
Evidence of Cooperation between Hippo Pathway and RAS Mutation in Thyroid Carcinomas


 and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
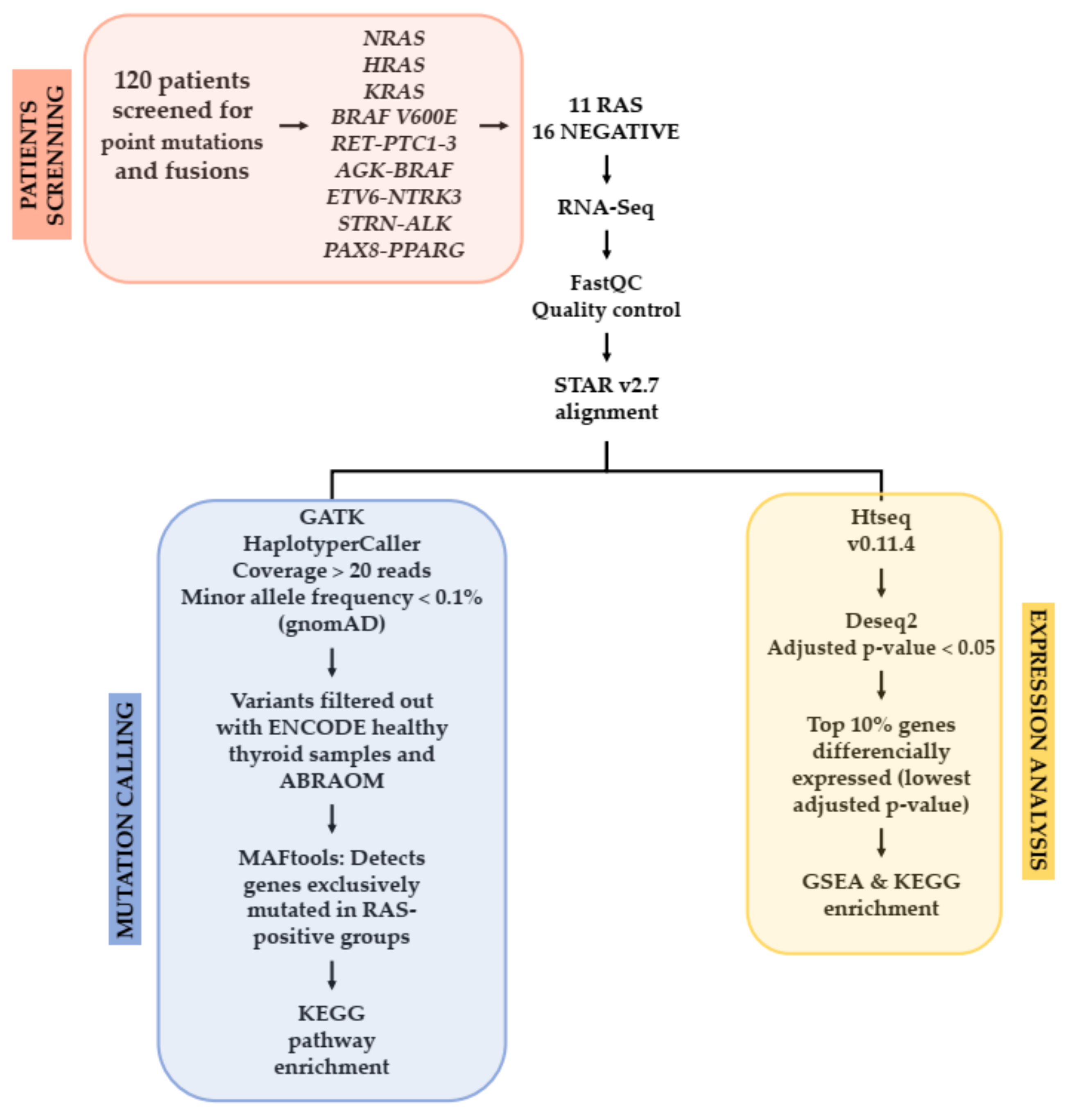
2.1. Patients
2.2. Study Design
2.3. RNA and DNA Isolation
2.4. RNA Library Preparation
2.5. RNA-Sequencing and Data Analysis
2.6. Experimental Validation by Sanger Sequencing
2.7. In Silico Analysis
2.8. Differentially Expressed Genes Analysis in a RAS-Positive Cohort
2.9. TCGA Cohort Mutation Analysis
2.10. TCGA Expression Analysis
2.11. Pathway Enrichment Analysis Using GSEA and KEGG
2.12. Expression Analysis by Quantitative RT-PCR (qRT-PCR)
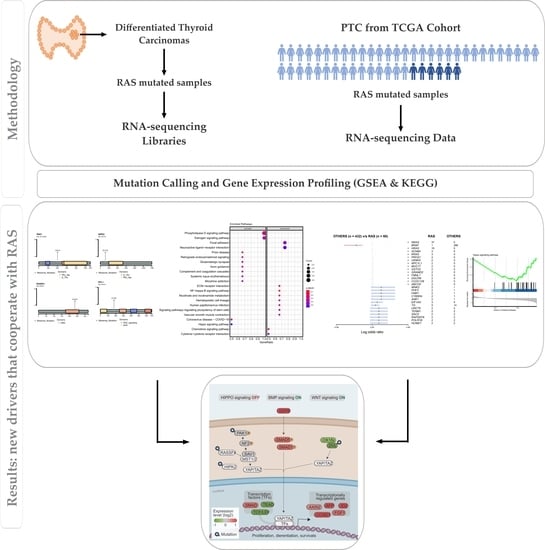
3. Results
3.1. Outline of RNA-Seq Data
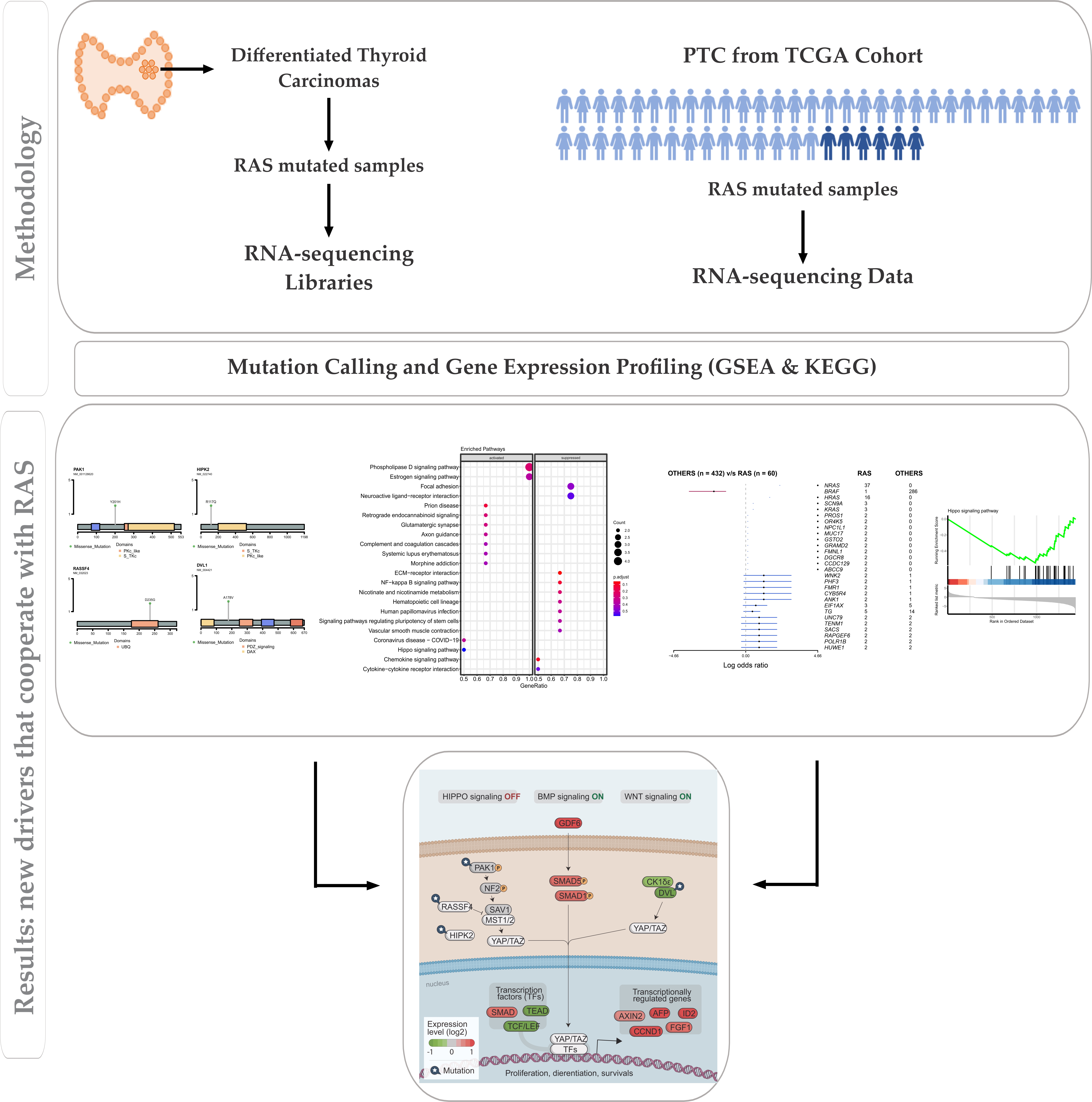
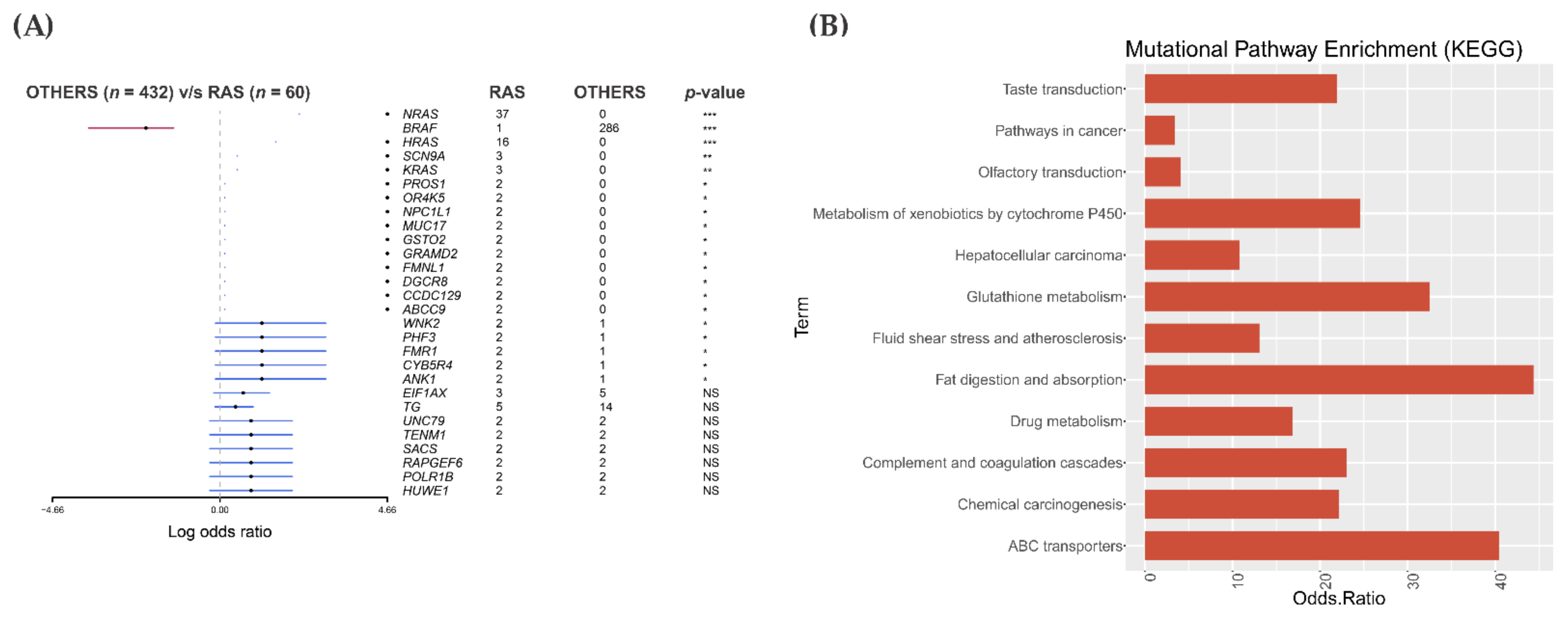
3.2. Somatic Single-Nucleotide Variants in the RAS-Positive Cohort
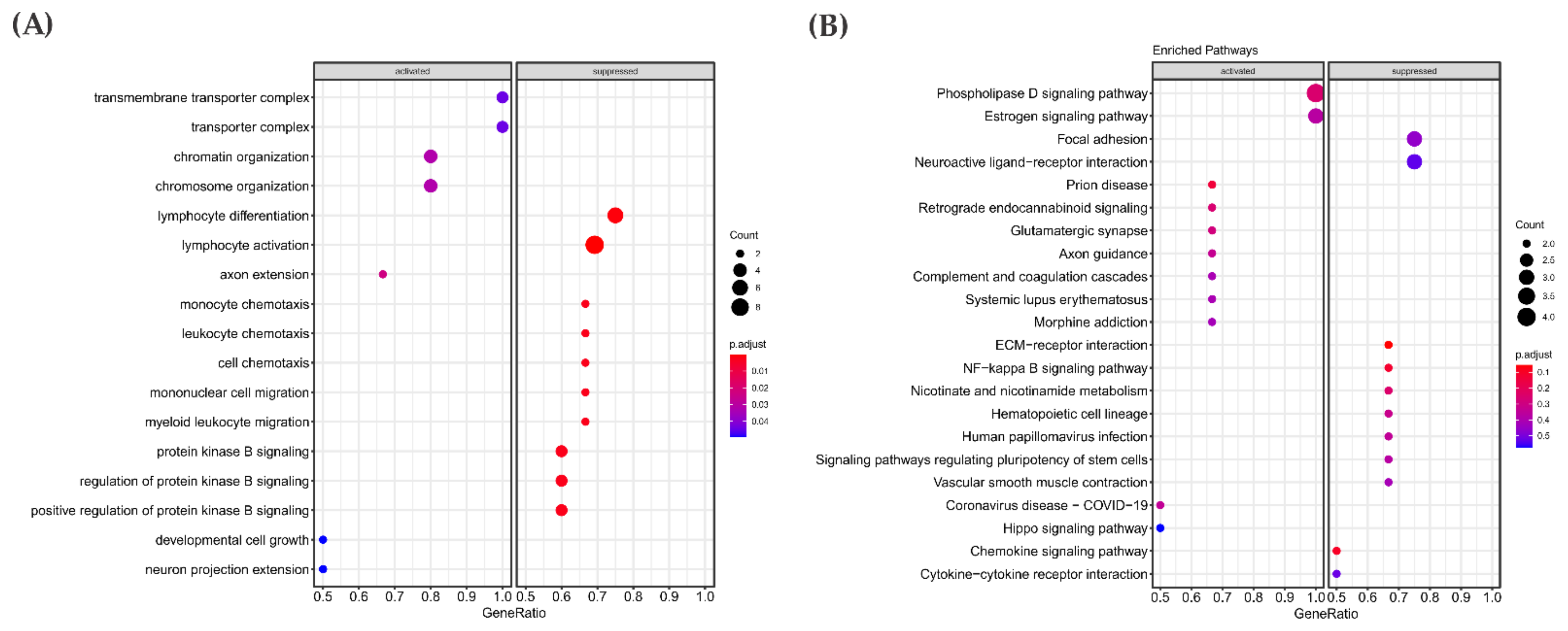
3.3. Pathway Enrichment Analysis in the RAS-Positive Cohort
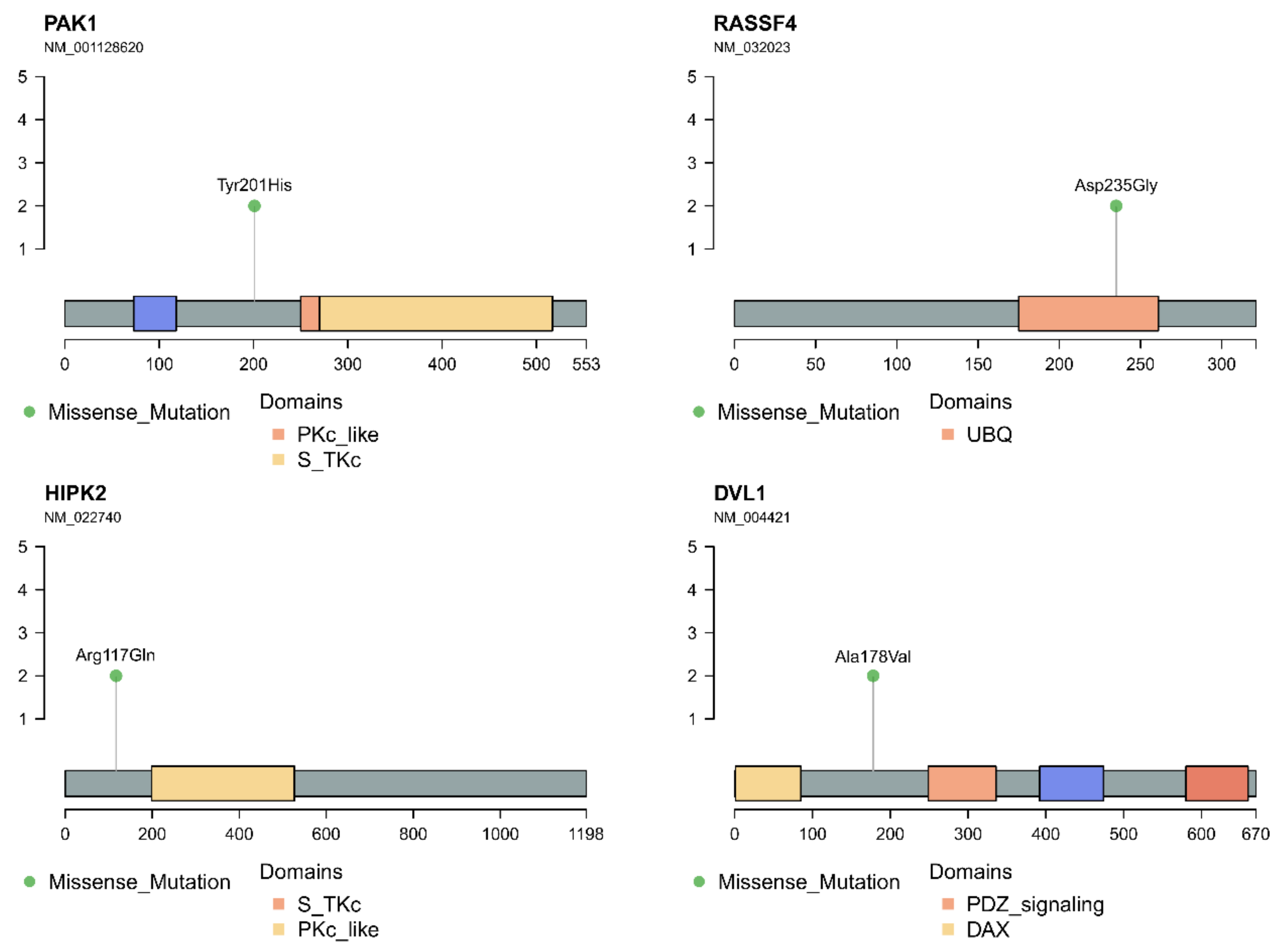
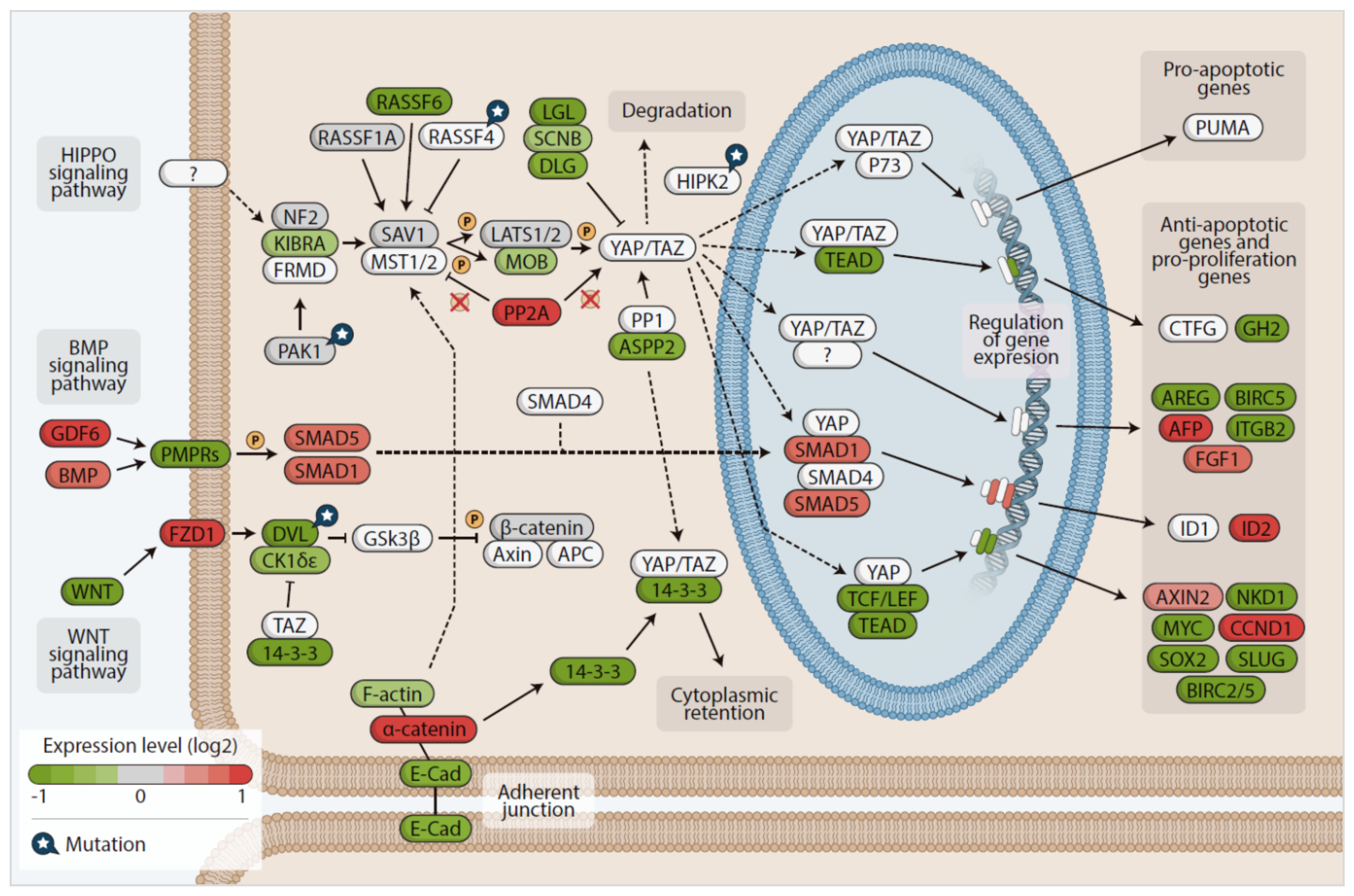
3.4. In Silico Analysis of Hippo Pathway Mutated Genes in RAS-Positive Samples and Its Mutational Impact
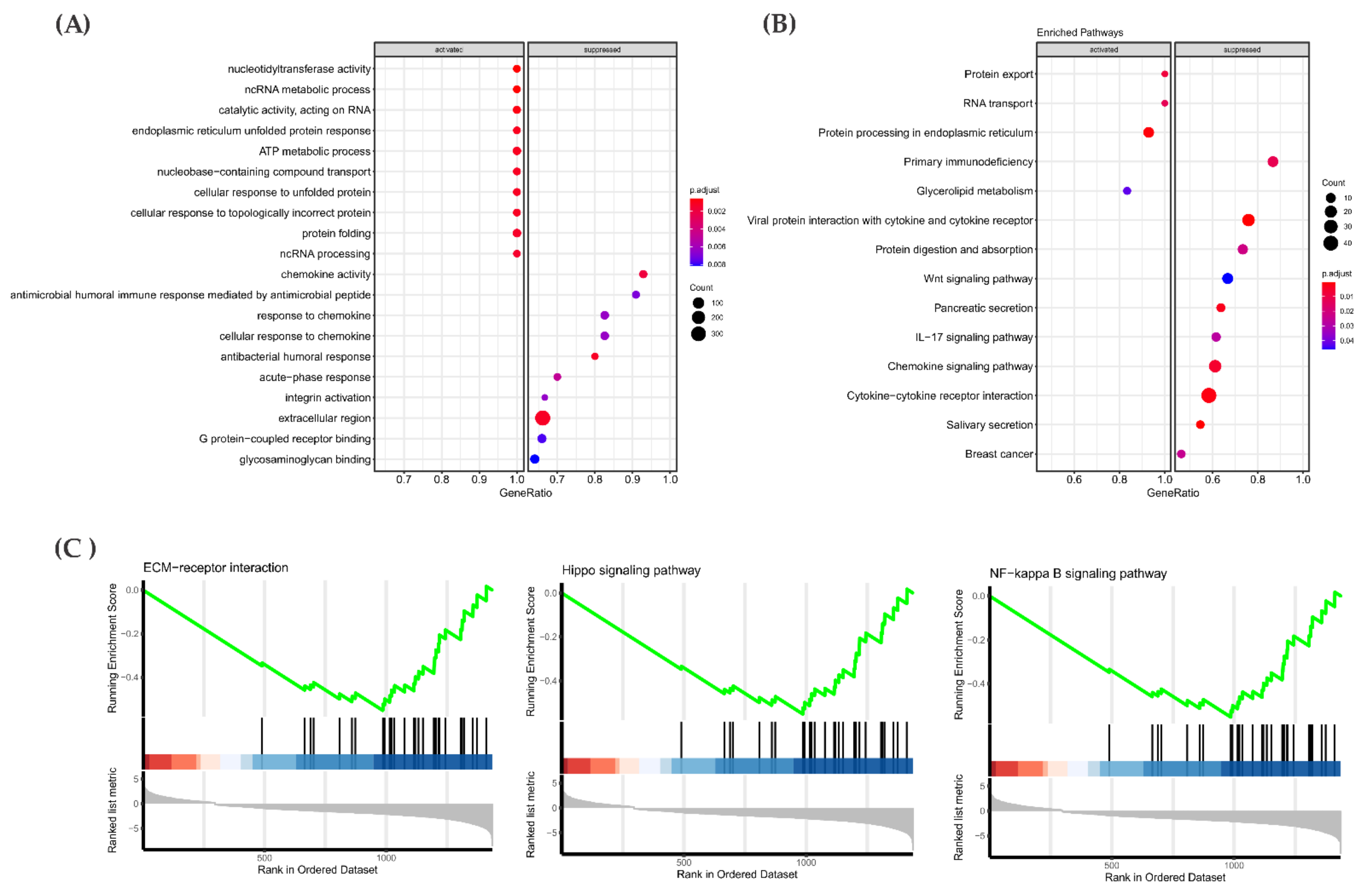
3.5. Mutational Landscape and Pathway Enrichment in the RAS-Positive Samples from TCGA Cohort
3.6. Gene Expression Pattern in the RAS-Positive Samples
3.7. Gene Expression Analysis: Proof of Concept
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef]
- Agrawal, N.; Akbani, R.; Aksoy, B.A.; Ally, A.; Arachchi, H.; Asa, S.L.; Auman, J.T.; Balasundaram, M.; Balu, S.; Baylin, S.B.; et al. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014, 159, 676–690. [Google Scholar] [CrossRef] [PubMed]
- Bastos, A.U.; De Jesus, A.C.; Cerutti, J.M. ETV6-NTRK3 and STRN-ALK kinase fusions are recurrent events in papillary thyroid cancer of adult population. Eur. J. Endocrinol. 2018, 178, 83–91. [Google Scholar] [CrossRef]
- Hemerly, J.P.; Bastos, A.U.; Cerutti, J.M. Identification of several novel non-p.R132 IDH1 variants in thyroid carcinomas. Eur. J. Endocrinol. 2010, 163, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Oler, G.; Cerutti, J.M. High prevalence of BRAF mutation in a Brazilian cohort of patients with sporadic papillary thyroid carcinomas: Correlation with more aggressive phenotype and decreased expression of iodide-metabolizing genes. Cancer 2009, 115, 972–980. [Google Scholar] [CrossRef]
