
Estetrol Combined to Progestogen for Menopause or Contraception Indication Is Neutral on Breast Cancer

 , , , ,
, , , ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Ethical Study Approval
2.2. Human Samples for PDX and Ethical Study Approval
2.3. Human Treatment Modeling in Mice: Dose Delineation
2.4. Steroids and Reagents
2.5. Cell Cultures
2.6. MMTV-PyMT, MCF7 Xenograft and PDX Mouse Models
2.7. Blood Sampling and Circulating E2, E4 and P4 Quantitation
2.8. Cell Proliferation
2.9. Lung Metastasis Quantification
2.10. Protein Extraction and Western Blot Analysis
2.11. RT-qPCR and RNAseq Analysis
2.12. Immunohistochemical Staining for Ki67, ERα, pS118-ERα and PR
2.13. MARCoNI Assay
2.14. Statistical Analysis
2.15. Other Methods
3. Results
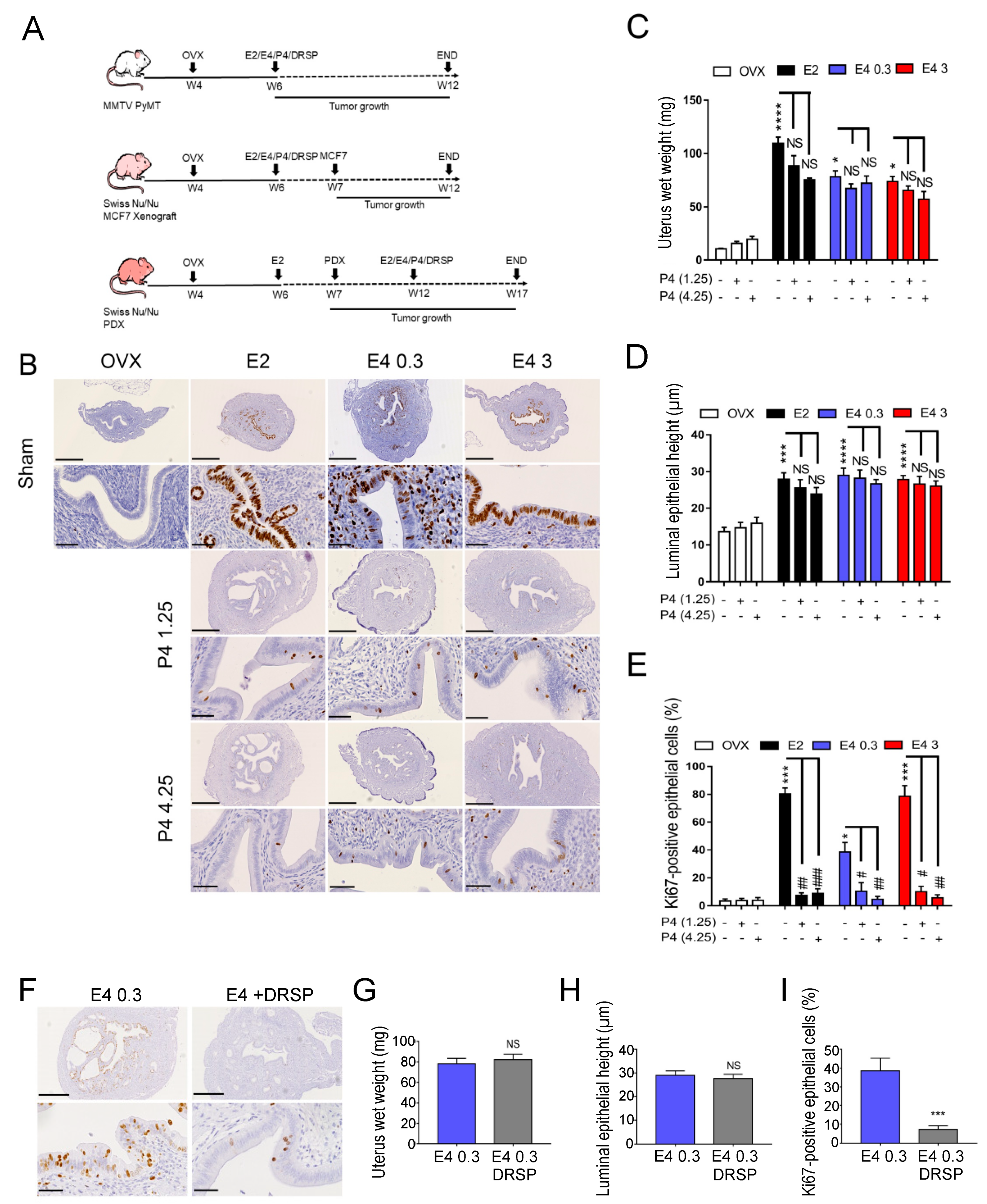
3.1. Therapeutic Dose of E4 Stimulates Endometrial Proliferation
3.2. Therapeutic Dose of E4 Is Neutral on Breast Cancer Progression
3.3. The Combination of a Therapeutic Dose of E4 with P4 Is Neutral on Breast Cancer Progression
3.4. E4 Is Less Potent Than E2 in Promoting ERα Signaling in ER+ Breast Cancer
3.5. The Therapeutic Dose of E4 Elicits No Transcriptional Activity, While a Supratherapeutic Dose of E4 Induces a Transcriptomic Profile Similar to E2 in Breast Cancer Cells
3.6. The Addition of P4 to E4 Induces Poor Transcriptional Activity in Breast Cancer Cells
3.7. E4 Is Less Potent Than E2 in Inducing the Recruitment of Co-Regulators to ERα in Breast Cancer Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Beral, V.; Peto, R.; Pirie, K. Type and timing of menopausal hormone therapy and breast cancer risk: Individual participant meta-analysis of the worldwide epidemiological evidence. Lancet 2019, 394, 1159–1168. [Google Scholar] [CrossRef]
- Department of Economic and Social Affairs. Contraceptive Use by Method 2019: Data Booklet; United Nations: New York, NY, USA, 2019. [Google Scholar]
- Mørch, L.S.; Skovlund, C.W.; Hannaford, P.C.; Iversen, L.; Fielding, S.; Lidegaard, Ø. Contemporary hormonal contraception and the risk of breast cancer. N. Engl. J. Med. 2017, 377, 2228–2239. [Google Scholar] [CrossRef]
- Calle, E.E.; Heath, C.W.; Miracle-McMahill, H.L.; Coates, R.J.; Liff, J.M.; Franceschi, S.; Talamini, R.; Chantarakul, N.; Koetsawang, S.; RachawatRachawat, D.; et al. Breast cancer and hormonal contraceptives: Collaborative reanalysis of individual data on 53 297 women with breast cancer and 100 239 women without breast cancer from 54 epidemiological studies. Lancet 1996, 347, 1713–1727. [Google Scholar]
