
Ubiquitin Proteasome Pathway Transcriptome in Epithelial Ovarian Cancer



Abstract
Simple Summary
Abstract
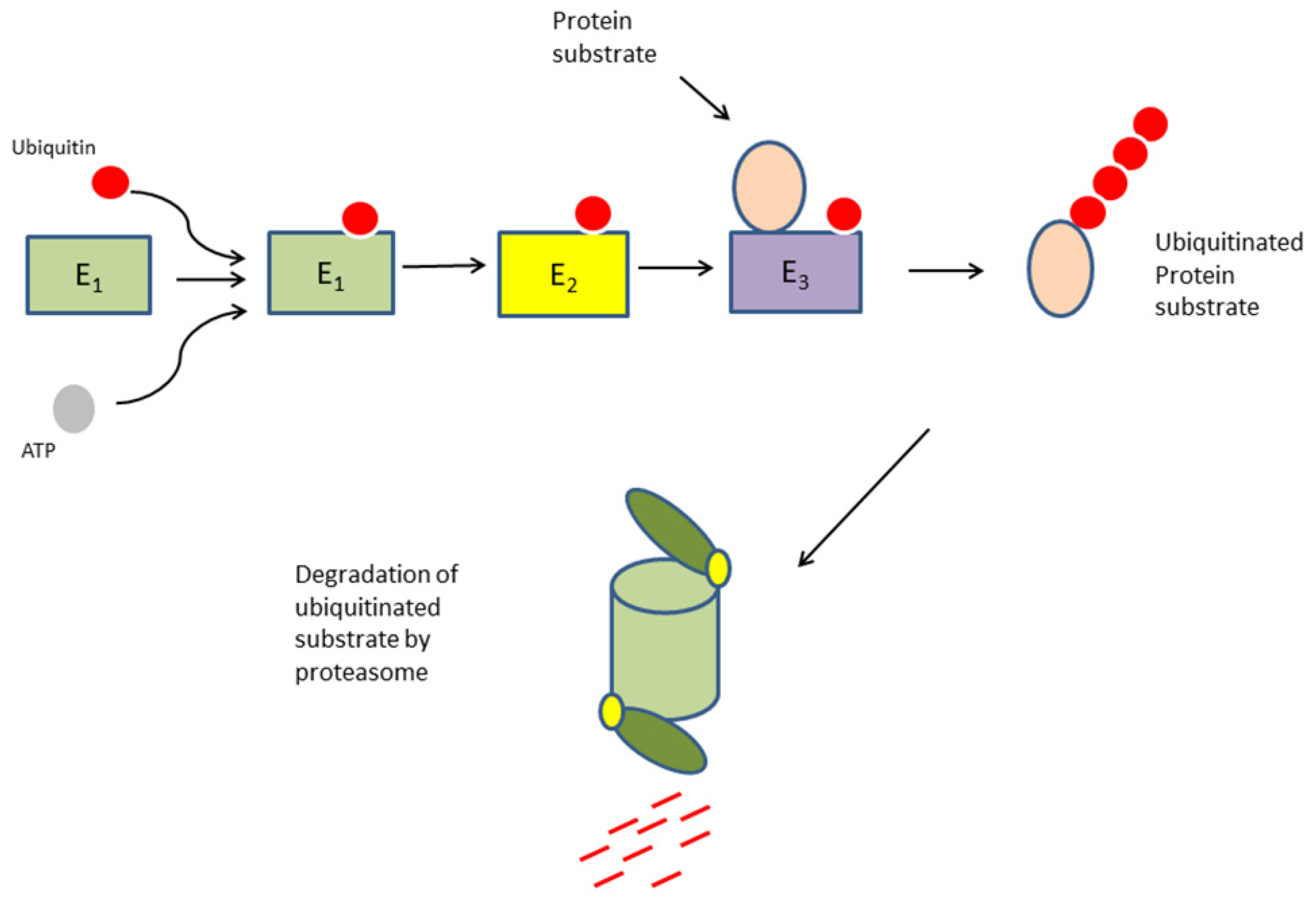
1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. KEGG Pathway Analysis Include Ubiquitin Ligase and Ubiquitin Ligase Adaptors
3.2. Expression of Genes Coding for Ubiquitin E1 Activators and E2 Conjugases
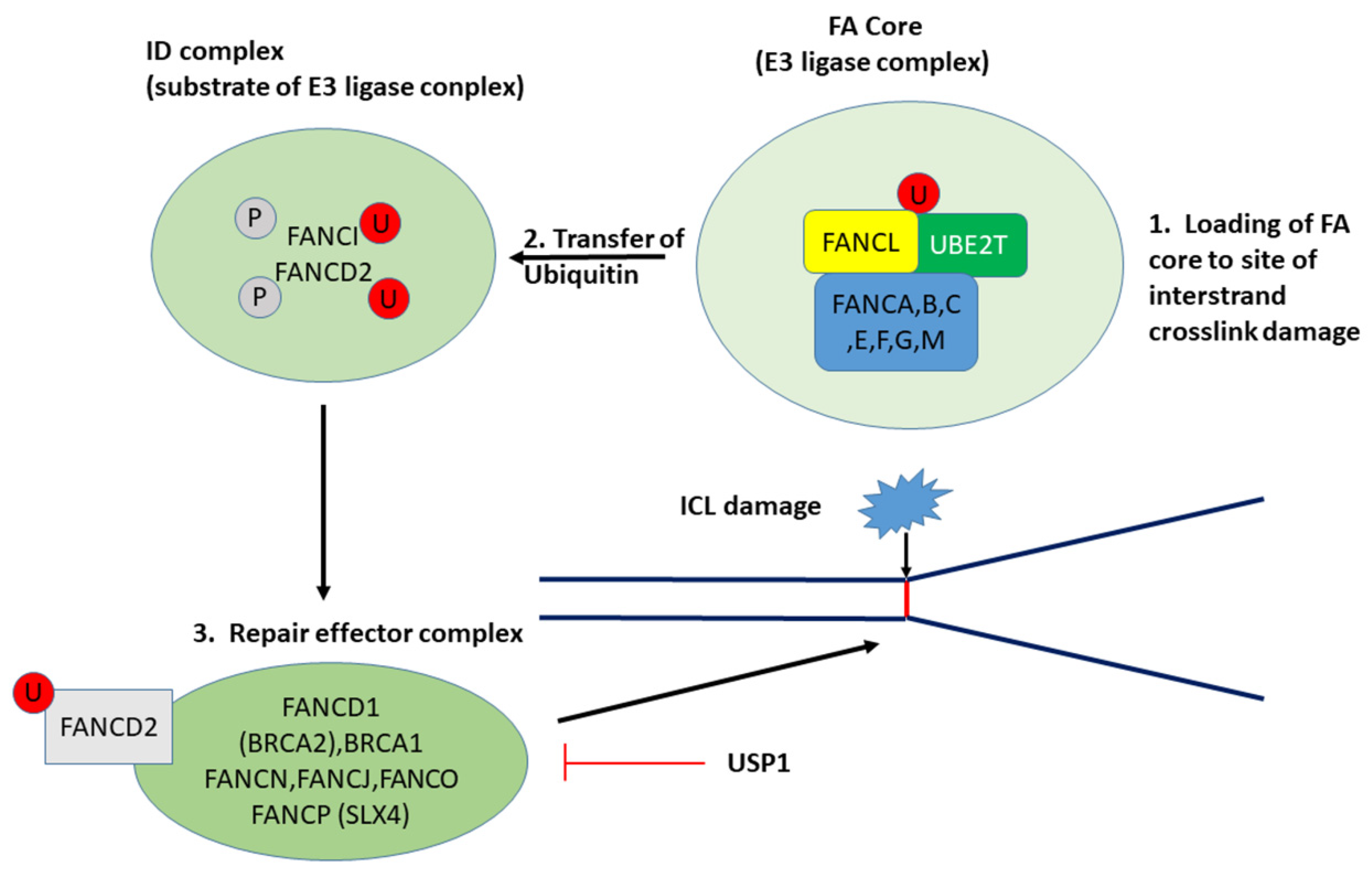
3.2.1. UBE2T and the Fanconi Anemia/BRCA Pathway
3.2.2. UBE2C, UBE2S, and the Cell Cycle
3.2.3. UBE2L6 and the Immunoproteasome
3.2.4. UBE2F and Neddylation
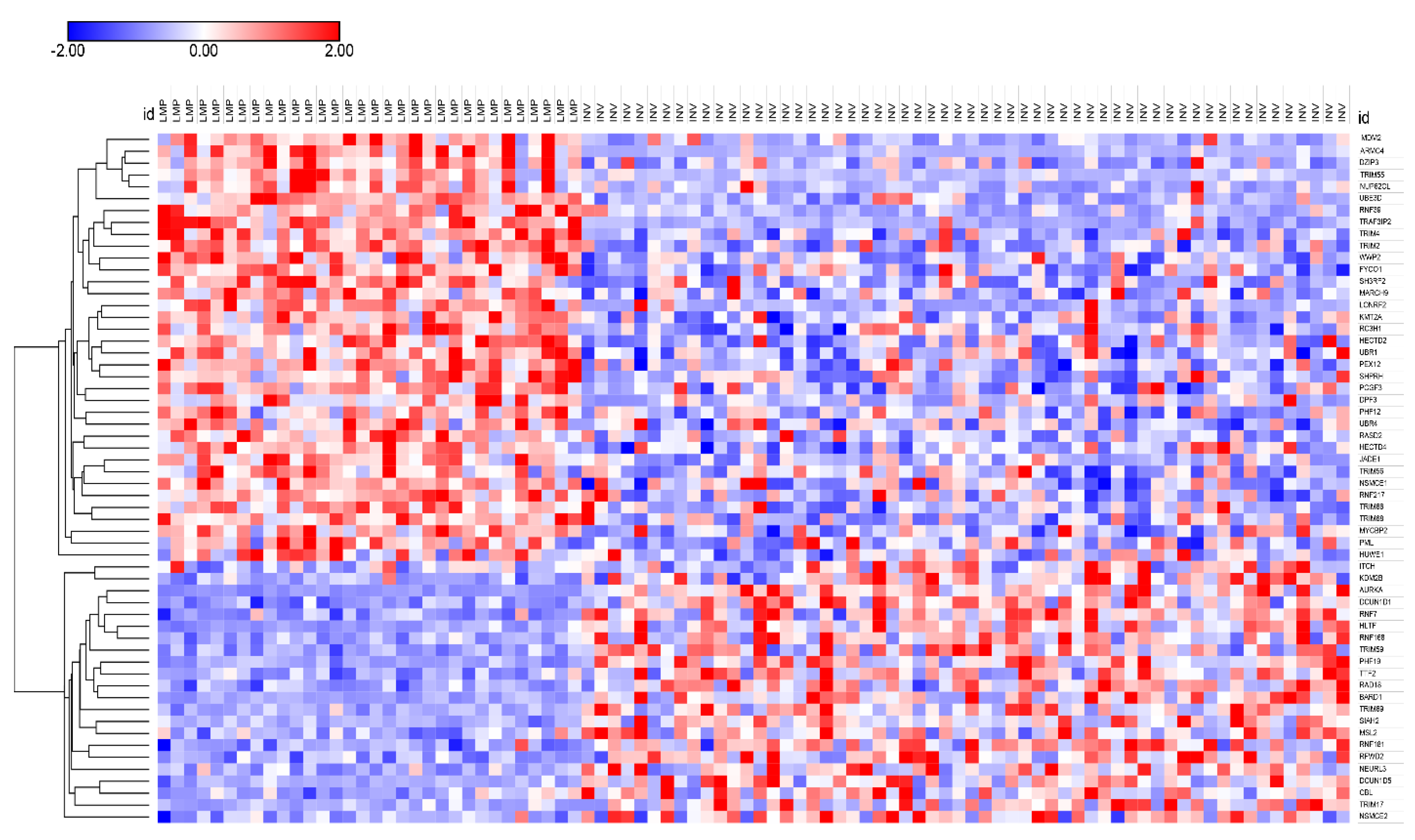
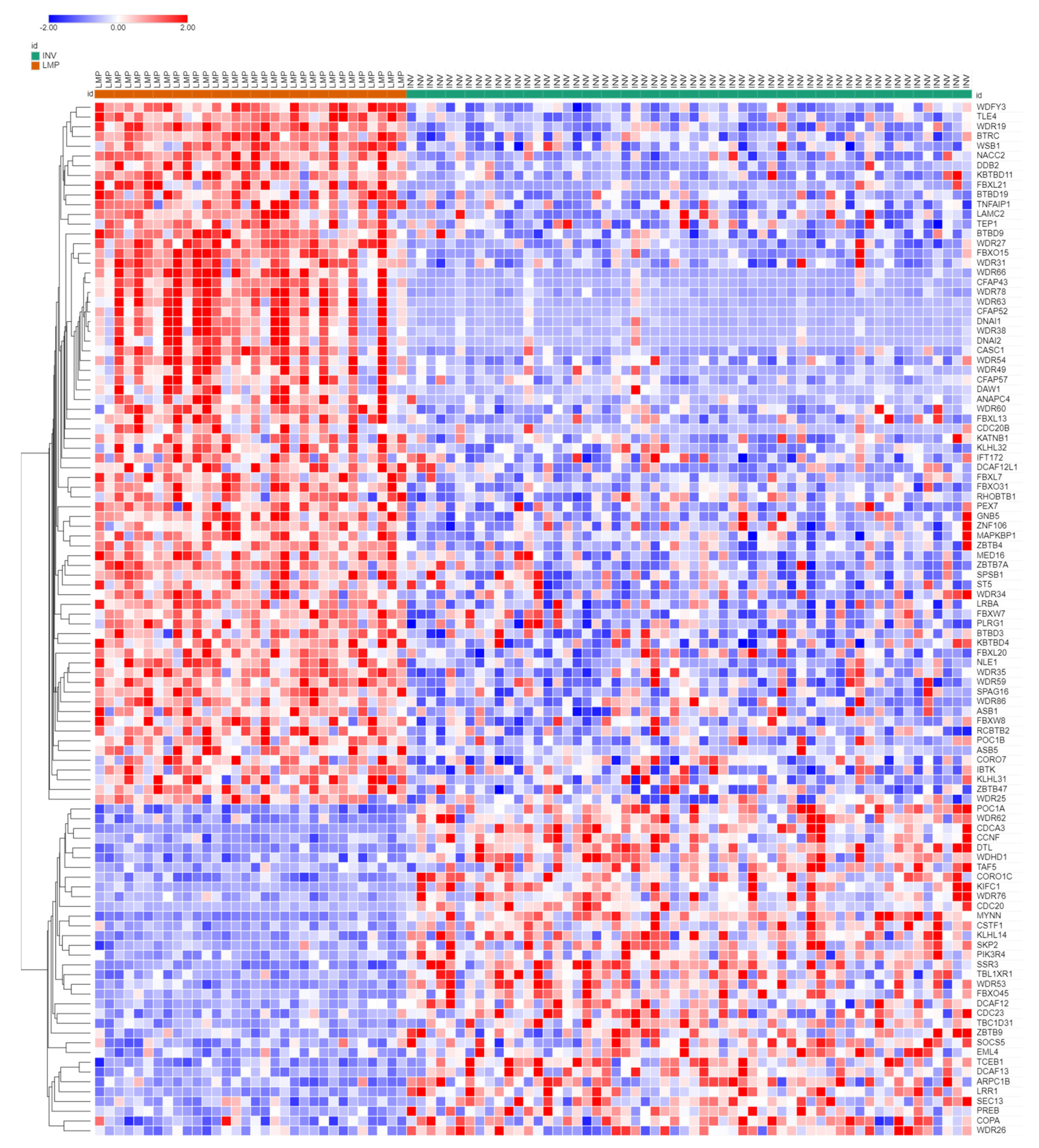
3.3. Ubiquitin Ligases and Adaptors Are Differentially Expressed between LMP and HGSOC
3.3.1. Heatmaps of Expression of E3 Ligases and E3 Adaptors
3.3.2. Neddylation and Cullin Ring Ubiquitin Ligases in EOC
3.3.3. The Cullin4 DCX/DWD E3 Subfamily in EOC
3.3.4. DDB2 and DNA Repair
3.4. BRCA1 Expression in EOC
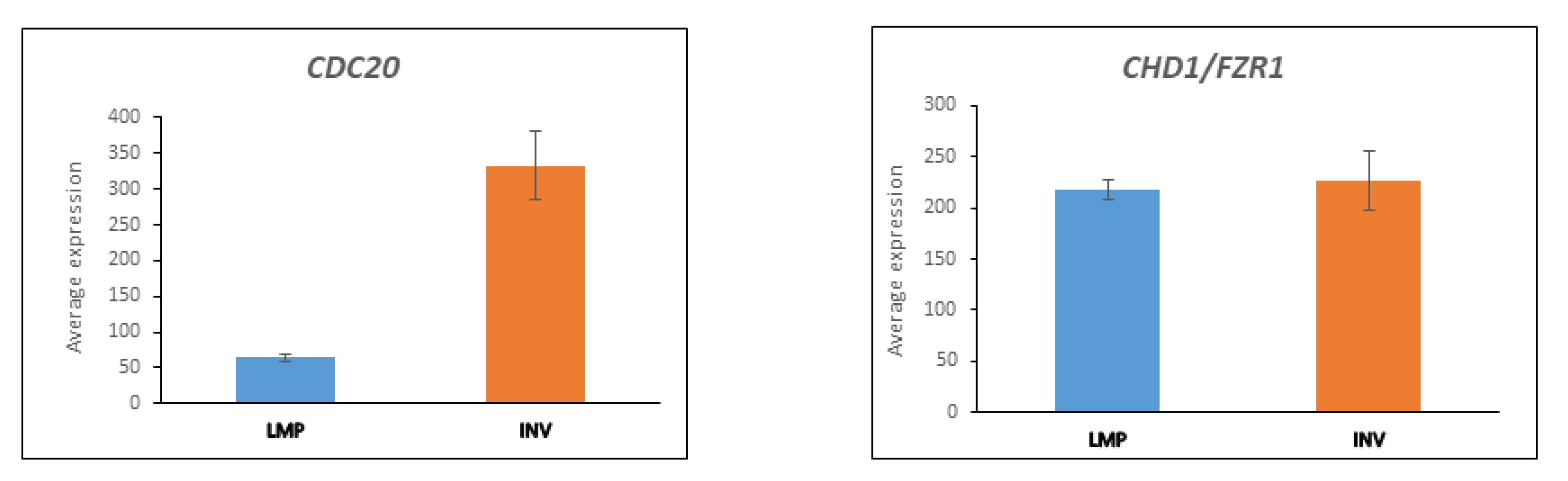
3.5. CDC20 and the APC/c Complex in Cell Division
3.5.1. Substrates of APC/c Are Differentially Expressed between LMP and HGSOC
3.5.2. UBE2C Expression in LMP and HGSOC
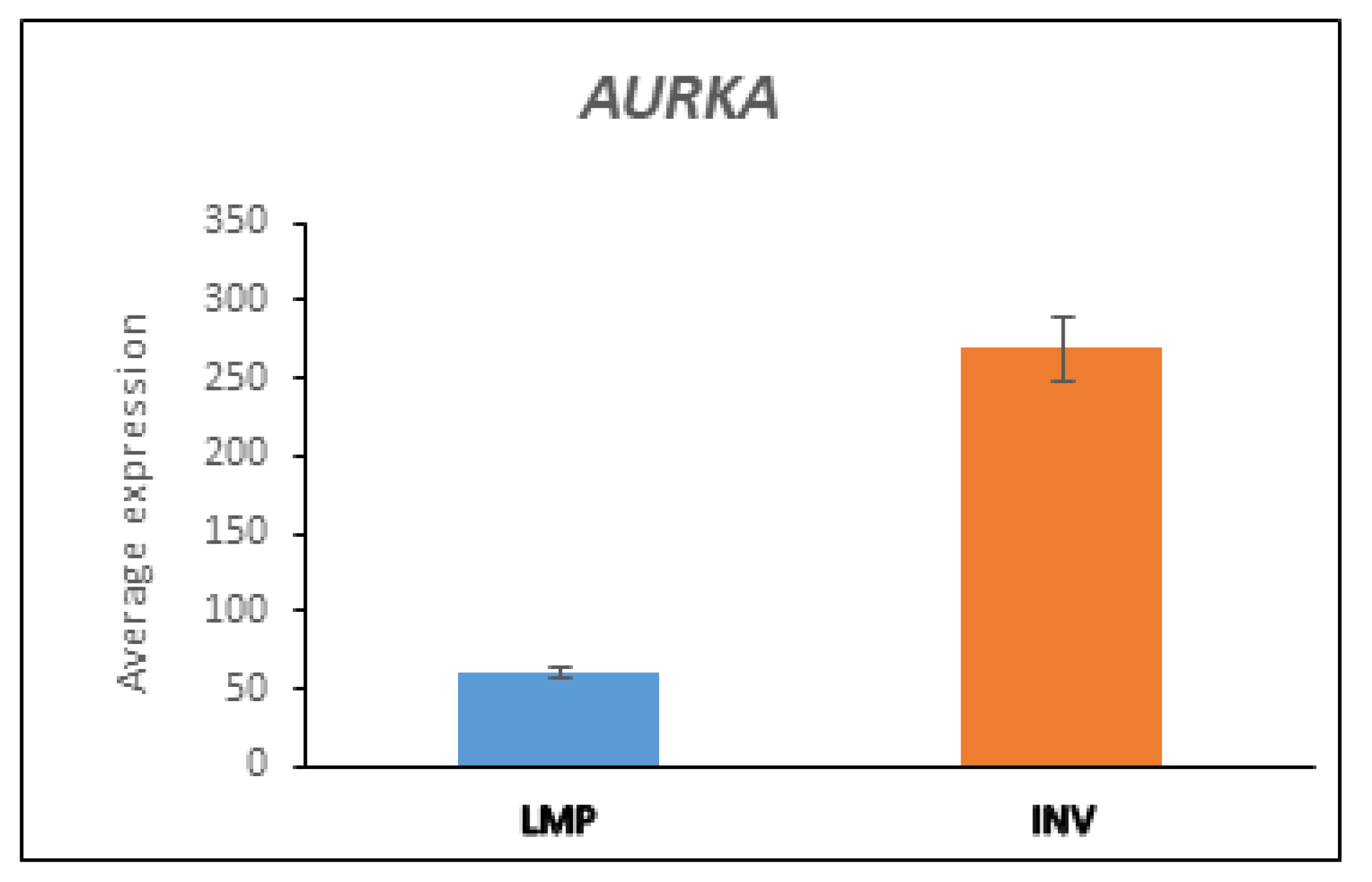
3.5.3. AURKA and BARD1 in LMP and HGSOC
3.6. Expression of Deubiquitinases (DUBs)
3.7. Transcriptome of Proteasome Subunits
3.8. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Tothill, R.W.; Tinker, A.V.; George, J.; Brown, R.; Fox, S.B.; Lade, S.; Johnson, D.S.; Trivett, M.K.; Etemadmoghadam, D.; Locandro, B.; et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin. Cancer Res. 2008, 14, 5198–5208. [Google Scholar] [CrossRef] [PubMed]
- Anglesio, M.S.; Arnold, J.M.; George, J.; Tinker, A.V.; Tothill, R.; Waddell, N.; Simms, L.; Locandro, B.; Fereday, S.; Traficante, N.; et al. Mutation of ERBB2 provides a novel alternative mechanism for the ubiquitous activation of RAS-MAPK in ovarian serous low malignant potential tumors. Mol. Cancer Res. 2008, 6, 1678–1690. [Google Scholar] [CrossRef]
- Hashizume, R.; Fukuda, M.; Maeda, I.; Nishikawa, H.; Oyake, D.; Yabuki, Y.; Ogata, H.; Ohta, T. The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J. Biol. Chem. 2001, 276, 14537–14540. [Google Scholar] [CrossRef]
- Chen, Y.; Hao, Q.; Wang, J.; Li, J.; Huang, C.; Zhang, Y.; Wu, X.; Lu, H.; Zhou, X. Ubiquitin ligase TRIM71 suppresses ovarian tumorigenesis by degrading mutant p53. Cell Death Dis. 2019, 10, 737. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Du, H.; Liu, H.; Hu, F.; Liu, G. SMAD specific E3 ubiquitin protein ligase 1 promotes ovarian cancer cell migration and invasion via the activation of the RhoA/ROCK signaling pathway. Oncol. Rep. 2019, 41, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; He, M.; Shah, A.A.; Wan, Y. Insights into APC/C: From cellular function to diseases and therapeutics. Cell Div. 2016, 11, 9. [Google Scholar] [CrossRef] [PubMed]
- Cole, A.J.; Dwight, T.; Gill, A.J.; Dickson, K.A.; Zhu, Y.; Clarkson, A.; Gard, G.B.; Maidens, J.; Valmadre, S.; Clifton-Bligh, R.; et al. Assessing mutant p53 in primary high-grade serous ovarian cancer using immunohistochemistry and massively parallel sequencing. Sci. Rep. 2016, 6, 26191. [Google Scholar] [CrossRef]
- Groettrup, M.; Pelzer, C.; Schmidtke, G.; Hofmann, K. Activating the ubiquitin family: UBA6 challenges the field. Trends Biochem. Sci. 2008, 33, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Barghout, S.H.; Schimmer, A.D. The ubiquitin-activating enzyme, UBA1, as a novel therapeutic target for AML. Oncotarget 2018, 9, 34198–34199. [Google Scholar] [CrossRef]
- Xu, W.; Lukkarila, J.L.; da Silva, S.R.; Paiva, S.L.; Gunning, P.T.; Schimmer, A.D. Targeting the ubiquitin E1 as a novel anti-cancer strategy. Curr. Pharm Des. 2013, 19, 3201–3209. [Google Scholar] [CrossRef]
- Hosseini, S.M.; Okoye, I.; Chaleshtari, M.G.; Hazhirkarzar, B.; Mohamadnejad, J.; Azizi, G.; Hojjat-Farsangi, M.; Mohammadi, H.; Shotorbani, S.S.; Jadidi-Niaragh, F. E2 ubiquitin-conjugating enzymes in cancer: Implications for immunotherapeutic interventions. Clin. Chim. Acta 2019, 498, 126–134. [Google Scholar] [CrossRef]
- Zou, R.; Xu, H.; Li, F.; Wang, S.; Zhu, L. Increased Expression of UBE2T Predicting Poor Survival of Epithelial Ovarian Cancer: Based on Comprehensive Analysis of UBE2s, Clinical Samples, and the GEO Database. DNA Cell Biol. 2021, 40, 36–60. [Google Scholar] [CrossRef]
- Machida, Y.J.; Machida, Y.; Chen, Y.; Gurtan, A.M.; Kupfer, G.M.; D’Andrea, A.D.; Dutta, A. UBE2T is the E2 in the Fanconi anemia pathway and undergoes negative autoregulation. Mol. Cell 2006, 23, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Duxin, J.P.; Walter, J.C. What is the DNA repair defect underlying Fanconi anemia? Curr. Opin. Cell Biol. 2015, 37, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Wen, M.; Kwon, Y.; Wang, Y.; Mao, J.H.; Wei, G. Elevated expression of UBE2T exhibits oncogenic properties in human prostate cancer. Oncotarget 2015, 6, 25226–25239. [Google Scholar] [CrossRef]
- Luo, C.; Yao, Y.; Yu, Z.; Zhou, H.; Guo, L.; Zhang, J.; Cao, H.; Zhang, G.; Li, Y.; Jiao, Z. UBE2T knockdown inhibits gastric cancer progression. Oncotarget 2017, 8, 32639–32654. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y.; Liu, B.; Song, A. Identifying Breast Cancer Subtypes Associated Modules and Biomarkers by Integrated Bioinformatics Analysis. Biosci Rep 2021, 41, BSR20203200. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, Y.; Yang, Z.; Liu, X.; Yang, P.; Wang, J.; Hu, K.; He, X.; Zhang, X.; Jing, H. High expression of UBE2T predicts poor prognosis and survival in multiple myeloma. Cancer Gene Ther. 2019, 26, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Rickman, K.A.; Lach, F.P.; Abhyankar, A.; Donovan, F.X.; Sanborn, E.M.; Kennedy, J.A.; Sougnez, C.; Gabriel, S.B.; Elemento, O.; Chandrasekharappa, S.C.; et al. Deficiency of UBE2T, the E2 Ubiquitin Ligase Necessary for FANCD2 and FANCI Ubiquitination, Causes FA-T Subtype of Fanconi Anemia. Cell Rep. 2015, 12, 35–41. [Google Scholar] [CrossRef]
