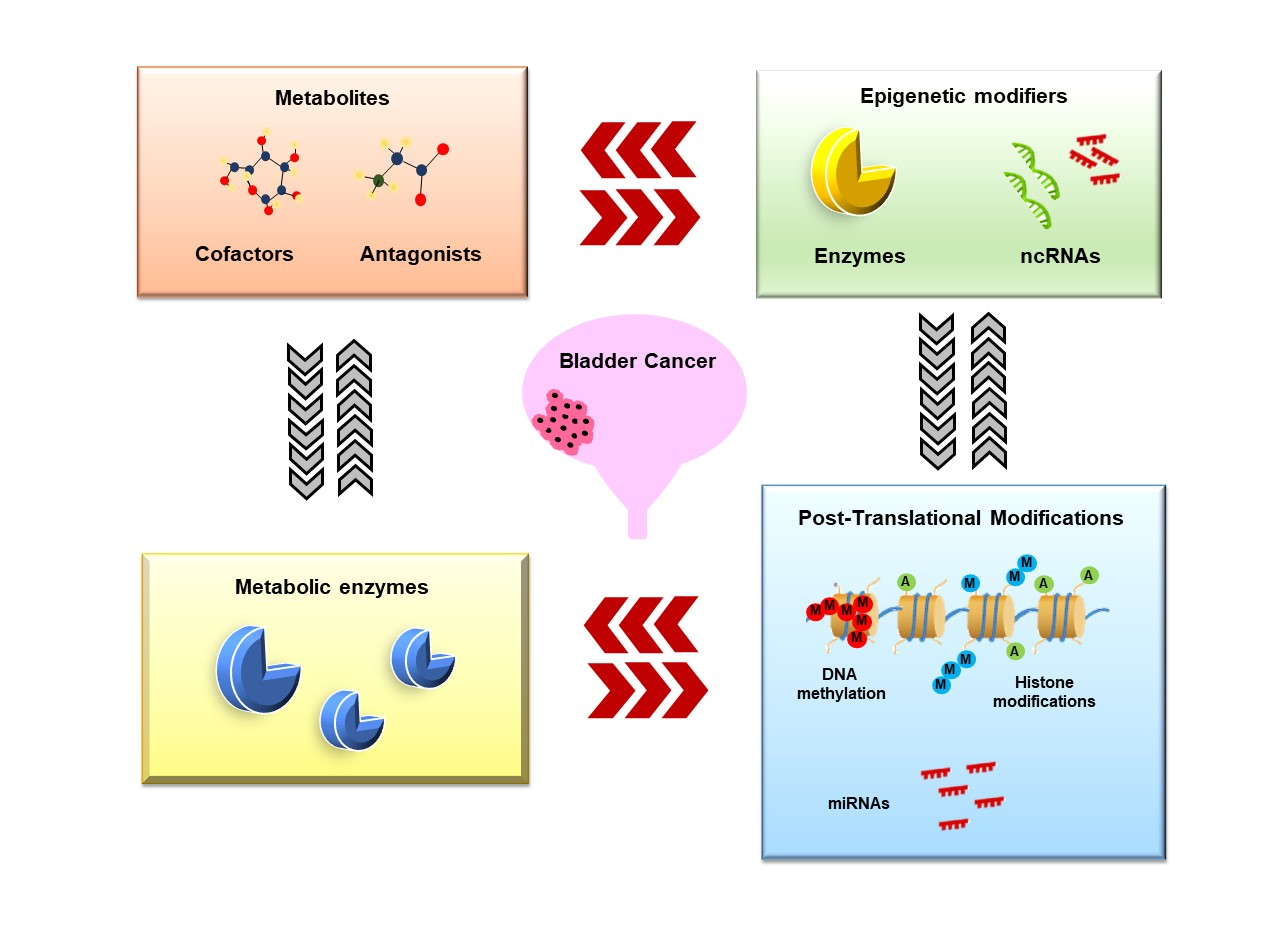
1. Introduction
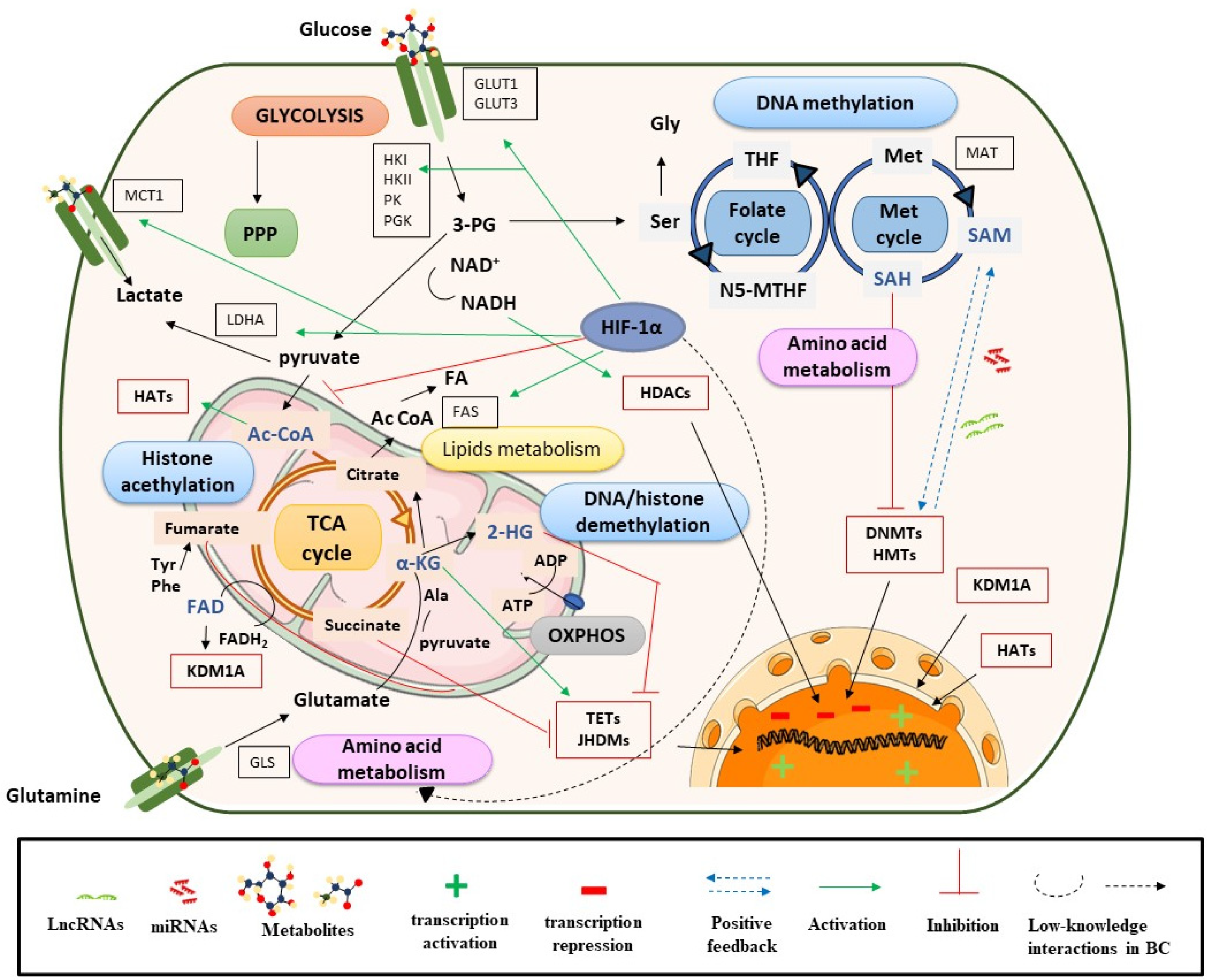
2. Metabolic Rewiring Controls the Epigenome in BC
2.1. Metabolites and DNA/Histone Methylation Processes
2.2. Metabolites, Histone Acetylation Processes and Sirtuins
2.3. Role of Metabolites in the Nucleus
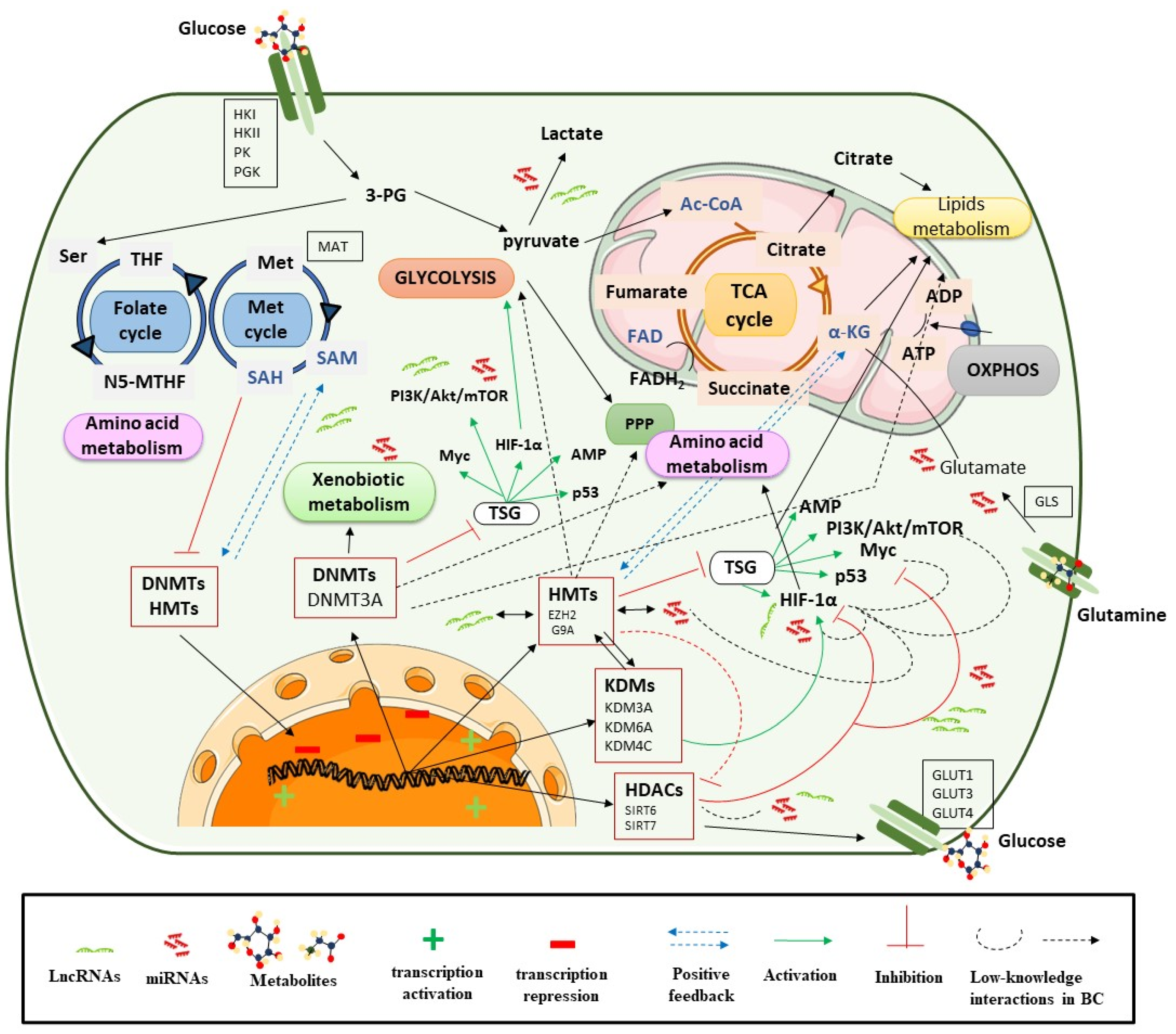
3. Epigenetics Control Metabolic Reprogramming
3.1. Epigenetic Regulation of Metabolic Enzymes and Oncogenic Pathways in Metabolism
3.1.1. DNA Methylation
3.1.2. Histone Modifications
3.1.3. ncRNAs
4. Non-Invasive Bladder Cancer Biomarkers
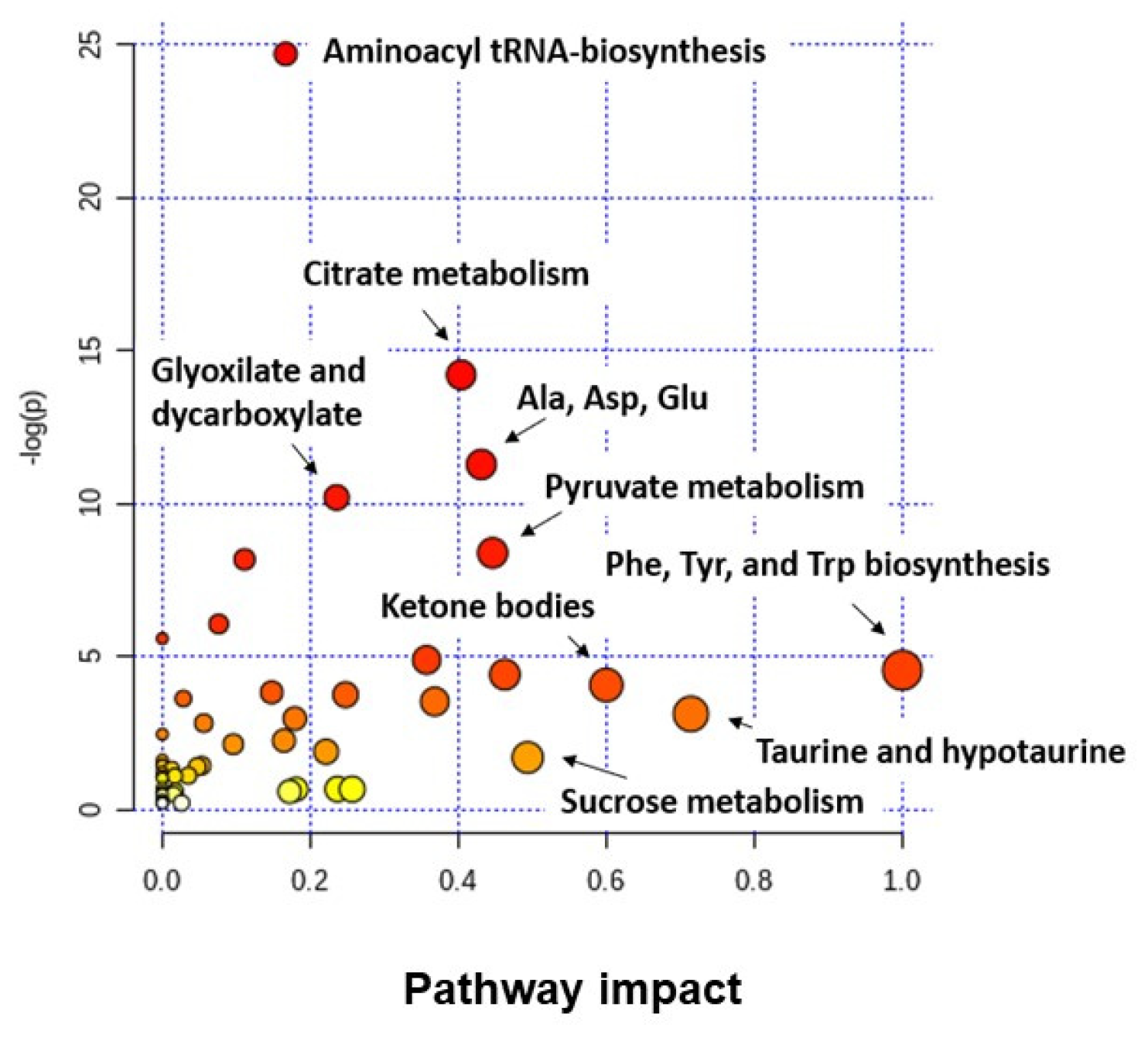
4.1. Metabolomic Studies in BC
4.2. Epigenetic Biomarkers in BC
5. Therapeutic Opportunities and Future Perspectives
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Burger, M.; Catto, J.W.F.; Dalbagni, G.; Grossman, H.B.; Herr, H.; Karakiewicz, P.; Kassouf, W.; Kiemeney, L.A.; La Vecchia, C.; Shariat, S.; et al. Epidemiology and risk factors of urothelial bladder cancer. Eur. Urol. 2013, 63, 234–241. [Google Scholar] [CrossRef]
- Kaseb, H.; Aeddula, N.R. Bladder Cancer. En: StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA. Available online: http://www.ncbi.nlm.nih.gov/books/NBK536923/ (accessed on 20 August 2020).
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Ca. Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Anastasiadis, A.; de Reijke, T.M. Best practice in the treatment of nonmuscle invasive bladder cancer. Adv. Urol. 2012, 4, 13–32. [Google Scholar] [CrossRef] [PubMed]
- Knowles, M.A.; Hurst, C.D. Molecular biology of bladder cancer: New insights into pathogenesis and clinical diversity. Nat. Publ. Gr. 2015, 15, 25–41. [Google Scholar] [CrossRef]
- Ghatalia, P.; Zibelman, M.; Geynisman, D.M.; Plimack, E. Approved checkpoint inhibitors in bladder cancer: Which drug should be used when? Adv. Med. Oncol. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Rosser, C.J.; Urquidi, V.; Goodison, S. Urinary biomarkers of bladder cancer: An update and future perspectives. Biomark. Med. 2013, 7, 779–790. [Google Scholar] [CrossRef]
- Cauberg Evelyne, C.; de Reijke, T.; de la Rosette, J.M.C. Emerging optical techniques in advanced cystoscopy for bladder cancer diagnosis: A review of the current literature. Indian J. Urol. 2011, 27, 245. [Google Scholar] [CrossRef] [PubMed]
- Babjuk, M.; Böhle, A.; Burger, M.; Capoun, O.; Cohen, D.; Compérat, E.M.; Hernández, V.; Kaasinen, E.; Palou, J.; Rouprêt, M.; et al. EAU Guidelines on Non–Muscle-invasive Urothelial Carcinoma of the Bladder: Update 2016. Eur. Urol. 2017, 71, 447–461. [Google Scholar] [CrossRef]