- Chen, H.; Luthra, R.; Routbort, M.J.; Patel, K.P.; Cabanillas, M.E.; Broaddus, R.R.; Williams, M.D. Molecular profile of advanced thyroid carcinomas by next-generation sequencing: Characterizing tumors beyond diagnosis for targeted therapy. Mol. Cancer Ther. 2018, 17, 1575–1584. [Google Scholar] [CrossRef]
- Duan, H.; Liu, X.; Ren, X.; Zhang, H.; Wu, H.; Liang, Z. Mutation profiles of follicular thyroid tumors by targeted sequencing. Diagn. Pathol. 2019, 14, 1–10. [Google Scholar] [CrossRef]
- Nicolson, N.G.; Murtha, T.D.; Dong, W.; O Paulsson, J.; Choi, J.; Barbieri, A.L.; Brown, T.C.; Kunstman, J.W.; Larsson, C.; Prasad, M.L.; et al. Comprehensive genetic analysis of follicular thyroid carcinoma predicts prognosis independent of histology. J. Clin. Endocrinol. Metab. 2018, 103, 2640–2650. [Google Scholar] [CrossRef]
- Ohba, K.; Mitsutake, N.; Matsuse, M.; Rogounovitch, T.; Nishino, N.; Oki, Y.; Goto, Y.; Kakudo, K. Encapsulated papillary thyroid tumor with delicate nuclear changes and a KRAS mutation as a possible novel subtype of borderline tumor. J. Pathol. Transl. Med. 2019, 53, 136–141. [Google Scholar] [CrossRef]
- Landa, I.; Ibrahimpasic, T.; Boucai, L.; Sinha, R.; Knauf, J.A.; Shah, R.H.; Dogan, S.; Ricarte-Filho, J.C.; Krishnamoorthy, G.P.; Xu, B.; et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J. Clin. Investig. 2016, 126, 1052–1066. [Google Scholar] [CrossRef] [PubMed]
- Fagin, J.A. Minireview: Branded from the start—Distinct oncogenic initiating events may determine tumor fate in the thyroid. Mol. Endocrinol. 2002, 16, 903–911. [Google Scholar] [CrossRef][Green Version]
- Nikiforova, M.N. Molecular testing of thyroid FNA samples. In Diagnostic Pathology and Molecular Genetics of the Thyroid; Nikiforov, Y.E., Biddinger, P.W., Thompson, L.D.R., Eds.; Lippincott Williams & Wilkins: Baltimore, MD, USA, 2009; pp. 94–102. [Google Scholar]
- Motoi, N.; Sakamoto, A.; Yamochi, T.; Horiuchi, H.; Motoi, T.; Machinami, R. Role of Ras mutation in the progression of thyroid carcinoma of follicular epithelial origin. Pathol. Res. Pract. 2000, 196, 1–7. [Google Scholar] [CrossRef]
- Nikiforov, Y.E.; Nikiforova, M.N. Molecular genetics and diagnosis of thyroid cancer. Nat. Rev. Endocrinol. 2011, 7, 569–580. [Google Scholar] [CrossRef]
- Patel, S.G.; Carty, S.E.; McCoy, K.L.; Ohori, N.P.; Lebeau, S.O.; Seethala, R.R.; Nikiforova, M.N.; Nikiforov, Y.E.; Yip, L. Preoperative detection of RAS mutation may guide extent of thyroidectomy. Surgery 2017, 161, 168–175. [Google Scholar] [CrossRef]
- Garcia-Rostan, G.; Zhao, H.; Camp, R.L.; Pollan, M.; Herrero, A.; Pardo, J.; Wu, R.; Carcangiu, M.L.; Costa, J.; Tallini, G. Ras mutations are associated with aggressive tumor phenotypes and poor prognosis in thyroid cancer. J. Clin. Oncol. 2003, 21, 3226–3235. [Google Scholar] [CrossRef] [PubMed]
