
AR Splicing Variants and Resistance to AR Targeting Agents



Abstract
Simple Summary
Abstract
1. Introduction
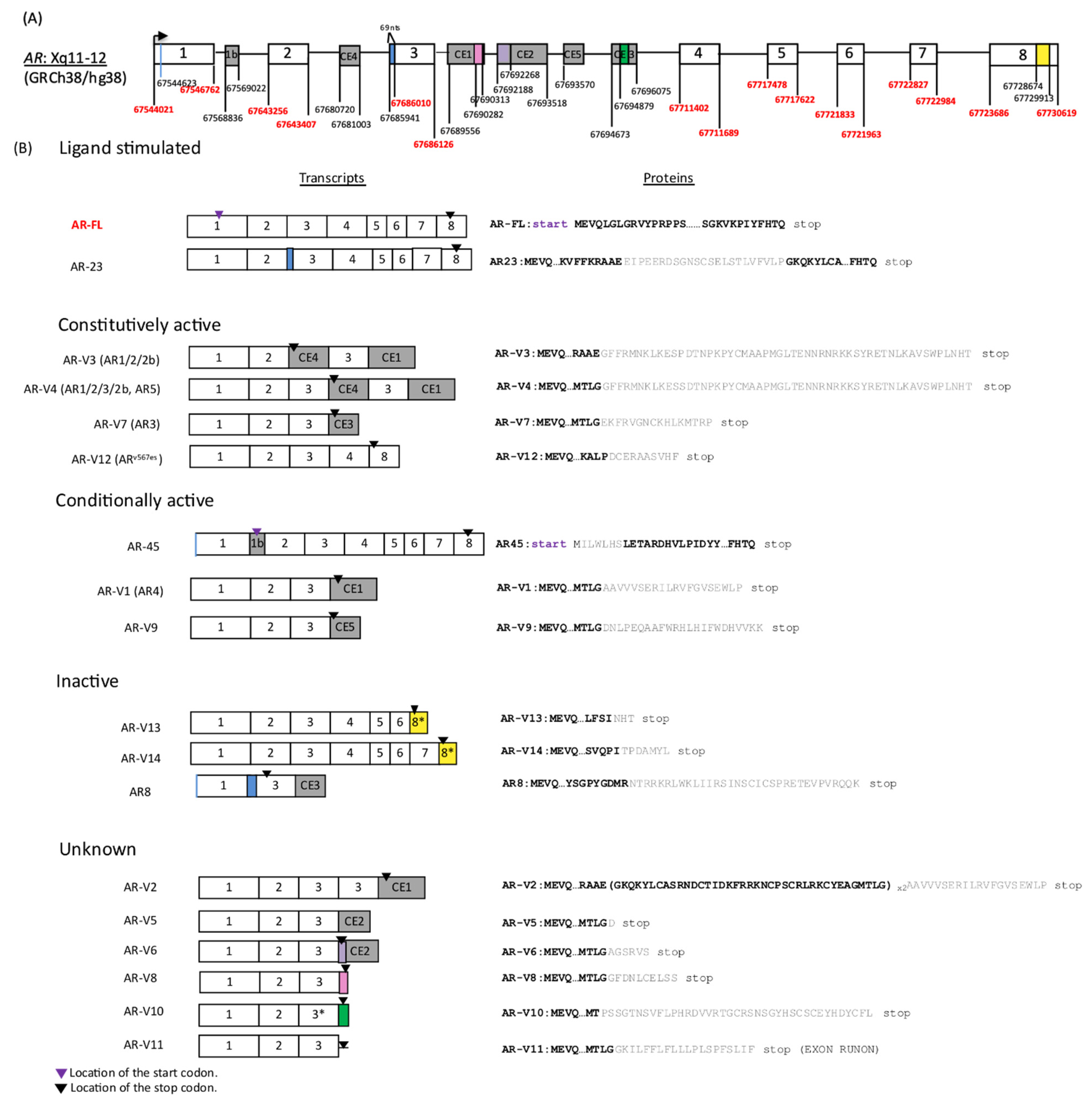
2. Structure of AR-Vs and Mechanisms Underlying Genesis of AR-Vs
3. AR-V Regulation, Dimerization and Transcriptional Activity
4. Detection of AR-Vs in Human Samples and Clinical Implications
5. AR-Vs as Therapeutic Targets
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hu, R.; Dunn, T.A.; Wei, S.; Isharwal, S.; Veltri, R.W.; Humphreys, E.; Han, M.; Partin, A.W.; Vessella, R.L.; Isaacs, W.B.; et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009, 69, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Dehm, S.M.; Schmidt, L.J.; Heemers, H.V.; Vessella, R.L.; Tindall, D.J. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008, 68, 5469–5477. [Google Scholar] [CrossRef] [PubMed]
- Gelmann, E.P. Molecular biology of the androgen receptor. J. Clin. Oncol. 2002, 20, 3001–3015. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Luo, J. Decoding the androgen receptor splice variants. Transl. Androl. Urol. 2013, 2, 178. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.C.; Li, Y.; Dehm, S.M. Androgen receptor splice variants activate androgen receptor target genes and support aberrant prostate cancer cell growth independent of canonical androgen receptor nuclear localization signal. J. Biol. Chem. 2012, 287, 19736–19749. [Google Scholar] [CrossRef] [PubMed]
- Abeshouse, A.; Ahn, J.; Akbani, R.; Ally, A.; Amin, S.; Andry, C.D.; Annala, M.; Aprikian, A.; Armenia, J.; Arora, A.; et al. The molecular taxonomy of primary prostate cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Henzler, C.; Li, Y.; Yang, R.; McBride, T.; Ho, Y.; Sprenger, C.; Liu, G.; Coleman, I.; Lakely, B.; Li, R.; et al. Truncation and constitutive activation of the androgen receptor by diverse genomic rearrangements in prostate cancer. Nat. Commun. 2016, 7, 13668. [Google Scholar] [CrossRef]
- Zhu, Y.; Luo, J. Regulation of androgen receptor variants in prostate cancer. Asian J. Urol. 2020, 7, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Chen, S.; Sowalsky, A.G.; Voznesensky, O.S.; Mostaghel, E.A.; Nelson, P.S.; Cai, C.; Balk, S.P. Rapid induction of androgen receptor splice variants by androgen deprivation in prostate cancer. Clin. Cancer Res. 2014, 20, 1590–1600. [Google Scholar] [CrossRef]
- Liu, L.; Xie, N.; Sun, S.; Plymate, S.; Mostaghel, E.; Dong, X. Mechanisms of the androgen receptor splicing in prostate cancer cells. Oncogene 2014, 33, 3140. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Lu, C.; Mostaghel, E.A.; Yegnasubramanian, S.; Gurel, M.; Tannahill, C.; Edwards, J.; Isaacs, W.B.; Nelson, P.S.; Bluemn, E.; et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012, 72, 3457–3462. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Brown, L.C.; Antonarakis, E.S.; Armstrong, A.J.; Luo, J. Androgen receptor variant-driven prostate cancer II: Advances in laboratory investigations. Prostate Cancer Prostatic Dis. 2020, 23, 381–397. [Google Scholar] [CrossRef] [PubMed]
- Van Bokhoven, A.; Varella-Garcia, M.; Korch, C.; Johannes, W.U.; Smith, E.E.; Miller, H.L.; Nordeen, S.K.; Miller, G.J.; Lucia, M.S. Molecular characterization of human prostate carcinoma cell lines. Prostate 2003, 57, 205–225. [Google Scholar] [CrossRef]
- Li, Y.; Hwang, T.H.; Oseth, L.; Hauge, A.; Vessella, R.L.; Schmechel, S.C.; Hirsch, B.; Beckman, K.B.; Silverstein, K.A.; Dehm, S.M.; et al. AR intragenic deletions linked to androgen receptor splice variant expression and activity in models of prostate cancer progression. Oncogene 2012, 31, 4759. [Google Scholar] [CrossRef]
- Li, Y.; Alsagabi, M.; Fan, D.; Bova, G.S.; Tewfik, A.H.; Dehm, S.M. Intragenic rearrangement and altered RNA splicing of the androgen receptor in a cell-based model of prostate cancer progression. Cancer Res. 2011, 71, 2108–2117. [Google Scholar] [CrossRef]
- Nyquist, M.D.; Li, Y.; Hwang, T.H.; Manlove, L.S.; Vessella, R.L.; Silverstein, K.A.; Voytas, D.F.; Dehm, S.M. TALEN-Engineered AR gene rearrangements reveal endocrine uncoupling of androgen receptor in prostate cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 17492–17497. [Google Scholar] [CrossRef]