- Iversen, L.; Sivasubramaniam, S.; Lee, A.J.; Fielding, S.; Hannaford, P.C. Lifetime cancer risk and combined oral contraceptives: The Royal College of General Practitioners’ Oral Contraception Study. Am. J. Obstet. Gynecol. 2017, 216, 580.e1–580.e9. [Google Scholar] [CrossRef] [PubMed]
- Hagen, A.A.; Barr, M.; Diczfalusy, E. Metabolism of 17-Beta-estradiol-4-14-C in early infancy. Acta Endocrinol. 1965, 49, 207–220. [Google Scholar] [CrossRef]
- Visser, M.; Foidart, J.M.; Coelingh Bennink, H.J.T. In vitro effects of estetrol on receptor binding, drug targets and human liver cell metabolism. Climacteric 2008, 11, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Gaspard, U.; Taziaux, M.; Mawet, M.; Jost, M.; Gordenne, V.; Coelingh Bennink, H.J.T.; Lobo, R.A.; Utian, W.H.; Foidart, J.-M. A multicenter, randomized study to select the minimum effective dose of estetrol (E4) in postmenopausal women (E4Relief). Menopause 2020, 27, 848–857. [Google Scholar] [CrossRef]
- Abot, A.; Fontaine, C.; Buscato, M.; Solinhac, R.; Flouriot, G.; Fabre, A.; Drougard, A.; Rajan, S.; Laine, M.; Milon, A.; et al. The uterine and vascular actions of estetrol delineate a distinctive profile of estrogen receptor α modulation, uncoupling nuclear and membrane activation. EMBO Mol. Med. 2014, 6, 1328–1346. [Google Scholar] [CrossRef]
- Coelingh Bennink, H.J.T.; Heegaard, A.M.; Visser, M.; Holinka, C.F.; Christiansen, C. Oral bioavailability and bone-sparing effects of estetrol in an osteoporosis model. Climacteric 2008, 11, 2–14. [Google Scholar] [CrossRef]
- Benoit, T.; Valera, M.-C.; Fontaine, C.; Buscato, M.; Lenfant, F.; Raymond-Letron, I.; Tremollieres, F.; Soulie, M.; Foidart, J.-M.; Game, X.; et al. Estetrol, a Fetal Selective Estrogen Receptor Modulator, Acts on the Vagina of Mice through Nuclear Estrogen Receptor α Activation. Am. J. Pathol. 2017, 187, 2499–2507. [Google Scholar] [CrossRef]
- Visser, M.; Holinka, C.F.; Coelingh Bennink, H.J.T. First human exposure to exogenous single-dose oral estetrol in early postmenopausal women. Climacteric 2008, 11 (Suppl. 1), 31–40. [Google Scholar] [CrossRef] [PubMed]
- Kuhl, H. Pharmacology of estrogens and progestogens: Influence of different routes of administration. Climacteric 2005, 8, 3–63. [Google Scholar] [CrossRef] [PubMed]
- Mawet, M.; Maillard, C.; Klipping, C.; Zimmerman, Y.; Foidart, J.M.; Bennink, H.J.T.C. Unique effects on hepatic function, lipid metabolism, bone and growth endocrine parameters of estetrol in combined oral contraceptives. Eur. J. Contracept. Reprod. Health Care 2015, 20, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Hammond, G.L.; Hogeveen, K.N.; Visser, M.; Coelingh Bennink, H.J.T. Estetrol does not bind sex hormone binding globulin or increase its production by human HepG2 cells. Climacteric 2008, 11, 41–46. [Google Scholar] [CrossRef]