- Svatek, R.S.; Hollenbeck, B.K.; Holmäng, S.; Lee, R.; Kim, S.P.; Stenzl, A.; Lotan, Y. The economics of bladder cancer: Costs and considerations of caring for this disease. Eur. Urol. 2014, 66, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Leal, J.; Luengo-Fernandez, R.; Sullivan, R.; Witjes, J.A. Economic Burden of Bladder Cancer across the European Union. Eur. Urol. 2016, 69, 438–447. [Google Scholar] [CrossRef]
- Sylvester, R.J.; Van Der Meijden, A.P.M.; Oosterlinck, W.; Witjes, J.A.; Bouffioux, C.; Denis, L.; Newling, D.W.W.; Kurth, K. Predicting recurrence and progression in individual patients with stage Ta T1 bladder cancer using EORTC risk tables: A combined analysis of 2596 patients from seven EORTC trials. Eur. Urol. 2006, 49, 466–475. [Google Scholar] [CrossRef]
- Lodewijk, I.; Dueñas, M.; Rubio, C.; Munera-Maravilla, E.; Segovia, C.; Bernardini, A.; Teijeira, A.; Paramio, J.M.; Suárez-Cabrera, C. Liquid Biopsy Biomarkers in Bladder Cancer: A Current Need for Patient Diagnosis and Monitoring. Int. J. Mol. Sci. 2019, 19, 2514. [Google Scholar] [CrossRef] [PubMed]
- Loras, A.; Su, C.; Mart, M.C. Integrative Metabolomic and Transcriptomic Analysis. Cancers 2019, 11, 686. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, M.C.; Caires, H.R.; Oliveira, M.J.; Fraga, A.; Vasconcelos, M.H.; Ribeiro, R. Urinary Biomarkers in Bladder Cancer: Where Do We Stand and Potential Role of Extracellular Vesicles. Cancers 2020, 12, 1400. [Google Scholar] [CrossRef]
- Armitage, E.G.; Ciborowski, M. Applications of Metabolomics in Cancer Studies. Adv. Exp. Med. Biol. 2017, 965, 209–234. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Holmes, E.; Wilson, I.D.; Nicholson, J.K. Metabolic phenotyping in health and disease. Cell 2008, 134, 714–717. [Google Scholar] [CrossRef] [PubMed]
- Smolinska, A.; Blanchet, L.; Buydens, L.M.; Wijmenga, S.S. NMR and pattern recognition methods in metabolomics: From data acquisition to biomarker discovery: A review. Anal. Chim. Acta 2012, 750, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Crispo, F.; Condelli, V.; Lepore, S.; Notarangelo, T.; Sgambato, A.; Esposito, F.; Maddalena, F.; Landriscina, M. Metabolic Dysregulations and Epigenetics: A Bidirectional Interplay that Drives Tumor Progression. Cells 2019, 8, 798. [Google Scholar] [CrossRef]
- Wong, C.C.; Qian, Y.; Yu, J. Interplay between epigenetics and metabolism in oncogenesis: Mechanisms and therapeutic approaches. Oncogene 2017, 36, 3359–3374. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357, eaal2380. [Google Scholar] [CrossRef] [PubMed]
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar]
- Frontiers-Metabolism and Transcription in Cancer: Merging Two Classic Tales-Cell and Developmental Biology [Internet]. Available online: https://www.frontiersin.org/articles/10.3389/fcell.00119/full (accessed on 20 August 2020).