- Stockley, J.; Markert, E.; Zhou, Y.; Robson, C.N.; Elliott, D.J.; Lindberg, J.; Leung, H.Y.; Rajan, P. The RNA-binding protein Sam68 regulates expression and transcription function of the androgen receptor splice variant AR-V7. Sci. Rep. 2015, 5, 1–13. [Google Scholar] [CrossRef]
- Takayama, K.-I.; Suzuki, T.; Fujimura, T.; Yamada, Y.; Takahashi, S.; Homma, Y.; Suzuki, Y.; Inoue, S. Dysregulation of spliceosome gene expression in advanced prostate cancer by RNA-binding protein PSF. Proc. Natl. Acad. Sci. USA 2017, 114, 10461–10466. [Google Scholar] [CrossRef]
- Kawamura, N.; Nimura, K.; Saga, K.; Ishibashi, A.; Kitamura, K.; Nagano, H.; Yoshikawa, Y.; Ishida, K.; Nonomura, N.; Arisawa, M.; et al. SF3B2-mediated RNA splicing drives human prostate cancer progression. Cancer Res. 2019, 79, 5204–5217. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, N.; Huang, J.; Ho, T.-T.; Zhu, Z.; Qiu, Z.; Zhou, X.; Bai, C.; Wu, F.; Xu, M.; et al. Regulation of androgen receptor splice variant AR3 by PCGEM1. Oncotarget 2016, 7, 15481. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.-I.; Fujimura, T.; Suzuki, Y.; Inoue, S. Identification of long non-coding RNAs in advanced prostate cancer associated with androgen receptor splicing factors. Commun. Biol. 2020, 3, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, C.E.; Sulpice, E.; Combe, S.; Shibakawa, A.; Leach, D.A.; Hamilton, M.P.; Chrysostomou, S.L.; Sharp, A.; Welti, J.; Yuan, W.; et al. Androgen receptor-modulatory microRNAs provide insight into therapy resistance and therapeutic targets in advanced prostate cancer. Oncogene 2019, 38, 5700–5724. [Google Scholar] [CrossRef]
- Yang, Y.; Jia, D.; Kim, H.; Abd Elmageed, Z.Y.; Datta, A.; Davis, R.; Srivastav, S.; Moroz, K.; Crawford, B.E.; Moparty, K.; et al. Dysregulation of miR-212 promotes castration resistance through hnRNPH1-mediated regulation of AR and AR-V7: Implications for racial disparity of prostate cancer. Clin. Cancer Res. 2016, 22, 1744–1756. [Google Scholar] [CrossRef]
- Cai, C.; He, H.H.; Chen, S.; Coleman, I.; Wang, H.; Fang, Z.; Chen, S.; Nelson, P.S.; Liu, X.S.; Brown, M.; et al. Androgen receptor gene expression in prostate cancer is directly suppressed by the androgen receptor through recruitment of lysine-specific demethylase 1. Cancer Cell 2011, 20, 457–471. [Google Scholar] [CrossRef]
- Centenera, M.M.; Harris, J.M.; Tilley, W.D.; Butler, L.M. Minireview: The contribution of different androgen receptor domains to receptor dimerization and signaling. Mol. Endocrinol. 2008, 22, 2373–2382. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Zhan, Y.; Qi, Y.; Cao, B.; Bai, S.; Xu, W.; Gambhir, S.S.; Lee, P.; Sartor, O.; Flemington, E.K.; et al. Androgen receptor splice variants dimerize to transactivate target genes. Cancer Res. 2015, 75, 3663–3671. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Sprenger, C.C.; Vessella, R.L.; Haugk, K.; Soriano, K.; Mostaghel, E.A.; Page, S.T.; Coleman, I.M.; Nguyen, H.M.; Sun, H.; et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J. Clin. Investig. 2010, 120, 2715–2730. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Qi, Y.; Zhang, G.; Xu, D.; Zhan, Y.; Alvarez, X.; Guo, Z.; Fu, X.; Plymate, S.R.; Sartor, O.; et al. Androgen receptor splice variants activating the full-length receptor in mediating resistance to androgen-directed therapy. Oncotarget 2014, 5, 1646. [Google Scholar] [CrossRef]
- Zhan, Y.; Zhang, G.; Wang, X.; Qi, Y.; Bai, S.; Li, D.; Ma, T.; Sartor, O.; Flemington, E.K.; Zhang, H.; et al. Interplay between cytoplasmic and nuclear androgen receptor splice variants mediates castration resistance. Mol. Cancer Res. 2017, 15, 59–68. [Google Scholar] [CrossRef]
- Hörnberg, E.; Ylitalo, E.B.; Crnalic, S.; Antti, H.; Stattin, P.; Widmark, A.; Bergh, A.; Wikström, P. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PLoS ONE 2011, 6, e19059. [Google Scholar] [CrossRef]
- Li, Y.; Chan, S.C.; Brand, L.J.; Hwang, T.H.; Silverstein, K.A.; Dehm, S.M. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013, 73, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wu, D.; Thomas-Ahner, J.M.; Lu, C.; Zhao, P.; Zhang, Q.; Geraghty, C.; Yan, P.S.; Hankey, W.; Sunkel, B.; et al. Diverse AR-V7 cistromes in castration-resistant prostate cancer are governed by HoxB13. Proc. Natl. Acad. Sci. USA 2018, 115, 6810–6815. [Google Scholar] [CrossRef] [PubMed]