- Hodson, C.; Cole, A.R.; Lewis, L.P.; Miles, J.A.; Purkiss, A.; Walden, H. Structural analysis of human FANCL, the E3 ligase in the Fanconi anemia pathway. J. Biol. Chem. 2011, 286, 32628–32637. [Google Scholar] [CrossRef]
- Cornwell, M.J.; Thomson, G.J.; Coates, J.; Belotserkovskaya, R.; Waddell, I.D.; Jackson, S.P.; Galanty, Y. Small-Molecule Inhibition of UBE2T/FANCL-Mediated Ubiquitylation in the Fanconi Anemia Pathway. ACS Chem. Biol. 2019, 14, 2148–2154. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; Tischkowitz, M.; Ameziane, N.; Hodgson, S.V.; Mathew, C.G.; Joenje, H.; Mok, S.C.; D’Andrea, A.D. Disruption of the Fanconi anemia-BRCA pathway in cisplatin-sensitive ovarian tumors. Nat. Med. 2003, 9, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Nijman, S.M.; Huang, T.T.; Dirac, A.M.; Brummelkamp, T.R.; Kerkhoven, R.M.; D’Andrea, A.D.; Bernards, R. The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol. Cell 2005, 17, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Ueki, T.; Park, J.H.; Nishidate, T.; Kijima, K.; Hirata, K.; Nakamura, Y.; Katagiri, T. Ubiquitination and downregulation of BRCA1 by ubiquitin-conjugating enzyme E2T overexpression in human breast cancer cells. Cancer Res. 2009, 69, 8752–8760. [Google Scholar] [CrossRef] [PubMed]
- Voter, A.F.; Manthei, K.A.; Keck, J.L. A High-Throughput Screening Strategy to Identify Protein-Protein Interaction Inhibitors That Block the Fanconi Anemia DNA Repair Pathway. J. Biomol. Screen 2016, 21, 626–633. [Google Scholar] [CrossRef]
- Gong, Y.Q.; Peng, D.; Ning, X.H.; Yang, X.Y.; Li, X.S.; Zhou, L.Q.; Guo, Y.L. UBE2T silencing suppresses proliferation and induces cell cycle arrest and apoptosis in bladder cancer cells. Oncol. Lett. 2016, 12, 4485–4492. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, X. UBE2T silencing inhibited non-small cell lung cancer cell proliferation and invasion by suppressing the wnt/beta-catenin signaling pathway. Int. J. Clin. Exp. Pathol. 2017, 10, 9482–9488. [Google Scholar]
- Shen, L.; Zhao, K.; Li, H.; Ning, B.; Wang, W.; Liu, R.; Zhang, Y.; Zhang, A. Downregulation of UBE2T can enhance the radiosensitivity of osteosarcoma in vitro and in vivo. Epigenomics 2019, 11, 1283–1305. [Google Scholar] [CrossRef]
- McCuaig, J.M.; Stockley, T.L.; Shaw, P.; Fung-Kee-Fung, M.; Altman, A.D.; Bentley, J.; Bernardini, M.Q.; Cormier, B.; Hirte, H.; Kieser, K.; et al. Evolution of genetic assessment for BRCA-associated gynaecologic malignancies: A Canadian multisociety roadmap. J. Med. Genet. 2018, 55, 571–577. [Google Scholar] [CrossRef]
- Chirnomas, D.; Taniguchi, T.; de la Vega, M.; Vaidya, A.P.; Vasserman, M.; Hartman, A.R.; Kennedy, R.; Foster, R.; Mahoney, J.; Seiden, M.V.; et al. Chemosensitization to cisplatin by inhibitors of the Fanconi anemia/BRCA pathway. Mol. Cancer Ther. 2006, 5, 952–961. [Google Scholar] [CrossRef]
- Wang, Z.; Li, M.; Lu, S.; Zhang, Y.; Wang, H. Promoter hypermethylation of FANCF plays an important role in the occurrence of ovarian cancer through disrupting Fanconi anemia-BRCA pathway. Cancer Biol. Ther. 2006, 5, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Synowiec, A.; Szenajch, J.; Wcislo, G.; Szczylik, C. The role of the Fanconi anemia pathway in the pathophysiology of ovarian cancer. Ginekol. Pol. 2015, 86, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Leveille, F.; Blom, E.; Medhurst, A.L.; Bier, P.; Laghmani, E.H.; Johnson, M.; Rooimans, M.A.; Sobeck, A.; Waisfisz, Q.; Arwert, F.; et al. The Fanconi anemia gene product FANCF is a flexible adaptor protein. J. Biol. Chem. 2004, 279, 39421–39430. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Jiang, X.; Qin, L.; Deng, H.; Wang, J.; Ren, W.; Li, H.; Zhao, L.; Liu, H.; Yan, H.; et al. A novel UBE2T inhibitor suppresses Wnt/beta-catenin signaling hyperactivation and gastric cancer progression by blocking RACK1 ubiquitination. Oncogene 2021, 40, 1027–1042. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, X.; Zhao, L.; Li, C.; Zhu, H.; Xu, L.; Shan, L.; Liao, X.; Guo, Z.; Huang, P. UBE2W interacts with FANCL and regulates the monoubiquitination of Fanconi anemia protein FANCD2. Mol. Cells 2011, 31, 113–122. [Google Scholar] [CrossRef]
- Christensen, D.E.; Brzovic, P.S.; Klevit, R.E. E2-BRCA1 RING interactions dictate synthesis of mono- or specific polyubiquitin chain linkages. Nat. Struct. Mol. Biol. 2007, 14, 941–948. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, X.; Yu, G.; Liu, L.; Wang, J.; Chen, X.; Bian, Y.; Ji, Y.; Zhou, X.; Chen, Y.; et al. UBE2C Is a Potential Biomarker of Intestinal-Type Gastric Cancer With Chromosomal Instability. Front. Pharmacol. 2018, 9, 847. [Google Scholar] [CrossRef]
- van Ree, J.H.; Jeganathan, K.B.; Malureanu, L.; van Deursen, J.M. Overexpression of the E2 ubiquitin-conjugating enzyme UbcH10 causes chromosome missegregation and tumor formation. J. Cell Biol. 2010, 188, 83–100. [Google Scholar] [CrossRef]
- Psyrri, A.; Kalogeras, K.T.; Kronenwett, R.; Wirtz, R.M.; Batistatou, A.; Bournakis, E.; Timotheadou, E.; Gogas, H.; Aravantinos, G.; Christodoulou, C.; et al. Prognostic significance of UBE2C mRNA expression in high-risk early breast cancer. A Hellenic Cooperative Oncology Group (HeCOG) Study. Ann. Oncol. 2012, 23, 1422–1427. [Google Scholar] [CrossRef]