- Paniza, A.C.D.J.; Mendes, T.B.; Vianna, M.D.B.; Thomaz, D.M.D.; O Chiappini, P.B.; Colazza-Gama, G.A.; Lindsey, S.C.; De Carvalho, M.B.; Alves, V.A.F.; Curione, O.; et al. Revised criteria for diagnosis of NIFTP reveals a better correlation with tumor biological behavior. Endocr. Connect. 2019, 8, 1529–1538. [Google Scholar] [CrossRef] [PubMed]
- Saavedra, H.I.; Knauf, J.A.; Shirokawa, J.M.; Wang, J.; Ouyang, B.; Elisei, R.; Stambrook, P.J.; Fagin, J.A. The RAS oncogene induces genomic instability in thyroid PCCL3 cells via the MAPK pathway. Oncogene 2000, 19, 3948–3954. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic Ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Tuveson, D.A.; Shaw, A.T.; Willis, N.A.; Silver, D.P.; Jackson, E.L.; Chang, S.; Mercer, K.L.; Grochow, R.; Hock, H.; Crowley, D.; et al. Endogenous oncogenic K-rasG12D stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell 2004, 5, 375–387. [Google Scholar] [CrossRef]
- Haigis, K.M.; Kendall, K.R.; Wang, Y.; Cheung, A.; Haigis, M.C.; Glickman, J.N.; Niwa-Kawakita, M.; Sweet-Cordero, A.; Sebolt-Leopold, J.; Shannon, K.M.; et al. Differential effects of oncogenic K-Ras and N-Ras on proliferation, differentiation and tumor progression in the colon. Nat. Genet. 2008, 40, 600–608. [Google Scholar] [CrossRef]
- Schuhmacher, A.J.; Guerra, C.; Sauzeau, V.; Cañamero, M.; Bustelo, X.R.; Barbacid, M. A mouse model for Costello syndrome reveals an Ang II-mediated hypertensive condition. J. Clin. Investig. 2008, 118, 2169–2179. [Google Scholar] [CrossRef]
- Franco, A.T.; Malaguarnera, R.; Refetoff, S.; Liao, X.-H.; Lundsmith, E.; Kimura, S.; Pritchard, C.; Marais, R.; Davies, T.F.; Weinstein, L.S.; et al. Thyrotrophin receptor signaling dependence of braf-induced thyroid tumor initiation in mice. Proc. Natl. Acad. Sci. USA 2011, 108, 1615–1620. [Google Scholar] [CrossRef]
- Miller, K.A.; Yeager, N.; Baker, K.; Liao, X.-H.; Refetoff, S.; Di Cristofano, A. Oncogenic Kras requires simultaneous PI3K signaling to induce ERK activation and transform thyroid epithelial cells in vivo. Cancer Res. 2009, 69, 3689–3694. [Google Scholar] [CrossRef]
