
Epigenomic Analysis of RAD51 ChIP-seq Data Reveals cis-regulatory Elements Associated with Autophagy in Cancer Cell Lines

 , and
, and 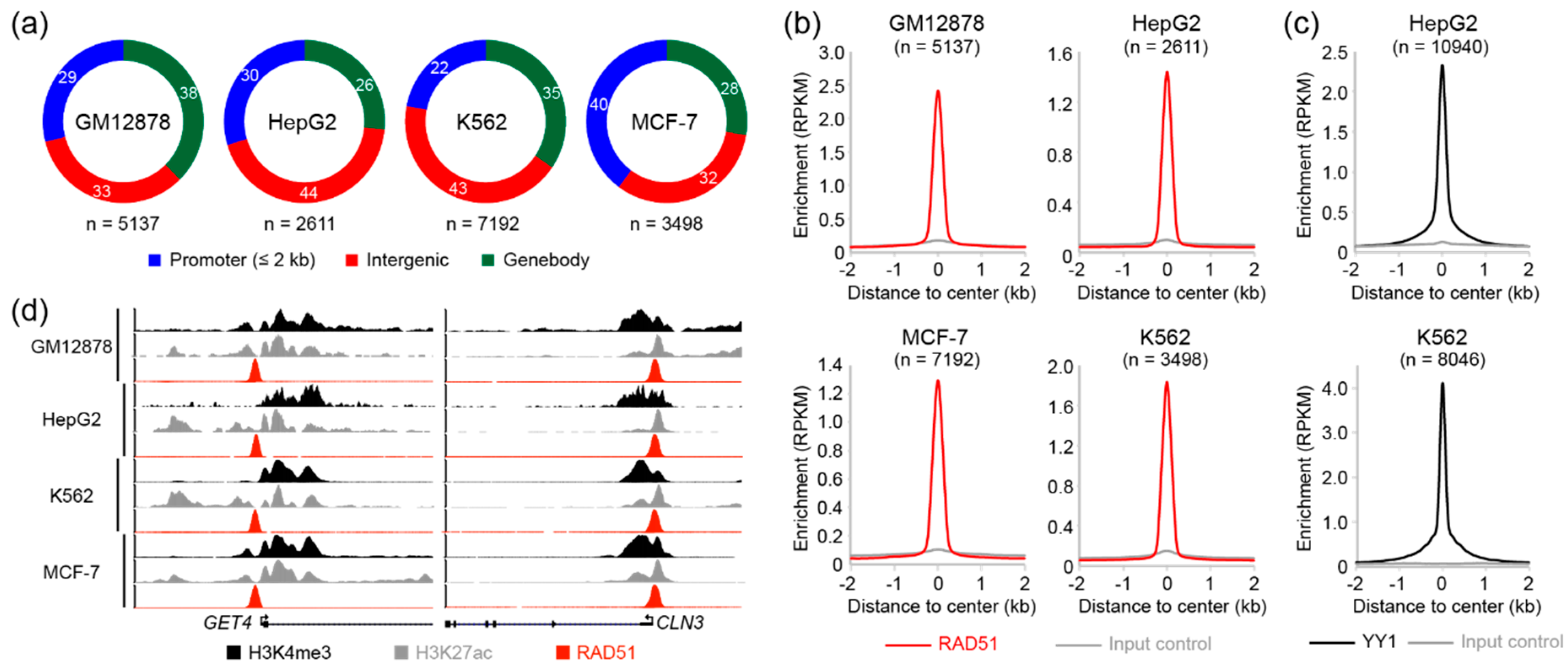{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. ChIP-seq Data Analysis
2.2. DNA Binding Motif Analysis
2.3. Gene Ontology Analysis
2.4. Data Visualization
2.5. Kaplan–Meier Survival Analysis
2.6. Cell Culture and Reagents
2.7. Real-Time Quantitative PCR
2.8. Statistical Analysis
3. Results
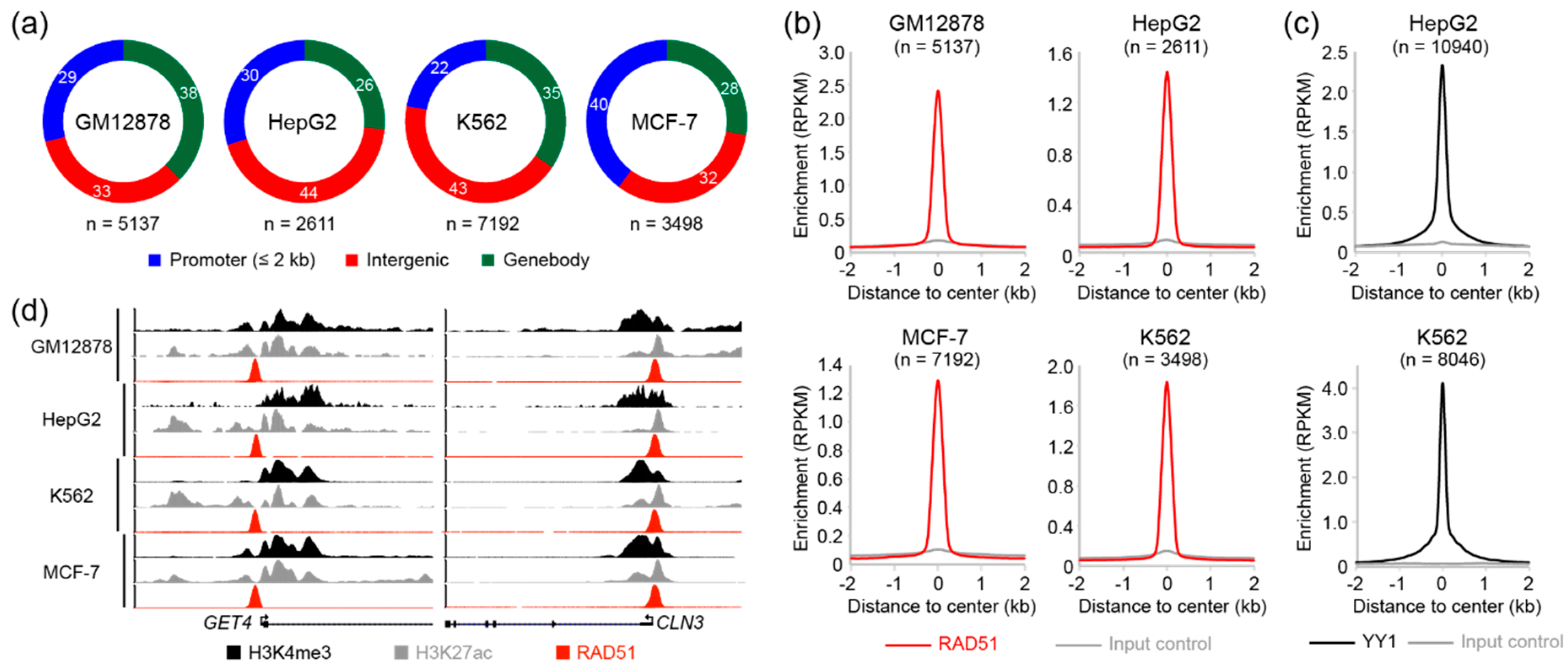
3.1. Analysis of Genome-Wide RAD51 Binding Sites in GM12878, HepG2, K562, and MCF-7 Cell Lines
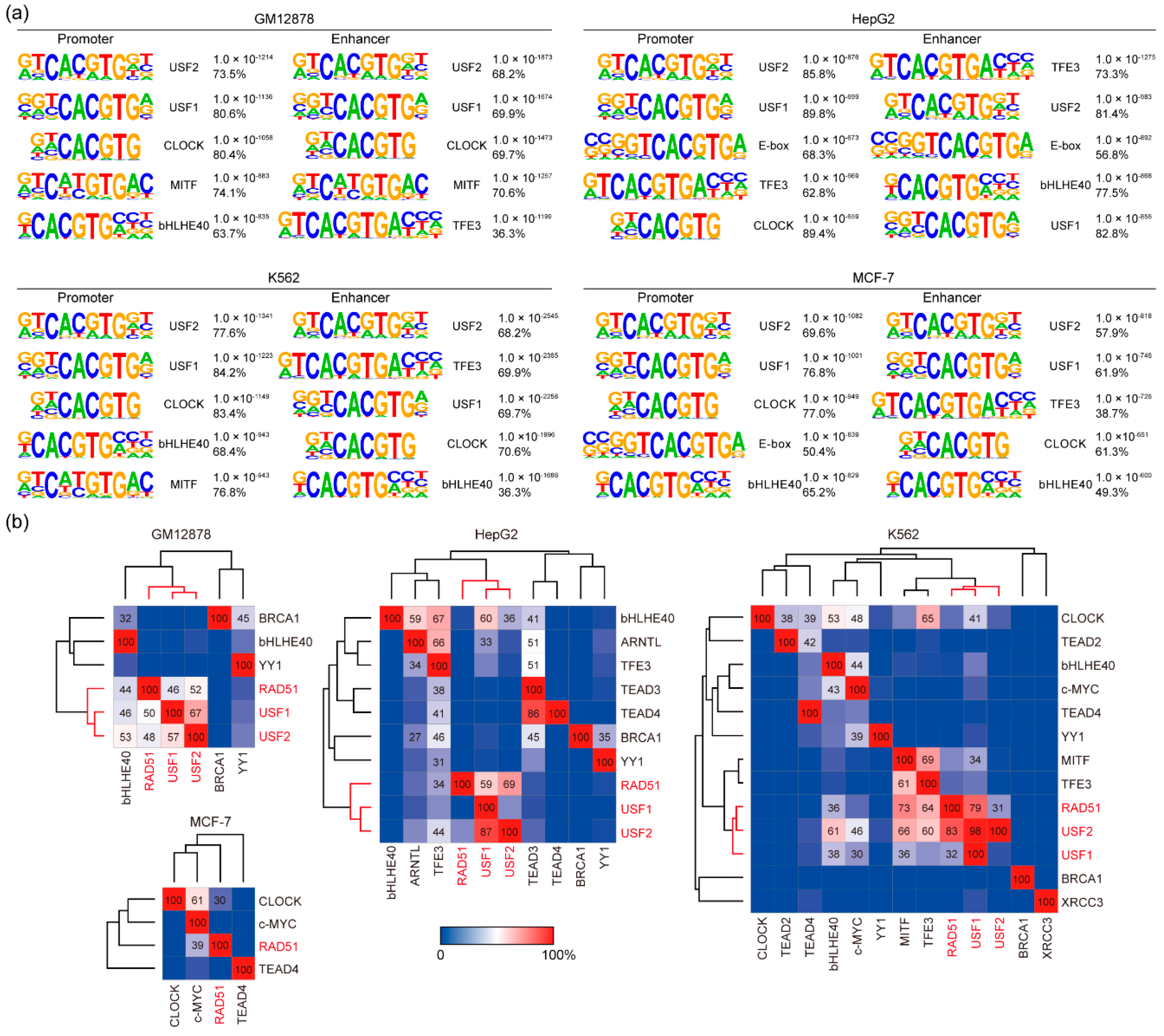
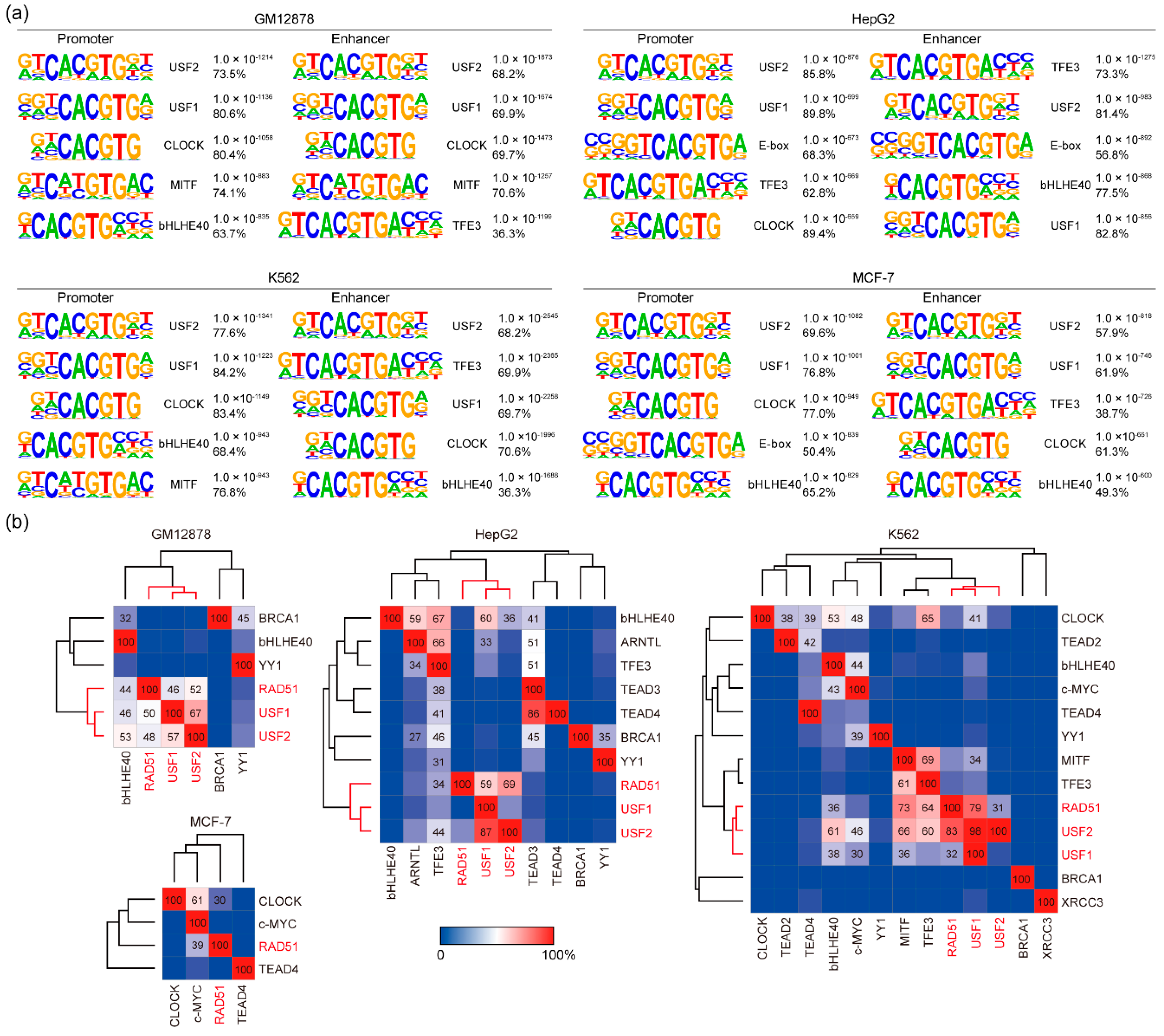
3.2. DNA Binding Motif Analysis of RAD51 Binding Sites in GM12878, HepG2, K562, and MCF-7 Cell Lines
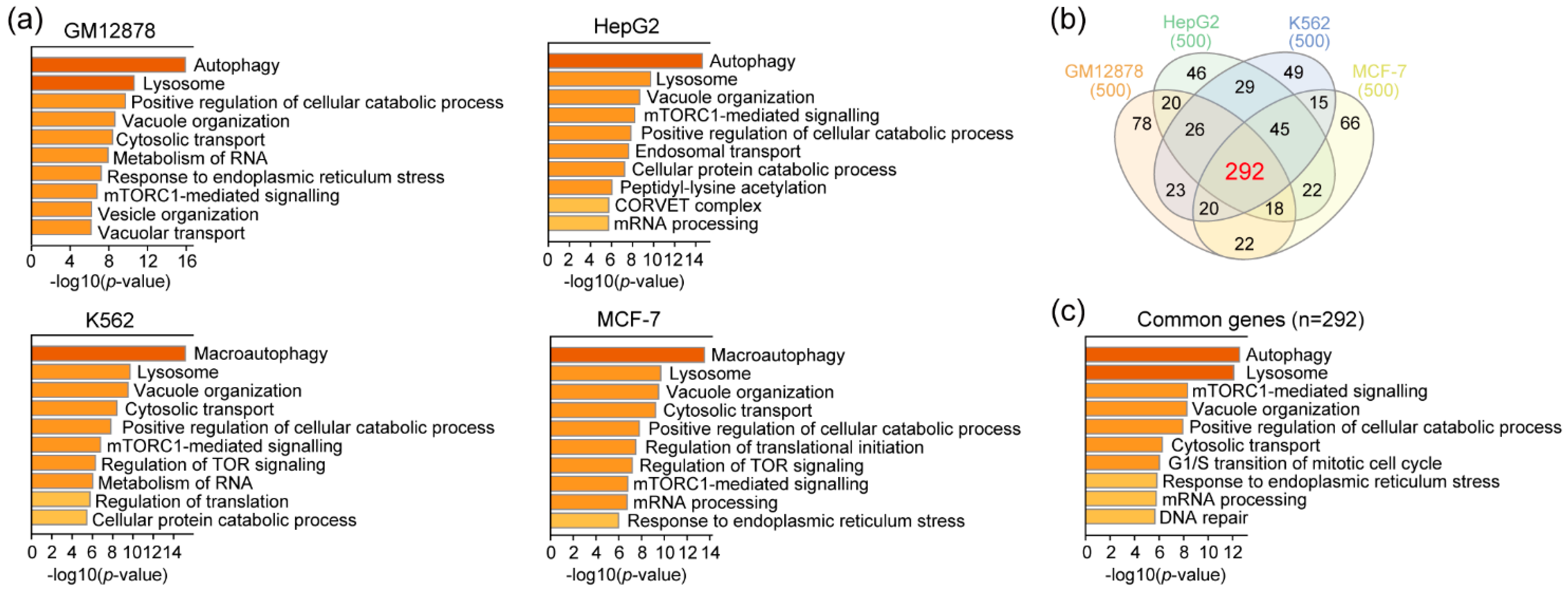
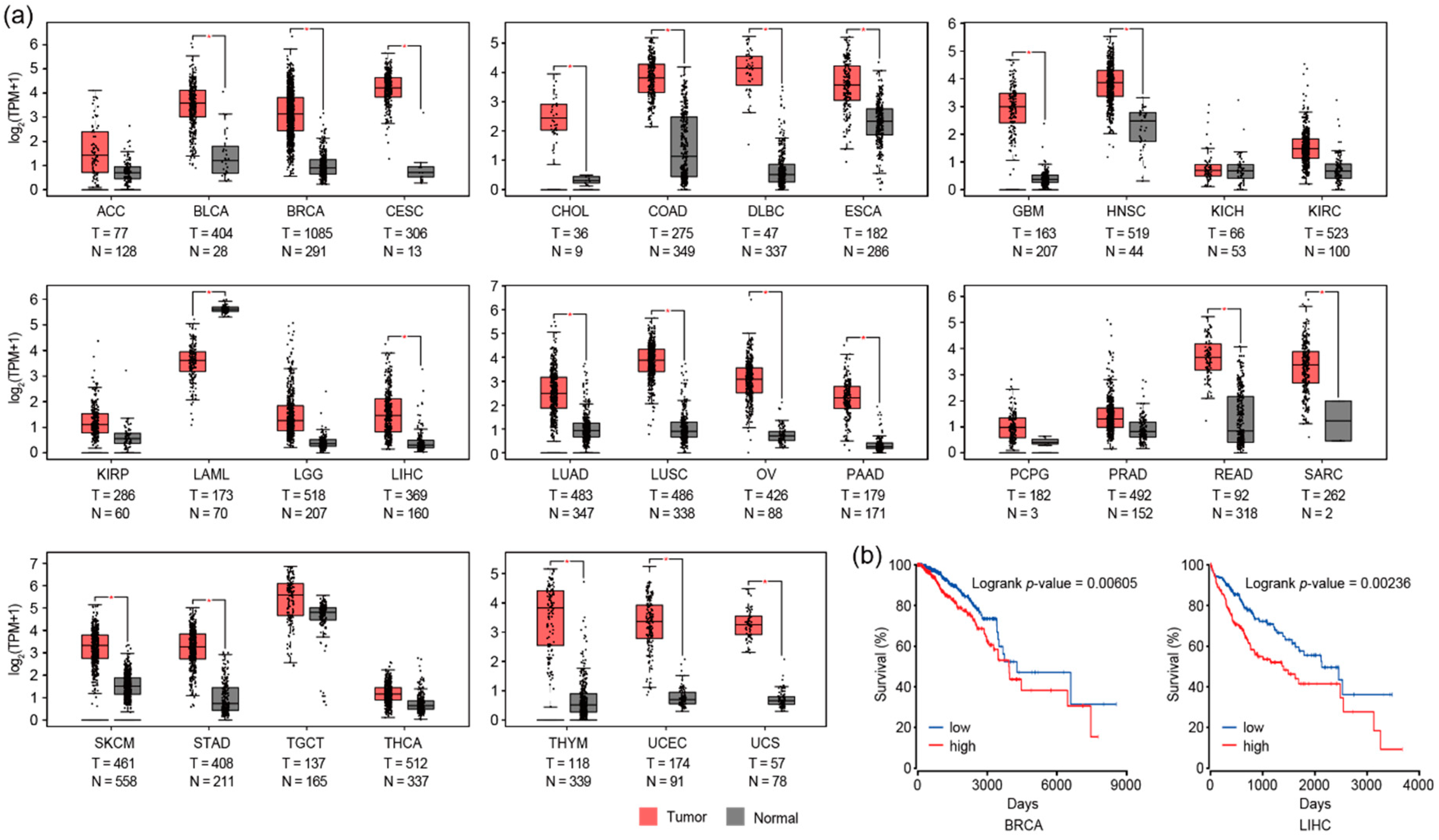
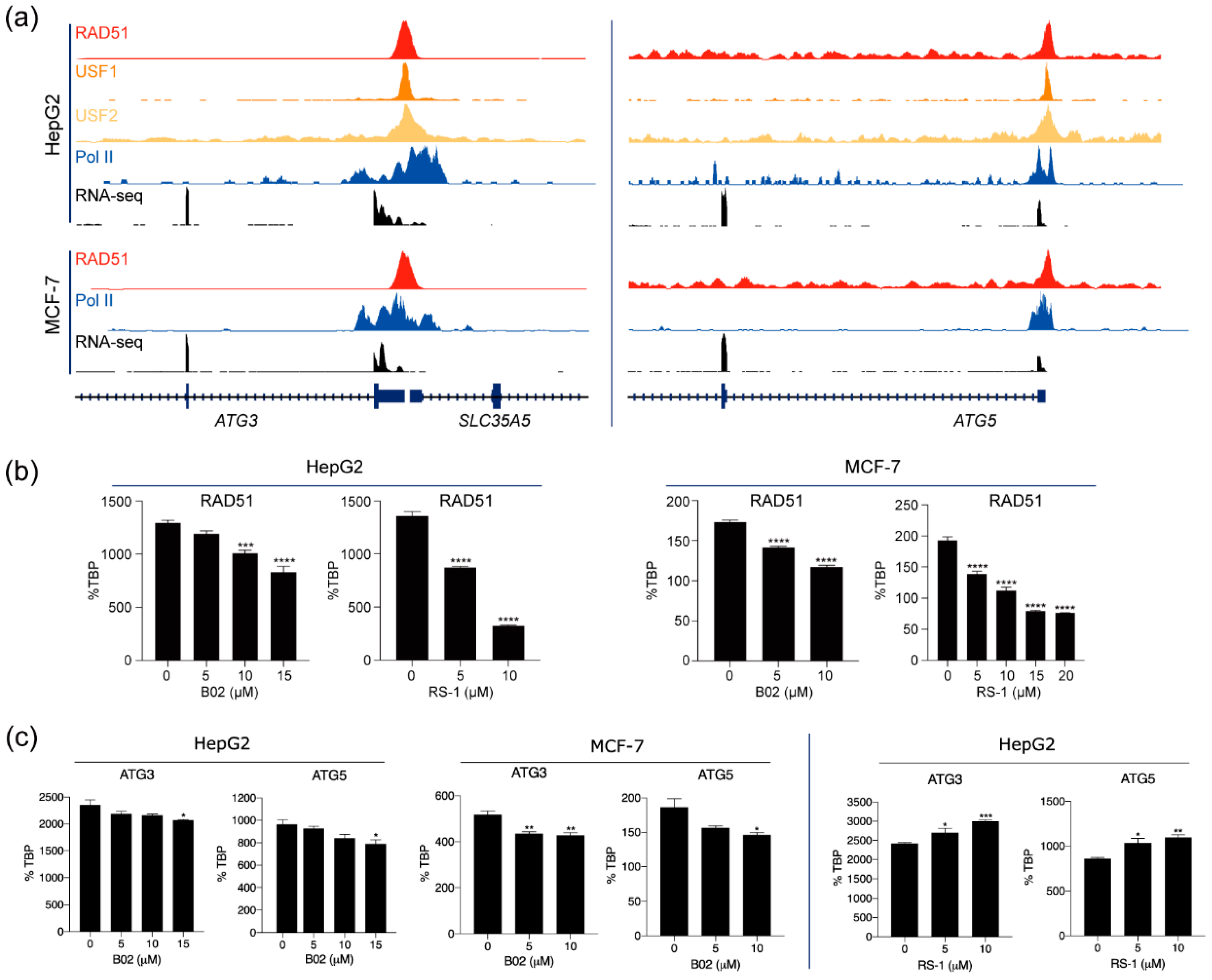
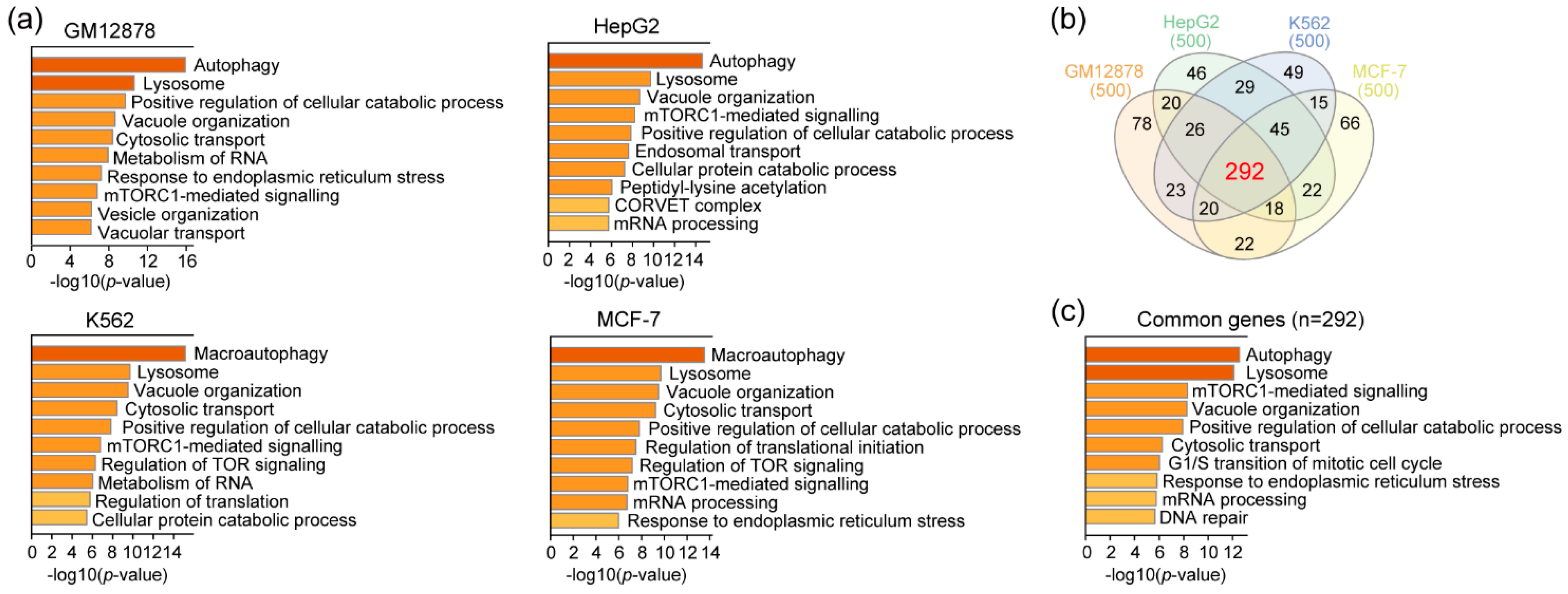
3.3. RAD51 and Autophagy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Baumann, P.; West, S.C. Role of the Human RAD51 Protein in Homologous Recombination and Double-Stranded-Break Repair. Trends Biochem. Sci. 1998, 23, 247–251. [Google Scholar] [CrossRef]
- Bindra, R.S.; Schaffer, P.J.; Meng, A.; Woo, J.; Lizardi, P.; Hedley, D.W.; Bristow, R.G.; Glazer, P.M. Down-Regulation of Rad51 and Decreased Homologous Recombination in Hypoxic Cancer Cells. Mol. Cell. Biol. 2004, 24, 8504–8518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vispe, S.; Cazaux, C.; Lesca, C.; Defais, M. Overexpression of Rad51 Protein Stimulates Homologous Recombination and Increases