- Ballman, K.V. Biomarker: Predictive or prognostic? J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 3968–3971. [Google Scholar] [CrossRef] [PubMed]
- Terada, N.; Akamatsu, S.; Kobayashi, T.; Inoue, T.; Ogawa, O.; Antonarakis, E.S. Prognostic and predictive biomarkers in prostate cancer: Latest evidence and clinical implications. Ther. Adv. Med. Oncol. 2017, 9, 565–573. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Luber, B.; Wang, H.; Chen, Y.; Zhu, Y.; Silberstein, J.L.; Taylor, M.N.; Maughan, B.L.; Denmeade, S.R.; et al. Clinical significance of androgen receptor splice variant-7 mRNA detection in circulating tumor cells of men with metastatic castration-resistant prostate cancer treated with first-and second-line abiraterone and enzalutamide. J. Clin. Oncol. 2017, 35, 2149. [Google Scholar] [CrossRef] [PubMed]
- Sharp, A.; Welti, J.C.; Lambros, M.B.; Dolling, D.; Rodrigues, D.N.; Pope, L.; Aversa, C.; Figueiredo, I.; Fraser, J.; Ahmad, Z.; et al. Clinical utility of circulating tumour cell androgen receptor splice variant-7 status in metastatic castration-resistant prostate cancer. Eur. Urol. 2019, 76, 676–685. [Google Scholar] [CrossRef]
- Sauerbrei, W.; Taube, S.E.; McShane, L.M.; Cavenagh, M.M.; Altman, D.G. Reporting recommendations for tumor marker prognostic studies (REMARK): An abridged explanation and elaboration. JNCI J. Natl. Cancer Inst. 2018, 110, 803–811. [Google Scholar] [CrossRef]
- Armstrong, A.J.; Halabi, S.; Luo, J.; Nanus, D.M.; Giannakakou, P.; Szmulewitz, R.Z.; Danila, D.C.; Healy, P.; Anand, M.; Rothwell, C.J.; et al. Prospective multicenter validation of androgen receptor splice variant 7 and hormone therapy resistance in high-risk castration-resistant prostate cancer: The PROPHECY study. J. Clin. Oncol. 2019, 37, 1120. [Google Scholar] [CrossRef]
- De Laere, B.; van Dam, P.-J.; Whitington, T.; Mayrhofer, M.; Diaz, E.H.; Van den Eynden, G.; Vandebroek, J.; Del-Favero, J.; Van Laere, S.; Dirix, L.; et al. Comprehensive profiling of the androgen receptor in liquid biopsies from castration-resistant prostate cancer reveals novel intra-AR structural variation and splice variant expression patterns. Eur. Urol. 2017, 72, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Fettke, H.; Kwan, E.M.; Docanto, M.M.; Bukczynska, P.; Ng, N.; Graham, L.-J.K.; Mahon, K.; Hauser, C.; Tan, W.; Wang, X.H.; et al. Combined cell-free DNA and RNA profiling of the androgen receptor: Clinical utility of a novel multianalyte liquid biopsy assay for metastatic prostate cancer. Eur. Urol. 2020, 78, 173–180. [Google Scholar] [CrossRef]
- Wyatt, A.W.; Azad, A.A.; Volik, S.V.; Annala, M.; Beja, K.; McConeghy, B.; Haegert, A.; Warner, E.W.; Mo, F.; Brahmbhatt, S.; et al. Genomic alterations in cell-free DNA and enzalutamide resistance in castration-resistant prostate cancer. JAMA Oncol. 2016, 2, 1598–1606. [Google Scholar] [CrossRef] [PubMed]
- Del Re, M.; Conteduca, V.; Crucitta, S.; Gurioli, G.; Casadei, C.; Restante, G.; Schepisi, G.; Lolli, C.; Cucchiara, F.; Danesi, R.; et al. Androgen receptor gain in circulating free DNA and splicing variant 7 in exosomes predict clinical outcome in CRPC patients treated with abiraterone and enzalutamide. Prostate Cancer Prostatic Dis. 2021, 1–8. [Google Scholar] [CrossRef]
- Lambros, M.B.; Seed, G.; Sumanasuriya, S.; Gil, V.; Crespo, M.; Fontes, M.; Chandler, R.; Mehra, N.; Fowler, G.; Ebbs, B.; et al. Single-cell analyses of prostate cancer liquid biopsies acquired by apheresis. Clin. Cancer Res. 2018, 24, 5635–5644. [Google Scholar] [CrossRef]
- Miyamoto, D.T.; Zheng, Y.; Wittner, B.S.; Lee, R.J.; Zhu, H.; Broderick, K.T.; Desai, R.; Fox, D.B.; Brannigan, B.W.; Trautwein, J.; et al. RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science 2015, 349, 1351–1356. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Luber, B.; Wang, H.; Chen, Y.; Nakazawa, M.; Nadal, R.; Paller, C.J.; Denmeade, S.R.; Carducci, M.A.; et al. Androgen receptor splice variant 7 and efficacy of taxane chemotherapy in patients with metastatic castration-resistant prostate cancer. JAMA Oncol. 2015, 1, 582–591. [Google Scholar] [CrossRef]
