
KRAS G12C Mutations in NSCLC: From Target to Resistance



Abstract
Simple Summary
Abstract
1. Introduction
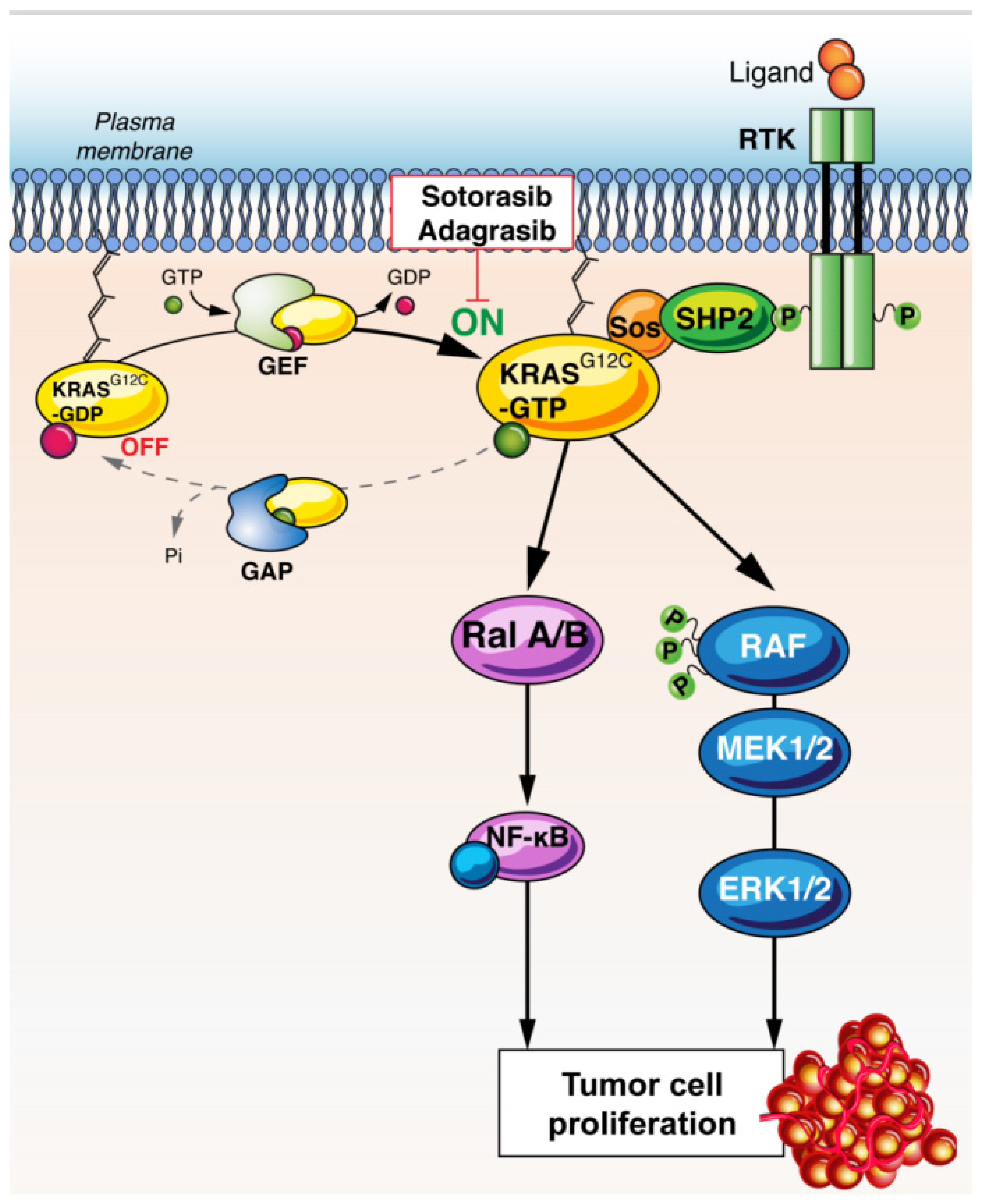
2. KRAS Biology
RAS-Mutant Biology and Heterogeneity
3. KRAS as a Therapeutic Target
Targeted Therapy
4. Mechanisms Underlying Resistance to K-RasG12C Inhibitors
5. Resistance to KRASG12C Inhibitors in Patients
6. How to Overcome Vertical Signal Resistance Pathways
7. Combining Current Treatments in KRAS G12C Mutant Cancers
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; Van Schil, P.E.; Hellmann, M.D.; et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv192–iv237. [Google Scholar] [CrossRef]
- Latest Global Cancer Data: Cancer Burden Rises to 18.1 million New Cases and 9.6 Million Cancer Deaths in 2018. Available online: https://www.iarc.who.int/featured-news/latest-global-cancer-data-cancer-burden-rises-to-18-1-million-new-cases-and-9-6-million-cancer-deaths-in-2018/ (accessed on 21 May 2021).