- Yamano, H. APC/C: Current understanding and future perspectives. F1000Research 2019, 8. [Google Scholar] [CrossRef]
- Martinez-Chacin, R.C.; Bodrug, T.; Bolhuis, D.L.; Kedziora, K.M.; Bonacci, T.; Ordureau, A.; Gibbs, M.E.; Weissmann, F.; Qiao, R.; Grant, G.D.; et al. Ubiquitin chain-elongating enzyme UBE2S activates the RING E3 ligase APC/C for substrate priming. Nat. Struct. Mol. Biol. 2020, 27, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Alfieri, C.; Chang, L.; Zhang, Z.; Yang, J.; Maslen, S.; Skehel, M.; Barford, D. Molecular basis of APC/C regulation by the spindle assembly checkpoint. Nature 2016, 536, 431–436. [Google Scholar] [CrossRef]
- Li, J.; Zhi, X.; Shen, X.; Chen, C.; Yuan, L.; Dong, X.; Zhu, C.; Yao, L.; Chen, M. Depletion of UBE2C reduces ovarian cancer malignancy and reverses cisplatin resistance via downregulating CDK1. Biochem. Biophys. Res. Commun 2020, 523, 434–440. [Google Scholar] [CrossRef]
- Martinez-Canales, S.; Lopez de Rodas, M.; Nuncia-Cantarero, M.; Paez, R.; Amir, E.; Gyorffy, B.; Pandiella, A.; Galan-Moya, E.M.; Ocana, A. Functional transcriptomic annotation and protein-protein interaction analysis identify EZH2 and UBE2C as key upregulated proteins in ovarian cancer. Cancer Med. 2018, 7, 1896–1907. [Google Scholar] [CrossRef]
- Liu, J.; Meng, H.; Li, S.; Shen, Y.; Wang, H.; Shan, W.; Qiu, J.; Zhang, J.; Cheng, W. Identification of Potential Biomarkers in Association With Progression and Prognosis in Epithelial Ovarian Cancer by Integrated Bioinformatics Analysis. Front. Genet. 2019, 10, 1031. [Google Scholar] [CrossRef]
- Gong, Y.; Wang, D.; Lin, L.; Dai, J.; Yu, L. The expression of ubiquitin-conjugating enzyme E2C and KAI1 in ovarian carcinoma and their clinical significance. Medicine 2019, 98, e17896. [Google Scholar] [CrossRef] [PubMed]
- Berlingieri, M.T.; Pallante, P.; Guida, M.; Nappi, C.; Masciullo, V.; Scambia, G.; Ferraro, A.; Leone, V.; Sboner, A.; Barbareschi, M.; et al. UbcH10 expression may be a useful tool in the prognosis of ovarian carcinomas. Oncogene 2007, 26, 2136–2140. [Google Scholar] [CrossRef] [PubMed][Green Version]
- George, S.H.; Greenaway, J.; Milea, A.; Clary, V.; Shaw, S.; Sharma, M.; Virtanen, C.; Shaw, P.A. Identification of abrogated pathways in fallopian tube epithelium from BRCA1 mutation carriers. J. Pathol. 2011, 225, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Lei, T.; Zheng, J.; Chen, S.; Du, L.; Xie, H. UBE2S mediates tumor progression via SOX6/beta-Catenin signaling in endometrial cancer. Int. J. Biochem. Cell Biol. 2019, 109, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.H.; Yang, M.; Liu, L.P.; Wu, D.C.; Li, M.Y.; Su, S.G. UBE2S enhances the ubiquitination of p53 and exerts oncogenic activities in hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2018, 503, 895–902. [Google Scholar] [CrossRef]
- Wang, L.; Liang, Y.; Li, P.; Liang, Q.; Sun, H.; Xu, D.; Hu, W. Oncogenic Activities Of UBE2S Mediated By VHL/HIF-1alpha/STAT3 Signal Via The Ubiquitin-Proteasome System In PDAC. OncoTargets Ther. 2019, 12, 9767–9781. [Google Scholar] [CrossRef]
- Ayesha, A.K.; Hyodo, T.; Asano, E.; Sato, N.; Mansour, M.A.; Ito, S.; Hamaguchi, M.; Senga, T. UBE2S is associated with malignant characteristics of breast cancer cells. Tumour Biol. 2016, 37, 763–772. [Google Scholar] [CrossRef]
- Presta, I.; Novellino, F.; Donato, A.; La Torre, D.; Palleria, C.; Russo, E.; Malara, N.; Donato, G. UbcH10 a Major Actor in Cancerogenesis and a Potential Tool for Diagnosis and Therapy. Int. J. Mol. Sci. 2020, 21, 2041. [Google Scholar] [CrossRef]
- Seifert, U.; Bialy, L.P.; Ebstein, F.; Bech-Otschir, D.; Voigt, A.; Schroter, F.; Prozorovski, T.; Lange, N.; Steffen, J.; Rieger, M.; et al. Immunoproteasomes preserve protein homeostasis upon interferon-induced oxidative stress. Cell 2010, 142, 613–624. [Google Scholar] [CrossRef]
- Falvey, C.M.; O’Donovan, T.R.; El-Mashed, S.; Nyhan, M.J.; O’Reilly, S.; McKenna, S.L. UBE2L6/UBCH8 and ISG15 attenuate autophagy in esophageal cancer cells. Oncotarget 2017, 8, 23479–23491. [Google Scholar] [CrossRef]
- Murakami, M.; Izumi, H.; Kurita, T.; Koi, C.; Morimoto, Y.; Yoshino, K. UBE2L6 is Involved in Cisplatin Resistance by Regulating the Transcription of ABCB6. Anticancer Agents Med. Chem. 2020, 20, 1487–1496. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Jiang, Y.; Luo, Q.; Li, L.; Jia, L. Neddylation: A novel modulator of the tumor microenvironment. Mol. Cancer 2019, 18, 77. [Google Scholar] [CrossRef] [PubMed]
- Merlet, J.; Burger, J.; Gomes, J.E.; Pintard, L. Regulation of cullin-RING E3 ubiquitin-ligases by neddylation and dimerization. Cell Mol. Life Sci. 2009, 66, 1924–1938. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhu, J.; Chen, W.; Jiang, Y.; Hu, T.; Wang, Y.; Ye, X.; Zhan, M.; Ji, C.; Xu, Z.; et al. Induction of NEDD8-conjugating enzyme E2 UBE2F by platinum protects lung cancer cells from apoptosis and confers to platinum-insensitivity. Cell Death Dis 2020, 11, 975. [Google Scholar] [CrossRef]
- Huang, D.T.; Ayrault, O.; Hunt, H.W.; Taherbhoy, A.M.; Duda, D.M.; Scott, D.C.; Borg, L.A.; Neale, G.; Murray, P.J.; Roussel, M.F.; et al. E2-RING expansion of the NEDD8 cascade confers specificity to cullin modification. Mol. Cell 2009, 33, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xiong, X.; Sun, Y. Cullin-RING Ligase 5: Functional characterization and its role in human cancers. Semin. Cancer Biol. 2020, 67, 61–79. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.M.; Redon, C.E.; Aladjem, M.I. Chromatin-Bound Cullin-Ring Ligases: Regulatory Roles in DNA Replication and Potential Targeting for Cancer Therapy. Front. Mol. Biosci. 2018, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Fouad, S.; Wells, O.S.; Hill, M.A.; D’Angiolella, V. Cullin Ring Ubiquitin Ligases (CRLs) in Cancer: Responses to Ionizing Radiation (IR) Treatment. Front. Physiol. 2019, 10, 1144. [Google Scholar] [CrossRef] [PubMed]