- Taplin, M.-E.; Antonarakis, E.S.; Ferrante, K.J.; Horgan, K.; Blumenstein, B.; Saad, F.; Luo, J.; de Bono, J.S. Androgen receptor modulation optimized for response—Splice variant: A phase 3, randomized trial of galeterone versus enzalutamide in androgen receptor splice variant-7—Expressing metastatic castration-resistant prostate cancer. Eur. Urol. 2019, 76, 843–851. [Google Scholar] [CrossRef]
- Armstrong, A.J.; Luo, J.; Nanus, D.M.; Giannakakou, P.; Szmulewitz, R.Z.; Danila, D.C.; Healy, P.; Anand, M.; Berry, W.R.; Zhang, T.; et al. Prospective multicenter study of circulating tumor cell AR-V7 and taxane versus hormonal treatment outcomes in metastatic castration-resistant prostate cancer. JCO Precis. Oncol. 2020, 4, 1285–1301. [Google Scholar] [CrossRef]
- Kohli, M.; Ho, Y.; Hillman, D.W.; Van Etten, J.L.; Henzler, C.; Yang, R.; Sperger, J.M.; Li, Y.; Tseng, E.; Hon, T.; et al. Androgen receptor variant AR-V9 is coexpressed with AR-V7 in prostate cancer metastases and predicts abiraterone resistance. Clin. Cancer Res. 2017, 23, 4704–4715. [Google Scholar] [CrossRef]
- Boudadi, K.; Suzman, D.L.; Anagnostou, V.; Fu, W.; Luber, B.; Wang, H.; Niknafs, N.; White, J.R.; Silberstein, J.L.; Sullivan, R.; et al. Ipilimumab plus nivolumab and DNA-repair defects in AR-V7-expressing metastatic prostate cancer. Oncotarget 2018, 9, 28561. [Google Scholar] [CrossRef] [PubMed]
- Shenderov, E.; Boudadi, K.; Fu, W.; Wang, H.; Sullivan, R.; Jordan, A.; Dowling, D.; Harb, R.; Schonhoft, J.; Jendrisak, A.; et al. Nivolumab plus ipilimumab, with or without enzalutamide, in AR-V7-expressing metastatic castration-resistant prostate cancer: A phase-2 nonrandomized clinical trial. Prostate 2021, 81, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Kessel, K.; Seifert, R.; Weckesser, M.; Roll, W.; Humberg, V.; Schlack, K.; Bögemann, M.; Bernemann, C.; Rahbar, K. Molecular analysis of circulating tumor cells of metastatic castration-resistant prostate cancer patients receiving 177Lu-PSMA-617 radioligand therapy. Theranostics 2020, 10, 7645. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Lu, D.; Schreiber, N.A.; Louw, J.; Graf, R.P.; Vargas, H.A.; Johnson, A.; Jendrisak, A.; Bambury, R.; Danila, D.; et al. Association of AR-V7 on circulating tumor cells as a treatment-specific biomarker with outcomes and survival in castration-resistant prostate cancer. JAMA Oncol. 2016, 2, 1441–1449. [Google Scholar] [CrossRef]
- Scher, H.I.; Graf, R.P.; Schreiber, N.A.; Jayaram, A.; Winquist, E.; McLaughlin, B.; Lu, D.; Fleisher, M.; Orr, S.; Lowes, L.; et al. Assessment of the validity of nuclear-localized androgen receptor splice variant 7 in circulating tumor cells as a predictive biomarker for castration-resistant prostate cancer. JAMA Oncol. 2018, 4, 1179–1186. [Google Scholar] [CrossRef]
- Del Re, M.; Biasco, E.; Crucitta, S.; Derosa, L.; Rofi, E.; Orlandini, C.; Miccoli, M.; Galli, L.; Falcone, A.; Jenster, G.W.; et al. The detection of androgen receptor splice variant 7 in plasma-derived exosomal RNA strongly predicts resistance to hormonal therapy in metastatic prostate cancer patients. Eur. Urol. 2017, 71, 680–687. [Google Scholar] [CrossRef]
- Chen, X.; Bernemann, C.; Tolkach, Y.; Heller, M.; Nientiedt, C.; Falkenstein, M.; Herpel, E.; Jenzer, M.; Grüllich, C.; Jäger, D.; et al. Overexpression of nuclear AR-V7 protein in primary prostate cancer is an independent negative prognostic marker in men with high-risk disease receiving adjuvant therapy. Urol. Oncol. Semin. Orig. Investig. 2018, 36, 161.e119–161.e130. [Google Scholar] [CrossRef]
- Zhu, Y.; Sharp, A.; Anderson, C.M.; Silberstein, J.L.; Taylor, M.; Lu, C.; Zhao, P.; De Marzo, A.M.; Antonarakis, E.S.; Wang, M.; et al. Novel junction-specific and quantifiable in situ detection of AR-V7 and its clinical correlates in metastatic castration-resistant prostate cancer. Eur. Urol. 2018, 73, 727–735. [Google Scholar] [CrossRef]