- Putluri, N.; Shojaie, A.; Vasu, V.T.; Vareed, S.K.; Nalluri, S.; Putluri, V.; Thangjam, G.S.; Panzitt, K.; Tallman, C.T.; Butler, C.; et al. Metabolomic profiling reveals potential markers and bioprocesses altered in bladder cancer progression. Cancer Res. 2011, 71, 7376–7386. [Google Scholar] [CrossRef]
- Wittmann, B.M.; Stirdivant, S.M.; Mitchell, M.W.; Wulff, J.E.; McDunn, J.E.; Li, Z.; Dennis-Barrie, A.; Neri, B.P.; Milburn, M.V.; Lotan, Y.; et al. Bladder cancer biomarker discovery using global metabolomic profiling of urine. PLoS ONE 2014, 9, e115870. [Google Scholar] [CrossRef]
- Miranda-Gonçalves, V.; Lameirinhas, A.; Henrique, R.; Jerónimo, C. Metabolism and epigenetic interplay in cancer: Regulation and putative therapeutic targets. Front. Genet. 2018, 9, 427. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Yun, S.J.; Jeong, P.; Kim, I.Y.; Kim, W.-J.; Park, S. Diagnosis of bladder cancer and prediction of survival by urinary metabolomics. Oncotarget 2014, 5, 1635–1645. [Google Scholar] [CrossRef]
- Rasmussen, K.D.; Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef]
- Knaap, J.A.; van der Verrijzer, C.P. Undercover: Gene control by metabolites and metabolic enzymes. Genes Dev. 11 Enero 2016, 30, 2345–2369. [Google Scholar] [CrossRef]
- Martínez-reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control. Nat. Commun. 2020, 1–11. [Google Scholar] [CrossRef]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia inducible factors and the response to hypoxic stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Zdzisin, B. Alpha-Ketoglutarate as a Molecule with Pleiotropic Activity: Well-Known and Novel Possibilities of Therapeutic Use. Arch. Immunol. Exp. 2017, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.C.; Rathore, A.; Younas, H.; Gilkes, D.; Polotsky, V.Y. Hypoxia-Inducible Factors and Cancer. Curr. Sleep Med. Rep. 2017, 3, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Theodoropoulos, V.E.; Lazaris, A.C.; Sofras, F.; Gerzelis, I.; Tsoukala, V.; Ghikonti, I.; Manikas, K.; Kastriotis, I. Hypoxia-inducible factor 1 alpha expression correlates with angiogenesis and unfavorable prognosis in bladder cancer. Eur. Urol. 2004, 46, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Lew, C.R.; Guin, S.; Theodorescu, D. Targeting glycogen metabolism in bladder cancer. Nat. Rev. Urol. 2015, 12, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Miao, P.; Sheng, S.; Sun, X.; Liu, J.; Huang, G. Lactate dehydrogenase A in cancer: A promising target for diagnosis and therapy. IUBMB Life 2013, 65, 904–910. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, Y.; Dong, D.; Wang, F.; Ma, X.; Guan, F. MCT1 regulates aggressive and metabolic phenotypes in bladder cancer. J. Cancer 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Conde, V.R.; Oliveira, P.F.; Nunes, A.R.; Rocha, C.S.; Ramalhosa, E.; Pereira, J.A.; Alves, M.G.; Silva, B.M. The progression from a lower to a higher invasive stage of bladder cancer is associated with severe alterations in glucose and pyruvate metabolism. Exp. Cell Res. 2015, 335, 91–98. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef]
- Mylonis, I.; Simos, G.; Paraskeva, E. Hypoxia-Inducible Factors and the Regulation of Lipid Metabolism. Cells 2019, 8, 214. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, L.; Mao, S.; Liu, M.; Zhang, W.; Zhang, Z.; Guo, Y.; Huang, B.; Yan, Y.; Huang, Y.; et al. MiR-1-3p contributes to cell proliferation and invasion by targeting glutaminase in bladder cancer cells. Cell. Physiol. Biochem. 2018, 51, 513–527. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhan, H.; Lin, F.; Liu, Y.; Yang, K.; Gao, Q.; Ding, M.; Liu, Y.; Huang, W.; Cai, Z. LincRNA-p21 suppresses glutamine catabolism and bladder cancer cell growth through inhibiting glutaminase expression. Biosci. Rep. 2019, 29. [Google Scholar] [CrossRef]
- Zhou, Y.; Song, R.; Zhang, Z.; Lu, X.; Zeng, Z.; Hu, C.; Liu, X.; Li, Y.; Hou, J.; Sun, Y.; et al. The development of plasma pseudotargeted GC-MS metabolic profiling and its application in bladder cancer. Anal. Bioanal. Chem. 2016, 408, 6741–6749. [Google Scholar] [CrossRef]
- Khatami, F.; Aghamir, S.M.K.; Tavangar, S.M. Oncometabolites: A new insight for oncology. Mol. Genet. Genom. Med. 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Sciacovelli, M.; Frezza, C. Oncometabolites: Unconventional triggers of oncogenic signalling cascades. Free Radic. Biol. Med. 2016, 100, 175–181. [Google Scholar] [CrossRef]
- Xiao, M.; Yang, H.; Xu, W.; Ma, S.; Lin, H.; Zhu, H.; Liu, L.; Liu, Y.; Yang, C.; Xu, Y.; et al. Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012, 26, 1326–1338. [Google Scholar] [CrossRef]
- Campbell, S.L.; Wellen, K.E. Metabolic Signaling to the Nucleus in Cancer. Mol. Cell. 2018, 71, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Rani, A.; Murphy, J.J. STAT5 in Cancer and Immunity. J. Interferon. Cytokine Res. 2016, 36, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Cheng, M.K.; Griffiths, T.R.; Mellon, J.K.; Kai, B.; Kriajevska, M.; Manson, M.M. Inhibition of STAT signalling in bladder cancer by diindolylmethane: Relevance to cell adhesion, migration and proliferation. Curr. Cancer Drug Targets 2013, 13, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Hindupur, S.V.; Schmid, S.C.; Koch, J.A.; Youssef, A.; Baur, E.M.; Wang, D.; Horn, T.; Slotta-Huspenina, J.; Gschwend, J.E.; Holm, P.S.; et al. STAT3/5 Inhibitors Suppress Proliferation in Bladder Cancer and Enhance Oncolytic Adenovirus Therapy. Int. J. Mol. Sci. 2020, 21, 1106. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Meissner, A.; Lander, E.S. The Mammalian Epigenome. Cell 2007, 128, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Lu, Z. Nuclear PKM2 regulates the Warburg effect. Cell Cycle 2013, 12, 3154–3158. [Google Scholar] [CrossRef]
- Zhu, Q.; Hong, B.; Zhang, L.; Wang, J. Pyruvate kinase M2 inhibits the progression of bladder cancer by targeting MAKP pathway. J. Cancer Res. 2018, 14, S616–S621. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, F.; Wu, X.R. Inhibition of Pyruvate Kinase M2 Markedly Reduces Chemoresistance of Advanced Bladder Cancer to Cisplatin. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Tao, T.; Tang, L.; Deng, J.; Darko, K.O.; Zhou, S.; Peng, M.; He, S.; Zeng, Q.; Chen, A.F.; et al. Down-regulation of PKM2 enhances anticancer efficiency of THP on bladder cancer. J. Cell. Mol. Med. 2018, 2774–2790. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Huang, Z.; Bai, P.; Luo, G.; Zhao, X.; Wang, X. Expression of pyruvate kinase M2 in human bladder cancer and its correlation with clinical parameters and prognosis. Oncol. Targets 2018, 11, 2075–2082. [Google Scholar] [CrossRef]
- Boukouris, A.E.; Zervopoulos, S.D.; Michelakis, E.D. Metabolic Enzymes Moonlighting in the Nucleus: Metabolic Regulation of Gene Transcription. Trends Biochem. Sci. 2016, 41, 712–730. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Liep, J.; Rabien, A.; Jung, K. Feedback networks between microRNAs and epigenetic modifications in urological tumors. Epigenetics 2012, 7, 315–325. [Google Scholar] [CrossRef]
- Tsai, H.-C.; Baylin, S.B. Cancer epigenetics: Linking basic biology to clinical medicine. Cell Res. 2011, 21, 502–517. [Google Scholar] [CrossRef]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.-C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.G.; Kim, J.; Al-Ahmadie, H.; Bellmunt, J.; Guo, G.; Cherniack, A.D.; Hinoue, T.; Laird, P.W.; Hoadley, K.A.; Akbani, R.; et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 2017, 171, 540–556.e25. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Akbani, R.; Broom, B.M.; Wang, W.; Verhaak, R.G.W.; McConkey, D.; Lerner, S.; Morgan, M.; Creighton, C.J.; Smith, C.; et al. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Hung, M.-C. Regulation and Role of EZH2 in Cancer. Cancer Res. Treat. 2014, 46, 209–222. [Google Scholar] [CrossRef]
- Santos, M.; Martínez-Fernández, M.; Dueñas, M.; García-Escudero, R.; Alfaya, B.; Villacampa, F.; Saiz-Ladera, C.; Costa, C.; Oteo, M.; Duarte, J.; et al. In vivo disruption of an Rb-E2F-Ezh2 signaling loop causes bladder cancer. Cancer Res. 2014, 74, 6565–6577. [Google Scholar] [CrossRef]
- Martínez-Fernández, M.; Feber, A.; Dueñas, M.; Segovia, C.; Rubio, C.; Fernandez, M.; Villacampa, F.; Duarte, J.; López-Calderón, F.F.; Gómez-Rodriguez, M.J.; et al. Analysis of the Polycomb-related lncRNAs HOTAIR and ANRIL in bladder cancer. Clin. Epigenetics 2015, 7, 109. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Fernández, M.; Dueñas, M.; Feber, A.; Segovia, C.; García-Escudero, R.; Rubio, C.; López-Calderón, F.F.; Díaz-García, C.; Villacampa, F.; Duarte, J.; et al. A Polycomb-mir200 loop regulates clinical outcome in bladder cancer. Oncotarget 2015, 6, 42258–42275. [Google Scholar] [CrossRef]
- Viré, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; Van Eynde, A.; Bernard, D.; Vanderwinden, J.-M.; et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006, 439, 871–874. [Google Scholar] [CrossRef]
- Kuzmichev, A.; Nishioka, K.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002, 16, 2893–2905. [Google Scholar] [CrossRef]
- Woolbright, B.L.; Ayres, M.; Taylor, J.A. Metabolic changes in bladder cancer. Urol. Oncol. Semin. Orig. Investig. 2018, 36, 327–337. [Google Scholar] [CrossRef]
- Ward, P.S.; Thompson, C.B. Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef]
- Thakur, C.; Chen, F. Connections between metabolism and epigenetics in cancers. Semin. Cancer Biol. 2019, 57, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; O’Donnell, K.A.; Zeller, K.I.; Nguyen, T.; Osthus, R.C.; Li, F. The c-Myc target gene network. Semin. Cancer Biol. 2006, 16, 253–264. [Google Scholar] [CrossRef]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. C-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer. 2011, 11, 85–95. [Google Scholar] [CrossRef]
- Trachootham, D.; Zhou, Y.; Zhang, H.; Demizu, Y.; Chen, Z.; Pelicano, H.; Chiao, P.J.; Achanta, G.; Arlinghaus, R.B.; Liu, J.; et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by β-phenylethyl isothiocyanate. Cancer Cell 2006, 10, 241–252. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef]