- Carlucci, A.; D’Angiolella, V. It is not all about BRCA: Cullin-Ring ubiquitin Ligases in ovarian cancer. Br. J. Cancer 2015, 112, 9–13. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Enchev, R.I.; Schulman, B.A.; Peter, M. Protein neddylation: Beyond cullin-RING ligases. Nat. Rev. Mol. Cell Biol. 2015, 16, 30–44. [Google Scholar] [CrossRef]
- Santonico, E. Old and New Concepts in Ubiquitin and NEDD8 Recognition. Biomolecules 2020, 10, 566. [Google Scholar] [CrossRef]
- Soucy, T.A.; Smith, P.G.; Rolfe, M. Targeting NEDD8-activated cullin-RING ligases for the treatment of cancer. Clin. Cancer Res. 2009, 15, 3912–3916. [Google Scholar] [CrossRef]
- Swords, R.T.; Kelly, K.R.; Smith, P.G.; Garnsey, J.J.; Mahalingam, D.; Medina, E.; Oberheu, K.; Padmanabhan, S.; O’Dwyer, M.; Nawrocki, S.T.; et al. Inhibition of NEDD8-activating enzyme: A novel approach for the treatment of acute myeloid leukemia. Blood 2010, 115, 3796–3800. [Google Scholar] [CrossRef]
- Zhou, L.; Zhang, W.; Sun, Y.; Jia, L. Protein neddylation and its alterations in human cancers for targeted therapy. Cell Signal. 2018, 44, 92–102. [Google Scholar] [CrossRef]
- Soucy, T.A.; Smith, P.G.; Milhollen, M.A.; Berger, A.J.; Gavin, J.M.; Adhikari, S.; Brownell, J.E.; Burke, K.E.; Cardin, D.P.; Critchley, S.; et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009, 458, 732–736. [Google Scholar] [CrossRef]
- Jiang, Y.; Jia, L. Neddylation Pathway as a Novel Anti-cancer Target: Mechanistic Investigation and Therapeutic Implication. Anticancer Agents Med. Chem. 2015, 15, 1127–1133. [Google Scholar] [CrossRef]
- Nawrocki, S.T.; Kelly, K.R.; Smith, P.G.; Espitia, C.M.; Possemato, A.; Beausoleil, S.A.; Milhollen, M.; Blakemore, S.; Thomas, M.; Berger, A.; et al. Disrupting protein NEDDylation with MLN4924 is a novel strategy to target cisplatin resistance in ovarian cancer. Clin. Cancer Res. 2013, 19, 3577–3590. [Google Scholar] [CrossRef] [PubMed]
- Schlierf, A.; Altmann, E.; Quancard, J.; Jefferson, A.B.; Assenberg, R.; Renatus, M.; Jones, M.; Hassiepen, U.; Schaefer, M.; Kiffe, M.; et al. Targeted inhibition of the COP9 signalosome for treatment of cancer. Nat. Commun. 2016, 7, 13166. [Google Scholar] [CrossRef] [PubMed]
- Chua, Y.S.; Boh, B.K.; Ponyeam, W.; Hagen, T. Regulation of cullin RING E3 ubiquitin ligases by CAND1 in vivo. PLoS ONE 2011, 6, e16071. [Google Scholar] [CrossRef] [PubMed]
- Che, Z.; Liu, F.; Zhang, W.; McGrath, M.; Hou, D.; Chen, P.; Song, C.; Yang, D. Targeting CAND1 promotes caspase-8/RIP1-dependent apoptosis in liver cancer cells. Am. J. Transl. Res. 2018, 10, 1357–1372. [Google Scholar] [PubMed]
- Biedermann, S.; Hellmann, H. WD40 and CUL4-based E3 ligases: Lubricating all aspects of life. Trends Plant. Sci. 2011, 16, 38–46. [Google Scholar] [CrossRef]
- Angers, S.; Li, T.; Yi, X.; MacCoss, M.J.; Moon, R.T.; Zheng, N. Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature 2006, 443, 590–593. [Google Scholar] [CrossRef]
- Higa, L.A.; Wu, M.; Ye, T.; Kobayashi, R.; Sun, H.; Zhang, H. CUL4-DDB1 ubiquitin ligase interacts with multiple WD40-repeat proteins and regulates histone methylation. Nat. Cell Biol. 2006, 8, 1277–1283. [Google Scholar] [CrossRef]
- Jackson, S.; Xiong, Y. CRL4s: The CUL4-RING E3 ubiquitin ligases. Trends Biochem. Sci. 2009, 34, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Higa, L.A.; Zhang, H. Stealing the spotlight: CUL4-DDB1 ubiquitin ligase docks WD40-repeat proteins to destroy. Cell Div. 2007, 2, 5. [Google Scholar] [CrossRef][Green Version]
- Bungsy, M.; Palmer, M.C.L.; Jeusset, L.M.; Neudorf, N.M.; Lichtensztejn, Z.; Nachtigal, M.W.; McManus, K.J. Reduced RBX1 expression induces chromosome instability and promotes cellular transformation in high-grade serous ovarian cancer precursor cells. Cancer Lett. 2021, 500, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Lepage, C.C.; Palmer, M.C.L.; Farrell, A.C.; Neudorf, N.M.; Lichtensztejn, Z.; Nachtigal, M.W.; McManus, K.J. Reduced SKP1 and CUL1 expression underlies increases in Cyclin E1 and chromosome instability in cellular precursors of high-grade serous ovarian cancer. Br. J. Cancer 2021. [Google Scholar] [CrossRef] [PubMed]
- Willems, A.R.; Schwab, M.; Tyers, M. A hitchhiker’s guide to the cullin ubiquitin ligases: SCF and its kin. Biochim. Biophys. Acta 2004, 1695, 133–170. [Google Scholar] [CrossRef]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.W.; Zhou, J.J.; Yu, C.; Xu, Y.; Guo, L.J.; Zhang, H.Y.; Zhou, D.; Song, F.Z.; Fan, H.Y. Ubiquitin E3 ligase CRL4(CDT2/DCAF2) as a potential chemotherapeutic target for ovarian surface epithelial cancer. J. Biol. Chem. 2013, 288, 29680–29691. [Google Scholar] [CrossRef] [PubMed]
- Barry, M.; Fruh, K. Viral modulators of cullin RING ubiquitin ligases: Culling the host defense. Sci. STKE 2006, 2006, pe21. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Robert, E.I.; van Breugel, P.C.; Strubin, M.; Zheng, N. A promiscuous alpha-helical motif anchors viral hijackers and substrate receptors to the CUL4-DDB1 ubiquitin ligase machinery. Nat. Struct. Mol. Biol. 2010, 17, 105–111. [Google Scholar] [CrossRef]