- Efstathiou, E.; Titus, M.; Wen, S.; Hoang, A.; Karlou, M.; Ashe, R.; Tu, S.M.; Aparicio, A.; Troncoso, P.; Mohler, J.; et al. Molecular characterization of enzalutamide-treated bone metastatic castration-resistant prostate cancer. Eur. Urol. 2015, 67, 53–60. [Google Scholar] [CrossRef]
- Markowski, M.C.; Wang, H.; Sullivan, R.; Rifkind, I.; Sinibaldi, V.; Schweizer, M.T.; Teply, B.A.; Ngomba, N.; Fu, W.; Carducci, M.A.; et al. A multicohort open-label phase II trial of bipolar androgen therapy in men with metastatic castration-resistant prostate cancer (RESTORE): A comparison of post-abiraterone versus post-enzalutamide cohorts. Eur. Urol. 2020, 79, 692–699. [Google Scholar] [CrossRef]
- Denmeade, S.R.; Wang, H.; Agarwal, N.; Smith, D.C.; Schweizer, M.T.; Stein, M.N.; Assikis, V.; Twardowski, P.W.; Flaig, T.W.; Szmulewitz, R.Z.; et al. TRANSFORMER: A randomized phase II study comparing bipolar androgen therapy versus enzalutamide in asymptomatic men with castration-resistant metastatic prostate cancer. J. Clin. Oncol. 2021, 39, 1371–1382. [Google Scholar] [CrossRef] [PubMed]
- Kanayama, M.; Luo, J. Predictive biomarkers in prostate cancer: Is it time to go “All In” on liquid biopsies? Eur. Urol. 2020, 78, 181–183. [Google Scholar] [CrossRef] [PubMed]
- Sharp, A.; Coleman, I.; Yuan, W.; Sprenger, C.; Dolling, D.; Rodrigues, D.N.; Russo, J.W.; Figueiredo, I.; Bertan, C.; Seed, G.; et al. Androgen receptor splice variant-7 expression emerges with castration resistance in prostate cancer. J. Clin. Investig. 2019, 129, 192–208. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, M.T.; Haugk, K.; McKiernan, J.S.; Gulati, R.; Cheng, H.H.; Maes, J.L.; Dumpit, R.F.; Nelson, P.S.; Montgomery, B.; McCune, J.S.; et al. A phase I study of niclosamide in combination with enzalutamide in men with castration-resistant prostate cancer. PLoS ONE 2018, 13, e0198389. [Google Scholar] [CrossRef]
- Parikh, M.; Liu, C.; Wu, C.-Y.; Evans, C.P.; Dall’Era, M.; Robles, D.; Lara, P.N.; Agarwal, N.; Gao, A.C.; Pan, C.-X.; et al. Phase Ib trial of reformulated niclosamide with abiraterone/prednisone in men with castration-resistant prostate cancer. Sci. Rep. 2021, 11, 1–7. [Google Scholar] [CrossRef]
- Le Moigne, R.; Pearson, P.; Lauriault, V.; Chi, K.; Ianotti, N.; Pachynski, R.; Vogelzang, N.; Hong, N.H.; Virsik, P.; Zhou, H.-J.; et al. Pre-Clinical and clinical pharmacology of EPI-7386, an androgen receptor N-terminal domain inhibitor for castration-resistant prostate cancer. J. Clin. Oncol. 2021, 39, 119. [Google Scholar] [CrossRef]
- Aggarwal, R.R.; Schweizer, M.T.; Nanus, D.M.; Pantuck, A.J.; Heath, E.I.; Campeau, E.; Attwell, S.; Norek, K.; Snyder, M.; Bauman, L.; et al. A phase Ib/IIa study of the pan-BET inhibitor ZEN-3694 in combination with enzalutamide in patients with metastatic castration-resistant prostate cancer. Clin. Cancer Res. 2020, 26, 5338–5347. [Google Scholar] [CrossRef]
- Welti, J.; Sharp, A.; Brooks, N.; Yuan, W.; McNair, C.; Chand, S.N.; Pal, A.; Figueiredo, I.; Riisnaes, R.; Gurel, B.; et al. Targeting p300/CBP axis in lethal prostate cancer. Cancer Discov. 2021. [Google Scholar] [CrossRef]
- Sharma, P.; Pachynski, R.K.; Narayan, V.; Fléchon, A.; Gravis, G.; Galsky, M.D.; Mahammedi, H.; Patnaik, A.; Subudhi, S.K.; Ciprotti, M.; et al. Nivolumab plus ipilimumab for metastatic castration-resistant prostate cancer: Preliminary analysis of patients in the CheckMate 650 trial. Cancer Cell 2020, 38, 489–499. [Google Scholar] [CrossRef]
- Liu, C.; Lou, W.; Zhu, Y.; Nadiminty, N.; Schwartz, C.T.; Evans, C.P.; Gao, A.C. Niclosamide inhibits androgen receptor variants expression and overcomes enzalutamide resistance in castration-resistant prostate cancer. Clin. Cancer Res. 2014, 20, 3198–3210. [Google Scholar] [CrossRef]
- Andersen, R.J.; Mawji, N.R.; Wang, J.; Wang, G.; Haile, S.; Myung, J.-K.; Watt, K.; Tam, T.; Yang, Y.C.; Bañuelos, C.A.; et al. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell 2010, 17, 535–546. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Chandhasin, C.; Osbourne, E.; Luo, J.; Sadar, M.D.; Perabo, F. Targeting the N-terminal domain of the androgen receptor: A new approach for the treatment of advanced prostate cancer. Oncologist 2016, 21, 1427. [Google Scholar] [CrossRef]