- Chen, X.; Mitsutake, N.; Laperle, K.; Akeno, N.; Zanzonico, P.; Longo, V.A.; Mitsutake, S.; Kimura, E.T.; Geiger, H.; Santos, E.; et al. Endogenous expression of HrasG12V induces developmental defects and neoplasms with copy number imbalances of the oncogene. Proc. Natl. Acad. Sci. USA 2009, 106, 7979–7984. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, G.P.; Davidson, N.R.; Leach, S.D.; Zhao, Z.; Lowe, S.W.; Lee, G.; Landa, I.; Nagarajah, J.; Saqcena, M.; Singh, K.; et al. EIF1AX and RAS mutations cooperate to drive thyroid tumorigenesis through ATF4 and c-MYC. Cancer Discov. 2019, 9, 264–281. [Google Scholar] [CrossRef] [PubMed]
- Ibrahimpasic, T.; Ghossein, R.; Shah, J.P.; Ganly, I. Poorly differentiated carcinoma of the thyroid gland: Current status and future prospects. Thyroid 2019, 29, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.-K.; Song, Y.S.; Lee, E.K.; Hwang, J.; Kim, H.H.; Jung, G.; A Kim, Y.; Kim, S.-J.; Cho, S.W.; Won, J.-K.; et al. Integrative analysis of genomic and transcriptomic characteristics associated with progression of aggressive thyroid cancer. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Owen, D.H.; Konda, B.; Sipos, J.; Liu, T.; Webb, A.; Ringel, M.D.; Timmers, C.D.; Shah, M.H. KRAS G12V mutation in acquired resistance to combined BRAF and MEK inhibition in papillary thyroid cancer. J. Natl. Compr. Cancer Netw. 2019, 17, 409–413. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Depristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; Del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76. [Google Scholar] [CrossRef] [PubMed]
- Naslavsky, M.S.; Yamamoto, G.L.; De Almeida, T.F.; Ezquina, S.A.M.; Sunaga, D.Y.; Pho, N.; Bozoklian, D.; Sandberg, T.O.M.; Brito, L.A.; Lazar, M.; et al. Exomic variants of an elderly cohort of Brazilians in the ABraOM database. Hum. Mutat. 2017, 38, 751–763. [Google Scholar] [CrossRef]
- Mayakonda, A.; Lin, D.-C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef]
- Venselaar, H.; Beek, T.A.H.T.; Kuipers, R.K.P.; Hekkelman, M.L.; Vriend, G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinform. 2010, 11, 548. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; He, Q.-Y. ReactomePA: An R/Bioconductor package for reactome pathway analysis and visualization. Mol. BioSyst. 2016, 12, 477–479. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. ClusterProfiler: An R Package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Luo, W.; Friedman, M.S.; Shedden, K.; Hankenson, K.D.; Woolf, P.J. GAGE: Generally applicable gene set enrichment for pathway analysis. BMC Bioinform. 2009, 10, 161. [Google Scholar] [CrossRef]
