1. Introduction
An emerging trend in recent years has been the development of targeted drug conjugates, where a targeting domain is coupled to a cytotoxic drug, allowing for selective killing of cancer cells. The most common format consists of a monoclonal antibody (mAb), decorated with multiple copies of a drug, a so-called antibody drug conjugate (ADC) [
1].
The human epidermal growth factor receptor 2 (HER2) is overexpressed in a sub-set of patients with e.g., breast, ovarian, and gastric cancer, and has limited expression on normal cells. Several drugs selectively targeting HER2 have been described and one of the first ADCs approved for clinical use by the US Food and Drug Administration, was trastuzumab emtansine (T-DM1), targeting HER2. It is approved for therapy of metastatic HER2-positive breast cancer, and has been found to significantly increase progression-free survival among patients compared to standard treatment [
2]. However, most patients treated with T-DM1 eventually progress [
2,
3], which motivates investigation and development of alternative or complimentary treatment options including novel drugs.
An important property of targeted drug conjugates is their ability to efficiently penetrate solid tumor mass to deliver a sufficiently high concentration of the cytotoxic payload to all neoplastic cells. In a pre-clinical mouse model of breast cancer, it was found that smaller JIMT-1 tumors (approximately 70 mm
3) were more sensitive to treatment with T-DM1 than larger tumors (approximately 350 mm
3) [
4]. One reason for the poorer response of the larger tumors could be inefficient tumor penetration. Due to insufficient lymphatic drainage, the rate of tumor penetration is typically diffusion dependent, and the relatively large size of a mAb hampers its diffusivity. The use of smaller proteins and peptides as targeting vehicles could be advantageous as their rate of diffusion is higher [
5,
6,
7,
8].
Moreover, the large size and complex structure of mAbs makes site-specific conjugation of the drug and control of the drug-to-antibody ratio difficult, which may affect the binding properties [
9] and the in vivo stability of the resulting ADC [
10]. Even though several strategies for site-specific conjugation have been described, many ADCs in clinical development still rely on non-site-specific conjugation of the drug to lysines or cysteines [
11].
Engineered scaffold proteins (ESPs) may be used as an alternative to mAbs for targeting tumor cells and have recently been investigated as carriers of cytotoxic drugs [
12,
13]. Affibody molecules constitute a class of ESPs, consisting of 58 amino acids arranged in an anti-parallel three-helix bundle [
14]. The use of affibody molecules for specific delivery of cytotoxic drugs to cancer cells may provide several advantages compared to mAbs. First, affibody molecules are much smaller (approx. 7 kDa), which ensures rapid extravasation into the extracellular space, as well as more efficient penetration of poorly perfused solid tumors. Second, since the affibody scaffold does not contain cysteines, one or more cysteines may be introduced at desired positions using genetic engineering techniques, which may be utilized for site-specific conjugation of cytotoxic drugs using thiol-directed chemistry. Clinical testing of affibody molecules targeting different cell surface receptors have shown that they are well tolerated by patients without causing immune reactions. For example, radionuclide molecular imaging of HER2-expressing primary tumors and metastases by PET and SPECT successfully met the trial’s endpoints without causing adverse effects [
15,
16], validating the clinical suitability of the affibody class of ESPs. In addition, repeated administration to rats of a HER2-targeting affibody tracer for molecular imaging showed that no anti-affibody antibodies were formed [
17]. Together, these features make affibody molecules promising carriers for tumor-targeted drug delivery.
The small size of affibody molecules provides fast clearance from blood through filtration in the kidneys. A strategy previously explored to extend the plasma half-life of affibody molecules was by fusion with an albumin binding domain (ABD) [
18]. The ABD binds with high affinity to serum albumin (SA) in blood, which increases the molecular weight by 67 kDa of the affibody-ABD/SA complex to above the cut-off of kidney filtration [
19].
Previously, anti-HER2 affibody-drug conjugates (AffiDCs) containing an ABD and the cytotoxic drug mcDM1 have been investigated [
13]. The AffiDCs demonstrated prolonged retention in circulation compared to HER2-binding affibody molecules alone, and increased survival of mice bearing HER2-expressing SKOV3 xenografts. It was found that the introduction of mcDM1 caused an increase in hydrophobicity, which resulted in an increase in hepatic uptake in vivo. Minimizing hepatic uptake is important because drug-induced liver injury (DILI) is a common cause of drug withdrawal from the market [
20], and is sometimes a difficulty also with ADCs [
11]. An effort to reduce the hepatic uptake of the HER2-targeting AffiDC was undertaken by introducing three or six glutamic acid residues next to the cysteine carrying mcDM1 [
21]. The resulting conjugates retained high tumor uptake while the unspecific uptake in liver was significantly reduced.
An essential aspect of molecular design is the binding valency of the cell targeting module. Previous studies on HER3-targeting, employing ABD-fused affibody molecules, have revealed that a bivalent format increases the rate of internalization [
22]. A high rate of internalization of AffiDCs is desirable since internalization is required for drug action. However, a bivalent format may not be advantageous for HER2-targeting affibody molecules armed with mcDM1, since mono- and divalent constructs have been found to have a similar cytotoxic potency in vitro [
13].
The cytotoxic payload mcDM1 is derived from the cytotoxic compound maytansine and acts as a microtubule polymerization inhibitor that disrupts the microtubule network, causing cell death [
23]. The maleimidocaproyl (mc) linker is non-cleavable and drug conjugates equipped with it first engage their intended target on the cell surface, after which the conjugate is internalized through receptor-mediated endocytosis. It is then transported to the lysosomes where the protein part is degraded, leading to release of mcDM1, which diffuses to the cytosol where the poisoning process occurs.