Resistance of Mammalian Cells to Ionizing Radiation. Nucleic Acids Res. 1998, 26, 2859–2864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazin, A.V.; Zaitseva, E.; Sung, P.; Kowalczykowski, S.C. Tailed Duplex DNA Is the Preferred Substrate for Rad51 Protein-Mediated Homologous Pairing. EMBO J. 2000, 19, 1148–1156. [Google Scholar] [CrossRef]
- Mason, J.M.; Chan, Y.-L.; Weichselbaum, R.W.; Bishop, D.K. Non-Enzymatic Roles of Human RAD51 at Stalled Replication Forks. Nat. Commun. 2019, 10, 4410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, S.; Srinivasan, K.; Abdisalaam, S.; Su, F.; Raj, P.; Dozmorov, I.; Mishra, R.; Wakeland, E.K.; Ghose, S.; Mukherjee, S.; et al. RAD51 Interconnects between DNA Replication, DNA Repair and Immunity. Nucleic Acids Res. 2017, 45, 4590–4605. [Google Scholar] [CrossRef] [Green Version]
- Ko, J.-C.; Hong, J.-H.; Wang, L.-H.; Cheng, C.-M.; Ciou, S.-C.; Lin, S.-T.; Jheng, M.-Y.; Lin, Y.-W. Role of Repair Protein Rad51 in Regulating the Response to Gefitinib in Human Non-Small Cell Lung Cancer Cells. Mol. Cancer Ther. 2008, 7, 3632–3641. [Google Scholar] [CrossRef] [Green Version]
- Nagathihalli, N.S.; Nagaraju, G. RAD51 as a Potential Biomarker and Therapeutic Target for Pancreatic Cancer. Biochim. Biophys. Acta 2011, 1816, 209–218. [Google Scholar] [CrossRef]
- Hine, C.M.; Seluanov, A.; Gorbunova, V. Use of the Rad51 Promoter for Targeted Anti-Cancer Therapy. Proc. Natl. Acad. Sci. USA 2008, 105, 20810–20815. [Google Scholar] [CrossRef] [Green Version]