- Cerutti, J.M.; Delcelo, R.; Amadei, M.J.; Nakabashi, C.; Maciel, R.M.; Peterson, B.; Shoemaker, J.; Riggins, G.J. A preoperative diagnostic test that distinguishes benign from malignant thyroid carcinoma based on gene expression. J. Clin. Investig. 2004, 113, 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Cordioli, M.I.C.V.; Moraes, L.; Alves, M.T.D.S.; Delcelo, R.; Monte, O.; Longui, C.A.; Cury, A.N.; Cerutti, J.M. Thyroid-specific genes expression uncovered age-related differences in pediatric thyroid carcinomas. Int. J. Endocrinol. 2016, 2016, 1–9. [Google Scholar] [CrossRef]
- The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’Ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Rider, L.; Shatrova, A.; Feener, E.P.; Webb, L.; Diakonova, M. JAK2 Tyrosine kinase phosphorylates PAK1 and regulates PAK1 activity and functions. J. Biol. Chem. 2007, 282, 30985–30996. [Google Scholar] [CrossRef]
- Martínez-Jiménez, F.; Muiños, F.; Sentís, I.; Deu-Pons, J.; Reyes-Salazar, I.; Arnedo-Pac, C.; Mularoni, L.; Pich, O.; Bonet, J.; Kranas, H.; et al. A compendium of mutational cancer driver genes. Nat. Rev. Cancer 2020, 20, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Umetani, N.; Sasaki, S.; Masaki, T.; Watanabe, T.; Matsuda, K.; Muto, T. Involvement of APC and K-Ras mutation in non-polypoid colorectal tumorigenesis. Br. J. Cancer 2000, 82, 9–15. [Google Scholar] [CrossRef]
- Stites, E.C.; Trampont, P.C.; Haney, L.B.; Walk, S.F.; Ravichandran, K.S. Cooperation between noncanonical Ras network mutations. Cell Rep. 2015, 10, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Jefferies, M.T.; Cox, A.C.; Shorning, B.Y.; Meniel, V.; Griffiths, D.; Kynaston, H.G.; Smalley, M.J.; Clarke, A.R. PTEN loss and activation of K-RAS and β-catenin cooperate to accelerate prostate tumourigenesis: PTEN, K-RAS and β-catenin in prostate cancer. J. Pathol. 2017, 243, 442–456. [Google Scholar] [CrossRef]
- Romero, R.; Sayin, V.I.; Davidson, S.M.; Bauer, M.R.; Singh, S.X.; Leboeuf, S.E.; Karakousi, T.R.; Ellis, D.C.; Bhutkar, A.; Sánchez-Rivera, F.J.; et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med. 2017, 23, 1362–1368. [Google Scholar] [CrossRef]
- Garcia-Rendueles, M.E.; Ricarte-Filho, J.C.; Untch, B.R.; Landa, I.; Knauf, J.A.; Voza, F.; Smith, V.E.; Ganly, I.; Taylor, B.S.; Persaud, Y.; et al. NF2 loss promotes oncogenic RAS-induced thyroid cancers via YAP-dependent transactivation of RAS proteins and sensitizes them to MEK inhibition. Cancer Discov. 2015, 5, 1178–1193. [Google Scholar] [CrossRef] [PubMed]