In this study, a monovalent HER2-binding affibody drug conjugate, Z
HER2:2891-ABD-E
3-mcDM1, was investigated, consisting of a HER2 binding affibody molecule (Z
HER2:2891) coupled to an ABD for in vivo half-life extension and a tri-glutamyl (E
3) linker to minimize hepatic uptake, conjugated with the maytansine derivate mcDM1. The properties of monovalent Z
HER2:2891-ABD-E
3-mcDM1 was compared to the properties of divalent (Z
HER2:2891)
2-ABD-E
3-mcDM1 [
21] to investigate the impact of targeting protein valency. As control of the impact of the tri-glutamyl linker, a construct where the E
3-linker was replaced with an uncharged and less hydrophilic tri-glycine linker (G
3), was created (Z
HER2:2891-ABD-G
3-mcDM1). A non-toxic control, Z
HER2:2891-ABD-E
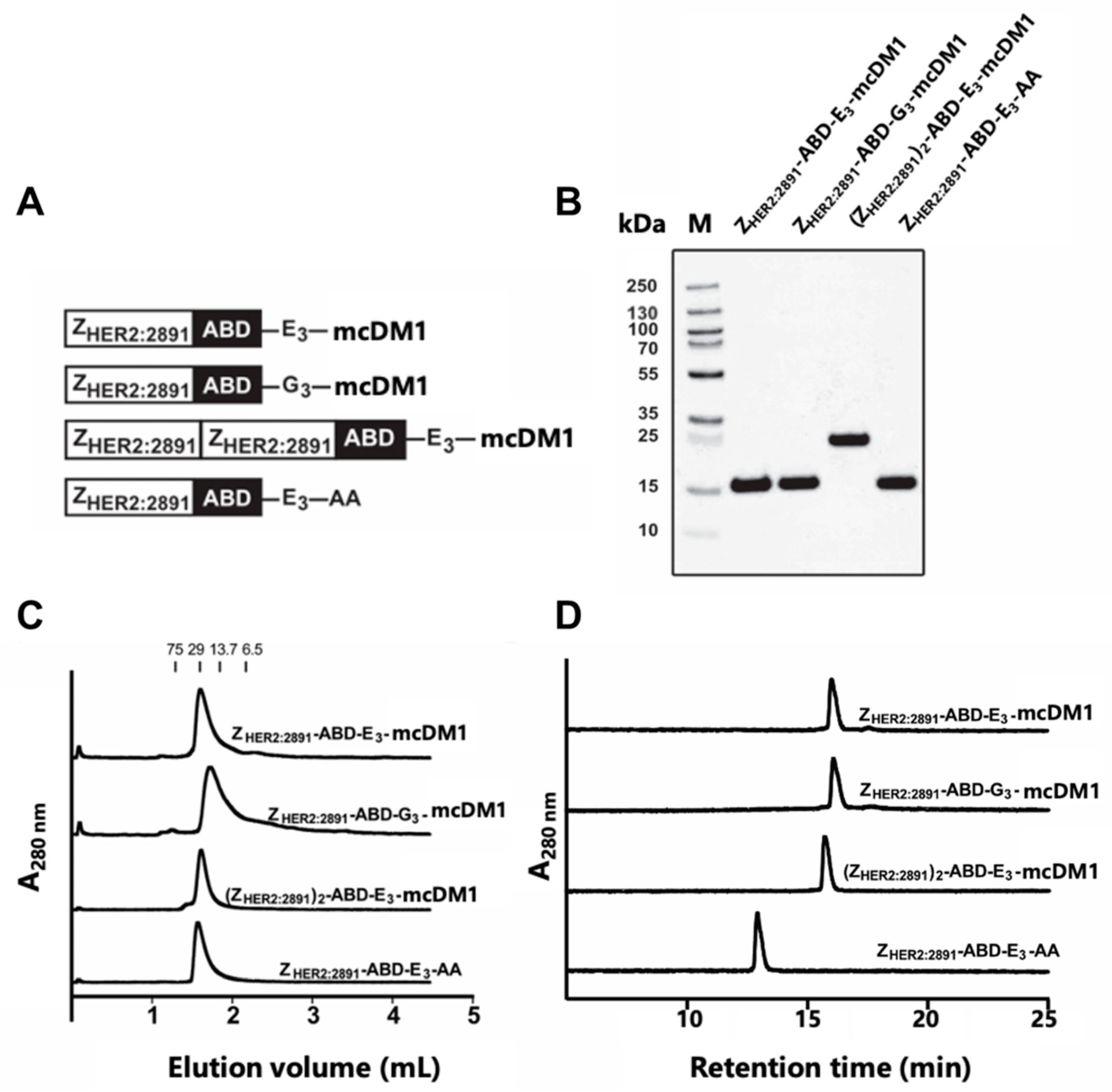
3-AA with the C-terminal cysteine capped by reaction with iodoacetamide was also included. The four constructs are schematically represented in
Figure 1A. After thorough in vitro characterization, we investigated biodistribution and potency for targeted therapy in a xenograft model of HER2-positive cancer in vivo.
3. Discussion
Patients with HER2-overexpressing metastatic breast cancer are normally prescribed treatment with HER2-targeting drugs. However, in most instances, this is not a curative treatment and patients eventually progress. Even though resistance may not be attributed to the mAb or ADC format, novel drugs are in need that may either be used in combination with standard drugs or even replace them. This motivated us to investigate the affibody-class of engineered affinity proteins (EAPs) as carriers of potent cytotoxic drugs, creating so called affibody drug conjugates (AffiDCs).
The AffiDCs generated in this study were relatively simple to produce, where recombinantly produced fusion proteins in
Escherichia coli could be purified with a single affinity chromatography step, before mcDM1 conjugation, followed by a high-resolution RP-HPLC purification step after conjugation. The resulting conjugates were highly pure and showed no tendency to aggregate (
Figure 1). A simple and efficient manufacturing process is desirable and is advantageous compared to the typical production processes of ADCs, which usually require more advanced host organisms and where the final high-resolution RP-HPLC purification step cannot be employed, since it would irreversibly denaturate the mAb-part of the ADC. In addition, many ADCs in clinical development employ random attachment of the drug to lysines or cysteines, in contrast to the AffiDCs in this study, where mcDM1 was site-specifically attached to the C-terminal cysteine in all cases, resulting in homogenous substances.
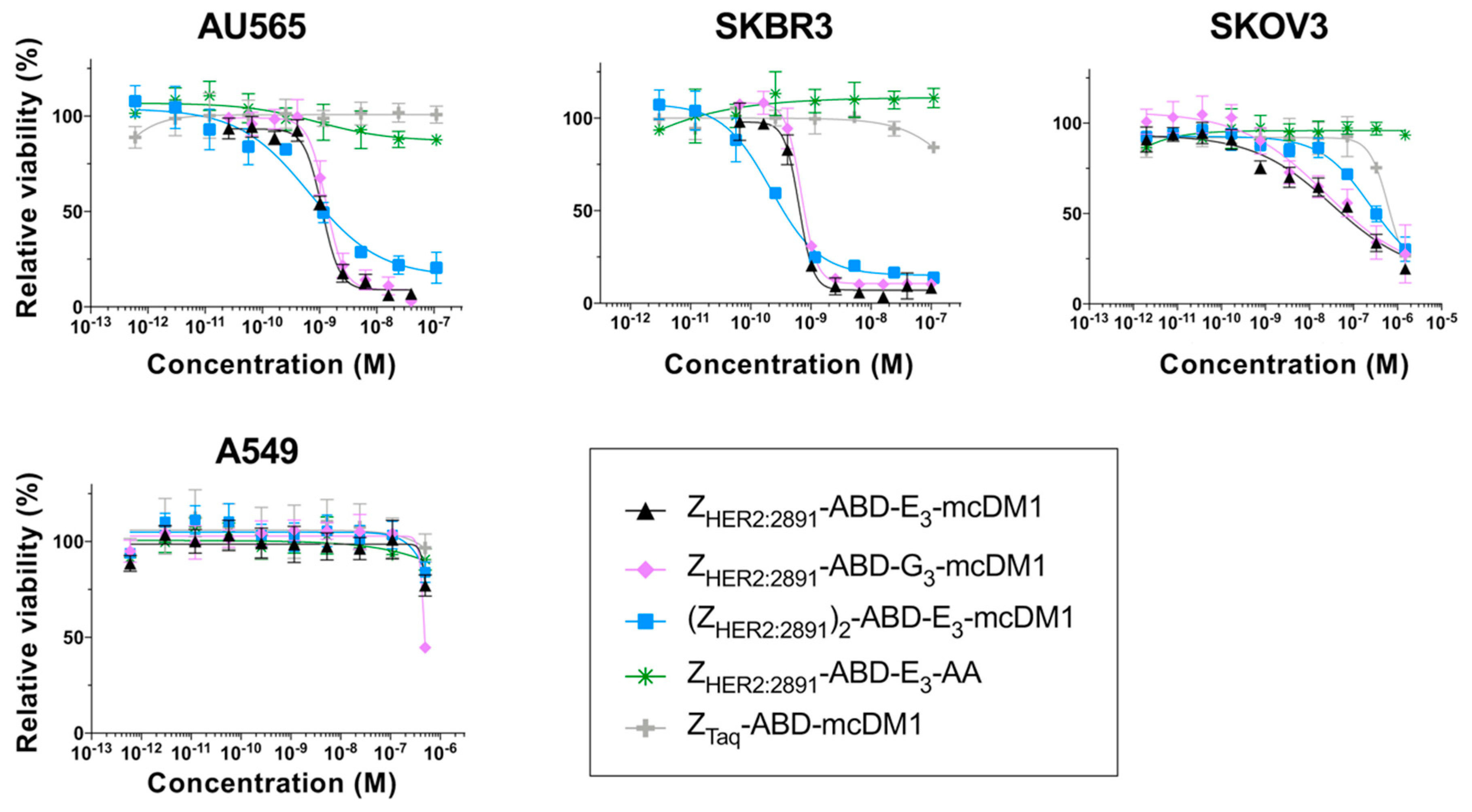
An interesting observation was the difference in Hill slopes between the di- and monovalent conjugates in the in vitro analysis of cytotoxic potential (