- Kluft, C.; Zimmerman, Y.; Mawet, M.; Klipping, C.; Duijkers, I.J.M.; Neuteboom, J.; Foidart, J.M.; Bennink, H.C. Reduced hemostatic effects with drospirenone-based oral contraceptives containing estetrol vs. ethinyl estradiol. Contraception 2017, 95, 140–147. [Google Scholar] [CrossRef]
- Apter, D.; Zimmerman, Y.; Beekman, L.; Mawet, M.; Maillard, C.; Foidart, J.M.; Coelingh Bennink, H.J.T. Bleeding pattern and cycle control with estetrol-containing combined oral contraceptives: Results from a phase II, randomised, dose-finding study (FIESTA). Contraception 2016, 94, 366–373. [Google Scholar] [CrossRef]
- Apter, D.; Zimmerman, Y.; Beekman, L.; Mawet, M.; Maillard, C.; Foidart, J.-M.; Coelingh Bennink, H.J.T. Estetrol combined with drospirenone: An oral contraceptive with high acceptability, user satisfaction, well-being and favourable body weight control. Eur. J. Contracept. Reprod. Health Care 2017, 22, 260–267. [Google Scholar] [CrossRef]
- Gérard, C.; Blacher, S.; Communal, L.; Courtin, A.; Tskitishvili, E.; Mestdagt, M.; Munaut, C.; Noel, A.; Gompel, A.; Péqueux, C.; et al. Estetrol is a weak estrogen antagonizing estradiol-dependent mammary gland proliferation. J. Endocrinol. 2014, 224, 85–95. [Google Scholar] [CrossRef]
- Visser, M.; Coelingh Bennink, H.J.T.; Kloosterboer, H.J. Estetrol prevents and suppresses mammary tumors induced by DMBA in a rat model. Horm. Mol. Biol. Clin. Investig. 2012, 9, 95–103. [Google Scholar] [CrossRef]
- Yue, W.; Verhoeven, C.; Bernnink, H.C.; Wang, J.P.; Santen, R.J. Pro-Apoptotic Effects of Estetrol on Long-Term Estrogen-Deprived Breast Cancer Cells and at Low Doses on Hormone-Sensitive Cells. Breast Cancer Basic Clin. Res. 2019, 13, 1–8. [Google Scholar] [CrossRef]
- Singer, C.F.; Bennink, H.J.T.C.; Natter, C.; Steurer, S.; Rudas, M.; Moinfar, F.; Appels, N.; Visser, M.; Kubista, E. Antiestrogenic effects of the fetal estrogen estetrol in women with estrogen-receptor positive early breast cancer. Carcinogenesis 2014, 35, 2447–2451. [Google Scholar] [CrossRef]
- Gérard, C.; Mestdagt, M.; Tskitishvili, E.; Communal, L.; Gompel, A.; Silva, E.; Arnal, J.-F.; Lenfant, F.; Noel, A.; Foidart, J.-M.; et al. Combined estrogenic and anti-estrogenic properties of estetrol on breast cancer may provide a safe therapeutic window for the treatment of menopausal symptoms. Oncotarget 2015, 6, 17621–17636. [Google Scholar] [CrossRef] [PubMed]