- Justice, R.W.; Zilian, O.; Woods, D.F.; Noll, M.; Bryant, P.J. The Drosophila tumor suppressor gene warts encodes a homolog of human myotonic dystrophy kinase and is required for the control of cell shape and proliferation. Genes Dev. 1995, 9, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Wang, W.; Zhang, S.; Stewart, R.A.; Yu, W. Identifying tumor suppressors in genetic mosaics: The Drosophila lats gene encodes a putative protein kinase. Development 1995, 121, 1053–1063. [Google Scholar] [PubMed]
- Harvey, K.F.; Zhang, X.; Thomas, D.M. The Hippo pathway and human cancer. Nat. Rev. Cancer 2013, 13, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Azad, T.; Rezaei, R.; Surendran, A.; Singaravelu, R.; Boulton, S.; Dave, J.; Bell, J.C.; Ilkow, C.S. Hippo signaling pathway as a central mediator of receptors tyrosine kinases (RTKs) in tumorigenesis. Cancers 2020, 12, 2042. [Google Scholar] [CrossRef]
- Grusche, F.A.; Richardson, H.E.; Harvey, K.F. Upstream regulation of the hippo size control pathway. Curr. Biol. 2010, 20, R574–R582. [Google Scholar] [CrossRef]
- Hansen, C.G.; Moroishi, T.; Guan, K.-L. YAP and TAZ: A nexus for Hippo signaling and beyond. Trends Cell Biol. 2015, 25, 499–513. [Google Scholar] [CrossRef]
- Piccolo, S.; Cordenonsi, M.; Dupont, S. Molecular pathways: YAP and TAZ take center stage in organ growth and tumorigenesis. Clin. Cancer Res. 2013, 19, 4925–4930. [Google Scholar] [CrossRef]
- Hong, X.; Nguyen, H.T.; Chen, Q.; Zhang, R.; Hagman, Z.; Voorhoeve, P.M.; Cohen, S.M. Opposing activities of the Ras and Hippo pathways converge on regulation of YAP protein turnover. EMBO J. 2014, 33, 2447–2457. [Google Scholar] [CrossRef]
- Volodko, N.; Gordon, M.; Salla, M.; Abu Ghazaleh, H.; Baksh, S. RASSF tumor suppressor gene family: Biological functions and regulation. FEBS Lett. 2014, 588, 2671–2684. [Google Scholar] [CrossRef]
- Sabra, H.; Brunner, M.; Mandati, V.; Wehrle-Haller, B.; Lallemand, D.; Ribba, A.-S.; Chevalier, G.; Guardiola, P.; Block, M.R.; Bouvard, D. β1 integrin-dependent Rac/group I PAK signaling mediates YAP activation of Yes-associated protein 1 (YAP1) via NF2/merlin. J. Biol. Chem. 2017, 292, 19179–19197. [Google Scholar] [CrossRef]
- Park, H.W.; Kim, Y.C.; Yu, B.; Moroishi, T.; Mo, J.-S.; Plouffe, S.W.; Meng, Z.; Lin, K.C.; Yu, F.-X.; Alexander, C.M.; et al. Alternative Wnt signaling activates YAP/TAZ. Cell 2015, 162, 780–794. [Google Scholar] [CrossRef]
- Kim, N.H.; Lee, Y.; Yook, J.I. Dishevelling Wnt and Hippo. BMB Rep. 2018, 51, 425–426. [Google Scholar] [CrossRef]
- Varelas, X.; Miller, B.W.; Sopko, R.; Song, S.; Gregorieff, A.; Fellouse, F.A.; Sakuma, R.; Pawson, T.; Hunziker, W.; McNeill, H.; et al. The Hippo pathway regulates Wnt/β-catenin signaling. Dev. Cell 2010, 18, 579–591. [Google Scholar] [CrossRef]