- Sites, A. SEER Cancer Statistics Review 1975–2011; National Cancer Institute: Bethesda, MD, USA, 2014. [Google Scholar]
- Askoxylakis, V.; Thieke, C.; Pleger, S.T.; Most, P.; Tanner, J.; Lindel, K.; Katus, H.A.; Debus, J.; Bischof, M. Long-term survival of cancer patients compared to heart failure and stroke: A systematic review. BMC Cancer 2010, 10, 105. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.-Y.; Huang, J.-Y.; Chen, H.-C.; Lin, C.-H.; Lin, S.-H.; Hung, W.-H.; Cheng, Y.-F. The comparison between adenocarcinoma and squamous cell carcinoma in lung cancer patients. J. Cancer Res. Clin. Oncol 2020, 146, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Addeo, A.; Banna, G.L.; Metro, G.; Di Maio, M. Chemotherapy in Combination with Immune Checkpoint Inhibitors for the First-Line Treatment of Patients with Advanced Non-small Cell Lung Cancer: A Systematic Review and Literature-Based Meta-Analysis. Front. Oncol. 2019, 9, 264. [Google Scholar] [CrossRef] [PubMed]
- Poulin, E.J.; Bera, A.K.; Lu, J.; Lin, Y.J.; Strasser, S.D.; Paulo, J.A.; Huang, T.Q.; Morales, C.; Yan, W.; Cook, J.; et al. Tissue-specific oncogenic activity of KRASA146T. Cancer Discov. 2019, 9, 738–755. [Google Scholar] [CrossRef]
- Chen, D.S.; Irving, B.A.; Hodi, F.S. Molecular Pathways: Next-Generation Immunotherapy—Inhibiting Programmed Death-Ligand 1 and Programmed Death-1. Clin. Cancer Res. 2012, 18, 6580–6587. [Google Scholar] [CrossRef]
- Dogan, S.; Shen, R.; Ang, D.C.; Johnson, M.L.; D’Angelo, S.P.; Paik, P.K.; Brzostowski, E.B.; Riely, G.J.; Kris, M.G.; Zakowski, M.F.; et al. Molecular Epidemiology of EGFR and KRAS Mutations in 3026 Lung Adenocarcinomas: Higher Susceptibility of Women to Smoking-Related KRAS-Mutant Cancers. Clin. Cancer Res. 2012, 18, 6169–6177. [Google Scholar] [CrossRef]
- Friedlaender, A.; Drilon, A.; Weiss, G.J.; Banna, G.L.; Addeo, A. KRAS as a druggable target in NSCLC: Rising like a phoenix after decades of development failures. Cancer Treat. Rev. 2020, 85, 101978. [Google Scholar] [CrossRef]
- Román, M.; Baraibar, I.; López, I.; Nadal, E.; Rolfo, C.; Vicent, S.; Gil-Bazo, I. KRAS oncogene in non-small cell lung cancer: Clinical perspectives on the treatment of an old target. Mol. Cancer 2018, 17, 33. [Google Scholar] [CrossRef]
- Seger, R.; Krebs, E.G. The MAPK signaling cascade. FASEB J. 1995, 9, 726–735. [Google Scholar] [CrossRef]
- Lambert, J.M.; Lambert, Q.T.; Reuther, G.W.; Malliri, A.; Siderovski, D.P.; Sondek, J.; Collard, J.G.; Der, C.J. Tiam1 mediates Ras activation of Rac by a PI(3)K-independent mechanism. Nat. Cell Biol. 2002, 4, 621–625. [Google Scholar] [CrossRef]
- Kitajima, S.; Thummalapalli, R.; Barbie, D.A. Inflammation as a driver and vulnerability of KRAS mediated oncogenesis. Semin. Cell Dev. Biol. 2016, 58, 127–135. [Google Scholar] [CrossRef]
- Golay, H.G.; Barbie, D.A. Targeting cytokine networks in KRAS-driven tumorigenesis. Expert Rev. Anticancer. Ther. 2014, 14, 869–871. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.J.; Neel, B.G.; Ikura, M. NMR-based functional profiling of RASopathies and oncogenic RAS mutations. Proc. Natl. Acad. Sci. USA 2013, 110, 4574–4579. [Google Scholar] [CrossRef]