- Abderrahman, B.; Maximov, P.Y.; Curpan, R.F.; Hanspal, J.S.; Fan, P.; Xiong, R.; Tonetti, D.A.; Thatcher, G.R.; Jordan, V.C. Pharmacology and Molecular Mechanisms of Clinically Relevant Estrogen Estetrol and Estrogen Mimic BMI-135 for the Treatment of Endocrine-Resistant Breast Cancer. Mol. Pharmacol. 2020, 98, 364–381. [Google Scholar] [CrossRef]
- Jozan, S.; Kreitmann, B.; Bayard, F. Different effects of oestradiol, oestriol, oestetrol and of oestrone on human breast cancer cells (MCF-7) in long term tissue culture. Acta Endocrinol. 1981, 98, 73–80. [Google Scholar] [CrossRef]
- Lin, E.Y.; Jones, J.G.; Li, P.; Zhu, L.; Whitney, K.D.; Muller, W.J.; Pollard, J.W. Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am. J. Pathol. 2003, 163, 2113–2126. [Google Scholar] [CrossRef]
- Guy, C.T.; Cardiff, R.D.; Muller, W.J. Induction of mammary tumors by expression of polyomavirus middle T oncogene: A transgenic mouse model for metastatic disease. Mol. Cell. Biol. 1992, 12, 954–961. [Google Scholar] [CrossRef]
- Coussy, F.; de Koning, L.; Lavigne, M.; Bernard, V.; Ouine, B.; Boulai, A.; El Botty, R.; Dahmani, A.; Montaudon, E.; Assayag, F.; et al. A large collection of integrated genomically characterized patient-derived xenografts highlighting the heterogeneity of triple-negative breast cancer. Int. J. Cancer 2019, 145, 1902–1912. [Google Scholar] [CrossRef] [PubMed]
- Byrne, A.T.; Alférez, D.G.; Amant, F.; Annibali, D.; Arribas, J.; Biankin, A.V.; Bruna, A.; Budinská, E.; Caldas, C.; Chang, D.K.; et al. Interrogating open issues in cancer precision medicine with patient-derived xenografts. Nat. Rev. Cancer 2017, 17, 254–268. [Google Scholar] [CrossRef]
- Montaudon, E.; Nikitorowicz-Buniak, J.; Sourd, L.; Morisset, L.; El Botty, R.; Huguet, L.; Dahmani, A.; Painsec, P.; Nemati, F.; Vacher, S.; et al. PLK1 inhibition exhibits strong anti-tumoral activity in CCND1-driven breast cancer metastases with acquired palbociclib resistance. Nat. Commun. 2020, 11, 4053. [Google Scholar] [CrossRef] [PubMed]
- Lobo, R.A.; Archer, D.F.; Kagan, R.; Kaunitz, A.M.; Constantine, G.D.; Pickar, J.H.; Graham, S.; Bernick, B.; Mirkin, S. A 17beta-Estradiol-Progesterone Oral Capsule for Vasomotor Symptoms in Postmenopausal Women: A Randomized Controlled Trial. Obstet. Gynecol. Surv. 2018, 132, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Pickar, J.H.; Bon, C.; Amadio, J.M.; Mirkin, S.; Bernick, B. Pharmacokinetics of the first combination 17beta-estradiol/progesterone capsule in clinical development for menopausal hormone therapy. Menopause 2015, 22, 2113–2126. [Google Scholar]