- Koukourakis, M.I.; Kakouratos, C.; Kalamida, D.; Bampali, Z.; Mavropoulou, S.; Sivridis, E.; Giatromanolaki, A. Hypoxia-inducible proteins HIF1α and lactate dehydrogenase LDH5, key markers of anaerobic metabolism, relate with stem cell markers and poor post-radiotherapy outcome in bladder cancer. Int. J. Radiat. Biol. 2016, 92, 353–363. [Google Scholar] [CrossRef]
- Plas, D.R.; Thompson, C.B. Akt-dependent transformation: There is more to growth than just surviving. Oncogene 2005, 24, 7435–7442. [Google Scholar] [CrossRef]
- Inoki, K.; Corradetti, M.N.; Guan, K.L. Dysregulation of the TSC-mTOR pathway in human disease. Nat. Genet. 2005, 37, 19–24. [Google Scholar] [CrossRef]
- Wolff, E.M.; Chihara, Y.; Pan, F.; Weisenberger, D.J.; Siegmund, K.D.; Sugano, K.; Kawashima, K.; Laird, P.W.; Jones, P.A.; Liang, G. Unique DNA Methylation Patterns Distinguish Noninvasive and Invasive Urothelial Cancers and Establish an Epigenetic Field Defect in Premalignant Tissue. Cancer Res. 2010, 70, 8169–8178. [Google Scholar] [CrossRef] [PubMed]
- Catto, J.W.F.; Azzouzi, A.-R.; Rehman, I.; Feeley, K.M.; Cross, S.S.; Amira, N.; Fromont, G.; Sibony, M.; Cussenot, O.; Meuth, M.; et al. Promoter Hypermethylation Is Associated With Tumor Location, Stage, and Subsequent Progression in Transitional Cell Carcinoma. J. Clin. Oncol. 2005, 23, 2903–2910. [Google Scholar] [CrossRef]
- Kandimalla, R.; van Tilborg, A.A.G.; Kompier, L.C.; Stumpel, D.J.P.M.; Stam, R.W.; Bangma, C.H.; Zwarthoff, E.C. Genome-wide Analysis of CpG Island Methylation in Bladder Cancer Identified TBX2, TBX3, GATA2, and ZIC4 as pTa-Specific Prognostic Markers. Eur. Urol. 2012, 61, 1245–1256. [Google Scholar] [CrossRef]
- Lameirinhas, A.; Miranda-Gonçalves, V.; Henrique, R.; Jerónimo, C. The complex interplay between metabolic reprogramming and epigenetic alterations in renal cell carcinoma. Genes 2019, 10, 264. [Google Scholar] [CrossRef]
- Kimura, F.; Florl, A.R.; Seifert, H.-H.; Louhelainen, J.; Maas, S.; Knowles, M.A.; Schulz, W.A. Destabilization of chromosome 9 in transitional cell carcinoma of the urinary bladder. Br. J. Cancer 2001, 85, 1887–1893. [Google Scholar] [CrossRef]
- Nakagawa, T.; Kanai, Y.; Ushijima, S.; Kitamura, T.; Kakizoe, T.; Hirohashi, S. DNA hypomethylation on pericentromeric satellite regions significantly correlates with loss of heterozygosity on chromosome 9 in urothelial carcinomas. J. Urol. 2005, 173, 243–246. [Google Scholar] [CrossRef]
- Lopez-Serra, P.; Marcilla, M.; Villanueva, A.; Ramos-Fernandez, A.; Palau, A.; Leal, L.; Wahi, J.E.; Setien-Baranda, F.; Szczesna, K.; Moutinho, C.; et al. A DERL3-associated defect in the degradation of SLC2A1 mediates the Warburg effect. Nat. Commun. 2014, 5, 3608. [Google Scholar] [CrossRef]
- Leung, J.Y.; Chia, K.; Ong, D.S.T.; Taneja, R. Interweaving Tumor Heterogeneity into the Cancer Epigenetic/Metabolic Axis. Antioxid. Redox Signal. 2020, 33, 946–965. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.; Agnihotri, S.; Munoz, D.; Guha, A. Developmental profile and regulation of the glycolytic enzyme hexokinase 2 in normal brain and glioblastoma multiforme. Neurobiol. Dis. 2011, 44, 84–91. [Google Scholar] [CrossRef]
- Desai, S.; Ding, M.; Wang, B.; Lu, Z.; Zhao, Q.; Shaw, K.; Alfred Yung, W.K.; Weinstein, J.N.; Tan, M.; Yao, J. Tissue-specific isoform switch and DNA hypomethylation of the pyruvate kinase PKM gene in human cancers. Oncotarget 2014, 5, 8202–8210. [Google Scholar] [CrossRef]
- Massari, F.; Ciccarese, C.; Santoni, M.; Iacovelli, R.; Mazzucchelli, R.; Piva, F.; Scarpelli, M.; Berardi, R.; Tortora, G.; Lopez-Beltran, A.; et al. Metabolic phenotype of bladder cancer. Cancer Treat. Rev. 2016, 45, 46–57. [Google Scholar] [CrossRef]
- Cheng, S.; Wang, G.; Wang, Y.; Cai, L.; Qian, K.; Ju, L.; Liu, X.; Xiao, Y.; Wang, X. Fatty acid oxidation inhibitor etomoxir suppresses tumor progression and induces cell cycle arrest via PPARγ-mediated pathway in bladder cancer. Clin. Sci. 2019, 133, 1745–1758. [Google Scholar] [CrossRef]
- Tian, Z.; Tian, Z.; Meng, L.; Meng, L.; Long, X.; Diao, T.; Hu, M.; Wang, M.; Liu, M.; Wang, J.; et al. DNA methylation-based classification and identification of bladder cancer prognosis-associated subgroups. Cancer Cell Int. 2020, 20, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Casagrande, V.; Mauriello, A.; Bischetti, S.; Mavilio, M.; Federici, M.; Menghini, R. Hepatocyte specific TIMP3 expression prevents diet dependent fatty liver disease and hepatocellular carcinoma. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Stöhr, R.; Kappel, B.A.; Carnevale, D.; Cavalera, M.; Mavilio, M.; Arisi, I.; Fardella, V.; Cifelli, G.; Casagrande, V.; Rizza, S.; et al. TIMP3 interplays with apelin to regulate cardiovascular metabolism in hypercholesterolemic mice. Mol. Metab. 2015, 4, 741–752. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gurioli, G.; Martignano, F.; Salvi, S.; Costantini, M.; Gunelli, R.; Casadio, V. GSTP1 methylation in cancer: A liquid biopsy biomarker? Clin. Chem. Lab. Med. 2018, 56, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Leygo, C.; Williams, M.; Jin, H.C.; Chan, M.W.Y.; Chu, W.K.; Grusch, M.; Cheng, Y.Y. DNA Methylation as a Noninvasive Epigenetic Biomarker for the Detection of Cancer. Dis. Markers 2017, 2017, 3726595. [Google Scholar] [CrossRef]
- Louie, S.M.; Grossman, E.A.; Crawford, L.A.; Ding, L.; Camarda, R.; Huffman, T.R.; Miyamoto, D.K.; Goga, A.; Weerapana, E.; Nomura, D.K. GSTP1 Is a Driver of Triple-Negative Breast Cancer Cell Metabolism and Pathogenicity. Cell Chem. Biol. 2016, 23, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Chuang, J.-J.; Dai, Y.-C.; Lin, Y.-L.; Chen, Y.-Y.; Lin, W.-H.; Chan, H.-L.; Liu, Y.-W. Downregulation of glutathione S-transferase M1 protein in N-butyl-N-(4-hydroxybutyl)nitrosamine-induced mouse bladder carcinogenesis. Toxicol. Appl. Pharm. 2014, 279, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-T.; Duymich, C.E.; Weisenberger, D.J.; Liang, G. Genetic and Epigenetic Alterations in Bladder Cancer. Int. Neurourol. J. 2016, 20, S84–S94. [Google Scholar] [CrossRef] [PubMed]
- Marques-Magalhães, Â.; Graça, I.; Henrique, R.; Jerónimo, C. Targeting DNA methyltranferases in urological tumors. Front. Pharm. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Martinez, V.G.; Munera-Maravilla, E.; Bernardini, A.; Rubio, C.; Suarez-Cabrera, C.; Segovia, C.; Lodewijk, I.; Dueñas, M.; Martínez-Fernández, M.; Paramio, J.M. Epigenetics of Bladder Cancer: Where Biomarkers and Therapeutic Targets Meet. Front. Genet. 2019, 10, 1–27. [Google Scholar] [CrossRef]
- Chan, M.W.Y.; Chan, L.W.; Tang, N.L.S.; Tong, J.H.M.; Lo, K.W.; Lee, T.L.; Cheung, H.Y.; Wong, W.S.; Chan, P.S.F.; Lai, F.M.M.; et al. Hypermethylation of multiple genes in tumor tissues and voided urine in urinary bladder cancer patients. Clin. Cancer Res. 2002, 8, 464–470. [Google Scholar]
- Dulaimi, E.; Uzzo, R.G.; Greenberg, R.E.; Al-Saleem, T.; Cairns, P. Detection of bladder cancer in urine by a tumor suppressor gene hypermethylation panel. Clin. Cancer Res. 2004, 10, 1887–1893. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, R.; Toyooka, S.; Toyooka, K.O.; Harada, K.; Virmani, A.K.; Zöchbauer-Müller, S.; Farinas, A.J.; Vakar-Lopez, F.; Minna, J.D.; Sagalowsky, A.; et al. Aberrant promoter methylation profile of bladder cancer and its relationship to clinicopathological features. Cancer Res. 2001, 61, 8659–8663. [Google Scholar] [PubMed]
- Yeon, A.; You, S.; Kim, M.; Gupta, A.; Park, M.H.; Weisenberger, D.J.; Liang, G.; Kim, J. Rewiring of cisplatin-resistant bladder cancer cells through epigenetic regulation of genes involved in amino acid metabolism. Theranostics 2018, 8, 4520–4534. [Google Scholar] [CrossRef] [PubMed]
- Tada, Y.; Wada, M.; Kuroiwa, K.; Kinugawa, N.; Harada, T.; Nagayama, J.; Nakagawa, M.; Naito, S.; Kuwano, M. MRD1 gene overexpression and altered degree of methylation at the promoter region in bladder cancer during chemotherapeutic treatment. Clin. Cancer Res. 2000, 6, 4618–4627. [Google Scholar]
- Peng, L.; Zhong, X. Epigenetic regulation of drug metabolism and transport. Acta Pharm. Sin. B 2015, 5, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Okino, S.T.; Pookot, D.; Li, L.C.; Zhao, H.; Urakami, S.; Shiina, H.; Igawa, M.; Dahiya, R. Epigenetic inactivation of the dioxin-responsive Cytochrome P4501A1 gene in human prostate cancer. Cancer Res. 2006, 66, 7420–7428. [Google Scholar] [CrossRef] [PubMed]
- Ieiri, I.; Hirota, T.; Takane, H.; Higuchi, S. Epigenetic Regulation of Genes Encoding Drug-Metabolizing Enzymes and Transporters; DNA Methylation and Other Mechanisms. Curr. Drug Metab. 2008, 9, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Karpf, A.R.; Deeb, K.K.; Muindi, J.R.; Morrison, C.D.; Johnson, C.S.; Trump, D.L. Epigenetic regulation of vitamin D 24-hydroxylase/CYP24A1 in human prostate cancer. Cancer Res. 2010, 70, 5953–5962. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Carey, M.; Workman, J.L. The Role of Chromatin during Transcription. Cell 2007, 128, 707–719. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Segovia, C.; San José-Enériz, E.; Munera-Maravilla, E.; Martínez-Fernández, M.; Garate, L.; Miranda, E.; Vilas-Zornoza, A.; Lodewijk, I.; Rubio, C.; Segrelles, C.; et al. Inhibition of a G9a/DNMT network triggers immune-mediated bladder cancer regression. Nat. Med. 2019, 25, 1073–1081. [Google Scholar] [CrossRef]
- Gupta, P.B.; Fillmore, C.M.; Jiang, G.; Shapira, S.D.; Tao, K.; Kuperwasser, C.; Lander, E.S. Stochastic State Transitions Give Rise to Phenotypic Equilibrium in Populations of Cancer Cells. Cell 2011, 146, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Gong, Y.; Meng, H.; Li, C.; Xue, L. Symphony of epigenetic and metabolic regulation—Interaction between the histone methyltransferase EZH2 and metabolism of tumor. Clin. Epigenetics 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Papathanassiu, A.E.; Ko, J.H.; Imprialou, M.; Bagnati, M.; Srivastava, P.K.; Vu, H.A.; Cucchi, D.; McAdoo, S.P.; Ananieva, E.A.; Mauro, C.; et al. BCAT1 controls metabolic reprogramming in activated human macrophages and is associated with inflammatory diseases. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Cha, T.-L.; Zhou, B.P.; Xia, W.; Wu, Y.; Yang, C.-C.; Chen, C.-T.; Ping, B.; Otte, A.P.; Hung, M.-C. Akt-Mediated Phosphorylation of EZH2 Suppresses Methylation of Lysine 27 in Histone H3. Science 2005, 310, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Vousden, K.H. Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer 2016, 16, 650–662. [Google Scholar] [CrossRef]