- Bournet, B.; Muscari, F.; Buscail, C.; Assenat, E.; Barthet, M.; Hammel, P.; Selves, J.; Guimbaud, R.; Cordelier, P.; Buscail, L. KRAS G12D mutation subtype is a prognostic factor for advanced pancreatic adenocarcinoma. Clin. Transl. Gastroenterol. 2016, 7, e157. [Google Scholar] [CrossRef]
- Blons, H.; Emile, J.-F.; Le Malicot, K.; Julié, C.; Zaanan, A.; Tabernero, J.; Mini, E.; Folprecht, G.; Van Laethem, J.-L.; Thaler, J. Prognostic value of KRAS mutations in stage III colon cancer: Post hoc analysis of the PETACC8 phase III trial dataset. Ann. Oncol. 2014, 25, 2378–2385. [Google Scholar] [CrossRef] [PubMed]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Balmain, A.; Counter, C.M. A model for RAS mutation patterns in cancers: Finding the sweet spot. Nat. Rev. Cancer 2018, 18, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Killoran, R.C.; Smith, M.J. Conformational resolution of nucleotide cycling and effector interactions for multiple small GTPases determined in parallel. J. Biol. Chem. 2019, 294, 9937–9948. [Google Scholar] [CrossRef]
- Pershing, N.L.; Lampson, B.L.; Belsky, J.A.; Kaltenbrun, E.; MacAlpine, D.M.; Counter, C.M. Rare codons capacitate Kras-driven de novo tumorigenesis. J. Clin. Investig. 2015, 125, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Burgess, M.R.; Hwang, E.; Mroue, R.; Bielski, C.M.; Wandler, A.M.; Huang, B.J.; Firestone, A.J.; Young, A.; Lacap, J.A.; Crocker, L. KRAS allelic imbalance enhances fitness and modulates MAP kinase dependence in cancer. Cell 2017, 168, 817–829.e815. [Google Scholar] [CrossRef]
- Potenza, N.; Vecchione, C.; Notte, A.; De Rienzo, A.; Rosica, A.; Bauer, L.; Affuso, A.; De Felice, M.; Russo, T.; Poulet, R. Replacement of K-Ras with H-Ras supports normal embryonic development despite inducing cardiovascular pathology in adult mice. EMBO Rep. 2005, 6, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, H.; Counter, C.M. Interrogating the protein interactomes of RAS isoforms identifies PIP5K1A as a KRAS-specific vulnerability. Nat. Commun. 2018, 9, 3646. [Google Scholar] [CrossRef]
- Johnson, C.W.; Reid, D.; Parker, J.A.; Salter, S.; Knihtila, R.; Kuzmic, P.; Mattos, C. The small GTPases K-Ras, N-Ras, and H-Ras have distinct biochemical properties determined by allosteric effects. J. Biol. Chem. 2017, 292, 12981–12993. [Google Scholar] [CrossRef] [PubMed]
- Terrell, E.M.; Durrant, D.E.; Ritt, D.A.; Sealover, N.E.; Sheffels, E.; Spencer-Smith, R.; Esposito, D.; Zhou, Y.; Hancock, J.F.; Kortum, R.L. Distinct binding preferences between ras and raf family members and the impact on oncogenic ras signaling. Mol. Cell 2019, 76, 872–884.e5. [Google Scholar] [CrossRef] [PubMed]
- Buhrman, G.; Kumar, V.S.; Cirit, M.; Haugh, J.M.; Mattos, C. Allosteric modulation of Ras-GTP is linked to signal transduction through RAF kinase. J. Biol. Chem. 2011, 286, 3323–3331. [Google Scholar] [CrossRef] [PubMed]
- Catalogue of Somatic Mutation in Cancer (Cosmic). 2019. Available online: https://cancer.sanger.ac.uk/cosmic (accessed on 21 May 2021).