- Devissaguet, J.P.; Brion, N.; Lhote, O.; Deloffre, P. Pulsed estrogen therapy: Pharmacokinetics of intranasal 17-beta-estradiol (S21400) in postmenopausal women and comparison with oral and transdermal formulations. Eur. J. Drug Metab. Pharmacokinet. 1999, 24, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Gérard, C.; Gallez, A.; Dubois, C.; Drion, P.; Delahaut, P.; Quertemont, E.; Noël, A.; Pequeux, C. Accurate Control of 17β-Estradiol Long-Term Release Increases Reliability and Reproducibility of Preclinical Animal Studies. J. Mammary Gland Biol. Neoplasia 2017, 22, 1–11. [Google Scholar] [CrossRef]
- Nys, G.; Gallez, A.; Kok, M.G.M.; Cobraiville, G.; Servais, A.-C.; Piel, G.; Pequeux, C.; Fillet, M. Whole blood microsampling for the quantitation of estetrol without derivatization by liquid chromatography-tandem mass spectrometry. J. Pharm. Biomed. Anal. 2017, 140, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Péqueux, C.; Raymond-Letron, I.; Blacher, S.; Boudou, F.; Adlanmerini, M.; Fouque, M.J.; Rochaix, P.; Noël, A.; Foidart, J.M.; Krust, A.; et al. Stromal estrogen receptor-α promotes tumor growth by normalizing an increased angiogenesis. Cancer Res. 2012, 72, 3010–3019. [Google Scholar] [CrossRef]
- Chantalat, E.; Boudou, F.; Laurell, H.; Palierne, G.; Houtman, R.; Melchers, D.; Rochaix, P.; Filleron, T.; Stella, A.; Burlet-Schiltz, O.; et al. The AF-1-deficient estrogen receptor ERα46 isoform is frequently expressed in human breast tumors. Breast Cancer Res. 2016, 18, 123. [Google Scholar] [CrossRef]
- Coelingh Bennink, H.J.T.; Verhoeven, C.; Zimmerman, Y.; Visser, M.; Foidart, J.-M.; Gemzell-Danielsson, K. Pharmacokinetics of the fetal estrogen estetrol in a multiple-rising-dose study in postmenopausal women. Climacteric 2017, 20, 285–289. [Google Scholar] [CrossRef]
- Horwitz, K.B.; Mcguire, W.L. Estrogen Control of Progesterone Receptor in Human Breast Cancer. Correlation with Nuclear Processing of Estrogen Receptor. J. Biol. Chem. 1978, 253, 2223–2228. [Google Scholar] [CrossRef]
- Purdie, C.A.; Quinlan, P.; Jordan, L.B.; Ashfield, A.; Ogston, S.; Dewar, J.A.; Thompson, A.M. Progesterone receptor expression is an independent prognostic variable in early breast cancer: A population-based study. Br. J. Cancer 2014, 110, 565–572. [Google Scholar] [CrossRef]
- Siersbæk, R.; Kumar, S.; Carroll, J.S. Signaling pathways and steroid receptors modulating estrogen receptor α function in breast cancer. Genes Dev. 2018, 32, 1141–1154. [Google Scholar] [CrossRef]
- Farcas, A.M.; Nagarajan, S.; Cosulich, S.; Carroll, J.S. Mini-Review Genome-Wide Estrogen Receptor Activity in Breast Cancer. Endocrinology 2021, 162, bqaa224. [Google Scholar] [CrossRef]