- Tao, T.; Chen, M.; Jiang, R.; Guan, H.; Huang, Y.; Su, H.; Hu, Q.; Han, X.; Xiao, J. Involvement of EZH2 in aerobic glycolysis of prostate cancer through miR-181b/HK2 axis. Oncol. Rep. 2017, 37, 1430–1436. [Google Scholar] [CrossRef]
- Wan, D.; Liu, C.; Sun, Y.; Wang, W.; Huang, K.; Zheng, L. MacroH2A1.1 cooperates with EZH2 to promote adipogenesis by regulating Wnt signaling. J. Mol. Cell Biol. 2017, 9, 1–13. [Google Scholar] [CrossRef]
- Yi, S.A.; Um, S.H.; Lee, J.; Yoo, J.H.; Bang, S.Y.; Park, E.K.; Lee, M.G.; Nam, K.H.; Jeon, Y.J.; Park, J.W.; et al. S6K1 Phosphorylation of H2B Mediates EZH2 Trimethylation of H3: A Determinant of Early Adipogenesis. Mol. Cell 2016, 62, 443–452. [Google Scholar] [CrossRef]
- Ahmad, F.; Patrick, S.; Sheikh, T.; Sharma, V.; Pathak, P.; Malgulwar, P.B.; Kumar, A.; Joshi, S.D.; Sarkar, C.; Sen, E. Telomerase reverse transcriptase (TERT)—enhancer of zeste homolog 2 (EZH2) network regulates lipid metabolism and DNA damage responses in glioblastoma. J. Neurochem. 2017, 143, 671–683. [Google Scholar] [CrossRef]
- Dong, Z.; Li, C.; Yin, C.; Xu, M.; Liu, S.; Gao, M. LncRNA PU.1 AS regulates arsenic-induced lipid metabolism through EZH2/Sirt6/SREBP-1c pathway. J. Env. Sci. 2019, 85, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Cormerais, Y.; Massard, P.A.; Vucetic, M.; Giuliano, S.; Tambutté, E.; Durivault, J.; Vial, V.; Endou, H.; Wempe, M.F.; Parks, S.K.; et al. The glutamine transporter ASCT2 (SLC1A5) promotes tumor growth independently of the amino acid transporter LAT1 (SLC7A5). J. Biol. Chem. 2018, 293, 2877–2887. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Liu, Y.; Cai, F.; Patrick, M.; Zmajkovic, J.; Cao, H.; Zhang, Y.; Tasdogan, A.; Chen, M.; Qi, L.; et al. Loss of EZH2 reprograms BCAA metabolism to drive leukemic transformation. Cancer Discov. 2019, 9, 1228–1247. [Google Scholar] [CrossRef] [PubMed]
- Vantaku, V.; Putluri, V.; Bader, D.A.; Maity, S.; Ma, J.; Arnold, J.M.; Rajapakshe, K.; Donepudi, S.R.; von Rundstedt, F.C.; Devarakonda, V.; et al. Epigenetic loss of AOX1 expression via EZH2 leads to metabolic deregulations and promotes bladder cancer progression. Oncogene 2020, 39, 6265–6285. [Google Scholar] [CrossRef]
- Martínez-Fernández, M.; Rubio, C.; Segovia, C.; López-Calderón, F.F.; Dueñas, M.; Paramio, J.M. EZH2 in Bladder Cancer, a Promising Therapeutic Target. Int. J. Mol. Sci. 2015, 16, 27107–27132. [Google Scholar] [CrossRef]
- Bracken, A.P.; Pasini, D.; Capra, M.; Prosperini, E.; Colli, E.; Helin, K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003, 22, 5323–5335. [Google Scholar] [CrossRef] [PubMed]
- Benetatos, L.; Voulgaris, E.; Vartholomatos, G.; Hatzimichael, E. Non-coding RNAs and EZH2 interactions in cancer: Long and short tales from the transcriptome. Int. J. Cancer 2013, 133, 267–274. [Google Scholar] [CrossRef]
- Ding, J.; Li, T.; Wang, X.; Zhao, E.; Choi, J.H.; Yang, L.; Zha, Y.; Dong, Z.; Huang, S.; Asara, J.M.; et al. The histone H3 methyltransferase G9A epigenetically activates the serine-glycine synthesis pathway to sustain cancer cell survival and proliferation. Cell Metab. 2013, 18, 896–907. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Ye, J.; Mancuso, A.; Tong, X.; Ward, P.S.; Fan, J.; Rabinowitz, J.D.; Thompson, C.B. Pyruvate kinase M2 promotes de novo serine synthesis to sustain mTORC1 activity and cell proliferation. Proc. Natl. Acad. Sci. USA 2012, 109, 6904–6909. [Google Scholar] [CrossRef] [PubMed]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81–120. [Google Scholar] [CrossRef]
- Lv, L.; Li, D.; Zhao, D.; Lin, R.; Chu, Y.; Zhang, H.; Zha, Z.; Liu, Y.; Li, Z.; Xu, Y.; et al. Acetylation Targets the M2 Isoform of Pyruvate Kinase for Degradation through Chaperone-Mediated Autophagy and Promotes Tumor Growth. Mol. Cell 2011, 42, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, A.; Schittenhelm, J.; Honegger, J.; Schluesener, H. Prognostic relevance of global histone 3 lysine 9 acetylation in ependymal tumors: Laboratory investigation. J. Neurosurg. 2013, 119, 1424–1431. [Google Scholar] [CrossRef]
- Ellinger, J.; Schneider, A.C.; Bachmann, A.; Kristiansen, G.; Müller, S.C.; Rogenhofer, S. Evaluation of Global Histone Acetylation Levels in Bladder Cancer Patients. Anticancer. Res. 2016, 36, 3961–3964. [Google Scholar] [PubMed]
- Giannopoulou, A.F.; Velentzas, A.D.; Konstantakou, E.G.; Avgeris, M.; Katarachia, S.A.; Papandreou, N.C.; Kalavros, N.I.; Mpakou, V.E.; Iconomidou, V.; Anastasiadou, E.; et al. Revisiting Histone Deacetylases in Human Tumorigenesis: The Paradigm of Urothelial Bladder Cancer. Int. J. Mol. Sci. 2019, 20, 1291. [Google Scholar] [CrossRef]
- Song, Y.; Wu, F.; Wu, J. Targeting histone methylation for cancer therapy: Enzymes, inhibitors, biological activity and perspectives. J. Hematol. Oncol. 2016, 9. [Google Scholar] [CrossRef]
- Wan, W.; Peng, K.; Li, M.; Qin, L.; Tong, Z.; Yan, J.; Shen, B.; Yu, C. Histone demethylase JMJD1A promotes urinary bladder cancer progression by enhancing glycolysis through coactivation of hypoxia inducible factor 1α. Oncogene 2017, 36, 3868–3877. [Google Scholar] [CrossRef]
- Mimura, I.; Nangaku, M.; Kanki, Y.; Tsutsumi, S.; Inoue, T.; Kohro, T.; Yamamoto, S.; Fujita, T.; Shimamura, T.; Suehiro, J.-I.; et al. Dynamic Change of Chromatin Conformation in Response to Hypoxia Enhances the Expression of GLUT3 (SLC2A3) by Cooperative Interaction of Hypoxia-Inducible Factor 1 and KDM3A. Mol. Cell. Biol. 2012, 32, 3018–3032. [Google Scholar] [CrossRef] [PubMed]
- Ezponda, T.; Dupéré-Richer, D.; Will, C.M.; Small, E.C.; Varghese, N.; Patel, T.; Nabet, B.; Popovic, R.; Oyer, J.; Bulic, M.; et al. UTX/KDM6A Loss Enhances the Malignant Phenotype of Multiple Myeloma and Sensitizes Cells to EZH2 inhibition. Cell Rep. 2017, 21, 628–640. [Google Scholar] [CrossRef]
- Chang, S.; Yim, S.; Park, H. The cancer driver genes IDH1/2, JARID1C/ KDM5C, and UTX/ KDM6A: Crosstalk between histone demethylation and hypoxic reprogramming in cancer metabolism. Exp. Mol. Med. 2019, 51, 1–17. [Google Scholar] [CrossRef]
- Miyake, H.; Nelson, C.; Rennie, P.S.; Gleave, M.E. Acquisition of chemoresistant phenotype by overexpression of the antiapoptotic gene Testosterone-repressed prostate message-2 in prostate cancer xenograft models. Cancer Res. 2000, 60, 2547–2554. [Google Scholar] [PubMed]
- Hurst, C.D.; Alder, O.; Platt, F.M.; Droop, A.; Stead, L.F.; Burns, J.E.; Burghel, G.J.; Jain, S.; Klimczak, L.J.; Lindsay, H.; et al. Genomic Subtypes of Non-invasive Bladder Cancer with Distinct Metabolic Profile and Female Gender Bias in KDM6A Mutation Frequency. Cancer Cell 2017, 32, 701–715.e7. [Google Scholar] [CrossRef] [PubMed]
- Ler, L.D.; Ghosh, S.; Chai, X.; Thike, A.A.; Heng, H.L.; Siew, E.Y.; Dey, S.; Koh, L.K.; Lim, J.Q.; Lim, W.K.; et al. Loss of tumor suppressor KDM6A amplifies PRC2-regulated transcriptional repression in bladder cancer and can be targeted through inhibition of EZH2. Sci. Transl. Med. 2017, 9, eaai8312. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Frydzińska, Z.; Owczarek, A.; Winiarska, K. Sirtuiny i ich rola w regulacji metabolizmu. Postepy Biochem. 2019, 65, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Niegisch, G.; Knievel, J.; Koch, A.; Hader, C.; Fischer, U.; Albers, P.; Schulz, W.A. Changes in histone deacetylase (HDAC) expression patterns and activity of HDAC inhibitors in urothelial cancers. Urol. Oncol. Semin. Orig. Investig. 2013, 31, 1770–1779. [Google Scholar] [CrossRef] [PubMed]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.A.B.; Otte, A.P.; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef]
- Monteiro-Reis, S.; Lameirinhas, A.; Miranda-Gonçalves, V.; Felizardo, D.; Dias, P.C.; Oliveira, J.; Graça, I.; Gonçalves, C.S.; Costa, B.M.; Henrique, R.; et al. Sirtuins’ deregulation in bladder cancer: Sirt7 is implicated in tumor progression through epithelial to mesenchymal transition promotion. Cancers 2020, 12, 1066. [Google Scholar] [CrossRef]
- Zhong, L.; Mostoslavsky, R. SIRT6: A master epigenetic gatekeeper of glucose metabolism. Transcription 2010, 1, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Hallows, W.C.; Yu, W.; Denu, J.M. Regulation of glycolytic enzyme phosphoglycerate mutase-1 by Sirt1 protein-mediated deacetylation. J. Biol. Chem. 2012, 287, 3850–3858. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.Y.; Xu, N.; Malyukova, A.; Scarlett, C.J.; Sun, Y.T.; Zhang, X.D.; Ling, D.; Su, S.P.; Nelson, C.; Chang, D.K.; et al. The histone deacetylase SIRT2 stabilizes Myc oncoproteins. Cell Death Differ. 2013, 20, 503–514. [Google Scholar] [CrossRef]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.L.; Emerling, B.M.; Ricoult, S.J.H.; Guarente, L. SirT3 suppresses hypoxia inducible factor 1α and tumor growth by inhibiting mitochondrial ROS production. Oncogene 2011, 30, 2986–2996. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.M.; Xiao, C.; Finley, L.W.S.; Lahusen, T.; Souza, A.L.; Pierce, K.; Li, Y.H.; Wang, X.; Laurent, G.; German, N.J.; et al. SIRT4 has tumor-suppressive activity and regulates the cellular metabolic response to dna damage by inhibiting mitochondrial glutamine metabolism. Cancer Cell 2013, 23, 450–463. [Google Scholar] [CrossRef]
- Wu, M.; Dickinson, S.I.; Wang, X.; Zhang, J. Expression and function of SIRT6 in muscle invasive urothelial carcinoma of the bladder. Int. J. Clin. Exp. Pathol. 2014, 7, 6504–6513. [Google Scholar]
- Shin, J.; He, M.; Liu, Y.; Paredes, S.; Villanova, L.; Brown, K.; Qiu, X.; Nabavi, N.; Mohrin, M.; Wojnoonski, K.; et al. SIRT7 represses myc activity to suppress er stress and prevent fatty liver disease. Cell Rep. 2013, 5, 654–665. [Google Scholar] [CrossRef]
- Barber, M.F.; Michishita-Kioi, E.; Xi, Y.; Tasselli, L.; Kioi, M.; Moqtaderi, Z.; Tennen, R.I.; Paredes, S.; Young, N.L.; Chen, K.; et al. SIRT7 links H3K18 deacetylation to maintenance of oncogenic transformation. Nature 2012, 487, 114–118. [Google Scholar] [CrossRef]