Figure 2). The shallower slopes of the divalent construct on AU565 and SKBR3 cells could indicate that there is negative cooperativity in binding to the cells compared to the monovalent constructs [
24]. However, for SKOV3 cells, the Hill slopes for the di- and the monovalent variants were similar. This shows that even though there could be negative or positive cooperativity in binding, the resulting cellular effect might be influenced by additional parameters. A possible reason could be that it is not only cell binding that is responsible for toxic action but that also other pathways are involved. This has been studied in some detail for ADC toxicity where differences in the rate of receptor internalization, expression level of multi-drug resistance proteins and impairment of lysosomal degradation are different between different cell lines [
4,
25,
26]. Such parameters likely affect the sensitivity to Z
HER2:2891-ABD-E
3-mcDM1 and this leads to differences in IC
50-values among the cell lines with high HER2 expression in this study. However, it is unclear why these parameters would affect the Hill slope. Differences in Hill slopes and sensitivity to ADCs have also previously been observed in dose-response curves for T-DM1 on different cell lines [
27].
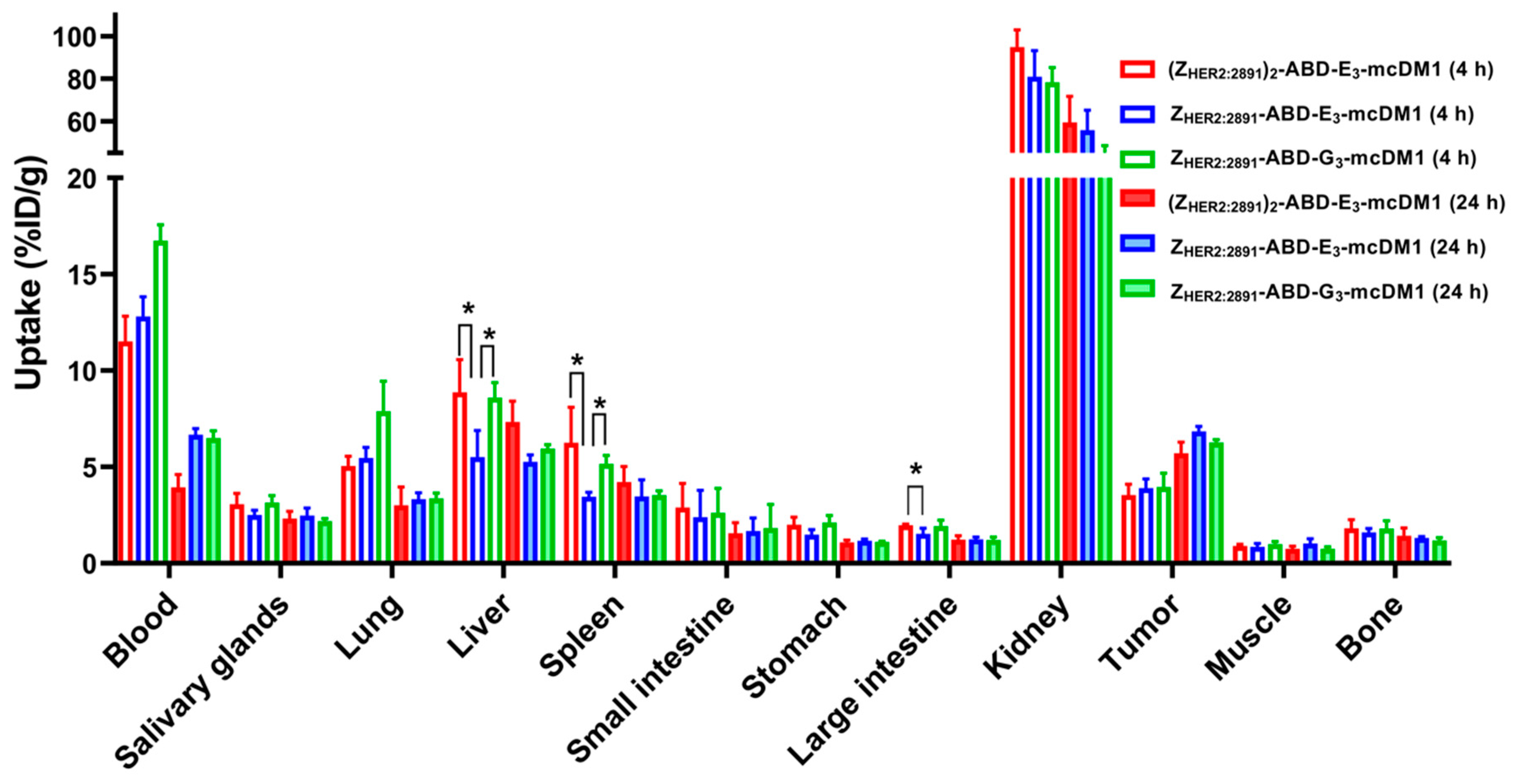
In vivo, the biodistribution experiment (
Figure 4) showed that even small changes in the carrier protein affect the uptake in different organs. Since Z
HER2:2891 does not cross-react with murine HER2, the uptake in normal organs is not mediated by HER2 and is only a consequence of non-specific uptake. At 4 h p.i., the uptake in liver of Z
HER2:2891-ABD-E
3-mcDM1 was significantly lower than the uptake of Z
HER2:2891-ABD-G
3-mcDM1. These conjugates were identical except for that the tri-glutamyl linker had been replaced with a tri-glycine linker. Interestingly, the uptake in liver of Z
HER2:2891-ABD-E
3-mcDM1 was also significantly lower than the divalent (Z
HER2:2891)
2-ABD-E
3-mcDM1, suggesting that the Z
HER2:2891 part promotes uptake. The plasma half-life was around 20 h for all three conjugates, which was lower than for example Z
HER2:342-ABD conjugated with
177Lu [
28]. Even though they cannot be directly compared since the mcDM1 and
177Lu parts may promote clearance differently, it would be interesting to investigate other methods for half-life extension of AffiDCs such as PEGylation [
29] and PASylation [
30] to learn if an even longer plasma half-life would lead to even more efficacious drug delivery to the tumor. In contrast to ADCs, the AffiDCs allow for detailed investigation of the impact of plasma half-life on drug candidate performance. ADCs, on the other hand, always have a relatively long plasma half-life, and for them a long half-life is generally regarded as beneficial [
31]. A translation of Z
HER2:2891-ABD-E
3-mcDM1 from mouse to human would likely lead to a longer plasma half-life since the plasma half-life of HSA is longer than MSA and the affinity of the ABD is stronger for HSA than for MSA.