- Myung, J.-K.; Banuelos, C.A.; Fernandez, J.G.; Mawji, N.R.; Wang, J.; Tien, A.H.; Yang, Y.C.; Tavakoli, I.; Haile, S.; Watt, K.; et al. An androgen receptor N-terminal domain antagonist for treating prostate cancer. J. Clin. Investig. 2013, 123, 2948–2960. [Google Scholar] [CrossRef] [PubMed]
- Asangani, I.A.; Dommeti, V.L.; Wang, X.; Malik, R.; Cieslik, M.; Yang, R.; Escara-Wilke, J.; Wilder-Romans, K.; Dhanireddy, S.; Engelke, C.; et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 2014, 510, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Ameratunga, M.; Braña, I.; Bono, P.; Postel-Vinay, S.; Plummer, R.; Aspegren, J.; Korjamo, T.; Snapir, A.; de Bono, J.S. First-in-human Phase 1 open label study of the BET inhibitor ODM-207 in patients with selected solid tumours. Br. J. Cancer 2020, 123, 1730–1736. [Google Scholar] [CrossRef] [PubMed]
- Faivre, E.J.; Wilcox, D.; Lin, X.; Hessler, P.; Torrent, M.; He, W.; Uziel, T.; Albert, D.H.; McDaniel, K.; Kati, W.; et al. Exploitation of castration-resistant prostate cancer transcription factor dependencies by the novel BET inhibitor ABBV-075. Mol. Cancer Res. 2017, 15, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Hupe, M.C.; Hoda, M.R.; Zengerling, F.; Perner, S.; Merseburger, A.S.; Cronauer, M.V. The BET-inhibitor PFI-1 diminishes AR/AR-V7 signaling in prostate cancer cells. World J. Urol. 2019, 37, 343–349. [Google Scholar] [CrossRef]
- Asangani, I.A.; Wilder-Romans, K.; Dommeti, V.L.; Krishnamurthy, P.M.; Apel, I.J.; Escara-Wilke, J.; Plymate, S.R.; Navone, N.M.; Wang, S.; Feng, F.Y.; et al. BET bromodomain inhibitors enhance efficacy and disrupt resistance to AR antagonists in the treatment of prostate cancer. Mol. Cancer Res. 2016, 14, 324–331. [Google Scholar] [CrossRef]
- Vaishampayan, U.N.; Narayan, V.; Wise, D.; Lang, J.M.; Lowentritt, B.H.; Mellado, B.; Carles, J.; Sáez, M.I.; Abida, W.; Taplin, M.-E.; et al. A phase Ib open-label, dose escalation and expansion study to investigate the safety, pharmacokinetics, pharmacodynamics and clinical activity of GSK525762 in combination with abiraterone or enzalutamide in metastatic castrate-resistant prostate cancer. J. Clin. Oncol. 2018, 36. [Google Scholar] [CrossRef]
- Björkman, M.; Mattila, E.; Riikonen, R.; Sekhar, C.; Jaleel, M.; Marappan, S.; Ikonen, T.; Nicorici, D.; Samajdar, S.; Ramachandra, M.; et al. ODM-207, a novel BET-bromodomain inhibitor as a therapeutic approach for the treatment of patients with castration resistant prostate cancer. Eur. J. Cancer 2016, 69, S89–S90. [Google Scholar] [CrossRef]
- Pan, W.; Zhang, Z.; Kimball, H.; Qu, F.; Berlind, K.; Stopsack, K.H.; Lee, G.-S.M.; Choueiri, T.K.; Kantoff, P.W. Abiraterone acetate induces CREB1 phosphorylation and enhances the function of the CBP-p300 complex, leading to resistance in prostate cancer cells. Clin. Cancer Res. 2021, 27, 2087–2099. [Google Scholar] [CrossRef] [PubMed]
- De Bono, J.S.; Cojocaru, E.; Plummer, E.R.; Knurowski, T.; Clegg, K.; Ashby, F.; Pegg, N.; West, W.; Brooks, A.N. An open label phase I/IIa study to evaluate the safety and efficacy of CCS1477 as monotherapy and in combination in patients with advanced solid/metastatic tumors. J. Clin. Oncol. 2019, 36. [Google Scholar] [CrossRef]
- Yan, Y.; Ma, J.; Wang, D.; Lin, D.; Pang, X.; Wang, S.; Zhao, Y.; Shi, L.; Xue, H.; Pan, Y.; et al. The novel BET-CBP/p300 dual inhibitor NEO2734 is active in SPOP mutant and wild-type prostate cancer. EMBO Mol. Med. 2019, 11, e10659. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Wei, T.; Ye, Z.; Orme, J.J.; Lin, D.; Sheng, H.; Fazli, L.; Karnes, R.J.; Jimenez, R.; Wang, L.; et al. A noncanonical AR addiction drives enzalutamide resistance in prostate cancer. Nat. Commun. 2021, 12, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Strebhardt, K. Multifaceted polo-like kinases: Drug targets and antitargets for cancer therapy. Nat. Rev. Drug Discov. 2010, 9, 643–660. [Google Scholar] [CrossRef]
- Zhang, Z.; Hou, X.; Shao, C.; Li, J.; Cheng, J.-X.; Kuang, S.; Ahmad, N.; Ratliff, T.; Liu, X. Plk1 inhibition enhances the efficacy of androgen signaling blockade in castration-resistant prostate cancer. Cancer Res. 2014, 74, 6635–6647. [Google Scholar] [CrossRef]