- Koppen, A.; Houtman, R.; Pijnenburg, D.; Jeninga, E.H.; Ruijtenbeek, R.; Kalkhoven, E. Nuclear receptor-coregulator interaction profiling identifies TRIP3 as a novel peroxisome proliferator-activated receptor gamma cofactor. Mol. Cell. Proteom. 2009, 8, 2212–2226. [Google Scholar] [CrossRef]
- Houtman, R.; de Leeuw, R.; Rondaij, M.; Melchers, D.; Verwoerd, D.; Ruijtenbeek, R.; Martens, J.W.M.; Neefjes, J.; Michalides, R. Serine-305 Phosphorylation Modulates Estrogen Receptor Alpha Binding to a Coregulator Peptide Array, with Potential Application in Predicting Responses to Tamoxifen. Mol. Cancer Ther. 2012, 11, 805–816. [Google Scholar] [CrossRef]
- Foulds, C.E.; Feng, Q.; Ding, C.; Bailey, S.; Hunsaker, T.L.; Malovannaya, A.; Hamilton, R.A.; Gates, L.A.; Zhang, Z.; Li, C.; et al. Proteomic analysis of coregulators bound to ERα on DNA and Nucleosomes reveals coregulator dynamics. Mol. Cell 2013, 51, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Dobrolecki, L.E.; Airhart, S.D.; Alferez, D.G.; Aparicio, S.; Behbod, F.; Bentires-Alj, M.; Brisken, C.; Bult, C.J.; Cai, S.; Clarke, R.B.; et al. Patient-derived xenograft (PDX) models in basic and translational breast cancer research. Cancer Metastasis Rev. 2016, 35, 547–573. [Google Scholar] [CrossRef]
- Giretti, M.S.; Montt Guevara, M.M.; Cecchi, E.; Mannella, P.; Palla, G.; Spina, S.; Bernacchi, G.; Di Bello, S.; Genazzani, A.R.; Genazzani, A.D.; et al. Effects of Estetrol on Migration and Invasion in T47-D Breast Cancer Cells through the Actin Cytoskeleton. Front. Endocrinol. 2014, 5, 80. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Han, F.; Liu, W.; Chen, H.; Hao, X.; Jiang, X.; Yin, L.; Huang, Y.; Cao, J.; Zhang, H.; et al. ALX4, an epigenetically down regulated tumor suppressor, inhibits breast cancer progression by interfering Wnt/β-catenin pathway. J. Exp. Clin. Cancer Res. 2017, 36, 170. [Google Scholar] [CrossRef]
- Mohammed, H.; Russell, I.A.; Stark, R.; Rueda, O.M.; Hickey, T.E.; Tarulli, G.A.; Serandour, A.A.A.; Birrell, S.N.; Bruna, A.; Saadi, A.; et al. Progesterone receptor modulates ERα action in breast cancer. Nature 2015, 523, 313–317. [Google Scholar] [CrossRef]
- Murayama, T.; Gotoh, N. Patient-Derived Xenograft Models of Breast Cancer and Their Application. Cells 2019, 8, 621. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| E2 | E4 | |||||
|---|---|---|---|---|---|---|
| Gene | Log2 Fc | Adj p-value | Gene | Log2 Fc | Adj p-value | |
| Upregulation | LINC01426 | 2.60402 | 0.00148 | ALX4 | 3.42629 | 0.00009 |