- Cannataro, V.L.; Gaffney, S.G.; Stender, C.; Zhao, Z.-M.; Philips, M.; Greenstein, A.E.; Townsend, J.P. Heterogeneity and mutation in KRAS and associated oncogenes: Evaluating the potential for the evolution of resistance to targeting of KRAS G12C. Oncogene 2018, 37, 2444–2455. [Google Scholar] [CrossRef]
- Nadal, E.; Chen, G.; Prensner, J.R.; Shiratsuchi, H.; Sam, C.; Zhao, L.; Kalemkerian, G.P.; Brenner, D.; Lin, J.; Reddy, R.M.; et al. KRAS-G12C Mutation Is Associated with Poor Outcome in Surgically Resected Lung Adenocarcinoma. J. Thorac. Oncol. 2014, 9, 1513–1522. [Google Scholar] [CrossRef]
- Ihle, N.T.; Byers, L.A.; Kim, E.S.; Saintigny, P.; Lee, J.J.; Blumenschein, G.R.; Tsao, A.; Liu, S.; Larsen, J.E.; Wang, J.; et al. Effect of KRAS Oncogene Substitutions on Protein Behavior: Implications for Signaling and Clinical Outcome. JNCI 2012, 104, 228–239. [Google Scholar] [CrossRef]
- Xu, S.; Long, B.N.; Boris, G.H.; Chen, A.; Ni, S.; Kennedy, M.A. Structural insight into the rearrangement of the switch I region in GTP-bound G12A K-Ras. Acta Crystallogr. Sect. D Struct. Biol. 2017, 73, 970–984. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Migoni, A.; Canning, P.; Quevedo, C.E.; Bataille, C.J.R.; Bery, N.; Miller, A.; Russell, A.J.; Phillips, S.E.V.; Carr, S.B.; Rabbitts, T.H. Structure-based development of new RAS-effector inhibitors from a combination of active and inactive RAS-binding compounds. Proc. Natl. Acad. Sci. USA 2019, 116, 2545–2550. [Google Scholar] [CrossRef]
- Zeng, M.; Lu, J.; Li, L.; Feru, F.; Quan, C.; Gero, T.W.; Ficarro, S.B.; Xiong, Y.; Ambrogio, C.; Paranal, R.M.; et al. Potent and Selective Covalent Quinazoline Inhibitors of KRAS G12C. Cell Chem. Biol. 2017, 24, 1005–1016.e1003. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef]
- Xiong, Y.; Lu, J.; Hunter, J.; Li, L.; Scott, D.; Choi, H.G.; Lim, S.M.; Manandhar, A.; Gondi, S.; Sim, T.; et al. Covalent Guanosine Mimetic Inhibitors of G12C KRAS. ACS Med. Chem. Lett. 2016, 8, 61–66. [Google Scholar] [CrossRef]
- Lito, P.; Solomon, M.; Li, L.-S.; Hansen, R.; Rosen, N. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science 2016, 351, 604–608. [Google Scholar] [CrossRef]
- Patricelli, M.P.; Janes, M.R.; Li, L.-S.; Hansen, R.; Peters, U.; Kessler, L.V.; Chen, Y.; Kucharski, J.M.; Feng, J.; Ely, T.; et al. Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discov. 2016, 6, 316–329. [Google Scholar] [CrossRef]
- Molina-Arcas, M.; Moore, C.; Rana, S.; van Maldegem, F.; Mugarza, E.; Romero-Clavijo, P.; Herbert, E.; Horswell, S.; Li, L.-S.; Janes, M.R. Development of combination therapies to maximize the impact of KRAS-G12C inhibitors in lung cancer. Sci. Transl. Med. 2019, 11, eaaw7999. [Google Scholar] [CrossRef]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Safety, G.P.; Sheets, S.D. Amgen Announces Positive Topline Phase 2 Results for Investigational KRAS G12C Inhibitor Sotorasib in Advanced Non-Small Cell Lung Cancer. Available online: https://pharmashots.com/press-releases/amgen-announces-positive-topline-phase-2-results-for-investigational-kras-g12c-inhibitor-sotorasib-in-advanced-non-small-cell-lung-cancer/ (accessed on 21 May 2021).