- Poon, C.L.; Zhang, X.; Lin, J.I.; Manning, S.A.; Harvey, K.F. Homeodomain-interacting protein kinase regulates hippo pathway-dependent tissue growth. Curr. Biol. 2012, 22, 1587–1594. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Verheyen, E.M. Homeodomain-interacting protein kinase regulates yorkie activity to promote tissue growth. Curr. Biol. 2012, 22, 1582–1586. [Google Scholar] [CrossRef] [PubMed]
- Mo, J.; Park, H.W.; Guan, K. The Hippo signaling pathway in stem cell biology and cancer. EMBO Rep. 2014, 15, 642–656. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Moroishi, T.; Guan, K.-L. Mechanisms of Hippo pathway regulation. Genes Dev. 2016, 30, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Deshmukh, H.; Gutmann, R.J.; Emnett, R.J.; Rodriguez, F.J.; Watson, M.A.; Nagarajan, R.; Gutmann, D.H. Alterations of BRAF and HIPK2 loci predominate in sporadic pilocytic astrocytoma. Neurology 2009, 73, 1526–1531. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, J.Z.; Vergara, I.A.; Zhang, Y.; Szeto, P.; Yang, L.; Mintoff, C.; Colebatch, A.; McIntosh, L.; Mitchell, K.A.; et al. Somatic hypermutation of the YAP oncogene in a human cutaneous melanoma. Mol. Cancer Res. 2019, 17, 1435–1449. [Google Scholar] [CrossRef] [PubMed]
- Blaquiere, J.A.; Wong, K.K.L.; Kinsey, S.D.; Wu, J.; Verheyen, E.M. Homeodomain-interacting protein kinase promotes tumorigenesis and metastatic cell behavior. Dis. Model. Mech. 2018, 11, 031146. [Google Scholar] [CrossRef] [PubMed]
- Knippschild, U.; Krüger, M.; Richter, J.; Xu, P.; Garcia-Reyes, B.; Peifer, C.; Halekotte, J.; Bakulev, V.; Bischof, J. The CK1 family: Contribution to cellular stress response and its role in carcinogenesis. Front. Oncol. 2014, 4, 96. [Google Scholar] [CrossRef] [PubMed]
- Fulcher, L.J.; Sapkota, G.P. Functions and regulation of the serine/threonine protein kinase CK1 family: Moving beyond promiscuity. Biochem. J. 2020, 477, 4603–4621. [Google Scholar] [CrossRef]
- Zeng, R.; Dong, J. The Hippo signaling pathway in drug resistance in cancer. Cancers 2021, 13, 318. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, C.-Y.; Zha, Z.-Y.; Zhao, B.; Yao, J.; Zhao, S.; Xiong, Y.; Lei, Q.-Y.; Guan, K.-L. TEAD transcription factors mediate the function of TAZ in cell growth and epithelial-mesenchymal transition. J. Biol. Chem. 2009, 284, 13355–13362. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, A.M.; Vyas, R.; Gramann, A.K.; Dresser, K.; Gujja, S.; Bhatnagar, S.; Chhangawala, S.; Gomes, C.B.F.; Xi, H.S.; Lian, C.G.; et al. Ligand-activated BMP signaling inhibits cell differentiation and death to promote melanoma. J. Clin. Investig. 2017, 128, 294–308. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