- Takedachi, A.; Saijo, M.; Tanaka, K. DDB2 complex-mediated ubiquitylation around DNA damage is oppositely regulated by XPC and Ku and contributes to the recruitment of XPA. Mol. Cell Biol. 2010, 30, 2708–2723. [Google Scholar] [CrossRef]
- Groisman, R.; Polanowska, J.; Kuraoka, I.; Sawada, J.; Saijo, M.; Drapkin, R.; Kisselev, A.F.; Tanaka, K.; Nakatani, Y. The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell 2003, 113, 357–367. [Google Scholar] [CrossRef]
- Tan, T.; Chu, G. p53 Binds and activates the xeroderma pigmentosum DDB2 gene in humans but not mice. Mol. Cell Biol. 2002, 22, 3247–3254. [Google Scholar] [CrossRef]
- Han, C.; Zhao, R.; Liu, X.; Srivastava, A.; Gong, L.; Mao, H.; Qu, M.; Zhao, W.; Yu, J.; Wang, Q.E. DDB2 suppresses tumorigenicity by limiting the cancer stem cell population in ovarian cancer. Mol. Cancer Res. 2014, 12, 784–794. [Google Scholar] [CrossRef]
- Crijns, A.P.; Fehrmann, R.S.; de Jong, S.; Gerbens, F.; Meersma, G.J.; Klip, H.G.; Hollema, H.; Hofstra, R.M.; te Meerman, G.J.; de Vries, E.G.; et al. Survival-related profile, pathways, and transcription factors in ovarian cancer. PLoS Med. 2009, 6, e24. [Google Scholar] [CrossRef]
- Hall, J.M.; Lee, M.K.; Newman, B.; Morrow, J.E.; Anderson, L.A.; Huey, B.; King, M.C. Linkage of early-onset familial breast cancer to chromosome 17q21. Science 1990, 250, 1684–1689. [Google Scholar] [CrossRef]
- Jazaeri, A.A.; Yee, C.J.; Sotiriou, C.; Brantley, K.R.; Boyd, J.; Liu, E.T. Gene expression profiles of BRCA1-linked, BRCA2-linked, and sporadic ovarian cancers. J. Natl. Cancer Inst. 2002, 94, 990–1000. [Google Scholar] [CrossRef]
- Pal, T.; Permuth-Wey, J.; Betts, J.A.; Krischer, J.P.; Fiorica, J.; Arango, H.; LaPolla, J.; Hoffman, M.; Martino, M.A.; Wakeley, K.; et al. BRCA1 and BRCA2 mutations account for a large proportion of ovarian carcinoma cases. Cancer 2005, 104, 2807–2816. [Google Scholar] [CrossRef]
- Tavassoli, M.; Ruhrberg, C.; Beaumont, V.; Reynolds, K.; Kirkham, N.; Collins, W.P.; Farzaneh, F. Whole chromosome 17 loss in ovarian cancer. Genes Chromosomes Cancer 1993, 8, 195–198. [Google Scholar] [CrossRef] [PubMed]
- Papp, J.; Csokay, B.; Bosze, P.; Zalay, Z.; Toth, J.; Ponder, B.; Olah, E. Allele loss from large regions of chromosome 17 is common only in certain histological subtypes of ovarian carcinomas. Br. J. Cancer 1996, 74, 1592–1597. [Google Scholar] [CrossRef]
- Lindon, C.; Grant, R.; Min, M. Ubiquitin-Mediated Degradation of Aurora Kinases. Front. Oncol. 2015, 5, 307. [Google Scholar] [CrossRef]
- Lukas, J.; Bartek, J. Cell division: The heart of the cycle. Nature 2004, 432, 564–567. [Google Scholar] [CrossRef]
- Qi, W.; Yu, H. KEN-box-dependent degradation of the Bub1 spindle checkpoint kinase by the anaphase-promoting complex/cyclosome. J. Biol. Chem. 2007, 282, 3672–3679. [Google Scholar] [CrossRef] [PubMed]
- Skaar, J.R.; Pagano, M. Cdh1: A master G0/G1 regulator. Nat. Cell Biol. 2008, 10, 755–757. [Google Scholar] [CrossRef]
- Engelbert, D.; Schnerch, D.; Baumgarten, A.; Wasch, R. The ubiquitin ligase APC(Cdh1) is required to maintain genome integrity in primary human cells. Oncogene 2008, 27, 907–917. [Google Scholar] [CrossRef]
- Li, M.; Zhang, P. The function of APC/CCdh1 in cell cycle and beyond. Cell Div. 2009, 4, 2. [Google Scholar] [CrossRef]
- Qiao, X.; Zhang, L.; Gamper, A.M.; Fujita, T.; Wan, Y. APC/C-Cdh1: From cell cycle to cellular differentiation and genomic integrity. Cell Cycle 2010, 9, 3904–3912. [Google Scholar] [CrossRef]
- Qiao, R.; Weissmann, F.; Yamaguchi, M.; Brown, N.G.; VanderLinden, R.; Imre, R.; Jarvis, M.A.; Brunner, M.R.; Davidson, I.F.; Litos, G.; et al. Mechanism of APC/CCDC20 activation by mitotic phosphorylation. Proc. Natl. Acad. Sci. USA 2016, 113, E2570–E2578. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.; Mark, K.G.; Mocciaro, A.; Watson, E.R.; Prabu, J.R.; Cha, D.D.; Kampmann, M.; Gamarra, N.; Zhou, C.Y.; Rape, M. Gene expression and cell identity controlled by anaphase-promoting complex. Nature 2020, 579, 136–140. [Google Scholar] [CrossRef]
- Ouellet, V.; Guyot, M.C.; Le Page, C.; Filali-Mouhim, A.; Lussier, C.; Tonin, P.N.; Provencher, D.M.; Mes-Masson, A.M. Tissue array analysis of expression microarray candidates identifies markers associated with tumor grade and outcome in serous epithelial ovarian cancer. Int. J. Cancer 2006, 119, 599–607. [Google Scholar] [CrossRef]
- Bonome, T.; Lee, J.Y.; Park, D.C.; Radonovich, M.; Pise-Masison, C.; Brady, J.; Gardner, G.J.; Hao, K.; Wong, W.H.; Barrett, J.C.; et al. Expression profiling of serous low malignant potential, low-grade, and high-grade tumors of the ovary. Cancer Res. 2005, 65, 10602–10612. [Google Scholar] [CrossRef]
- Karra, H.; Repo, H.; Ahonen, I.; Loyttyniemi, E.; Pitkanen, R.; Lintunen, M.; Kuopio, T.; Soderstrom, M.; Kronqvist, P. Cdc20 and securin overexpression predict short-term breast cancer survival. Br. J. Cancer 2014, 110, 2905–2913. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wan, L.; Zhong, J.; Inuzuka, H.; Liu, P.; Sarkar, F.H.; Wei, W. Cdc20: A potential novel therapeutic target for cancer treatment. Curr Pharm Des. 2013, 19, 3210–3214. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, J.; Wan, L.; Zhou, X.; Wang, Z.; Wei, W. Targeting Cdc20 as a novel cancer therapeutic strategy. Pharmacol. Ther. 2015, 151, 141–151. [Google Scholar] [CrossRef]
- Sackton, K.L.; Dimova, N.; Zeng, X.; Tian, W.; Zhang, M.; Sackton, T.B.; Meaders, J.; Pfaff, K.L.; Sigoillot, F.; Yu, H.; et al. Synergistic blockade of mitotic exit by two chemical inhibitors of the APC/C. Nature 2014, 514, 646–649. [Google Scholar] [CrossRef]