- Einstein, D.J.; Choudhury, A.D.; Saylor, P.J.; Patterson, J.C.; Croucher, P.; Ridinger, M.; Erlander, M.G.; Yaffe, M.B.; Bubley, G. A phase II study of onvansertib in combination with abiraterone and prednisone in patients with metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2021, 38. [Google Scholar] [CrossRef]
- Shao, C.; Wang, Y.; Yue, H.H.; Zhang, Y.T.; Shi, C.H.; Liu, F.; Bao, T.Y.; Yang, Z.Y.; Yuan, J.L.; Shao, G.X.; et al. Biphasic effect of androgens on prostate cancer cells and its correlation with androgen receptor coactivator dopa decarboxylase. J. Androl. 2007, 28, 804–812. [Google Scholar] [CrossRef]
- Litvinov, I.V.; Vander Griend, D.J.; Antony, L.; Dalrymple, S.; De Marzo, A.M.; Drake, C.G.; Isaacs, J.T. Androgen receptor as a licensing factor for DNA replication in androgen-sensitive prostate cancer cells. Proc. Natl. Acad. Sci. USA 2006, 103, 15085–15090. [Google Scholar] [CrossRef]
- Chuu, C.-P.; Kokontis, J.M.; Hiipakka, R.A.; Fukuchi, J.; Lin, H.-P.; Lin, C.-Y.; Huo, C.; Su, L.-C. Androgens as therapy for androgen receptor-positive castration-resistant prostate cancer. J. Biomed. Sci. 2011, 18, 63. [Google Scholar] [CrossRef]
- Chatterjee, P.; Schweizer, M.T.; Lucas, J.M.; Coleman, I.; Nyquist, M.D.; Frank, S.B.; Tharakan, R.; Mostaghel, E.; Luo, J.; Pritchard, C.C. Supraphysiological androgens suppress prostate cancer growth through androgen receptor–mediated DNA damage. J. Clin. Investig. 2019, 129, 4245–4260. [Google Scholar] [CrossRef] [PubMed]
- Markowski, M.C.; Shenderov, E.; Eisenberger, M.A.; Kachhap, S.; Pardoll, D.M.; Denmeade, S.R.; Antonarakis, E.S. Extreme responses to immune checkpoint blockade following bipolar androgen therapy and enzalutamide in patients with metastatic castration resistant prostate cancer. Prostate 2020, 80, 407–411. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Ciuleanu, T.-E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef]
- Kyriakopoulos, C.E.; Eickhoff, J.C.; Ferrari, A.C.; Schweizer, M.T.; Wargowski, E.; Olson, B.M.; McNeel, D.G. Multicenter phase I trial of a DNA vaccine encoding the androgen receptor ligand-binding domain (pTVG-AR, MVI-118) in patients with metastatic prostate cancer. Clin. Cancer Res. 2020, 26, 5162–5171. [Google Scholar] [CrossRef]
- Olson, B.M.; Gamat, M.; Seliski, J.; Sawicki, T.; Jeffery, J.; Ellis, L.; Drake, C.G.; Weichert, J.; McNeel, D.G. Prostate cancer cells express more androgen receptor (AR) following androgen deprivation, improving recognition by AR-specific T cells. Cancer Immunol. Res. 2017, 5, 1074–1085. [Google Scholar] [CrossRef] [PubMed]
- Rahbar, K.; Ahmadzadehfar, H.; Kratochwil, C.; Haberkorn, U.; Schäfers, M.; Essler, M.; Baum, R.P.; Kulkarni, H.R.; Schmidt, M.; Drzezga, A.; et al. German multicenter study investigating 177Lu-PSMA-617 radioligand therapy in advanced prostate cancer patients. J. Nucl. Med. 2017, 58, 85–90. [Google Scholar] [CrossRef]
- Vlachostergios, P.J.; Puca, L.; Beltran, H. Emerging variants of castration-resistant prostate cancer. Curr. Oncol. Rep. 2017, 19, 32. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Sample Type | AR-Vs and Other AR Structural Aberrations | |
|---|---|---|
| Plasma | cfDNA | Intra-AR structural variation [41], Point mutations [41,42,43], Amplification [43], Copy number gain [42,43,44] |
| cfRNA | AR-V7 [42], AR-V9 [42] | |
| CTC | DNA | Amplification [45], Copy number loss [45], Copy number gain [45], Deletion [45] |
| RNA | AR-45 [41], AR-V1 [41,46], AR-V2 [41], AR-V3 [41,46], AR-V4 [46], AR-V5 [41], AR-V7 [36,37,38,40,41,46,47,48,49,50,51,52,53], AR-V9 [41,50], AR-V12 [46], Point mutations [46] | |
| Protein | AR-V7 [40,49,54,55] | |
| PBMC | DNA and RNA | PBMC is used to detect germline mutations. Because AR mutations in PCa are somatic, PBMC is not used for AR mutation detection. Instead, PBMC is utilized to detect mutations in genes where germline mutations are common such as in BRCA1/2. |
| Exosome | RNA | AR-V7 [44,56] |
| Tissue | DNA | Amplification [7], Missense mutation [6,7], In-frame indels [7] |