- Makino, E.; Fröhlich, L.M.; Sinnberg, T.; Kosnopfel, C.; Sauer, B.; Garbe, C.; Schittek, B. Targeting Rad51 as a Strategy for the Treatment of Melanoma Cells Resistant to MAPK Pathway Inhibition. Cell Death Dis. 2020, 11, 581. [Google Scholar] [CrossRef]
- Tsai, M.-S.; Kuo, Y.-H.; Chiu, Y.-F.; Su, Y.-C.; Lin, Y.-W. Down-Regulation of Rad51 Expression Overcomes Drug Resistance to Gemcitabine in Human Non–Small-Cell Lung Cancer Cells. J. Pharmacol. Exp. Ther. 2010, 335, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Qiao, G.-B.; Wu, Y.-L.; Yang, X.-N.; Zhong, W.-Z.; Xie, D.; Guan, X.-Y.; Fischer, D.; Kolberg, H.-C.; Kruger, S.; Stuerzbecher, H.-W. High-Level Expression of Rad51 Is an Independent Prognostic Marker of Survival in Non-Small-Cell Lung Cancer Patients. Br. J. Cancer 2005, 93, 137–143. [Google Scholar] [CrossRef] [Green Version]
- Maacke, H.; Jost, K.; Opitz, S.; Miska, S.; Yuan, Y.; Hasselbach, L.; Lüttges, J.; Kalthoff, H.; Stürzbecher, H.-W. DNA Repair and Recombination Factor Rad51 Is Over-Expressed in Human Pancreatic Adenocarcinoma. Oncogene 2000, 19, 2791–2795. [Google Scholar] [CrossRef] [PubMed]
- Maacke, H.; Opitz, S.; Jost, K.; Hamdorf, W.; Henning, W.; Ger, S.K.; Feller, A.C.; Lopens, A.; Diedrich, K.; Schwinger, E.; et al. Over-Expression of Wild-Type Rad51 Correlates with Histological Grading of Invasive Ductal Breast Cancer. Int. J. Cancer 2000, 88, 907–913. [Google Scholar] [CrossRef]
- Chen, J.-J.; Silver, D.; Cantor, S.; Livingston, D.M.; Scully, R. BRCA1, BRCA2, and Rad51 Operate in a Common DNA Damage Response Pathway. Cancer Res. 1999, 59, 1752–1756. [Google Scholar]
- Schild, D.; Wiese, C. Overexpression of RAD51 Suppresses Recombination Defects: A Possible Mechanism to Reverse Genomic Instability. Nucleic Acids Res. 2010, 38, 1061–1070. [Google Scholar] [CrossRef] [Green Version]