- Lara-Gonzalez, P.; Moyle, M.W.; Budrewicz, J.; Mendoza-Lopez, J.; Oegema, K.; Desai, A. The G2-to-M Transition Is Ensured by a Dual Mechanism that Protects Cyclin B from Degradation by Cdc20-Activated APC/C. Dev. Cell 2019, 51, 313–325.e310. [Google Scholar] [CrossRef]
- Song, L.; Rape, M. Regulated degradation of spindle assembly factors by the anaphase-promoting complex. Mol. Cell 2010, 38, 369–382. [Google Scholar] [CrossRef]
- Rape, M.; Kirschner, M.W. Autonomous regulation of the anaphase-promoting complex couples mitosis to S-phase entry. Nature 2004, 432, 588–595. [Google Scholar] [CrossRef]
- Berlingieri, M.T.; Pallante, P.; Sboner, A.; Barbareschi, M.; Bianco, M.; Ferraro, A.; Mansueto, G.; Borbone, E.; Guerriero, E.; Troncone, G.; et al. UbcH10 is overexpressed in malignant breast carcinomas. Eur. J. Cancer 2007, 43, 2729–2735. [Google Scholar] [CrossRef]
- Tanner, M.M.; Grenman, S.; Koul, A.; Johannsson, O.; Meltzer, P.; Pejovic, T.; Borg, A.; Isola, J.J. Frequent amplification of chromosomal region 20q12-q13 in ovarian cancer. Clin. Cancer Res. 2000, 6, 1833–1839. [Google Scholar]
- Jazaeri, A.A.; Lu, K.; Schmandt, R.; Harris, C.P.; Rao, P.H.; Sotiriou, C.; Chandramouli, G.V.; Gershenson, D.M.; Liu, E.T. Molecular determinants of tumor differentiation in papillary serous ovarian carcinoma. Mol. Carcinog 2003, 36, 53–59. [Google Scholar] [CrossRef]
- Wang, C.; Yan, Q.; Hu, M.; Qin, D.; Feng, Z. Effect of AURKA Gene Expression Knockdown on Angiogenesis and Tumorigenesis of Human Ovarian Cancer Cell Lines. Target. Oncol. 2016, 11, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, F.; Formicola, D.; Capasso, M. Dualistic Role of BARD1 in Cancer. Genes 2017, 8, 375. [Google Scholar] [CrossRef]
- Baer, R.; Ludwig, T. The BRCA1/BARD1 heterodimer, a tumor suppressor complex with ubiquitin E3 ligase activity. Curr. Opin. Genet. Dev. 2002, 12, 86–91. [Google Scholar] [CrossRef]
- Mallery, D.L.; Vandenberg, C.J.; Hiom, K. Activation of the E3 ligase function of the BRCA1/BARD1 complex by polyubiquitin chains. EMBO J. 2002, 21, 6755–6762. [Google Scholar] [CrossRef]
- Starita, L.M.; Parvin, J.D. Substrates of the BRCA1-dependent ubiquitin ligase. Cancer Biol. Ther. 2006, 5, 137–141. [Google Scholar] [CrossRef][Green Version]
- Sato, K.; Hayami, R.; Wu, W.; Nishikawa, T.; Nishikawa, H.; Okuda, Y.; Ogata, H.; Fukuda, M.; Ohta, T. Nucleophosmin/B23 is a candidate substrate for the BRCA1-BARD1 ubiquitin ligase. J. Biol. Chem. 2004, 279, 30919–30922. [Google Scholar] [CrossRef]
- Yu, X.; Baer, R. Nuclear localization and cell cycle-specific expression of CtIP, a protein that associates with the BRCA1 tumor suppressor. J. Biol. Chem. 2000, 275, 18541–18549. [Google Scholar] [CrossRef]
- Yu, X.; Chen, J. DNA damage-induced cell cycle checkpoint control requires CtIP, a phosphorylation-dependent binding partner of BRCA1 C-terminal domains. Mol. Cell Biol. 2004, 24, 9478–9486. [Google Scholar] [CrossRef]
- Yu, X.; Fu, S.; Lai, M.; Baer, R.; Chen, J. BRCA1 ubiquitinates its phosphorylation-dependent binding partner CtIP. Genes Dev. 2006, 20, 1721–1726. [Google Scholar] [CrossRef]
- Yuan, J.; Chen, J. N terminus of CtIP is critical for homologous recombination-mediated double-strand break repair. J. Biol. Chem. 2009, 284, 31746–31752. [Google Scholar] [CrossRef]
- Atipairin, A.; Canyuk, B.; Ratanaphan, A. The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by the platinum-based anticancer drugs. Breast Cancer Res. Treat. 2011, 126, 203–209. [Google Scholar] [CrossRef]
- Saskova, A.; Solc, P.; Baran, V.; Kubelka, M.; Schultz, R.M.; Motlik, J. Aurora kinase A controls meiosis I progression in mouse oocytes. Cell Cycle 2008, 7, 2368–2376. [Google Scholar] [CrossRef]
- Bertolin, G.; Tramier, M. Insights into the non-mitotic functions of Aurora kinase A: More than just cell division. Cell Mol. Life Sci. 2020, 77, 1031–1047. [Google Scholar] [CrossRef]
- Eyers, P.A.; Erikson, E.; Chen, L.G.; Maller, J.L. A novel mechanism for activation of the protein kinase Aurora A. Curr. Biol. 2003, 13, 691–697. [Google Scholar] [CrossRef]
- Abdelbaki, A.; Akman, H.B.; Poteau, M.; Grant, R.; Gavet, O.; Guarguaglini, G.; Lindon, C. AURKA destruction is decoupled from its activity at mitotic exit but is essential to suppress interphase activity. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef]
- Nikonova, A.S.; Astsaturov, I.; Serebriiskii, I.G.; Dunbrack, R.L., Jr.; Golemis, E.A. Aurora A kinase (AURKA) in normal and pathological cell division. Cell Mol. Life Sci. 2013, 70, 661–687. [Google Scholar] [CrossRef]
- Yu, X.; Minter-Dykhouse, K.; Malureanu, L.; Zhao, W.M.; Zhang, D.; Merkle, C.J.; Ward, I.M.; Saya, H.; Fang, G.; van Deursen, J.; et al. Chfr is required for tumor suppression and Aurora A regulation. Nat. Genet. 2005, 37, 401–406. [Google Scholar] [CrossRef]
- den Hollander, J.; Rimpi, S.; Doherty, J.R.; Rudelius, M.; Buck, A.; Hoellein, A.; Kremer, M.; Graf, N.; Scheerer, M.; Hall, M.A.; et al. Aurora kinases A and B are up-regulated by Myc and are essential for maintenance of the malignant state. Blood 2010, 116, 1498–1505. [Google Scholar] [CrossRef]
- Willems, E.; Dedobbeleer, M.; Digregorio, M.; Lombard, A.; Lumapat, P.N.; Rogister, B. The functional diversity of Aurora kinases: A comprehensive review. Cell Div. 2018, 13, 7. [Google Scholar] [CrossRef]
- Tanaka, M.; Ueda, A.; Kanamori, H.; Ideguchi, H.; Yang, J.; Kitajima, S.; Ishigatsubo, Y. Cell-cycle-dependent regulation of human aurora A transcription is mediated by periodic repression of E4TF1. J. Biol. Chem. 2002, 277, 10719–10726. [Google Scholar] [CrossRef]
- Pugacheva, E.N.; Jablonski, S.A.; Hartman, T.R.; Henske, E.P.; Golemis, E.A. HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell 2007, 129, 1351–1363. [Google Scholar] [CrossRef]