- Available online: https://www.mirati.com/wp-content/uploads/2020/10/Janne-849-001_NSCLC-ENA-Presentation_25Oct2020_FINAL.pdf (accessed on 21 May 2021).
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef]
- Singh, A.; Greninger, P.; Rhodes, D.; Koopman, L.; Violette, S.; Bardeesy, N.; Settleman, J. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell 2009, 15, 489–500. [Google Scholar] [CrossRef]
- Castellano, E.; Downward, J. RAS interaction with PI3K: More than just another effector pathway. Genes Cancer 2011, 2, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K pathway in human disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef]
- Muzumdar, M.D.; Chen, P.-Y.; Dorans, K.J.; Chung, K.M.; Bhutkar, A.; Hong, E.; Noll, E.M.; Sprick, M.R.; Trumpp, A.; Jacks, T. Survival of pancreatic cancer cells lacking KRAS function. Nat. Commun. 2017, 8, 1090. [Google Scholar] [CrossRef]
- Garraway, L.A.; Jänne, P.A. Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discov. 2012, 2, 214–226. [Google Scholar] [CrossRef]
- Kordiak, J.; Szemraj, J.; Grabska-Kobylecka, I.; Bialasiewicz, P.; Braun, M.; Kordek, R.; Nowak, D. Intratumor heterogeneity and tissue distribution of KRAS mutation in non-small cell lung cancer: Implications for detection of mutated KRAS oncogene in exhaled breath condensate. J. Cancer Res. Clin. Oncol. 2019, 145, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Poulikakos, P.I.; Solit, D.B. Resistance to MEK inhibitors: Should we co-target upstream? Sci. Signal. 2011, 4, pe16. [Google Scholar] [CrossRef]
- Xue, J.Y.; Zhao, Y.; Aronowitz, J.; Mai, T.T.; Vides, A.; Qeriqi, B.; Kim, D.; Li, C.; de Stanchina, E.; Mazutis, L. Rapid non-uniform adaptation to conformation-specific KRAS (G12C) inhibition. Nature 2020, 577, 421–425. [Google Scholar] [CrossRef]
- Ryan, M.B.; de la Cruz, F.F.; Phat, S.; Myers, D.T.; Wong, E.; Shahzade, H.A.; Hong, C.B.; Corcoran, R.B. Vertical pathway inhibition overcomes adaptive feedback resistance to KRASG12C inhibition. Clin. Cancer Res. 2020, 26, 1633–1643. [Google Scholar] [CrossRef]
- Franke, T. PI3K/Akt: Getting it right matters. Oncogene 2008, 27, 6473–6488. [Google Scholar] [CrossRef]
- Shao, D.D.; Xue, W.; Krall, E.B.; Bhutkar, A.; Piccioni, F.; Wang, X.; Schinzel, A.C.; Sood, S.; Rosenbluh, J.; Kim, J.W. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell 2014, 158, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Infante, J.R.; Fecher, L.A.; Falchook, G.S.; Nallapareddy, S.; Gordon, M.S.; Becerra, C.; DeMarini, D.J.; Cox, D.S.; Xu, Y.; Morris, S.R. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: A phase 1 dose-escalation trial. Lancet Oncol. 2012, 13, 773–781. [Google Scholar] [CrossRef]
- Jänne, P.A.; Van Den Heuvel, M.M.; Barlesi, F.; Cobo, M.; Mazieres, J.; Crinò, L.; Orlov, S.; Blackhall, F.; Wolf, J.; Garrido, P. Selumetinib plus docetaxel compared with docetaxel alone and progression-free survival in patients with kras-mutant advanced non–small cell lung cancer: The select-1 randomized clinical trial. JAMA 2017, 317, 1844–1853. [Google Scholar] [CrossRef]
- Blumenschein Jr, G.; Smit, E.; Planchard, D.; Kim, D.-W.; Cadranel, J.; De Pas, T.; Dunphy, F.; Udud, K.; Ahn, M.-J.; Hanna, N. A randomized phase II study of the MEK1/MEK2 inhibitor trametinib (GSK1120212) compared with docetaxel in KRAS-mutant advanced non-small-cell lung cancer (NSCLC). Ann. Oncol. 2015, 26, 894–901. [Google Scholar] [CrossRef]