| PCDHB7 | 2.31027 | 0.00546 | IGF2 | 2.46035 | 0.00923 | |
| CTB-178M22.2 | 2.08914 | 0.00193 | DCLK2 | 1.91340 | 0.00299 | |
| OTOF | 2,33653 | 0.00589 | AARD | 1.89141 | 0.00188 | |
| KLRG1 | 1.86945 | 0.00994 | EREG | 1.87651 | 0.00002 | |
| MIR3153 | 1.81026 | 0.00603 | F5 | 1.81722 | 0.00770 | |
| KCNRG | 1.72432 | 0.00064 | CCDC73 | 1.81317 | 0.00605 | |
| CFAP58-AS1 | 1.67121 | 0.00079 | HOXB2 | 1.77432 | 0.00005 | |
| C4orf47 | 1.60288 | 0.00488 | NAALADL2-AS2 | 1.72651 | 0.00017 | |
| CDKL1 | 1.56006 | 0.00018 | TEX15 | 1.66229 | 0.00932 | |
| Downregulation | LMO3 | −2.80268 | 0.00128 | ODAM | −2.59037 | 0.00098 |
| ABCC6P1 | −2.80138 | 0.00244 | ABHD11-AS1 | −2.53907 | 0.00021 | |
| GUCY1B3 | −2.80121 | 0.00211 | MAGEC1 | −2.36747 | 0.00118 | |
| TSPEAR-AS1 | −2.63897 | 0.00351 | LRRC39 | −2.35825 | 0.00007 | |
| CEMP1 | −2.63778 | 0.00374 | NCALD | −2.25073 | 0.00003 | |
| LOC101929584 | −2.48102 | 0.00399 | CD4 | −2.23406 | 0.00861 | |
| CFAP57 | −2.02884 | 0.00452 | CECR1 | −2.01936 | 0.00307 | |
| MIR24-1 | −1.98172 | 0.00827 | PAX7 | −1.95793 | 0.00975 | |
| LOC102724450 | −1.97767 | 0.00351 | HAPLN1 | −1.95591 | 0.00021 | |
| PLA2G10 | −1.93472 | 0.00159 | RPLP0P2 | −1.84793 | 0.00017 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gallez, A.; Blacher, S.; Maquoi, E.; Konradowski, E.; Joiret, M.; Primac, I.; Gérard, C.; Taziaux, M.; Houtman, R.; Geris, L.; et al. Estetrol Combined to Progestogen for Menopause or Contraception Indication Is Neutral on Breast Cancer. Cancers 2021, 13, 2486. https://doi.org/10.3390/cancers13102486
Gallez A, Blacher S, Maquoi E, Konradowski E, Joiret M, Primac I, Gérard C, Taziaux M, Houtman R, Geris L, et al. Estetrol Combined to Progestogen for Menopause or Contraception Indication Is Neutral on Breast Cancer. Cancers. 2021; 13(10):2486. https://doi.org/10.3390/cancers13102486
Chicago/Turabian StyleGallez, Anne, Silvia Blacher, Erik Maquoi, Erika Konradowski, Marc Joiret, Irina Primac, Céline Gérard, Mélanie Taziaux, René Houtman, Liesbet Geris, and et al. 2021. "Estetrol Combined to Progestogen for Menopause or Contraception Indication Is Neutral on Breast Cancer" Cancers 13, no. 10: 2486. https://doi.org/10.3390/cancers13102486
APA StyleGallez, A., Blacher, S., Maquoi, E., Konradowski, E., Joiret, M., Primac, I., Gérard, C., Taziaux, M., Houtman, R., Geris, L., Lenfant, F., Marangoni, E., Sounni, N. E., Foidart, J.-M., Noël, A., & Péqueux, C. (2021). Estetrol Combined to Progestogen for Menopause or Contraception Indication Is Neutral on Breast Cancer. Cancers, 13(10), 2486. https://doi.org/10.3390/cancers13102486