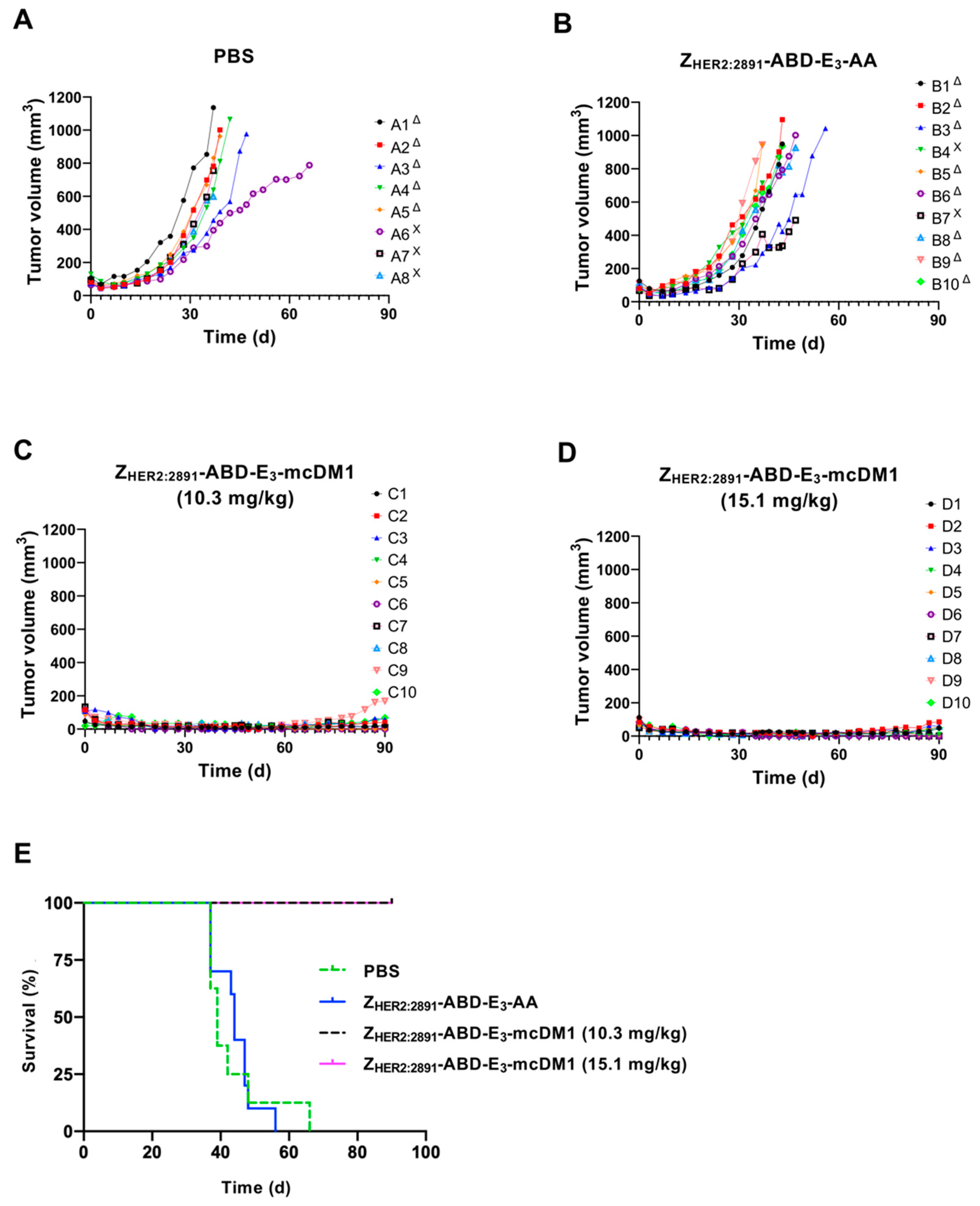
Previously, we found that a divalent format of an AffiDC, (Z
HER2:2891)
2-ABD-mcDM1, was able to slow tumor growth and as a consequence increase survival of mice with implanted HER2-overexpressing SKOV3 tumors [
13]. However, when determining the cytotoxic potential on HER2-overexpressing cells in vitro in a dose-response experiment, there was no significant difference of the IC
50 values of the di- and monovalent constructs. Similarly, in this study the difference in IC
50 values between di- and monovalent variants was not significant. Since a smaller construct will theoretically penetrate solid tumors better than a larger one [
6], one of the aims of this study was to evaluate the efficacy of the smaller monovalent conjugate. The experimental therapy data cannot be directly compared between the divalent (Z
HER2:2891)
2-ABD-mcDM1 and the monovalent Z
HER2:2891-ABD-E
3-mcDM1 in present study, since the monovalent construct included an E
3 linker next to the cysteine onto which mcDM1 was attached. The injected dose was also higher in the present study (15.1 and 10.3 mg/kg), which corresponds to 1.11 μmol/kg or 0.76 μmol/kg compared to the previous study where the dose was 0.41 μmol/kg. However, a strikingly more efficient therapy was detected in the present study, with efficient regression of all tumors and complete disappearance of the xenografts in some animals treated with Z
HER2:2891-ABD-E
3-mcDM1. At the highest dose (15.1 mg/kg), we found that 2 out of 10 animals started to lose weight after three weekly injections, which suggested that this was close to the maximum tolerated dose. One interesting aspect of this and our previous study [
13] is that the histology investigation did not reveal pathologic changes in the kidneys although the renal uptake was higher than the tumor uptake. There are similar observations with other engineered scaffold proteins, conjugated with microtubule polymerization inhibitory drugs, e.g., conjugates of PASylated DARPin with MMAF [
30,
32] and Bicycles conjugate with MMAE [
12]. In these cases, a strong anti-tumor effect was observed without a pronounced effect on the kidneys, even though the renal uptake was higher that the tumor uptake. It has to be noted that these drugs are inhibiting microtubule polymerization and are likely more toxic to rapidly dividing cells in the tumor than to slower dividing cells in normal tissues.