- Jia, Y.; Song, Y.; Dong, G.; Hao, C.; Zhao, W.; Li, S.; Tong, Z. Aberrant Regulation of RAD51 Promotes Resistance of Neoadjuvant Endocrine Therapy in ER-Positive Breast Cancer. Sci. Rep. 2019, 9, 12939. [Google Scholar] [CrossRef] [PubMed]
- Kimmelman, A.C. The Dynamic Nature of Autophagy in Cancer. Genes Dev. 2011, 25, 1999–2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, E. Deconvoluting the Context-Dependent Role for Autophagy in Cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Howell, M.E.A.; Sparks-Wallace, A.; Hawkins, C.; Nicksic, C.A.; Kohne, C.; Hall, K.H.; Moorman, J.P.; Yao, Z.Q.; Ning, S. P62-Mediated Selective Autophagy Endows Virus-Transformed Cells with Insusceptibility to DNA Damage under Oxidative Stress. PLoS Pathog. 2019, 15, e1007541. [Google Scholar] [CrossRef] [Green Version]
- Hewitt, G.; Korolchuk, V.I. Repair, Reuse, Recycle: The Expanding Role of Autophagy in Genome Maintenance. Trends Cell Biol. 2017, 27, 340–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Pan, Q.; Huang, N.; Wu, J.; Zhen, N.; Sun, F.; Li, Z.; Yang, Q. RAD51 Regulates CHK1 Stability via Autophagy to Promote Cell Growth in Esophageal Squamous Carcinoma Cells. Tumor Biol. 2016, 37, 16151–16161. [Google Scholar] [CrossRef]
- Mo, N.; Lu, Y.-K.; Xie, W.-M.; Liu, Y.; Zhou, W.-X.; Wang, H.-X.; Nong, L.; Jia, Y.-X.; Tan, A.-H.; Chen, Y.; et al. Inhibition of Autophagy Enhances the Radiosensitivity of Nasopharyngeal Carcinoma by Reducing Rad51 Expression. Oncol. Rep. 2014, 32, 1905–1912. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; Li, N.; Wang, H.; He, C.; Hu, Y.; Peng, C.; Ouyang, Y.; Wang, D.; Xie, Y.; Chen, J.; et al. Inhibition of Autophagy Aggravates DNA Damage Response and Gastric Tumorigenesis via Rad51 Ubiquitination in Response to H. Pylori Infection. Gut Microbes 2020, 11, 1567–1589. [Google Scholar] [CrossRef]
- Zai, W.; Chen, W.; Han, Y.; Wu, Z.; Fan, J.; Zhang, X.; Luan, J.; Tang, S.; Jin, X.; Fu, X.; et al. Targeting PARP and Autophagy Evoked Synergistic Lethality in Hepatocellular Carcinoma. Carcinogenesis 2020, 41, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Wu, J.; Luo, Y.; Huang, N.; Zhen, N.; Zhou, Y.; Sun, F.; Li, Z.; Pan, Q.; Li, Y. (−)-Guaiol Regulates RAD51 Stability via Autophagy to Induce Cell Apoptosis in Non-Small Cell Lung Cancer. Oncotarget 2016, 7, 62585–62597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.S.; Vats, S.; Chia, A.Y.-Q.; Tan, T.Z.; Deng, S.; Ong, M.S.; Arfuso, F.; Yap, C.T.; Goh, B.C.; Sethi, G.; et al. Dual Role of Autophagy in Hallmarks of Cancer. Oncogene 2018, 37, 1142–1158. [Google Scholar] [CrossRef]
- Aymard, F.; Bugler, B.; Schmidt, C.K.; Guillou, E.; Caron, P.; Briois, S.; Iacovoni, J.S.; Daburon, V.; Miller, K.M.; Jackson, S.P.; et al. Transcriptionally Active Chromatin Recruits Homologous Recombination at DNA Double-Strand Breaks. Nat. Struct. Mol. Biol. 2014, 21, 366–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazan, I.; Monin, J.; Bouwman, B.A.M.; Crosetto, N.; Aqeilan, R.I. Activation of Oncogenic Super-Enhancers Is Coupled with DNA Repair by RAD51. Cell Rep. 2019, 29, 560–572.