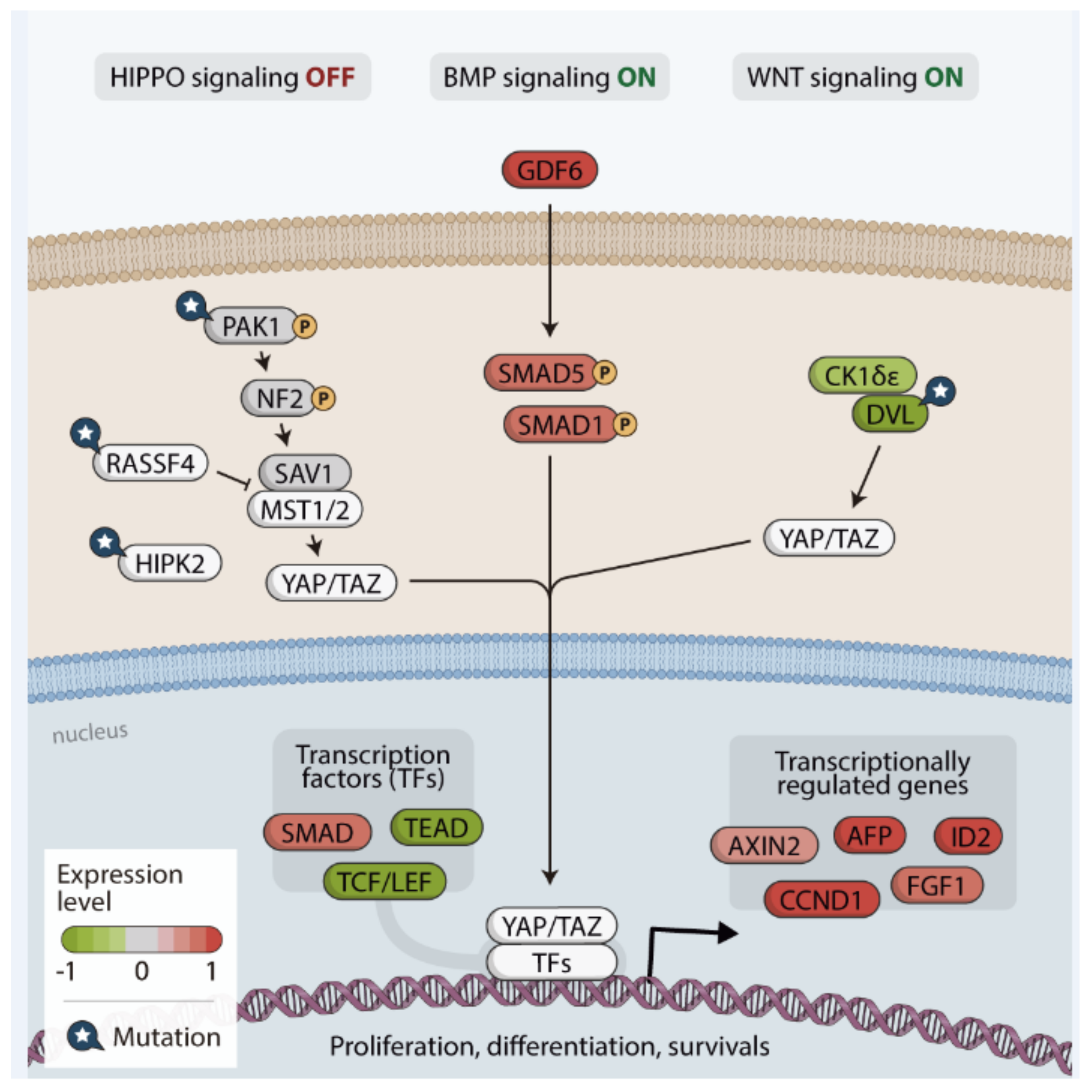{kind=link}
| Sample | RAS Mutation | No. of Reads (Millions) | Reads Mapping Mutant Allele (%) | Tumor Type * | Tumor Size (cm) | Multifocal | Extrathyroidal Extension | Vascular Invasion | Metastases | Recurrence | Age at Diagnosis | Gender |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | N Q61R | 136 | 66 | FVPTC | 5.0 | N | Y | N | N | N | 32 | M |
| 2 | N Q61K | 113 | 62 | FVPTC | 4.0 | N | N | N | N | N | 40 | M |
| 3 | N Q61R | 75 | 78 | FVPTC | 5.0 | Y | N | N | N | N | 28 | F |
| 4 | N Q61R | 106 | 33 | FVPTC | 1.7 | Y | N | N | N | N | 55 | F |
| 5 | N Q61R | 118 | 56 | FVPTC | 3.0 | N | N | N | N | N | 36 | F |
| 6 | H Q61R | 242 | 71 | FVPTC | 1.1 | N | N | N | N | N | 45 | F |
| 7 | N Q61R | 93 | 70 | FTC | 3.5 | Y | Y | N | Y | N | 45 | F |
| 8 | K Q61R | 168 | 22 | FTC | 3.2 | N | N | Y | N | N | 48 | F |
| 9 | N Q61R | 131 | 50 | FTC | 1.6 | N | Y | Y | N | N | 48 | F |
| 10 | H Q61K | 91 | 59 | FTC | 7.5 | N | Y | Y | Y | N | 76 | F |
| 11 | H A11G | 117 | 13 | FTC | 10.0 | N | Y | Y | Y | Y | 70 | M |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carneiro, T.N.R.; Bim, L.V.; Buzatto, V.C.; Galdeno, V.; Asprino, P.F.; Lee, E.A.; Galante, P.A.F.; Cerutti, J.M. Evidence of Cooperation between Hippo Pathway and RAS Mutation in Thyroid Carcinomas. Cancers 2021, 13, 2306. https://doi.org/10.3390/cancers13102306
Carneiro TNR, Bim LV, Buzatto VC, Galdeno V, Asprino PF, Lee EA, Galante PAF, Cerutti JM. Evidence of Cooperation between Hippo Pathway and RAS Mutation in Thyroid Carcinomas. Cancers. 2021; 13(10):2306. https://doi.org/10.3390/cancers13102306
Chicago/Turabian StyleCarneiro, Thaise Nayane Ribeiro, Larissa Valdemarin Bim, Vanessa Candiotti Buzatto, Vanessa Galdeno, Paula Fontes Asprino, Eunjung Alice Lee, Pedro Alexandre Favoretto Galante, and Janete Maria Cerutti. 2021. "Evidence of Cooperation between Hippo Pathway and RAS Mutation in Thyroid Carcinomas" Cancers 13, no. 10: 2306. https://doi.org/10.3390/cancers13102306
APA StyleCarneiro, T. N. R., Bim, L. V., Buzatto, V. C., Galdeno, V., Asprino, P. F., Lee, E. A., Galante, P. A. F., & Cerutti, J. M. (2021). Evidence of Cooperation between Hippo Pathway and RAS Mutation in Thyroid Carcinomas. Cancers, 13(10), 2306. https://doi.org/10.3390/cancers13102306