Z
HER2:2891-ABD-E
3-mcDM1 could be interesting to use in combination with already established drugs, such as T-DM1. One reason is that the pharmacokinetic profile is different with faster clearance of Z
HER2:2891-ABD-E
3-mcDM1 compared to T-DM1. A second reason is that the uptake in normal organs is different. For example,
89Zr-labeled T-DM1 has a higher uptake in spleen and bone [
33] compared to
99mTc-labeled Z
HER2:2891-ABD-E
3-mcDM1, whereas
99mTc-labeled Z
HER2:2891-ABD-E
3-mcDM1 has a higher uptake in kidneys. A combination of T-DM1 and Z
HER2:2891-ABD-E
3-mcDM1 would therefore likely allow for delivery of a higher total dose of DM1 to the tumor, while the unspecific uptake in spleen, bone, and kidney could be kept on a manageable level. However, side effects associated with T-DM1 such as severe thrombocytopenia (grade ≥ 3) [
34] requires careful studies to elucidate if a combination of Z
HER2:2891-ABD-E
3-mcDM1 and T-DM1 is superior to either of the conjugates alone. It should also be noted that the antibody part of an ADC delivers some of its therapeutic effect by acting as a HER2 antagonist and by antibody-dependent cellular cytotoxicity (ADCC) [
35]. These therapeutic mechanisms are not available to the monomeric affibody carrier in this article.
ADCs are often predominantly cleared through the liver and the second most common grade 3 adverse effect in patients receiving T-DM1 is elevation of liver enzymes including aspartate and alanine aminotransferases [
36]. Our results show that clearance through the kidneys is the predominant route for Z
HER2:2891-ABD-E
3-mcDM1, which could be beneficial if the kidneys are more resistant to the cytotoxic effect of DM1. Radiolabeled compounds could prove to be beneficial in a combination treatment strategy including Z
HER2:2891-ABD-E
3-mcDM1 and e.g., the analogous Z
HER2:342-ABD conjugate labeled with
177Lu [
28] or a pre-targeting concept involving
177Lu labeled PNA [
37]. Since Z
HER2:342 interacts with the same epitope as Z
HER2:2891, the injected dose of Z
HER2:2891-ABD-E
3-mcDM1 has to be lower than the saturating concentration (15.3 mg/kg in the experiment presented in
Figure 5).
The composition of the linker is an important part of drug conjugates and for ADCs, the linkage is often a weak link in the design. The maleimide-thiol bond employed in T-DM1 as well as the AffiDCs in the present study has an in vivo half-life of only 24 h, and the lability of this bond may cause more release of free mcDM1 and cross-transfer to endogenous proteins [
38] for the ADC than for the AffiDC due to its longer plasma half-life. However, strategies to increase linker stability have been described, such as hydrolytic opening of the thiosuccinimide ring [
38].
Z
HER2:2891-ABD-E
3-mcDM1 is a promising prototype where every affibody molecule is conjugated with a single drug moiety. One of the interesting directions for further improvement of AffiDC would be development of constructs capable of delivering several drugs. One of the challenges with this approach is that coupling of multiple hydrophobic drugs to a relatively small protein might have an adverse effect on its biodistribution. It has been demonstrated that an increase of lipophilicity of affibody molecules is associated with significantly elevated hepatic uptake [
39], particularly when a lipophilic moiety is placed at the N-terminus. The finding in this study that a drug conjugate consisting of a monomeric affibody carrier in combination with a lipophilic drug, connected by a negatively charged and hydrophilic linker, has reduced hepatic uptake, is an essential input information for molecular design of the next-generation of AffiDCs. The next-generation AffiDCs might include more potent payloads at a higher multiplicity and would be interesting to compare head-to-head with T-DM1 and other clinically approved HER2 targeting drugs.
A big potential of radionuclide molecular imaging for enhancing efficacy of HER2-targeting therapies is well-recognized [
40]. In the case of targeted delivery of cytotoxic drugs, the most apparent application is the stratification of patients according to HER2 expression level in tumors, to identify patients that would benefit from targeted therapy. Clinical studies suggest that trastuzumab emtansine is the most efficient in the case of tumors with high HER2 expression [
41,
42] and it is thus important to identify those patients. It has been demonstrated that a combination of SPECT/CT imaging using [
18F]2-fluoro2-deoxy-D-glucose (FDG) and
89Zr-labeled trastuzumab can predict the radiologic response and the median time to treatment failure for T-DM1 therapy [
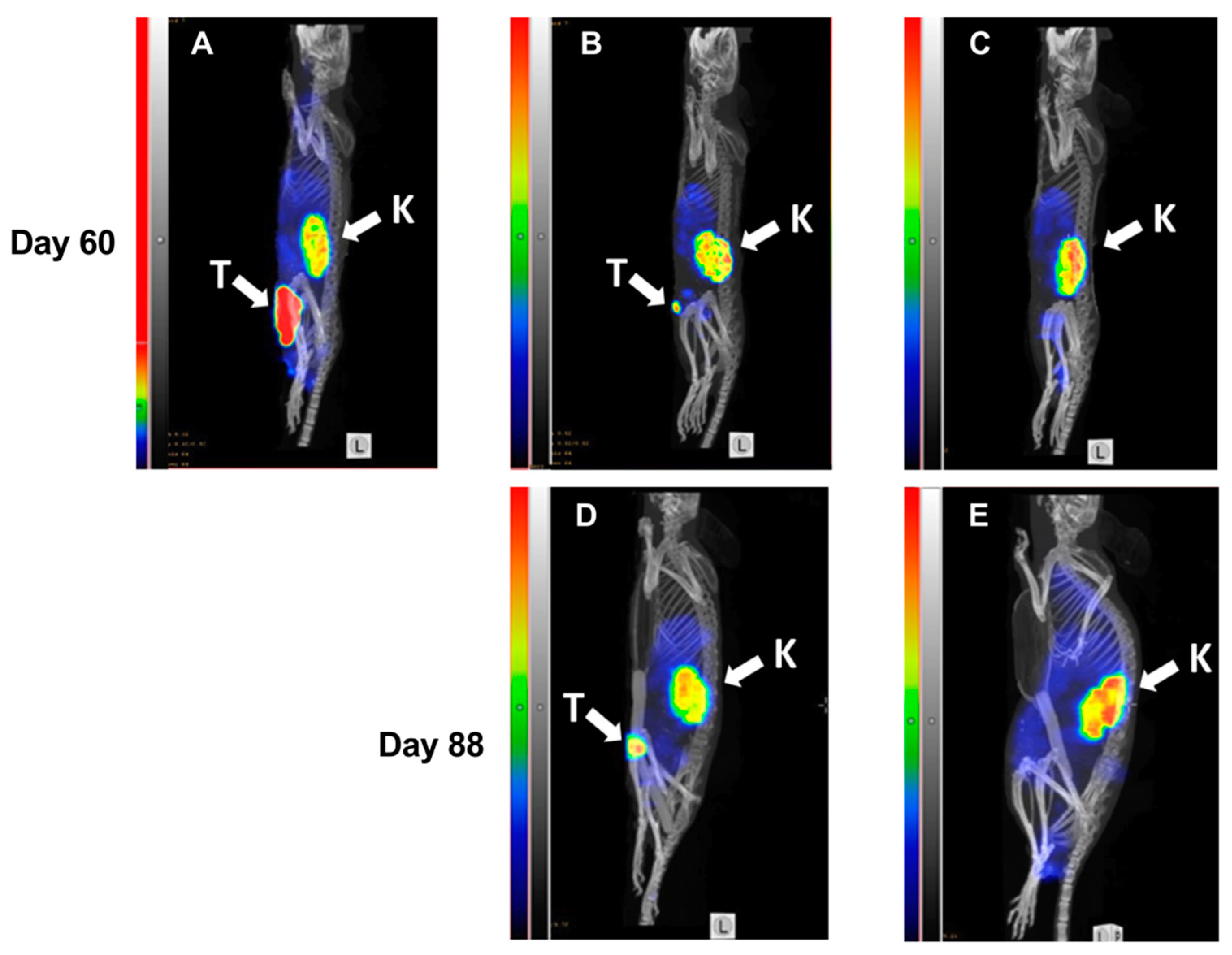
43]. Our study indicated that radionuclide imaging may be used for monitoring of response to Z
HER2:2891-ABD-E
3-mcDM1-mediated therapy (
Figure 7), and that it can also be utilized for stratification purposes. The use of an affibody molecule as a theranostic companion to Z
HER2:2891-ABD-E
3-mcDM1 is preferable to the use of radiolabeled antibodies, as it also indicates an accessibility of the targeted epitope.