- Battistelli, C.; Cicchini, C.; Santangelo, L.; Tramontano, A.; Grassi, L.; Gonzalez, F.J.; De Nonno, V.; Grassi, G.; Amicone, L.; Tripodi, M. The Snail repressor recruits EZH2 to specific genomic sites through the enrollment of the lncRNA HOTAIR in epithelial-to-mesenchymal transition. Oncogene 2017, 36, 942–955. [Google Scholar] [CrossRef]
- Han, Y.; Liu, Y.; Zhang, H.; Wang, T.; Diao, R.; Jiang, Z.; Gui, Y.; Cai, Z. Hsa-miR-125b suppresses bladder cancer development by down-regulating oncogene SIRT7 and oncogenic long noncoding RNA MALAT1. Febs Lett. 2013, 587, 3875–3882. [Google Scholar] [CrossRef]
- Xie, H.; Liao, X.; Chen, Z.; Fang, Y.; He, A.; Zhong, Y.; Gao, Q.; Xiao, H.; Li, J.; Huang, W.; et al. LncRNA MALAT1 inhibits apoptosis and promotes invasion by antagonizing miR-125b in bladder cancer cells. J. Cancer 2017, 8, 3803–3811. [Google Scholar] [CrossRef] [PubMed]
- Beltrán-Anaya, F.O.; Cedro-Tanda, A.; Hidalgo-Miranda, A.; Romero-Cordoba, S.L. Insights into the regulatory role of non-coding RNAs in cancer metabolism. Front. Physiol. 2016, 7, 1–21. [Google Scholar] [CrossRef]
- Pedroza-Torres, A.; Romero-Córdoba, S.L.; Justo-Garrido, M.; Salido-Guadarrama, I.; Rodríguez-Bautista, R.; Montaño, S.; Muñiz-Mendoza, R.; Arriaga-Canon, C.; Fragoso-Ontiveros, V.; Álvarez-Gómez, R.M.; et al. MicroRNAs in Tumor Cell Metabolism: Roles and Therapeutic Opportunities. Front. Oncol. 2019, 9, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Peschiaroli, A.; Giacobbe, A.; Formosa, A.; Markert, E.K.; Bongiorno-Borbone, L.; Levine, A.J.; Candi, E.; D’Alessandro, A.; Zolla, L.; Finazzi Agrò, A.; et al. MiR-143 regulates hexokinase 2 expression in cancer cells. Oncogene 2013, 32, 797–802. [Google Scholar] [CrossRef]
- Yoshino, H.; Seki, N.; Itesako, T.; Chiyomaru, T.; Nakagawa, M.; Enokida, H. Aberrant expression of microRNAs in bladder cancer. Nat. Rev. Urol. 2013, 10, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Enokida, H.; Yoshino, H.; Matsushita, R.; Nakagawa, M. The role of microRNAs in bladder cancer. Investig. Clin. Urol. 2016, 57, S60. [Google Scholar] [CrossRef] [PubMed]
- Woodford, M.R.; Chen, V.Z.; Backe, S.J.; Bratslavsky, G.; Mollapour, M. Structural and functional regulation of lactate dehydrogenase-A in cancer. Future Med. Chem. 2020, 12, 439–455. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Yang, X.; Cheng, Y.; Zhang, X.; Yang, C.; Deng, X.; Li, P.; Tao, J.; Yang, H.; Wei, J.; et al. MicroRNA-218 Increases the Sensitivity of Bladder Cancer to Cisplatin by Targeting Glut1. Cell. Physiol. Biochem. 2017, 41, 921–932. [Google Scholar] [CrossRef]
- Chen, Y.H.; Heneidi, S.; Lee, J.M.; Layman, L.C.; Stepp, D.W.; Gamboa, G.M.; Chen, B.S.; Chazenbalk, G.; Azziz, R. Mirna-93 inhibits glut4 and is overexpressed in adipose tissue of polycystic ovary syndrome patients and women with insulin resistance. Diabetes 2013, 62, 2278–2286. [Google Scholar] [CrossRef] [PubMed]
- Fei, X.; Qi, M.; Wu, B.; Song, Y.; Wang, Y.; Li, T. MicroRNA-195-5p suppresses glucose uptake and proliferation of human bladder cancer T24 cells by regulating GLUT3 expression. FEBS Lett. 2012, 586, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Zeng, Q.; Xu, W.; Jiao, L.; Chen, Y.; Zhang, Z.; Wu, C.; Jin, T.; Pan, A.; Wei, R.; et al. miRNA-100 inhibits human bladder urothelial carcinogenesis by directly targeting mTOR. Mol. Cancer 2013, 12, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Yu, S.L.; Wang, Y.; Guo, G.Y.; Ding, Q.; An, R.H. MicroRNA-dependent regulation of PTEN after arsenic trioxide treatment in bladder cancer cell line T24. Tumor Biol. 2011, 32, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Cheng, Y.; Li, P.; Tao, J.; Deng, X.; Zhang, X.; Gu, M.; Lu, Q.; Yin, C. A lentiviral sponge for miRNA-21 diminishes aerobic glycolysis in bladder cancer T24 cells via the PTEN/PI3K/AKT/mTOR axis. Tumor Biol. 2014, 36, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Peng, M.; Zhang, Y.; Xu, W.; Darko, K.O.; Tao, T.; Huang, Y.; Tao, X.; Yang, X. Quercetin induces bladder cancer cells apoptosis by activation of AMPK signaling pathway. Am. J. Cancer Res. 2016, 6, 498–508. [Google Scholar] [PubMed]
- Li, F.; Yang, C.; Zhang, H.B.; Ma, J.; Jia, J.; Tang, X.; Zeng, J.; Chong, T.; Wang, X.; He, D.; et al. BET inhibitor JQ1 suppresses cell proliferation via inducing autophagy and activating LKB1/AMPK in bladder cancer cells. Cancer Med. 2019, 8, 4792–4805. [Google Scholar] [CrossRef]
- English, S.G.; Hadj-Moussa, H.; Storey, K.B. MicroRNAs regulate survival in oxygen-deprived environments. J. Exp. Biol. 2018, 221. [Google Scholar] [CrossRef] [PubMed]
- Blick, C.; Ramachandran, A.; Mccormick, R.; Wigfield, S.; Cranston, D.; Catto, J.; Harris, A.L. Identification of a hypoxia-regulated MIRNA signature in bladder cancer and a role for MIR-145 in hypoxia-dependent apoptosis. Br. J. Cancer 2015, 113, 634–644. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Lu, Z.; Takwi, A.A.L.; Chen, W.; Callander, N.S.; Ramos, K.S.; Young, K.H.; Li, Y. Negative regulation of the tumor suppressor p53 gene by microRNAs. Oncogene 2011, 30, 843–853. [Google Scholar] [CrossRef]
- Lee, H.; Jun, S.Y.; Lee, Y.S.; Lee, H.J.; Lee, W.S.; Park, C.S. Expression of miRNAs and ZEB1 and ZEB2 correlates with histopathological grade in papillary urothelial tumors of the urinary bladder. Virchows Arch. 2014, 464, 213–220. [Google Scholar] [CrossRef]
- Wiklund, E.D.; Bramsen, J.B.; Hulf, T.; Dyrskjøt, L.; Ramanathan, R.; Hansen, T.B.; Villadsen, S.B.; Gao, S.; Ostenfeld, M.S.; Borre, M.; et al. Coordinated epigenetic repression of the miR-200 family and miR-205 in invasive bladder cancer. Int. J. Cancer 2011, 128, 1327–1334. [Google Scholar] [CrossRef] [PubMed]
- Geiger, J.; Dalgaard, L.T. Interplay of mitochondrial metabolism and microRNAs. Cell. Mol. Life Sci. 2017, 74, 631–646. [Google Scholar] [CrossRef]
- Lim, Y.Y.; Wright, J.A.; Attema, J.L.; Gregory, P.A.; Bert, A.G.; Smith, E.; Thomas, D.; Lopez, A.F.; Drew, P.A.; Khew-Goodall, Y.; et al. Epigenetic modulation of the miR-200 family is associated with transition to a breast cancer stem-celllike state. J. Cell Sci. 2013, 126, 2256–2266. [Google Scholar] [CrossRef] [PubMed]
- Segovia, C.; Martínez-Fernández, M.; Dueñas, M.; Rubio, C.; López-Calderón, F.F.; Costa, C.; Saiz-Ladera, C.; Fernández-Grajera, M.; Duarte, J.; Muñoz, H.G.; et al. Opposing roles of PIK3CA gene alterations to EZH2 signaling in non-muscle invasive bladder cancer. Oncotarget 2017, 8, 10531–10542. [Google Scholar] [CrossRef]
- Bu, Q.; Fang, Y.; Cao, Y.; Chen, Q.; Liu, Y. Enforced expression of miR-101 enhances cisplatin sensitivity in human bladder cancer cells by modulating the cyclooxygenase-2 pathway. Mol. Med. Rep. 2014, 10, 2203–2209. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.K.; Wang, J.M.; Zhang, P.; Wang, Y.Q. MicroRNA-138 regulates metastatic potential of bladder cancer through ZEB2. Cell. Physiol. Biochem. 2015, 37, 2366–2374. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, W.; Zuo, Y.; Ding, M.; Ke, C.; Yan, R.; Zhan, H.; Liu, J.; Wang, J. miR-9 promotes cell proliferation and inhibits apoptosis by targeting LASS2 in bladder cancer. Tumor Biol. 2015, 36, 9631–9640. [Google Scholar] [CrossRef] [PubMed]
- Dyrskjot, L.; Ostenfeld, M.S.; Bramsen, J.B.; Silahtaroglu, A.N.; Lamy, P.; Ramanathan, R.; Fristrup, N.; Jensen, J.L.; Andersen, C.L.; Zieger, K.; et al. Genomic Profiling of MicroRNAs in Bladder Cancer: miR-129 Is Associated with Poor Outcome and Promotes Cell Death In vitro. Cancer Res. 2009, 69, 4851–4860. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Huang, Z.; Sheng, W.; Xu, M.D. Emerging roles of long non-coding RNAs in tumor metabolism. J. Hematol. Oncol. 2018, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Luo, J.; Luan, S.; He, C.; Li, Z. Long non-coding RNAs involved in cancer metabolic reprogramming. Cell. Mol. Life Sci. 2019, 76, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Zhou, Q.; Wang, C.Q.; Zhu, L.; Bi, C.; Zhang, S.; Wang, X.; Jin, H. LncRNAs regulate metabolism in cancer. Int. J. Biol. Sci. 2020, 16, 1194–1206. [Google Scholar] [CrossRef]
- Yang, F.; Zhang, H.; Mei, Y.; Wu, M. Reciprocal Regulation of HIF-1α and LincRNA-p21 Modulates the Warburg Effect. Mol. Cell 2014, 53, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.R.; Xiang, S.; Song, Z.; Wu, M. The p53-inducible long noncoding RNA TRINGS protects cancer cells from necrosis under glucose starvation. EMBO J. 2017, 36, 3483–3500. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xiao, Z.D.; Han, L.; Zhang, J.; Lee, S.W.; Wang, W.; Lee, H.; Zhuang, L.; Chen, J.; Lin, H.K.; et al. LncRNA NBR2 engages a metabolic checkpoint by regulating AMPK under energy stress. Nat. Cell Biol. 2016, 18, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Li, X.; Song, Y.; Zhang, P.; Xiao, Y.; Xing, Y. Long non-coding RNA ANRIL is up-regulated in bladder cancer and regulates bladder cancer cell proliferation and apoptosis through the intrinsic pathway. Biochem. Biophys. Res. Commun. 2015, 467, 223–228. [Google Scholar] [CrossRef]
- Kong, Y.; Hsieh, C.H.; Alonso, L.C. ANRIL: A lncRNA at the CDKN2A/B locus with roles in cancer and metabolic disease. Front. Endocrinol. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Mao, Y.; Chen, K.; He, W.; Shi, W.; Han, Y. The long noncoding RNA ANRIL acts as an oncogene and contributes to paclitaxel resistance of lung adenocarcinoma A549 cells. Oncotarget 2017, 8, 39177–39184. [Google Scholar] [CrossRef]
- Lan, W.G.; Xu, D.H.; Xu, C.; Ding, C.L.; Ning, F.L.; Zhou, Y.L.; Ma, L.B.; Liu, C.M.; Han, X. Silencing of long non-coding RNA ANRIL inhibits the development of multidrug resistance in gastric cancer cells. Oncol. Rep. 2016, 36, 263–270. [Google Scholar] [CrossRef]
- Yu, G.; Liu, G.; Yuan, D.; Dai, J.; Cui, Y.; Tang, X. Long non-coding RNA ANRIL is associated with a poor prognosis of osteosarcoma and promotes tumorigenesis via PI3K/Akt pathway. J. Bone Oncol. 2018, 11, 51–55. [Google Scholar] [CrossRef]
- Lu, Y.; Zhou, X.H.; Xu, L.; Rong, C.H.; Shen, C.; Bian, W. Long noncoding RNA ANRIL could be transactivated by c-Myc and promote tumor progression of non-small-cell lung cancer. OncoTargets Ther. 2016, 9, 3077–3084. [Google Scholar] [CrossRef][Green Version]