- Do, T.V.; Xiao, F.; Bickel, L.E.; Klein-Szanto, A.J.; Pathak, H.B.; Hua, X.; Howe, C.; O’Brien, S.W.; Maglaty, M.; Ecsedy, J.A.; et al. Aurora kinase A mediates epithelial ovarian cancer cell migration and adhesion. Oncogene 2014, 33, 539–549. [Google Scholar] [CrossRef]
- Fukui, S.; Nagasaka, K.; Miyagawa, Y.; Kikuchi-Koike, R.; Kawata, Y.; Kanda, R.; Ichinose, T.; Sugihara, T.; Hiraike, H.; Wada-Hiraike, O.; et al. The proteasome deubiquitinase inhibitor bAP15 downregulates TGF-beta/Smad signaling and induces apoptosis via UCHL5 inhibition in ovarian cancer. Oncotarget 2019, 10, 5932–5948. [Google Scholar] [CrossRef]
- Livneh, I.; Cohen-Kaplan, V.; Cohen-Rosenzweig, C.; Avni, N.; Ciechanover, A. The life cycle of the 26S proteasome: From birth, through regulation and function, and onto its death. Cell Res. 2016, 26, 869–885. [Google Scholar] [CrossRef]
- Ao, Q.; Su, W.; Guo, S.; Cai, L.; Huang, L. SENP1 desensitizes hypoxic ovarian cancer cells to cisplatin by up-regulating HIF-1alpha. Sci. Rep. 2015, 5, 16396. [Google Scholar] [CrossRef]
- Zhao, X.; Su, X.; Cao, L.; Xie, T.; Chen, Q.; Li, J.; Xu, R.; Jiang, C. OTUD4: A Potential Prognosis Biomarker for Multiple Human Cancers. Cancer Manag. Res. 2020, 12, 1503–1512. [Google Scholar] [CrossRef]
- Young, M.J.; Hsu, K.C.; Lin, T.E.; Chang, W.C.; Hung, J.J. The role of ubiquitin-specific peptidases in cancer progression. J. Biomed. Sci. 2019, 26, 42. [Google Scholar] [CrossRef]
- Basters, A.; Knobeloch, K.P.; Fritz, G. USP18—A multifunctional component in the interferon response. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef]
- Jeusset, L.M.; McManus, K.J. Developing Targeted Therapies That Exploit Aberrant Histone Ubiquitination in Cancer. Cells 2019, 8, 165. [Google Scholar] [CrossRef]
- Melo-Cardenas, J.; Zhang, Y.; Zhang, D.D.; Fang, D. Ubiquitin-specific peptidase 22 functions and its involvement in disease. Oncotarget 2016, 7, 44848–44856. [Google Scholar] [CrossRef]
- Jeusset, L.M.; McManus, K.J. Ubiquitin Specific Peptidase 22 Regulates Histone H2B Mono-Ubiquitination and Exhibits Both Oncogenic and Tumor Suppressor Roles in Cancer. Cancers 2017, 9, 167. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Ramachandran, S.; Haddad, D.; Li, C.; Le, M.X.; Ling, A.K.; So, C.C.; Nepal, R.M.; Gommerman, J.L.; Yu, K.; Ketela, T.; et al. The SAGA Deubiquitination Module Promotes DNA Repair and Class Switch Recombination through ATM and DNAPK-Mediated gammaH2AX Formation. Cell Rep. 2016, 15, 1554–1565. [Google Scholar] [CrossRef]
- Li, C.; Irrazabal, T.; So, C.C.; Berru, M.; Du, L.; Lam, E.; Ling, A.K.; Gommerman, J.L.; Pan-Hammarstrom, Q.; Martin, A. The H2B deubiquitinase Usp22 promotes antibody class switch recombination by facilitating non-homologous end joining. Nat. Commun. 2018, 9, 1006. [Google Scholar] [CrossRef]
- Arlt, A.; Bauer, I.; Schafmayer, C.; Tepel, J.; Muerkoster, S.S.; Brosch, M.; Roder, C.; Kalthoff, H.; Hampe, J.; Moyer, M.P.; et al. Increased proteasome subunit protein expression and proteasome activity in colon cancer relate to an enhanced activation of nuclear factor E2-related factor 2 (Nrf2). Oncogene 2009, 28, 3983–3996. [Google Scholar] [CrossRef]
- Morozov, A.V.; Karpov, V.L. Proteasomes and Several Aspects of Their Heterogeneity Relevant to Cancer. Front. Oncol. 2019, 9, 761. [Google Scholar] [CrossRef]
- Saulle, E.; Petronelli, A.; Pasquini, L.; Petrucci, E.; Mariani, G.; Biffoni, M.; Ferretti, G.; Scambia, G.; Benedetti-Panici, P.; Cognetti, F.; et al. Proteasome inhibitors sensitize ovarian cancer cells to TRAIL induced apoptosis. Apoptosis 2007, 12, 635–655. [Google Scholar] [CrossRef]
- Mohammad, R.M.; Muqbil, I.; Lowe, L.; Yedjou, C.; Hsu, H.Y.; Lin, L.T.; Siegelin, M.D.; Fimognari, C.; Kumar, N.B.; Dou, Q.P.; et al. Broad targeting of resistance to apoptosis in cancer. Semin. Cancer Biol. 2015, 35, S78–S103. [Google Scholar] [CrossRef]
- Motosugi, R.; Murata, S. Dynamic Regulation of Proteasome Expression. Front. Mol. Biosci. 2019, 6, 30. [Google Scholar] [CrossRef] [PubMed]
- Baird, L.; Tsujita, T.; Kobayashi, E.H.; Funayama, R.; Nagashima, T.; Nakayama, K.; Yamamoto, M. A Homeostatic Shift Facilitates Endoplasmic Reticulum Proteostasis through Transcriptional Integration of Proteostatic Stress Response Pathways. Mol. Cell Biol. 2017, 37. [Google Scholar] [CrossRef]
- Metzbower, S.R.; Joo, Y.; Benavides, D.R.; Blanpied, T.A. Properties of Individual Hippocampal Synapses Influencing NMDA-Receptor Activation by Spontaneous Neurotransmission. eNeuro 2019, 6. [Google Scholar] [CrossRef]
- Koizumi, S.; Irie, T.; Hirayama, S.; Sakurai, Y.; Yashiroda, H.; Naguro, I.; Ichijo, H.; Hamazaki, J.; Murata, S. The aspartyl protease DDI2 activates Nrf1 to compensate for proteasome dysfunction. Elife 2016, 5, e18357. [Google Scholar] [CrossRef]
- Hamazaki, J.; Murata, S. ER-Resident Transcription Factor Nrf1 Regulates Proteasome Expression and Beyond. Int. J. Mol. Sci. 2020, 21, 3683. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, Y.; Morita, T.; Kim, M.; Iemura, S.; Natsume, T.; Yamamoto, M.; Kobayashi, A. Dual regulation of the transcriptional activity of Nrf1 by beta-TrCP- and Hrd1-dependent degradation mechanisms. Mol. Cell Biol. 2011, 31, 4500–4512. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Overrepresented KEGG Pathway * | Number of Genes | p-Value for Pathway | Genes Coding for Components of the Ubiquitin Proteasome Pathway | |
|---|---|---|---|---|
| Cell cycle | 49 | 1.1 × 10−11 | ANAPC4 3, CDC20 3, SKP2 3 | LMP > INV INV > LMP INV > LMP |
| DNA replication | 19 | 4.1 × 10−8 | ||