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, C.A.; Hitz, B.C.; Sloan, C.A.; Chan, E.T.; Davidson, J.M.; Gabdank, I.; Hilton, J.A.; Jain, K.; Baymuradov, U.K.; Narayanan, A.K.; et al. The Encyclopedia of DNA Elements (ENCODE): Data Portal Update. Nucleic Acids Res. 2018, 46, D794–D801. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarasov, A.; Vilella, A.J.; Cuppen, E.; Nijman, I.J.; Prins, P. Sambamba: Fast Processing of NGS Alignment Formats. Bioinformatics 2015, 31, 2032–2034. [Google Scholar] [CrossRef] [PubMed]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple Combinations of Lineage-Determining Transcription Factors Prime Cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape Provides a Biologist-Oriented Resource for the Analysis of Systems-Level Datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Heberle, H.; Meirelles, G.V.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A Web-Based Tool for the Analysis of Sets through Venn Diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramírez, F.; Dündar, F.; Diehl, S.; Grüning, B.A.; Manke, T. DeepTools: A Flexible Platform for Exploring Deep-Sequencing Data. Nucleic Acids Res. 2014, 42, W187–W191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anaya, J. OncoLnc: Linking TCGA Survival Data to MRNAs, MiRNAs, and LncRNAs. PeerJ Comput. Sci. 2016, 2, e67. [Google Scholar] [CrossRef] [Green Version]
- Kang, K.; Choi, Y.; Kim, H.H.; Yoo, K.H.; Yu, S. Predicting FOXM1-Mediated Gene Regulation through the Analysis of Genome-Wide FOXM1 Binding Sites in MCF-7, K562, SK-N-SH, GM12878 and ECC-1 Cell Lines. Int. J. Mol. Sci. 2020, 21, 6141. [Google Scholar] [CrossRef]