4. Materials and Methods
4.1. General
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA) or Merck (Darmstadt, Germany) unless otherwise stated. Restriction enzymes were from New England Biolabs (Ipswitch, MA, USA).
4.2. Construction of Genes
Genes encoding Z
HER2:2891-ABD-E
3-Cys and Z
HER2:2891-ABD-G
3-Cys, flanked by NdeI and BamHI restriction sites, were amplified from a plasmid encoding (Z
HER2:2891)
2-ABD-Cys [
13] using Phusion polymerase (New England Biolabs). Likewise, the gene encoding Z
Taq-ABD-Cys was amplified from a plasmid encoding (Z
Taq)
2-ABD-Cys [
13]. During PCR amplification, a tag with the amino acid sequence His-Glu-His-Glu-His-Glu (HEHEHE) was placed at the N-terminus of all constructs allowing for radionuclide labeling. The linker connecting the affibody molecules and ABD had the amino acid sequence Gly-Gly-Gly-Gly-Ser. The genes were sub-cloned into the pET-21a(+) plasmid vector (Novagen, Madison, WI, USA) using NdeI and BamHI restriction enzymes. The expression vector encoding (Z
HER2:2891)
2-ABD-E
3-Cys was constructed previously [
21]. The sequences of all genes were confirmed by DNA sequencing.
4.3. Expression and Purification
The affibody constructs were expressed at 37 °C in shake flask cultures of
Escherichia coli BL21 Star (DE3) (New England Biolabs) in tryptic soy broth containing 100 µg/mL ampicillin. When OD
600 was between 0.6 and 1, protein expression was induced by addition of 1 mM isopropyl-β-D-1-thiogalactopyranoside (Appolo Scientific, Stockport, UK). Protein production was carried out for 3 h, after which the cells were harvested by centrifugation and lysed by sonication. The supernatants were clarified by centrifugation and filtration through a 0.45 µm Acrodisc syringe filter (Pall, Port Washington, NY, USA). The recombinantly expressed affibody constructs were purified by affinity chromatography on a HiTrap NHS sepharose column (GE Healthcare, Uppsala, Sweden) with immobilized human serum albumin (HSA) using an ÄKTA system (GE Healthcare), essentially as previously described [
44] including elution with 500 mM acetic acid, pH 2.6. The fractions containing affibody constructs were pooled and lyophilized.
Z
HER2:V2 was produced and purified as previously described [
45].
4.4. Conjugation with mcDM1
The lyophilized proteins were dissolved in 100 mM Tris-HCl buffer (pH 7.85) to a final concentration of 0.1 mM. Subsequently, the proteins were incubated with 5 mM tris (2-carboxyethyl) phosphine (TCEP) for 30 min at 37 °C to reduce the thiol group on the C-terminal cysteine of the constructs, which could potentially have been oxidized during protein production and purification. Before mcDM1 was added, the pH of protein solution was adjusted to 6.5 using 1 M HCl. A freshly prepared solution of mcDM1 (Levena Biopharma, San Diego, CA, USA), in DMSO (20 mM), was mixed with the affibody constructs at a molar ratio of 2:1 (drug: protein), and the conjugation mixture was incubated overnight at room temperature. The conjugation reaction mixture was diluted with HPLC buffer A (0.1% trifluoroacetic acid in H2O) and then loaded on a Zorbax SB-C18 column (Agilent, Santa Clara, CA, USA). Bound material was eluted by a 30 min gradient from 30% to 60% buffer B (0.1% trifluoroacetic acid in acetonitrile) at a flowrate of 3 mL/min. The fractions containing affibody-mcDM1 conjugates were pooled followed by lyophilization. Capping of the C-terminal cysteine to create the non-toxic control ZHER2:2891-ABD-E3-AA was carried out with 2-iodoacetamide. Lyophilized ZHER2:2891-ABD-E3-Cys was dissolved in alkylation buffer (0.2 M NH4HCO3, pH 8.0) potentially oxidized cysteine residues were reduced with TCEP as above. 2-iodoacetamide was added to a final concentration of 10 mM followed by incubation for 30 min at room temperature in the dark to alkylate cysteines. The capped proteins were purified by RP-HPLC as described above for the AffiDCs, followed by lyophilization. The AffiDCs and the non-toxic control were dissolved in phosphate buffered saline (PBS, 10 mM Na-phosphate, 2.7 mM KCl, 137 mM NaCl, pH 7.4) and stored at −80 °C until use.
Purified conjugates (5 µg in each sample) were analyzed by SDS-PAGE (Biorad, Hercules, CA, USA) under reducing conditions. The oligomeric state of the conjugates was determined by size exclusion chromatography, by passage through a 5/150 column (GE Healthcare), packed with Superdex-75, at a flowrate of 0.45 mL/min in PBS. The molecular weight of purified affibody-mcDM1 conjugates was measured by ESI-TOF mass spectrometry (Agilent). Analysis of purity was measured by RP-HPLC using a Zorbax 300SB-C18 column (Agilent) using a gradient from 30% to 60% of HPLC buffer B during 20 min at a flow rate of 1 mL/min.
