Relation of Metal-Binding Property and Selective Toxicity of 8-Hydroxyquinoline Derived Mannich Bases Targeting Multidrug Resistant Cancer Cells

 ,
,  ,
,  , and
, and
Abstract
Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Culture
2.3. MTT Viability Assay
2.4. Total-Reflection X-ray Fluorescence (TXRF) Measurements
2.5. Reactive Oxygen Species (ROS) Determination Using DCFDA
3. Results
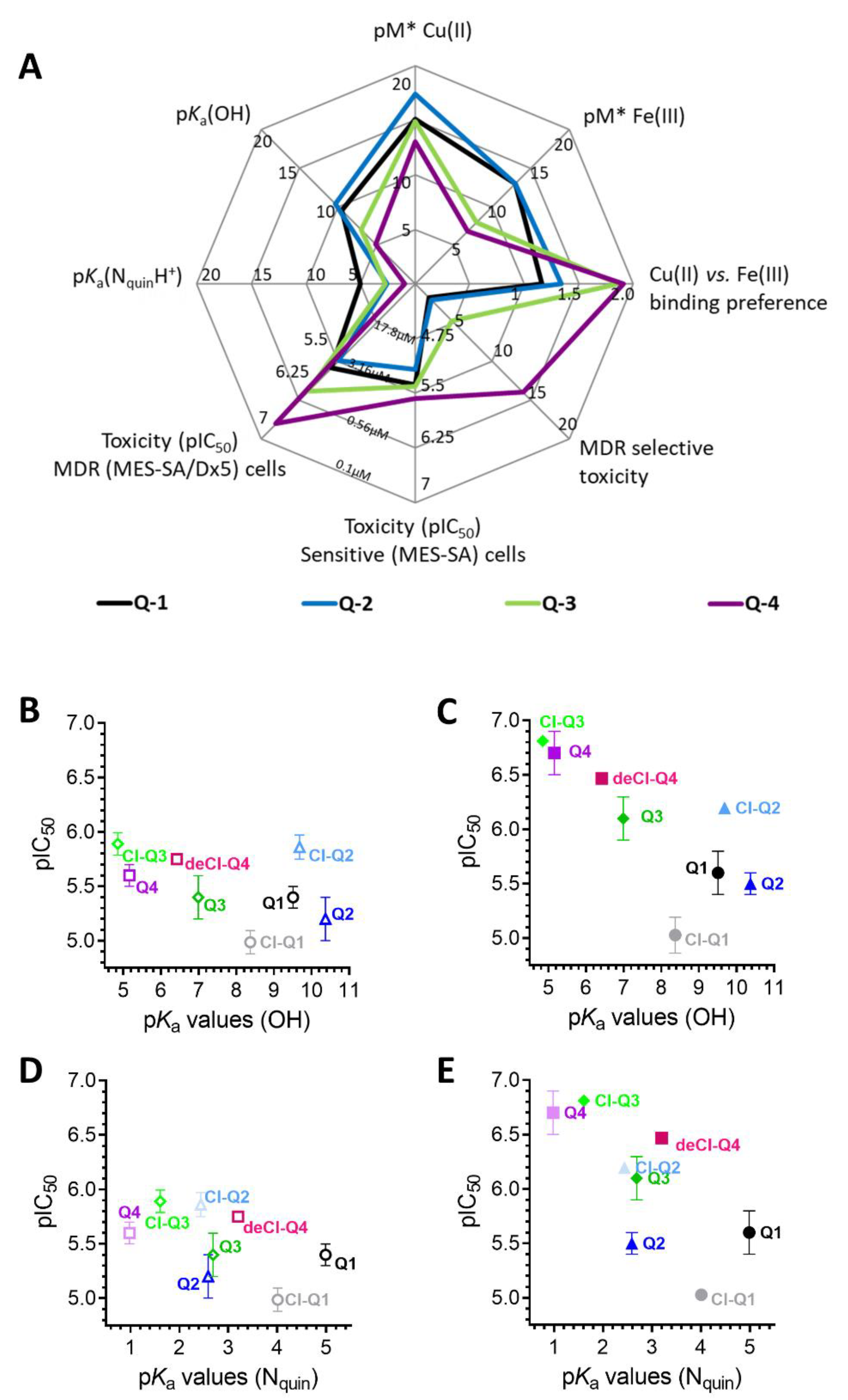
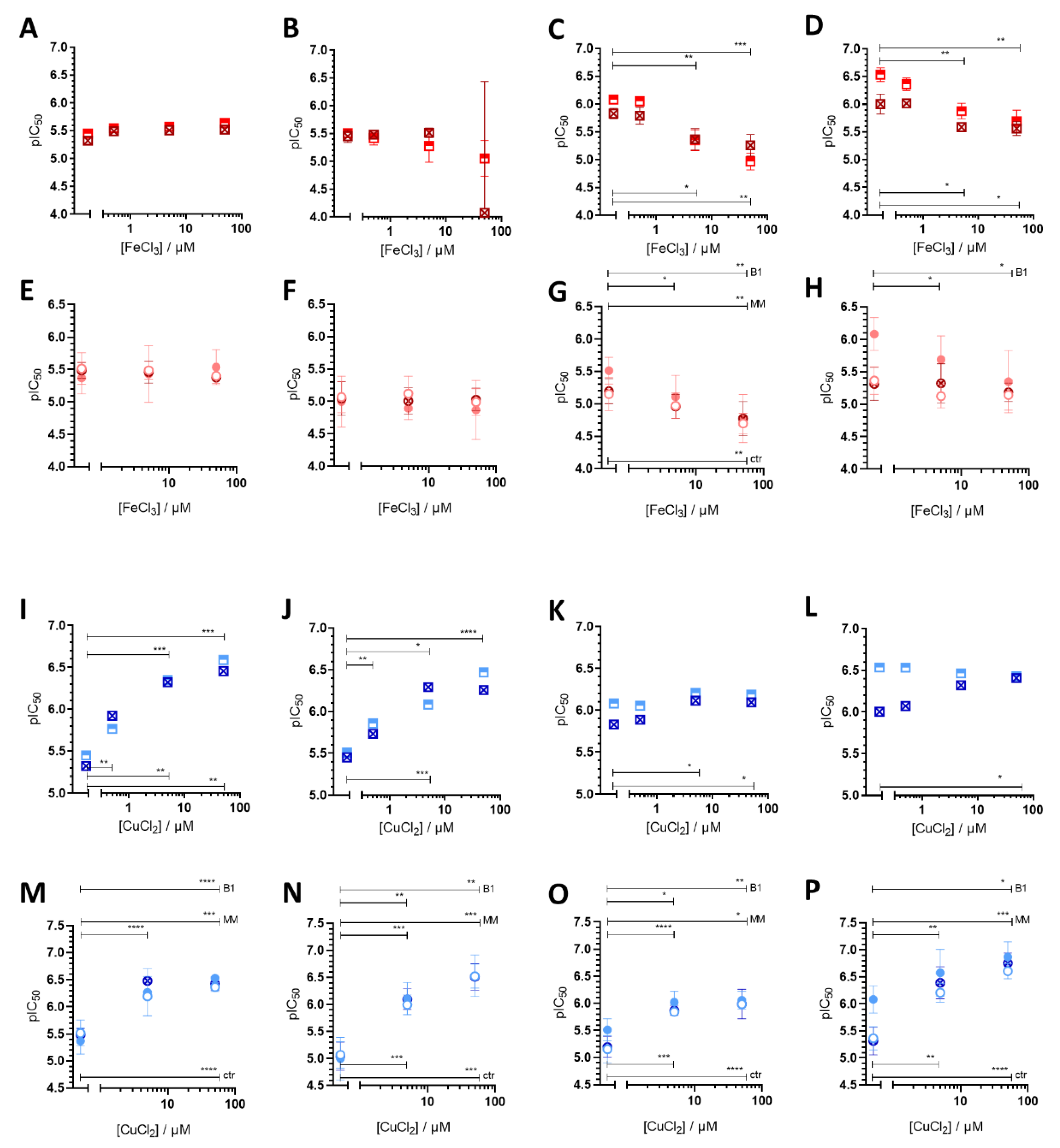
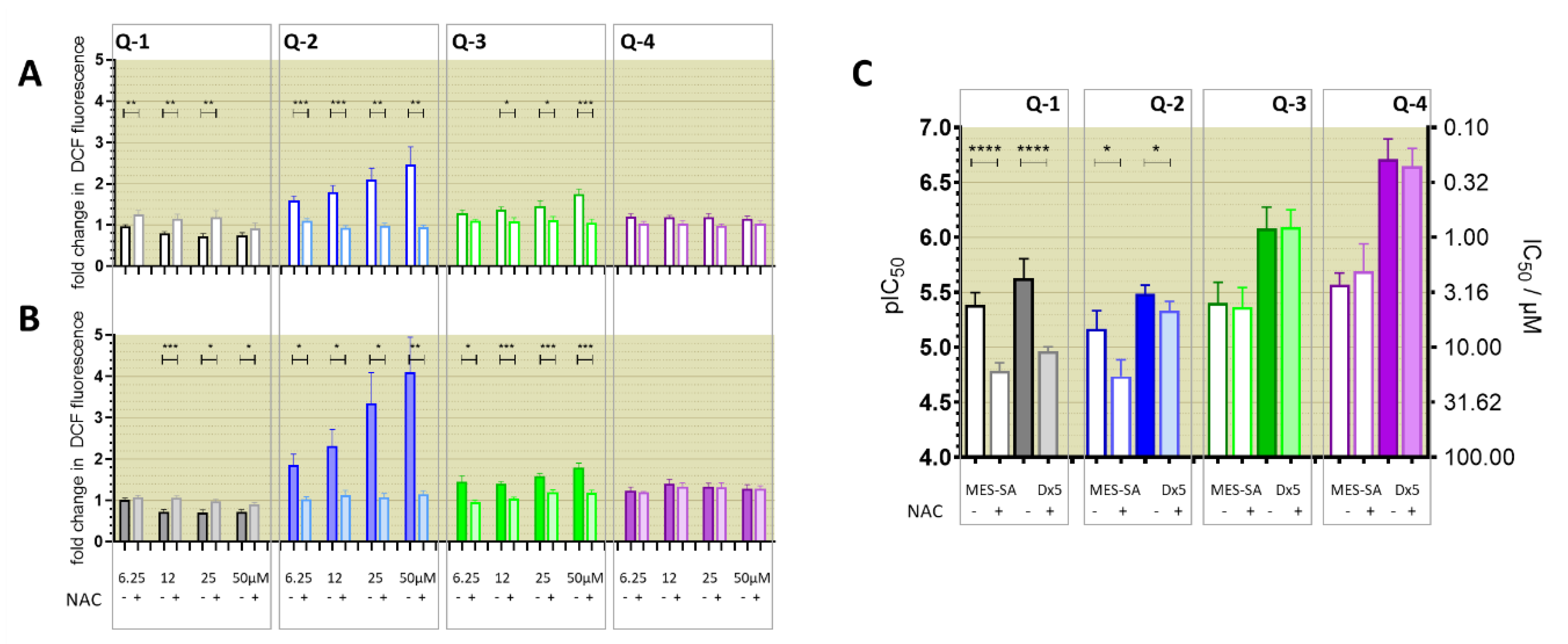