- Ristic, D. Human Rad51 Filaments on Double- and Single-Stranded DNA: Correlating Regular and Irregular Forms with Recombination Function. Nucleic Acids Res. 2005, 33, 3292–3302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Zhao, L.; Xu, Y.; Zhao, W.; Sung, P.; Wang, H.-W. Cryo-EM Structures of Human RAD51 Recombinase Filaments during Catalysis of DNA-Strand Exchange. Nat. Struct. Mol. Biol. 2017, 24, 40–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, Z.; Redding, S.; Lee, J.Y.; Gibb, B.; Kwon, Y.; Niu, H.; Gaines, W.A.; Sung, P.; Greene, E.C. DNA Sequence Alignment by Microhomology Sampling during Homologous Recombination. Cell 2015, 160, 856–869. [Google Scholar] [CrossRef] [Green Version]
- Gomes, L.; Menck, C.; Leandro, G. Autophagy Roles in the Modulation of DNA Repair Pathways. Int. J. Mol. Sci. 2017, 18, 2351. [Google Scholar] [CrossRef] [Green Version]
- Dikic, I.; Elazar, Z. Mechanism and Medical Implications of Mammalian Autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Feitelson, M.A.; Arzumanyan, A.; Kulathinal, R.J.; Blain, S.W.; Holcombe, R.F.; Mahajna, J.; Marino, M.; Martinez-Chantar, M.L.; Nawroth, R.; Sanchez-Garcia, I.; et al. Sustained Proliferation in Cancer: Mechanisms and Novel Therapeutic Targets. Semin. Cancer Biol. 2015, 35, S25–S54. [Google Scholar] [CrossRef] [PubMed]
- White, E. The Role for Autophagy in Cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting Autophagy in Cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Sonoda, E. Rad51-Deficient Vertebrate Cells Accumulate Chromosomal Breaks Prior to Cell Death. EMBO J. 1998, 17, 598–608. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.M.; Ko, J.H.; Hu, L.; Kim, S.