4.5. Binding Specificity and Affinity Determination
A Biacore 3000 instrument (GE Healthcare) was used for biosensor analysis. HER2-Fc chimeric protein (R&D Systems, Minneapolis, MN, USA) was immobilized to 696 RUs on a CM5 chip by amine coupling in sodium acetate buffer, pH 4.5. On a second CM5 chip, HSA (Novozymes, Bagsvaerd, Denmark) and MSA (Sigma-Aldrich) were immobilized in the same way. The final immobilization levels were 395 and 457 RUs, respectively. A reference flow cell was created by activation and deactivation on both chips. HBS-EP (10 mM HEPES, 150 mM NaCl, 3 mM EDTA, 0.005% v/v surfactant P20, pH 7.4) was used as running buffer and for dilution of the analytes. All experiments were performed at 25 °C at a flow rate of 50 µL/min. The chips were regenerated by injection of 10 mM HCl for 30 s. The binding kinetics were analyzed by Biacore evaluation software using the one-to-one kinetics model.
4.6. Cell Culture
AU565, SKBR3, SKOV3, BT474, and A549 cell lines were obtained from American Type Culture Collection (ATCC, via LGC Promochem, Borås, Sweden) and were grown in McCoy’s 5A (SKOV3, SKBR3), RPMI-1640 (AU565, BT474) or Dulbecco’s modified Eagle medium (A549) (Cytiva Hyclone, Uppsala, Sweden) supplemented with 10% FBS (20% FBS for BT474) (Sigma-Aldrich) in a humidified incubator at 37 °C in 5% CO2 atmosphere.
4.7. In Vitro Cytotoxicity Analysis
Approximately 5000 cells/well (2000 cells/well for SKOV3) were seeded in a 96-well plate and allowed to attach overnight. Subsequently, the medium was replaced with fresh medium containing serial dilutions of AffiDCs or controls, followed by incubation for 72 h. Cell viability was determined using Cell Counting Kit-8 (CCK-8, Sigma-Aldrich) according to the manufacturer’s protocol with measurement of A450 in each well. The obtained absorbance values were analyzed by GraphPad Prism using a log(inhibitor) vs. response-variable slope (four parameters) model (GraphPad Software, La Jolla, CA, USA).
4.8. Radiolabeling
Radiolabeling of the AffiDCs with 99mTc, using [99mTc(CO)3(H2O)3]+ precursor, was performed using a CRS kit (PSI, Villigen, Switzerland) according to manufacturer’s instructions. In brief, technetium-99m was freshly eluted as pertechnetate, 99mTcO4−, with 0.9% NaCl from a commercial 99Mo/99mTc generator. To generate the [99mTc(CO)3(H2O)3]+ (tricarbonyl technetium) precursor, 500 μL 99mTcO4− was added to a CRS kit vial, followed by incubation at 100 °C for 30 min. A solution containing 60 μg of an AffiDC in 100 μL PBS was mixed with 60 MBq of [99mTc(CO)3(H2O)3]+ and incubated at 60 °C for 60 min.
The radiolabeled AffiDCs were separated from [99mTc(CO)3(H2O)3]+ by passage through a NAP-5 size exclusion chromatography column (GE Healthcare) eluted with 2% BSA in PBS. To evaluate the stability of labeling, the radiolabeled conjugates were challenged with 5000-fold molar excess of histidine in PBS for 4 h at room temperature. The radiochemical yield and purity were measured using iTLC analysis where the iTLC strips were eluted with PBS. Analysis of the iTLC strips were done using a Cyclone Storage Phosphor System (PerkinElmer, Waltham, MA, USA). The activity was measured using an automatic gamma spectrometer equipped with a 3-inch NaI(Tl) well detector (1480 Wizard, Wallac, Finland).
Labeling of Z
HER2:V2 with
99mTc was performed as previously described [
45].
4.9. In Vitro Characterization of 99mTc-Labeled AffiDCs
An in vitro specificity test was performed as described earlier [
46]. In brief, SKOV3 and BT474 cells were seeded in 6-well plates at a density of 4 to 6 × 10
5 cells/well, 1 or 2 days before the experiment. Non-radiolabeled AffiDC (1000 nM) was then added to each well or an equal volume of medium only to control wells followed by incubation for 15 min at room temperature. Then a solution of a radiolabeled AffiDC was added to each well to reach a final concentration of 2 nM followed by incubation for 1 h at 37 °C. Then, the medium was collected, and the cells were washed once with 1 mL PBS, detached from the well, and collected. The radioactivity of medium and cells was measured using a gamma spectrometer.
Evaluation of cellular processing was performed according to a method described earlier [
46]. In brief, SKOV3 and BT474 cells were seeded (5 × 10
5 cells/well) in 35 mm dishes 1 or 2 days before the experiment. The radiolabeled compound (2 nM) in medium was added and the dishes were incubated at 37 °C. At determined time points (1, 2, 4, 6 and 24 h after addition), the dishes were analyzed for membrane-bound and internalized radioactivity. Briefly, the medium from cells was collected followed by a wash with PBS. The cells were then treated with acid wash buffer (4 M urea in 0.2 M glycine, pH 2.0), for 5 min on ice to provide the membrane-bound fraction. Then the cells were incubated with 1 M NaOH at 37 °C for 30 min to release internalized radioactivity. The activity in every fraction was measured using an automatic gamma spectrometer.