- Dong, X.; Jin, Z.; Chen, Y.; Xu, H.; Ma, C.; Hong, X.; Li, Y.; Zhao, G. Knockdown of long non-coding RNA ANRIL inhibits proliferation, migration, and invasion but promotes apoptosis of human glioma cells by upregulation of miR-34a. J. Cell. Biochem. 2018, 119, 2708–2718. [Google Scholar] [CrossRef]
- Zhang, J.J.; Wang, D.D.; Du, C.X.; Wang, Y. Long noncoding RNA ANRIL promotes cervical cancer development by acting as a sponge of miR-186. Oncol. Res. 2018, 26, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.W.; Ma, C.; Medoro, L.; Chen, L.; Wang, B.; Gupta, R.; Liu, T.; Yang, X.Z.; Chen, T.T.; Wang, R.Z.; et al. LncRNA ANRIL is up-regulated in nasopharyngeal carcinoma and promotes the cancer progression via increasing proliferation, reprograming cell glucose metabolism and inducing sidepopulation stem-like cancer cells. Oncotarget 2016, 7, 61741–61754. [Google Scholar] [CrossRef] [PubMed]
- Bochenek, G.; Häsler, R.; El Mokhtari, N.E.; König, I.R.; Loos, B.G.; Jepsen, S.; Schreiber, P.R.; Schaefer, A.S. The large non-coding RNA ANRIL, which is associated with atherosclerosis, periodontitis and several forms of cancer, regulates ADIPOR1, VAMP3 and C11ORF10. Hum. Mol. Genet. 2013, 22, 4516–4527. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Fan, Q.; Liang, Y.; Xiaodong, Z.; Ma, Y.; Zhihong, Z.; Hua, X.; Su, D.; Sun, H.; Li, H.; et al. Promotion of glycolysis by HOTAIR through GLUT1 upregulation via mTOR signaling. Oncol. Rep. 2017, 38, 1902–1908. [Google Scholar] [CrossRef]
- Cai, B.; Song, X.Q.; Cai, J.P.; Zhang, S. HOTAIR: A cancer-related long non-coding RNA. Neoplasma 2014, 61, 379–391. [Google Scholar] [CrossRef]
- Li, Z.; Li, X.; Wu, S.; Xue, M.; Chen, W. Long non-coding RNA UCA1 promotes glycolysis by upregulating hexokinase 2 through the mTOR-STAT3/microRNA143 pathway. Cancer Sci. 2014, 105, 951–955. [Google Scholar] [CrossRef]
- Li, H.J.; Li, X.; Pang, H.; Pan, J.J.; Xie, X.J.; Chen, W. Long non-coding RNA UCA1 promotes glutamine metabolism by targeting miR-16 in human bladder cancer. Jpn. J. Clin. Oncol. 2015, 45, 1055–1063. [Google Scholar] [CrossRef]
- Li, H.J.; Sun, X.M.; Li, Z.K.; Yin, Q.W.; Pang, H.; Pan, J.J.; Li, X.; Chen, W. LncRNA UCA1 Promotes Mitochondrial Function of Bladder Cancer via the MiR-195/ARL2 Signaling Pathway. Cell. Physiol. Biochem. 2017, 43, 2548–2561. [Google Scholar] [CrossRef]
- Xue, M.; Li, X.; Li, Z.; Chen, W. Urothelial carcinoma associated 1 is a hypoxia-inducible factor-1α-targeted long noncoding RNA that enhances hypoxic bladder cancer cell proliferation, migration, and invasion. Tumor Biol. 2014, 35, 6901–6912. [Google Scholar] [CrossRef]
- Xue, M.; Pang, H.; Li, X.; Li, H.; Pan, J.; Chen, W. Long non-coding RNA urothelial cancer-associated 1 promotes bladder cancer cell migration and invasion by way of the hsa-miR-145-ZEB1/2-FSCN1 pathway. Cancer Sci. 2016, 107, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Chen, J.; Li, H.; Yang, Y.; Yun, H.; Yang, S.; Mao, X. LncRNA UCA1 promotes the invasion and EMT of bladder cancer cells by regulating the miR-143/HMGB1 pathway. Oncol. Lett. 2017, 14, 5556–5562. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Li, X.; Wu, W.; Xue, M.; Hou, H.; Zhai, W.; Chen, W. Long non-coding RNA UCA1 promotes cisplatin/gemcitabine resistance through CREB modulating miR-196a-5p in bladder cancer cells. Cancer Lett. 2016, 382, 64–76. [Google Scholar] [CrossRef]
- Wang, T.; Yuan, J.; Feng, N.; Li, Y.; Lin, Z.; Jiang, Z.; Gui, Y. Hsa-miR-1 downregulates long non-coding RNA urothelial cancer associated 1 in bladder cancer. Tumor Biol. 2014, 35, 10075–10084. [Google Scholar] [CrossRef] [PubMed]
- Logotheti, S.; Marquardt, S.; Gupta, S.K.; Richter, C.; Edelhäuser, B.A.H.; Engelmann, D.; Brenmoehl, J.; Söhnchen, C.; Murr, N.; Alpers, M.; et al. LncRNA-SLC16A1-AS1 induces metabolic reprogramming during Bladder Cancer progression as target and co-activator of E2F1. Theranostics 2020, 10, 9620–9643. [Google Scholar] [CrossRef]
- Strimbu, K.; Tavel, J.A. What are biomarkers? Curr. Opin. HIV AIDS 2010, 5, 463–466. [Google Scholar] [CrossRef]
- Patel, S.; Ahmed, S. Emerging field of metabolomics: Big promise for cancer biomarker identification and drug discovery. J. Pharm. Biomed. Anal. 2015, 107, 63–74. [Google Scholar] [CrossRef]
- Bujak, R.; Struck-Lewicka, W.; Markuszewski, M.J.; Kaliszan, R. Metabolomics for laboratory diagnostics. J. Pharm. Biomed. Anal. 2015, 113, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Issaq, H.J.; Nativ, O.; Waybright, T.; Luke, B.; Veenstra, T.D.; Issaq, E.J.; Kravstov, A.; Mullerad, M. Detection of bladder cancer in human urine by metabolomic profiling using high performance liquid chromatography/mass spectrometry. J. Urol. 2008, 179, 2422–2426. [Google Scholar] [CrossRef]
- Pasikanti, K.K.; Esuvaranathan, K.; Hong, Y.; Ho, P.C.; Mahendran, R.; Raman Nee Mani, L.; Chiong, E.; Chan, E.C.Y. Urinary metabotyping of bladder cancer using two-dimensional gas chromatography time-of-flight mass spectrometry. J. Proteome Res. 2017, 8, 20719–20728. [Google Scholar] [CrossRef]
- Srivastava, S.; Roy, R.; Singh, S.; Kumar, P.; Dalela, D.; Sankhwar, S.N.; Goel, A.; Sonkar, A.A. Taurine—a possible fingerprint biomarker in non-muscle invasive bladder cancer: A pilot study by 1H NMR spectroscopy. Cancer Biomark. 2010, 6, 11–20. [Google Scholar] [CrossRef]
- Duskova, K.; Vesely, S.; Do, J.; Silva, C.; Cernei, N. Differences in Urinary Amino Acid Patterns in Individuals with Different Types of Urological Tumor Urinary Amino Acid Patterns as Markers of Urological Tumors. In Vivo 2018, 429, 425–429. [Google Scholar] [CrossRef]
- Alberice, J.V.; Amaral, A.F.S.; Armitage, E.G.; Lorente, J.A.; Algaba, F.; Carrilho, E.; Márquez, M.; García, A.; Malats, N.; Barbas, C. Searching for urine biomarkers of bladder cancer recurrence using a liquid chromatography-mass spectrometry and capillary electrophoresis-mass spectrometry metabolomics approach. J. Chromatogr. 2013, A 1318, 163–170. [Google Scholar] [CrossRef]
- Loras, A.; Trassierra, M.; Castell, J. V Bladder cancer recurrence surveillance by urine metabolomics analysis. Sci. Rep. 2018, 1–10. [Google Scholar] [CrossRef]
- Huang, Z.; Lin, L.; Gao, Y.; Chen, Y.; Yan, X.; Xing, J.; Hang, W. Bladder cancer determination via two urinary metabolites: A biomarker pattern approach. Mol. Cell. Proteom. 2011, 10, M111.007922. [Google Scholar] [CrossRef] [PubMed]
- Reinert, T.; Modin, C.; Castano, F.M.; Lamy, P.; Wojdacz, T.K.; Hansen, L.L.; Wiuf, C.; Borre, M.; Dyrskjot, L.; Orntoft, T.F. Comprehensive Genome Methylation Analysis in Bladder Cancer: Identification and Validation of Novel Methylated Genes and Application of These as Urinary Tumor Markers. Clin. Cancer Res. 2011, 17, 5582–5592. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Chen, Y.; Hang, W.; Gao, Y.; Lin, L.; Li, D.Y.; Xing, J.; Yan, X. Holistic metabonomic profiling of urine affords potential early diagnosis for bladder and kidney cancers. Metabolomics 2013, 9, 119–129. [Google Scholar] [CrossRef]
- Kim, J.-W.; Lee, G.; Moon, S.-M.; Park, M.-J.; Hong, S.K.; Ahn, Y.-H.; Kim, K.-R.; Paik, M.-J. Metabolomic screening and star pattern recognition by urinary amino acid profile analysis from bladder cancer patients. Metabolomics 2010, 6, 202–206. [Google Scholar] [CrossRef]
- Gamagedara, S.; Shi, H.; Ma, Y. Quantitative determination of taurine and related biomarkers in urine by liquid chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 2012, 402, 763–770. [Google Scholar] [CrossRef]
- Cao, M.; Zhao, L.; Chen, H.; Xue, W.; Lin, D. NMR-based metabolomic analysis of human bladder cancer. Anal. Sci. 2012, 28, 451–456. [Google Scholar] [CrossRef]
- Hoque, M.O.; Begum, S.; Topaloglu, O.; Chatterjee, A.; Rosenbaum, E.; Van Criekinge, W.; Westra, W.H.; Schoenberg, M.; Zahurak, M.; Goodman, S.N.; et al. Quantitation of Promoter Methylation of Multiple Genes in Urine DNA and Bladder Cancer Detection. JNCI J. Natl. Cancer Inst. 2006, 98, 996–1004. [Google Scholar] [CrossRef]
- Tan, G.; Wang, H.; Yuan, J.; Qin, W.; Dong, X.; Wu, H.; Meng, P. Three serum metabolite signatures for diagnosing low-grade and high-grade bladder cancer. Sci. Rep. 2017, 7, 46176. [Google Scholar] [CrossRef] [PubMed]
- Bansal, N.; Gupta, A.; Mitash, N.; Shakya, P.S.; Mandhani, A.; Mahdi, A.A.; Sankhwar, S.N.; Mandal, S.K. Low- and high-grade bladder cancer determination via human serum-based metabolomics approach. J. Proteome Res. 2013, 12, 5839–5850. [Google Scholar] [CrossRef]
- Loras, A.; Martínez-Bisbal, M.C.; Quintás, G.; Gil, S.; Martínez-Máñez, R.; Ruiz-Cerdá, J.L. Urinary metabolic signatures detect recurrences in non-muscle invasive bladder cancer. Cancers 2019, 11, 914. [Google Scholar] [CrossRef]
- Hauser, S.; Kogej, M.; Fechner, G.; Pezold Von, J.; Vorreuther, R.; Lümmen, G.; Müller, S.; Ellinger, J. Serum DNA hypermethylation in patients with bladder cancer: Results of a prospective multicenter study. Anticancer Res. 2013, 33, 779–784. [Google Scholar] [PubMed]
- Hegi, M.E.; Liu, L.; Herman, J.G.; Stupp, R.; Wick, W.; Weller, M.; Mehta, M.P.; Gilbert, M.R. Correlation of O6-methylguanine methyltransferase (MGMT) promoter methylation with clinical outcomes in glioblastoma and clinical strategies to modulate MGMT activity. J. Clin. Oncol. 2008, 26, 4189–4199. [Google Scholar] [CrossRef]
- Eissa, S.; Matboli, M.; Essawy, N.O.E.; Kotb, Y.M. Integrative functional genetic-epigenetic approach for selecting genes as urine biomarkers for bladder cancer diagnosis. Tumor Biol. 2015, 36, 9545–9552. [Google Scholar] [CrossRef] [PubMed]
- Berrondo, C.; Flax, J.; Kucherov, V.; Siebert, A.; Osinski, T.; Rosenberg, A.; Fucile, C.; Richheimer, S.; Beckham, C.J. Expression of the long non-coding RNA HOTAIR correlates with disease progression in bladder cancer and is contained in bladder cancer patient urinary exosomes. PLoS ONE 2016, 11, e0147236. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, M.; Lee, W.; Santarelli, L.; Neuzil, J. Exosome-derived microRNAs in cancer metabolism: Possible implications in cancer diagnostics and therapy. Exp. Mol. Med. 2017, 49, e285. [Google Scholar] [CrossRef]
- Faleiro, I.; Leão, R.; Binnie, A.; Andrade De Mello, R.; Maia, A.-T.; Castelo-Branco, P. Epigenetic therapy in urologic cancers: An update on clinical trials. Oncotarget 2017, 8, 12484–12500. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, C.H.; Bountra, C.; Fish, P.V.; Lee, K.; Schapira, M. Epigenetic protein families: A new frontier for drug discovery. Nat. Rev. Drug Discov. 2012, 11, 384–400. [Google Scholar] [CrossRef]