- Little, A.S.; Balmanno, K.; Sale, M.J.; Newman, S.; Dry, J.R.; Hampson, M.; Edwards, P.A.; Smith, P.D.; Cook, S.J. Amplification of the driving oncogene, KRAS or BRAF, underpins acquired resistance to MEK1/2 inhibitors in colorectal cancer cells. Sci. Signal. 2011, 4, ra17. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Ebi, H.; Turke, A.B.; Coffee, E.M.; Nishino, M.; Cogdill, A.P.; Brown, R.D.; Della Pelle, P.; Dias-Santagata, D.; Hung, K.E. EGFR-mediated reactivation of MAPK signaling contributes to insensitivity of BRAF-mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012, 2, 227–235. [Google Scholar] [CrossRef]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E–Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef] [PubMed]
- Ambrogio, C.; Köhler, J.; Zhou, Z.W.; Wang, H.; Paranal, R.; Li, J.; Capelletti, M.; Caffarra, C.; Li, S.; Lv, Q.; et al. KRAS Dimerization Impacts MEK Inhibitor Sensitivity and Oncogenic Activity of Mutant KRAS. Cell 2018, 172, 857–868.e815. [Google Scholar] [CrossRef]
- Misale, S.; Fatherree, J.P.; Cortez, E.; Li, C.; Bilton, S.; Timonina, D.; Myers, D.T.; Lee, D.; Gomez-Caraballo, M.; Greenberg, M. KRAS G12C NSCLC models are sensitive to direct targeting of KRAS in combination with PI3K inhibition. Clin. Cancer Res. 2019, 25, 796–807. [Google Scholar] [CrossRef]
- Yao, Y.; Ye, H.; Qi, Z.; Mo, L.; Yue, Q.; Baral, A.; Hoon, D.S.; Vera, J.C.; Heiss, J.D.; Chen, C.C. B7-H4 (B7x)–Mediated Cross-talk between glioma-initiating cells and macrophages via the IL6/JAK/STAT3 pathway lead to poor prognosis in glioma patients. Clin. Cancer Res. 2016, 22, 2778–2790. [Google Scholar] [CrossRef]
- Dance, M.; Montagner, A.; Salles, J.-P.; Yart, A.; Raynal, P. The molecular functions of Shp2 in the Ras/Mitogen-activated protein kinase (ERK1/2) pathway. Cell. Signal. 2008, 20, 453–459. [Google Scholar] [CrossRef]
- Tanaka, N.; Lin, J.J.; Li, C.; Ryan, M.B.; Zhang, J.; Kiedrowski, L.A.; Michel, A.G.; Syed, M.U.; Fella, K.A.; Sakhi, M.; et al. Clinical acquired resistance to KRASG12C inhibition through a novel KRAS switch-II pocket mutation and polyclonal alterations converging on RAS-MAPK reactivation. Cancer Discov. 2021. [Google Scholar] [CrossRef] [PubMed]
- Koga, T.; Suda, K.; Fujino, T.; Ohara, S.; Hamada, A.; Nishino, M.; Chiba, M.; Shimoji, M.; Takemoto, T.; Arita, T.; et al. KRAS secondary mutations that confer acquired resistance to KRAS G12C inhibitors, sotorasib and adagrasib, and overcoming strategies: Insights from the in vitro experiment. J. Thorac. Oncol. 2021, in press. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.S.; Whittle, M.C.; Nakamura, K.; Abell, A.N.; Midland, A.A.; Zawistowski, J.S.; Johnson, N.L.; Granger, D.A.; Jordan, N.V.; Darr, D.B. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell 2012, 149, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Wang, Y.; Shao, C.; Huang, J.; Gan, J.; Huang, X.; Bucci, E.; Piacentini, M.; Ippolito, G.; Melino, G. COVID-19 infection: The perspectives on immune responses. Cell Death Differ. 2020, 27, 1451–1454. [Google Scholar] [CrossRef]