The binding affinity of radiolabeled AffiDCs to HER2-expressing SKOV3 cells was evaluated using a LigandTracer Yellow instrument (Ridgeview Instruments, Vänge, Sweden) as described previously [
47]. Approximately 2 × 10
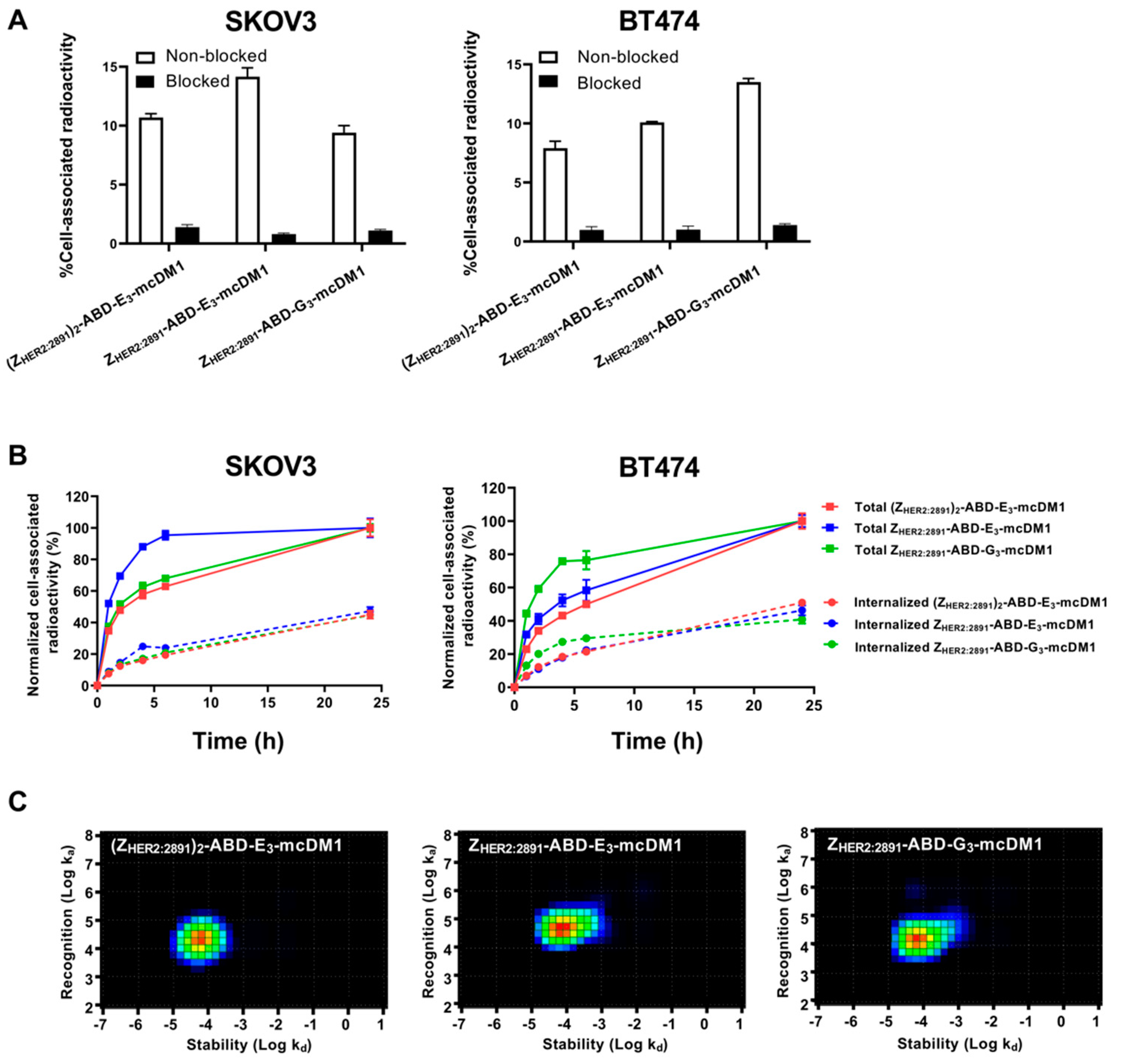
6 cells were seeded in 10 cm dishes 1 or 2 days before the experiment. To measure binding kinetics during the association phase, a stepwise addition of concentrations (1, 2 and 5 nM) of the AffiDCs in complete medium was performed. To measure dissociation, the medium was exchanged to complete medium lacking the AffiDC. Data were collected and analyzed using TraceDrawer software (Ridgeview Instruments), assuming a one-to-one kinetic model. The signal was corrected for nuclide decay. Analysis by InteractionMap software (Ridgeview Diagnostics, Uppsala, Sweden) was performed to estimate the interaction heterogeneity.
4.10. Biodistribution in Tumor Bearing Mice
SKOV3 xenografts were established in 22 female BALB/c nu/nu mice by implanting 12 × 106 cells subcutaneously in a hind leg. The mice were randomized into 6 groups, 3 to 4 mice per group. Biodistribution of radiolabeled AffiDCs was measured 3 weeks after implantation. Mice were intravenously (i.v.) injected with 6 μg of 99mTc-labeled conjugates in 2% BSA in PBS. The injected activity was calculated to be 60 kBq per mouse at the dissection time point. Before dissection, the mice were weighed and euthanized by an intraperitoneal injection of a ketamine-xylazine solution (30 μL of solution per gram body weight; ketamine 10 mg/mL; xylazine 1 mg/mL) and sacrificed by heart punctation. Blood, organs, and tissues were collected and weighed, and their radioactivity was measured.
An initial experiment to determine the dose of ZHER2:2891-ABD-E3-mcDM1 to be used for experimental therapy was performed. Briefly, SKOV3 xenografts were established in 28 female BALB/c nu/nu mice as described above. The mice were randomized and intraperitoneally injected with 2.4, 4.8, 10.3, 15.1, or 30.3 mg/kg of radiolabeled ZHER2:2891-ABD-E3-mcDM (5.12 MBq per animal) in 150–200 μL PBS. The biodistribution was measured after 48 h, as described above.
4.11. Therapy
An experiment to evaluate the therapeutic potential of Z
HER2:2891-ABD-E
3-mcDM1 was performed as follows: Female BALB/c nu/nu mice were subcutaneously implanted with 1×10
7 SKOV3 cells in the abdomen. One week later, the mice were randomized to 4 groups (
n = 8–10) and subcutaneously (scruff of the neck) injected with a test substance. This injection route was selected to avoid multiple injections in the same tail vein. Our previous studies have demonstrated that biodistribution of affibody molecules and their tumor uptake do not differ significantly already 4 h after subcutaneous or intravenous injections [
48,
49,
50]. In treatment groups, either 10.3 or 15.1 mg/kg of Z
HER2:2891-ABD-E
3-mcDM1 in PBS were injected. Mice in control groups were injected with either PBS or with Z
HER2:2891-ABD-E
3-AA (15.1 mg/kg). The tumor volumes at the start of treatment were 86 ± 39, 80 ± 18, 96 ± 33, and 91 ± 22, mm
3, for mice treated with PBS, Z
HER2:2891-ABD-E
3-AA, Z
HER2:2891-ABD-E
3-mcDM1 (10.3 mg/kg), and Z
HER2:2891-ABD-E
3-mcDM1 (15.1 mg/kg), respectively. Injection was done once a week for 4 consecutive weeks. Throughout the experiment, tumor volumes and body weights were monitored twice per week. The tumor volumes were determined by caliper measurement of the largest longitudinal (length) and transverse (width) diameter and calculated by the formula: Tumor volume = 1/2(length × width
2).
Mice were euthanized when the subcutaneous tumor volume exceeded 1000 mm3, bleeding ulcers on the tumor were observed or 15% overall weight loss or 10% weight loss within one week was measured. The study was terminated 90 days after tumor implantation. After the mice were sacrificed, livers and kidneys were collected, fixated, and tested for histopathological changes. The histological examination was performed at the Department of Pathology and Wildlife Diseases, National Veterinary Institute, Uppsala, Sweden.
4.12. Imaging during Experimental Therapy
Three mice were selected for imaging using 99mTc-labeled ZHER2:V2. Whole body micro-single photon emission computed tomography (SPECT)/CT scans were performed using nanoScan SPECT/CT (Mediso Medical Imaging Systems, Budapest, Hungary) at days 60 and 88. Mice were injected with 99mTc labeled ZHER2:V2 (5 µg, 15–19 MBq) and a whole-body CT scan followed by a 10 min SPECT scan were performed 4 h after injection.
4.13. Statistics
Prism (version 8.4.3) was used for statistical analysis (GraphPad Software). Comparison of variation between multiple groups were carried out by one-way ANOVA with Bonferroni’s post hoc multiple comparisons test. Differences were considered significant when p < 0.05.

 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}