- Lavery, H.J.; Stensland, K.D.; Niegisch, G.; Albers, P.; Droller, M.J. Pathological T0 following radical cystectomy with or without neoadjuvant chemotherapy: A useful surrogate. J. Urol. 2014, 191, 898–906. [Google Scholar] [CrossRef]
- Jiang, X.; Lim, C.Z.H.; Li, Z.; Lee, P.L.; Yatim, S.M.J.M.; Guan, P.; Li, J.; Zhou, J.; Pan, J.; Chng, W.-J.; et al. Functional Characterization of D9, a Novel Deazaneplanocin A (DZNep) Analog, in Targeting Acute Myeloid Leukemia (AML). PLoS ONE 2015, 10, e0122983. [Google Scholar] [CrossRef]
- Sweis, R.F.; Pliushchev, M.; Brown, P.J.; Guo, J.; Li, F.; Maag, D.; Petros, A.M.; Soni, N.B.; Tse, C.; Vedadi, M.; et al. Discovery and Development of Potent and Selective Inhibitors of Histone Methyltransferase G9a. Acs Med. Chem. Lett. 2014, 5, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.T.; So, C.W.E. Epigenetic therapies by targeting aberrant histone methylome in AML: Molecular mechanisms, current preclinical and clinical development. Oncogene 2017, 36, 1753–1759. [Google Scholar] [CrossRef] [PubMed]
- Konze, K.D.; Ma, A.; Li, F.; Barsyte-Lovejoy, D.; Parton, T.; MacNevin, C.J.; Liu, F.; Gao, C.; Huang, X.-P.; Kuznetsova, E.; et al. An Orally Bioavailable Chemical Probe of the Lysine Methyltransferases EZH2 and EZH1. Acs Chem. Biol. 2013, 8, 1324–1334. [Google Scholar] [CrossRef]
- San José-Enériz, E.; Agirre, X.; Rabal, O.; Vilas-Zornoza, A.; Sanchez-Arias, J.A.; Miranda, E.; Ugarte, A.; Roa, S.; Paiva, B.; Estella-Hermoso de Mendoza, A.; et al. Discovery of first-in-class reversible dual small molecule inhibitors against G9a and DNMTs in hematological malignancies. Nat. Commun. 2017, 8, 15424. [Google Scholar] [CrossRef] [PubMed]
- Garzon, R.; Marcucci, G.; Croce, C.M. Targeting microRNAs in cancer: Rationale, strategies and challenges. Nat. Rev. Drug Discov. 2010, 9, 775–789. [Google Scholar] [CrossRef] [PubMed]
- Hatziapostolou, M.; Polytarchou, C.; Iliopoulos, D. MiRNAs link metabolic reprogramming to oncogenesis. Trends Endocrinol. Metab. 2013, 24, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Kunze, D.; Erdmann, K.; Froehner, M.; Wirth, M.; Fuessel, S. Enhanced Inhibition of Bladder Cancer Cell Growth by Simultaneous Knockdown of Antiapoptotic Bcl-xL and Survivin in Combination with Chemotherapy. Int. J. Mol. Sci. 2013, 14, 12297–12312. [Google Scholar] [CrossRef] [PubMed]
- Rieger, C.; Huebner, D.; Temme, A.; Wirth, M.P.; Fuessel, S. Antisense- and siRNA-mediated inhibition of the anti-apoptotic gene Bcl-xL for chemosensitization of bladder cancer cells. Int. J. Oncol. 2015, 47, 1121–1130. [Google Scholar] [CrossRef]
- Vinall, R.L.; Ripoll, A.Z.; Wang, S.; Pan, C.X.; Devere White, R.W. MiR-34a chemosensitizes bladder cancer cells to cisplatin treatment regardless of p53-Rb pathway status. Int. J. Cancer 2012, 130, 2526–2538. [Google Scholar] [CrossRef]
- Costa-Pinheiro, P.; Montezuma, D. Diagnostic and prognostic epigenetic. Epigenomics 2015, 7, 1003–1015. [Google Scholar] [CrossRef] [PubMed]
- Duquesne, I.; Weisbach, L.; Aziz, A.; Kluth, L.A.; Xylinas, E. The contemporary role and impact of urine-based biomarkers in bladder cancer. Transl. Urol. 2017, 6, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Pasikanti, K.K.; Esuvaranathan, K.; Ho, P.C.; Mahendran, R.; Kamaraj, R.; Wu, Q.H.; Chiong, E.; Chan, E.C.Y. Noninvasive urinary metabonomic diagnosis of human bladder cancer. J. Proteome Res. 2010, 9, 2988–2995. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolic Biomarkers in BC | |||
|---|---|---|---|
| Urine | |||
| Perturbed Biochemical Pathway |
Levels (BC/Control) | Metabolites |
Clinical Application |
| Glycolysis | High | Fructose [222], lactic acid [26,223] | Diagnosis |
| Low | Fructose [26] | Diagnosis | |
| TCA cycle | High | -- | -- |
| Low | Citric acid [222,223,224], succinate [26] | Diagnosis | |
| Amino acid metabolism | High | Val [20,26,222], Phe, Met, S−Adenosylmethionine, Lys [225], Leu [26,225,226], Ile, His, Ser [26], Tyr, Trp, hydroxyphenylalanine, phenylacetilglutamine, homophenylalanine, phenylglycoxylyc acid, kynurenine, hydroxyhippuric acid [221,227], hydroxytryptophan, indolacetic acid, minohippuric acid [227] | Diagnosis |
| Low | Ala, PAGN, Pro, Arg [226], Asp [225,226], hippuric acid [224,227,228], creatine [26,229,230] | Diagnosis | |
| GSH metabolism | High | -- | -- |
| Low | Pyroglutamic acid [231] | Diagnosis | |
| Taurine and hypotaurine | High | Taurine [224,230,232] | Diagnosis |
| Low | -- | -- | |
| Lipid metabolism | High | Carnitine [225], acetylcarnitine [26,227,230] | Diagnosis |
| Low | Glycerol [222], palmitic acid [225] | Diagnosis | |
| Nucleotide/nucleoside metabolism | High | Thymine [227], hypoxanthine, uridine [222] | Diagnosis |
| Low | Adenosine [26] | Diagnosis | |
| NAD cycle | High | -- | |
| Low | Trigonelline [229] | Diagnosis | |
| Metabolic Biomarkers in BC | |||
|---|---|---|---|
| Serum | |||
| Altered Biochemical Pathway | Levels (BC/Control) | Metabolites |
Clinical Application |
| Glycolysis | High | Glucose [232], erythritol, D-lyxosylamine, ribonic acid [44] | Diagnosis |
| Low | Lactate [232] | -- | |
| PPP | High | Ribose, gluconic acid, 2-keto-gluconic acid, xylitol, arabitol [44] | Diagnosis |
| Low | -- | -- | |
| Sucrose metabolism | High | Galacturonic acid, D-cellobiose, maltose [44] | Diagnosis |
| Low | -- | -- | |
| TCA cycle | High | Succinate, pyruvate, oxalacetate, phosphoenolpyruvate, acetyl-CoA [28], Cis-aconitic acid, fumaric acid, malic acid [44] | Diagnosis |
| Low | Citrate [232] | Diagnosis | |
| Amino acid metabolism | High | Gln, His, Malonate, Val [233], creatinine, kynurenine, norleucine [44] | Diagnosis |
| Low | Tyr, Ile, Phe, Leu, Gly [232] | Diagnosis | |
| Taurine and hypotaurine | High | Hypotaurine [44] | Diagnosis |
| Low | -- | -- | |
| Lipid metabolism | High | Carnitine [28] | Diagnosis |
| Low | -- | -- | |
| Nucleotide/nucleoside metabolism | High | Uridine, hypoxanthine [44] | Diagnosis |
| Low | -- | -- | |
| Organic acid | High | 2-hydroxyglutaric acid, (R,R)-2,3-dihydroxybutanoic acid, 2,3,4-trihydroxybutyric acid, 2,4-dihydroxybutanoic acid, 3,4,5-trihydroxypentanoic acid, 3,4-Dihydroxybutanoic acid [44] | Diagnosis |
| Low | -- | -- | |
| Choline | High | Choline [232] | Diagnosis |
| Low | -- | -- | |
| Ketone metabolism | High | Acetoacetate [232] | Diagnosis |
| Low | -- | -- | |
| ctDNA (Urine and Serum) | ||||
|---|---|---|---|---|
| Metabolic Gene/Pathway Related | Gene Status | Urine | Serum | Clinical Application |
| Xenobiotic metabolism | Hipermet. | CDKN2A, MGMT ARF, GSPT1 | -- | diagnosis |
| Xenobiotic metabolism, lipid metabolism and β-oxidation fatty acids | Hipermet. | -- | TIMP3, APC, RAR-β2, TIG1, p16INK4a, PTGS2, p14ARF, RASSF1A, GSTP1 | diagnosis |
| Unknown | Hipermet. | GDF15, TMEFF2, VIM | -- | diagnosis |
| Unknown | Hipermet. | PCDH10, PCDH17, APC | -- | prognosis |
| Unknown | Hipermet. | TWIST, NID2 | -- | diagnosis |
| Unknown | Hipermet. | -- | p16INK4a, p14ARF, CDH1, PCDH10, PCDH17 | diagnosis |
| Urine | |||
|---|---|---|---|
| Metabolic Gene/Pathway Related | Levels | Biomarker Panels | Clinical Application |
| GLUT1, GLUT4LDHA, HIF1 | High | miR-652, miR-199a-3p, miR-140-5p, miR-93, miR-142-5p, miR-1305, miR-30a, miR-224, miR-96, miR-766, miR-223, miR-99b, miR-140-3p, let-7b, miR-141, miR-191, miR-146b-5p, miR-491-5p, miR-339-3p, miR-200c, miR-106b, miR-143, miR-429, miR-222 and miR-200a | Diagnosis |
| Unknown | High | miR-7-5p, miR-22-3p, miR-29a-3p, miR-126-5p, miR-200a-3p, miR-375 and miR-423-5p | Diagnosis |
| GLS, LHDA, HIF1,GLUT1, GLUT3, LHD, PKM2, HK2LDHA, HK1 (p53) | High | UCA1-miR-16, miR-200c, miR-205, miR-21, miR-221 and miR-34a | Recurrence and surveillance |
| Unknown | High/low | NMIBC: miR-30a-5p, let-7c-5p, miR-486-5p, miR-205-5p and let-7i-5p NMIBC (high grade): miR-30a-5p, let-7c-5p, miR-486-5p, miR-21-5p, miR-106b-3p, miR-151a-3p, miR-200c-3p, miR-183-5p, miR-185-5p, miR-224-5p, miR-30c-2-5p and miR-10b-5p MIBC: miR-30a-5p, let-7c-5p, miR-486-5p, miR-205-5p, miR-451a, miR-25-3p, miR-30a-5p and miR-7-1-5 | Diagnosis/prognosis |
| p53, HIF1 | Low | miR-125b, miR-204, miR-99a, miR-30b, and miR-532-3p. | Diagnosis |
| Glutamine metabolism, xenobiotic metabolism, mitochondrial activity, HIF1 | High | hyal, lncRNA UCA1, microRNA-210, microRNA-96 | Diagnosis (MIBC) |
| Serum | |||
|---|---|---|---|
| Metabolic Gene/Pathway Related | Level | Biomarker Panels | Clinical Application |
| Unknown | High | miR-422a-3p, miR-486-3p, miR-103a-3p and miR-27a-3p | Prognosis (MIBC) |
| Unknown | High | miR-152, miR-148b-3p, miR-3187-3p, miR-15b-5p, miR-27a-3p and miR-30a-5p | Prognosis |
| Unknown | High | miR-422a-3p, miR-486-3p, miR-103a-3p and miR-27a-3p | Prognosis (MIBC) |
| Unknown | High | miR-541, miR-200b, miR-566, miR-487 and miR-148b | Diagnosis |
| Unknown | Low | miR-25, miR-92a, -92b, miR-302 and miR-33b | Diagnosis |
| Unknown | High | miR-152 | Prognosis |
| lncRNA-Derived Exosomes (Urine) | |||
|---|---|---|---|
| Metabolic Gene/Pathway Related | Levels | Biomarker Panels | Clinical Application |
| GLUT1 | High | HOTAIR, HOX-AS-2, MALAT1, HYMAI, LINC00477 | Diagnosis (MIBC) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loras, A.; Segovia, C.; Ruiz-Cerdá, J.L. Epigenomic and Metabolomic Integration Reveals Dynamic Metabolic Regulation in Bladder Cancer. Cancers 2021, 13, 2719. https://doi.org/10.3390/cancers13112719
Loras A, Segovia C, Ruiz-Cerdá JL. Epigenomic and Metabolomic Integration Reveals Dynamic Metabolic Regulation in Bladder Cancer. Cancers. 2021; 13(11):2719. https://doi.org/10.3390/cancers13112719
Chicago/Turabian StyleLoras, Alba, Cristina Segovia, and José Luis Ruiz-Cerdá. 2021. "Epigenomic and Metabolomic Integration Reveals Dynamic Metabolic Regulation in Bladder Cancer" Cancers 13, no. 11: 2719. https://doi.org/10.3390/cancers13112719
APA StyleLoras, A., Segovia, C., & Ruiz-Cerdá, J. L. (2021). Epigenomic and Metabolomic Integration Reveals Dynamic Metabolic Regulation in Bladder Cancer. Cancers, 13(11), 2719. https://doi.org/10.3390/cancers13112719