| p53 signaling | 28 | 1.6 × 10−7 | DDB2 3, MDM2 2, RFWD2 3 | LMP > INV LMP > INV INV > LMP |
| Huntington’s disease | 54 | 3.06 × 10−6 | DNAI13, DNAI2 3, | LMP > INV LMP > INV |
| Fanconi anemia | 45 | 4.6 × 10−4 | USP1 4, UBE2T 1 | INV > LMP INV > LMP |
| Gene | LMP (Means ± se) | HGSOC (Means ± se) | F | p |
|---|---|---|---|---|
| N = 32 | N = 58 | |||
| UBE2T | 57.42 ± 5.11 | 278.59 ± 19.41 | 69.806 | 8.70 × 10−13 |
| UBE2C | 101.08 ± 6.31 | 560.62 ± 57.31 | 35.172 | 5.81 × 10−8 |
| UBE2W | 133.59 ± 4.89 | 226.82 ± 9.81 | 46.139 | 1.25 × 10−9 |
| UBE2L6 | 461.86 ± 23.54 | 967.39 ± 60.35 | 36.832 | 3.17 × 10−8 |
| UBE2K | 318.68 ± 7.98 | 498.06 ± 22.97 | 32.321 | 1.67 × 10−7 |
| UBE2S | 117.97 ± 11.14 | 345.04 ± 34.57 | 22.971 | 6.63 × 10−6 |
| FA Component Gene | LMP Means ± se | HGSOC (INV) Means ± se | F | p |
|---|---|---|---|---|
| UBE2T | 57.42 ± 5.11 | 278.59 ± 19.41 | 69.806 | 8.70 × 10−13 |
| RAD51 * | 31.12 ± 2.36 | 93.94 ± 6.44 | 50.158 | 3.32 × 10−10 |
| BRCA2 * | 18.12 ± 1.32 | 47.57 ± 3.20 | 44.357 | 2.28 × 10−9 |
| FANCD2 | 55.21 ± 3.07 | 153.36 ± 11.46 | 39.392 | 1.27 × 10−8 |
| FANCA | 29.68 ± 2.13 | 64.47 ± 4.31 | 33.334 | 1.15 × 10−7 |
| FANCG | 125.63 ± 4.13 | 189.16 ± 7.99 | 32.145 | 1.78 × 10−7 |
| FANCI | 62.02 ± 4.56 | 185.60 ± 16.63 | 29.659 | 4.62 × 10−7 |
| FANCF | 211.81 ± 8.02 | 150.65 ± 7.13 | 29.293 | 5.31 × 10−7 |
| BRIP1 * | 36.78 ± 2.53 | 105.43 ± 11.57 | 19.041 | 3.47 × 10−5 |
| FANCB | 3.04 ± 0.60 | 8.84 ± 1.04 | 15.45 | 1.68 × 10−4 |
| MAD2L2 | 104.18 ± 5.53 | 152.97 ± 8.80 | 15.087 | 1.98 × 10−4 |
| FANCC | 45.02 ± 1.91 | 57.64 ± 2.59 | 11.231 | 1.19 × 10−3 |
| RFWD3 | 46.58 ± 2.83 | 64.12 ± 3.80 | 10.013 | 2.14 × 10−3 |
| XRCC2 * | 97.42 ± 3.97 | 114.13 ± 4.22 | 6.801 | 1.10 × 10−2 |
| PALB2 | 146.16 ± 5.05 | 167.84 ± 5.99 | 5.932 | 1.70 × 10−2 |
| BRCA1 * | 51.21 ± 3.50 | 75.88 ± 7.46 | 5.62 | 2.00 × 10−2 |
| Prometaphase | Metaphase | Anaphase–Telophase | G1 Phase |
|---|---|---|---|
| NEK2A | CYCLIN B | AURKA | CDC25A |
| CYCLIN A2 | SECURIN | AURKB | SKP2 |
| CDC20 | GEMININ | ||
| PLK1 | CDC6 | ||
| TPX2 | UBE2C | ||
| HEC1 | TK1 | ||
| BARD1 | RRM2 | ||
| HMMR | FOXM1 | ||
| HURP | ORC1 | ||
| NUSAP | ID2 | ||
| GEMININ | CDCA3 | ||
| ANLN | CCNB1 | ||
| PRC1 | CLSPN | ||
| SGO1 | EMI1 |
| LMP (Means ± S.E.) | HGSOC (INV) (Means ± S.E.) | F | p | |
|---|---|---|---|---|
| UCHL5 | 132.01 ± 4.26 | 213.58 ± 7.37 | 61.097 | 1.10 × 10−11 |
| USP40 | 236.20 ± 10.03 | 146.94 ± 6.45 | 60.862 | 1.18 × 10−11 |
| EIF3F | 3205.01 ± 113.3 | 2174.67 ± 89.43 | 49.178 | 4.57 × 10−10 |
| PSMD14 | 650.33 ± 19.43 | 981.20 ± 35.20 | 44.434 | 2.22 × 10−9 |
| SENP2 | 134.95 ± 3.55 | 216.18 ± 9.20 | 40.930 | 7.40 × 10−9 |
| USP53 | 603.97 ± 60.66 | 244.64 ± 29.83 | 35.550 | 5.06 × 10−8 |
| COPS5 | 578.00 ± 22.98 | 793.79 ± 29.47 | 24.893 | 3.03 × 10−8 |
| UFSP2 | 275.43 ± 9.83 | 203.58 ± 8.32 | 28.872 | 6.26 × 10−7 |
| OTUD4 | 476.54 ± 11.25 | 373.88 ± 15.00 | 22.001 | 9.91 × 10−8 |
| COX8A | 1488.84 ± 52.18 | 2044 ± 87.17 | 20.127 | 2.18 × 10−5 |
| USP1 | 699.23 ± 20.99 | 1102.74 ± 62.22 | 22.325 | 8.66 × 10−6 |
| USP18 | 58.20 ± 4.38 | 174.79 ± 17.73 | 23.319 | 5.75 × 10−6 |
| USP43 | 42.40 ± 2.62 | 28.57 ± 2.62 | 30.408 | 3.46 × 10−7 |
| USP22 | 731.28 ± 24.50 | 558.52 ± 20.31 | 27.635 | 1.01 × 10−6 |
| USP39 | 106.97 ± 3.81 | 146.08 ± 5.86 | 21.693 | 1.13 × 10−5 |
| FAM63A | 374.76 ± 19.69 | 287.18 ± 12.70 | 15.171 | 1.91 × 10−4 |
| USP14 | 560.93 ± 13.07 | 808.09 ± 38.81 | 21.528 | 1.21 × 10−5 |
| USP47 | 381.45 ± 12.84 | 314.94 ± 11.83 | 12.828 | 5.59 × 10−4 |
| Proteasome Gene | Chromosome Location | LMP (Means ± S.E.) | HGSOC (INV) (Means ± S.E.) | F Value | p Value |
|---|---|---|---|---|---|
| PSMD2 | 3 | 554.91 ± 17.11 | 993.61 ± 35.51 | 77.929 | 9.29 × 10−14 |
| PSME4 | 2 | 233.77 ± 10.04 | 409.82 ± 17.64 | 49.812 | 3.71 × 10−10 |
| PSMD1 | 2 | 385.60 ± 18.30 | 623.56 ± 25.11 | 45.516 | 4.27 × 10−9 |
| PSMD14 | 2 | 650.33 ± 19.43 | 981.20 ± 35.20 | 44.434 | 2.22 × 10−9 |
| PSMC2 | 7 | 954.96 ± 34.12 | 1502.52 ± 59.53 | 42.276 | 4.64 × 10−9 |
| PSMA7 | 20 | 833.01 ± 36.19 | 1403.95 ± 64.94 | 38.811 | 1.56 × 10−8 |
| PSMB2 | 1 | 810.30 ± 24.63 | 1566.96 ± 106.75 | 27.144 | 1.23 × 10−6 |
| PSMD12 | 17 | 275.32 ± 6.80 | 470.16 ± 32.54 | 19.430 | 2.94 × 10−5 |
| PSMB9 | 6 | 198.19 ± 17.51 | 525.31 ± 54.63 | 19.095 | 3.39 × 10−5 |
| PSMB3 | 17 | 924.33 ± 38.58 | 1252.30 ± 57.68 | 15.648 | 1.54 × 10−4 |
| PSMB4 | 1 | 1769.29 ± 34.96 | 2274.05 ± 93.15 | 15.465 | 1.67 × 10−4 |
| PSMA3 | 14 | 931.61 ± 29.36 | 1177.96 ± 43.63 | 15.406 | 1.72 × 10−4 |
| PSMA2 | 7 | 1118.16 ± 44.05 | 1396.67 ± 46.99 | 15.283 | 1.82 × 10−4 |
| PSMD8 | 19 | 625.55 ± 25.73 | 965.88 ± 63.49 | 15.033 | 2.03 × 10−4 |
| PSMC4 | 19 | 246.34 ± 8.33 | 432.46 ± 36.56 | 14.008 | 3.24 × 10−4 |
| PSMD4 | 1 | 887.14 ± 24.65 | 1175.07 ± 56.63 | 13.424 | 4.24 × 10−4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vriend, J.; Nachtigal, M.W. Ubiquitin Proteasome Pathway Transcriptome in Epithelial Ovarian Cancer. Cancers 2021, 13, 2659. https://doi.org/10.3390/cancers13112659
Vriend J, Nachtigal MW. Ubiquitin Proteasome Pathway Transcriptome in Epithelial Ovarian Cancer. Cancers. 2021; 13(11):2659. https://doi.org/10.3390/cancers13112659
Chicago/Turabian StyleVriend, Jerry, and Mark W. Nachtigal. 2021. "Ubiquitin Proteasome Pathway Transcriptome in Epithelial Ovarian Cancer" Cancers 13, no. 11: 2659. https://doi.org/10.3390/cancers13112659
APA StyleVriend, J., & Nachtigal, M. W. (2021). Ubiquitin Proteasome Pathway Transcriptome in Epithelial Ovarian Cancer. Cancers, 13(11), 2659. https://doi.org/10.3390/cancers13112659