-A.; Bishop, A.J.R.; Vijg, J.; Montagna, C.; Hasty, P. RAD51 Mutants Cause Replication Defects and Chromosomal Instability. Mol. Cell. Biol. 2012, 32, 3663–3680. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.-W.; Kim, D.-K.; Kim, K.P.; Park, K.-S. Rad51 Regulates Cell Cycle Progression by Preserving G2/M Transition in Mouse Embryonic Stem Cells. Stem Cells Dev. 2014, 23, 2700–2711. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, K.; Choi, Y.; Moon, H.; You, C.; Seo, M.; Kwon, G.; Yun, J.; Beck, B.; Kang, K. Epigenomic Analysis of RAD51 ChIP-seq Data Reveals cis-regulatory Elements Associated with Autophagy in Cancer Cell Lines. Cancers 2021, 13, 2547. https://doi.org/10.3390/cancers13112547
Kang K, Choi Y, Moon H, You C, Seo M, Kwon G, Yun J, Beck B, Kang K. Epigenomic Analysis of RAD51 ChIP-seq Data Reveals cis-regulatory Elements Associated with Autophagy in Cancer Cell Lines. Cancers. 2021; 13(11):2547. https://doi.org/10.3390/cancers13112547
Chicago/Turabian StyleKang, Keunsoo, Yoonjung Choi, Hyeonjin Moon, Chaelin You, Minjin Seo, Geunho Kwon, Jahyun Yun, Boram Beck, and Kyuho Kang. 2021. "Epigenomic Analysis of RAD51 ChIP-seq Data Reveals cis-regulatory Elements Associated with Autophagy in Cancer Cell Lines" Cancers 13, no. 11: 2547. https://doi.org/10.3390/cancers13112547
APA StyleKang, K., Choi, Y., Moon, H., You, C., Seo, M., Kwon, G., Yun, J., Beck, B., & Kang, K. (2021). Epigenomic Analysis of RAD51 ChIP-seq Data Reveals cis-regulatory Elements Associated with Autophagy in Cancer Cell Lines. Cancers, 13(11), 2547. https://doi.org/10.3390/cancers13112547