- Torres-Ayuso, P.; Brognard, J. Shipping out MEK inhibitor resistance with SHP2 inhibitors. Cancer Discov. 2018, 8, 1210–1212. [Google Scholar] [CrossRef]
- Mainardi, S.; Mulero-Sánchez, A.; Prahallad, A.; Germano, G.; Bosma, A.; Krimpenfort, P.; Lieftink, C.; Steinberg, J.D.; De Wit, N.; Gonçalves-Ribeiro, S. SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer in vivo. Nat. Med. 2018, 24, 961–967. [Google Scholar] [CrossRef]
- Wong, G.S.; Zhou, J.; Liu, J.B.; Wu, Z.; Xu, X.; Li, T.; Xu, D.; Schumacher, S.E.; Puschhof, J.; McFarland, J. Targeting wild-type KRAS-amplified gastroesophageal cancer through combined MEK and SHP2 inhibition. Nat. Med. 2018, 24, 968–977. [Google Scholar] [CrossRef]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A. The KRASG12C inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef]
- Boriack-Sjodin, P.A.; Margarit, S.M.; Bar-Sagi, D.; Kuriyan, J. The structural basis of the activation of Ras by Sos. Nature 1998, 394, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Hillig, R.C.; Sautier, B.; Schroeder, J.; Moosmayer, D.; Hilpmann, A.; Stegmann, C.M.; Werbeck, N.D.; Briem, H.; Boemer, U.; Weiske, J. Discovery of potent SOS1 inhibitors that block RAS activation via disruption of the RAS–SOS1 interaction. Proc. Natl. Acad. Sci. USA 2019, 116, 2551–2560. [Google Scholar] [CrossRef]
- Yaeger, R.; Solit, D.B. Overcoming adaptive resistance to KRAS inhibitors through vertical pathway targeting. Clin. Cancer Res. 2020, 26, 1538–1540. [Google Scholar] [CrossRef]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N. The clinical KRAS (G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Cullis, J.; Das, S.; Bar-Sagi, D. Kras and tumor immunity: Friend or foe? Cold Spring Harb. Perspect. Med. 2018, 8, a031849. [Google Scholar] [CrossRef]
- Briere, D.M.; Calinisan, A.; Aranda, R.; Sudhakar, N.; Hargis, L.; Gatto, S.; Fernandez-Banet, J.; Pavlicek, A.; Engstrom, L.D.; Hallin, J. Abstract LB-C09: The KRASG12C inhibitor MRTX849 reconditions the tumor immune microenvironment and leads to durable complete responses in combination with anti-PD-1 therapy in a syngeneic mouse model. AACR 2019. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Drug | Sponsor | Stage | ClinicalTrials.gov Identifier |
|---|---|---|---|
| Sotorasib | Amgen | Phase I–III | NCT04185883 |
| Adagrasib | Mirati | Phase I–III | NCT03785249 |
| GDC-6036 | Genentech/Roche | Phase I | NCT04449874 |
| JNJ-74699157 | Janssen | Phase I | NCT04006301 |
| D-1553 | Inventis Bio | Phase I | NCT04585035 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Addeo, A.; Banna, G.L.; Friedlaender, A. KRAS G12C Mutations in NSCLC: From Target to Resistance. Cancers 2021, 13, 2541. https://doi.org/10.3390/cancers13112541
Addeo A, Banna GL, Friedlaender A. KRAS G12C Mutations in NSCLC: From Target to Resistance. Cancers. 2021; 13(11):2541. https://doi.org/10.3390/cancers13112541
Chicago/Turabian StyleAddeo, Alfredo, Giuseppe Luigi Banna, and Alex Friedlaender. 2021. "KRAS G12C Mutations in NSCLC: From Target to Resistance" Cancers 13, no. 11: 2541. https://doi.org/10.3390/cancers13112541
APA StyleAddeo, A., Banna, G. L., & Friedlaender, A. (2021). KRAS G12C Mutations in NSCLC: From Target to Resistance. Cancers, 13(11), 2541. https://doi.org/10.3390/cancers13112541