| RNA | AR-45 [50], AR-23 [50], AR-V1 [6,7], AR-V3 [6,7,50], AR-V5 [6,7], AR-V6 [6,7], AR-V7 [6,7,50,57], AR-V7 (RISH) [58], AR-V8 [6,7], AR-V9 [6,7,50], AR-v5es [6,7], AR-v56es [6,7], AR-v7es [6,7], AR-V13 [6,7], AR-V14 [6,7], | |
| Protein | AR-V7 [19,57,59] | |
| Types of AR-Vs | Significance | Reference |
|---|---|---|
| AR-V7 | CTC AR-V7 is associated with resistance to ABI and ENZ. | [36,37,40,49] |
| Nuclear AR-V7 in CTC is associated with superior survival on taxane chemotherapy over AR-targeted therapy. | [54,55] | |
| CTC AR-V7 is associated with CTC counts and disease burden. There is sometimes discordance between CTC AR-V7 and tissue AR-V7. | [38] | |
| CTC AR-V7 is not associated with resistance to taxanes. | [47,49] | |
| CTC AR-V7 is associated with advanced disease. The ability of AR-V7 to serve as a treatment-selection marker for galeterone could not be evaluated. | [48] | |
| The presence of any AR-V in CTC is associated with shorter PFS after 2nd hormonal treatment. | [41] | |
| Exosomal AR-V7 is associated with resistance to ABI and ENZ. | [44,56] | |
| AR-V7 in biopsies detected by RISH is associated with a shorter PFS. | [58] | |
| High AR-V7 and AR-V7/ AR-FL ratio in nuclear of PCa tissues are associated with shorter BCR-free survival. | [57] | |
| AR-V7 in bone marrow is associated with resistance to ENZ. | [59] | |
| CTC AR-V7 is associated with shorter PFS and OS on BAT and enzalutamide, but not predictive of treatment effects. | [60,61] | |
| Nivolumab plus ipilimumab showed modest efficacy in CTC AR-V7(+) patients irrespective of enzalutamide addition. | [51,52] | |
| CTC AR-V7 is not associated with PFS and OS after 177Lu-PSMA-617 therapy. | [53] | |
| AR-V1, AR-V2, AR-V3, AR-V5, AR-V7, AR-V9 and AR-45 | AR gain and cumulative number of AR aberrations including AR-V7 and AR-V9 are associated with shorter PFS and OS. | [42] |
| AR-V7 and AR-V9 | AR-V9 in tissue is associated with resistance to ABI. | [50] |
| Agent | Description | Compound Name, NCT Number and Reference | |
|---|---|---|---|
| Directly Targeting AR-Vs | Niclosamide | Inhibit AR-Vs activity by protein degradation. Refomulated niclosamide has improved oral bioavailability. | Niclosamide (NCT02532114) [64] Refomulated niclosamide (NCT02807805) [65] |
| EPI compounds | Target N-terminal domain of AR-FL and AR-Vs and suppress transcriptional activity by inhibiting protein–protein interactions of AR-FL and AR-Vs with other co-activators. | EPI-506 (NCT02606123) EPI-7386 (NCT04421222) [66] | |
| Indirectly Targeting AR-Vs | BET inhibitors | Disrupt the interaction between BRD4 and AR-FL and AR-Vs. | ZEN-3694 (NCT02711956) [67] |
| CBP/p300 inhibitors | Suppress AR and AR-V7 signaling by inhibiting CBP/p300 (AR coactivators). NEO2734 simultaneously targets BET and CBP/p300. | CCS1477 (NCT03568656) [68] | |
| PLK1 inhibitors | Inhibit cell cycle progression. Suppress cholesterol biosynthesis and downregulate AR-FL and AR-Vs. | Onvansertib (NCT03414034) | |
| Bipolar androgen therapy | Supraphysiological exogenous androgen inhibits PCa growth and re-sensitizes CRPC to AR-targeted drugs. | NCT02090114 [60] NCT02286921 [61] | |
| Immune checkpoint inhibitors | Nivolumab plus Ipilimumab may have modest activity in AR-V7–expressing CRPC patients and/or in those with high TMB. | NCT02601014 [51,52] NCT02985957 [69] | |
| 177Lu-PSMA-617 | PSMA ligands labeled with β-radiating lutetium-177 target PSMA-expressing PCa cells. | NCT03511664 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanayama, M.; Lu, C.; Luo, J.; Antonarakis, E.S. AR Splicing Variants and Resistance to AR Targeting Agents. Cancers 2021, 13, 2563. https://doi.org/10.3390/cancers13112563
Kanayama M, Lu C, Luo J, Antonarakis ES. AR Splicing Variants and Resistance to AR Targeting Agents. Cancers. 2021; 13(11):2563. https://doi.org/10.3390/cancers13112563
Chicago/Turabian StyleKanayama, Mayuko, Changxue Lu, Jun Luo, and Emmanuel S. Antonarakis. 2021. "AR Splicing Variants and Resistance to AR Targeting Agents" Cancers 13, no. 11: 2563. https://doi.org/10.3390/cancers13112563
APA StyleKanayama, M., Lu, C., Luo, J., & Antonarakis, E. S. (2021). AR Splicing Variants and Resistance to AR Targeting Agents. Cancers, 13(11), 2563. https://doi.org/10.3390/cancers13112563