3.1. Impact of iron(III) and copper(II) Ions on MDR-Selective Cytotoxicity
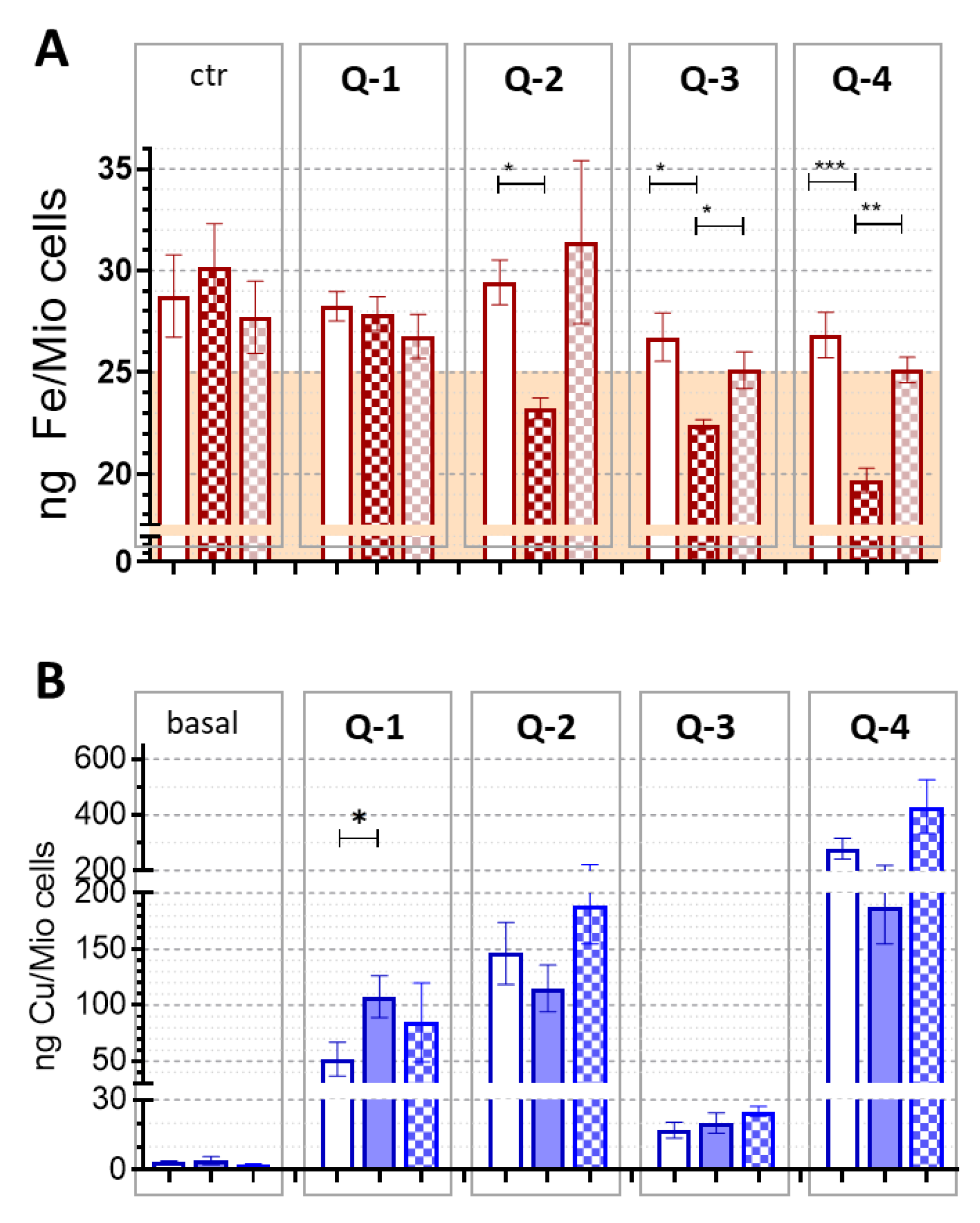
3.2. Intracellular Metal Levels
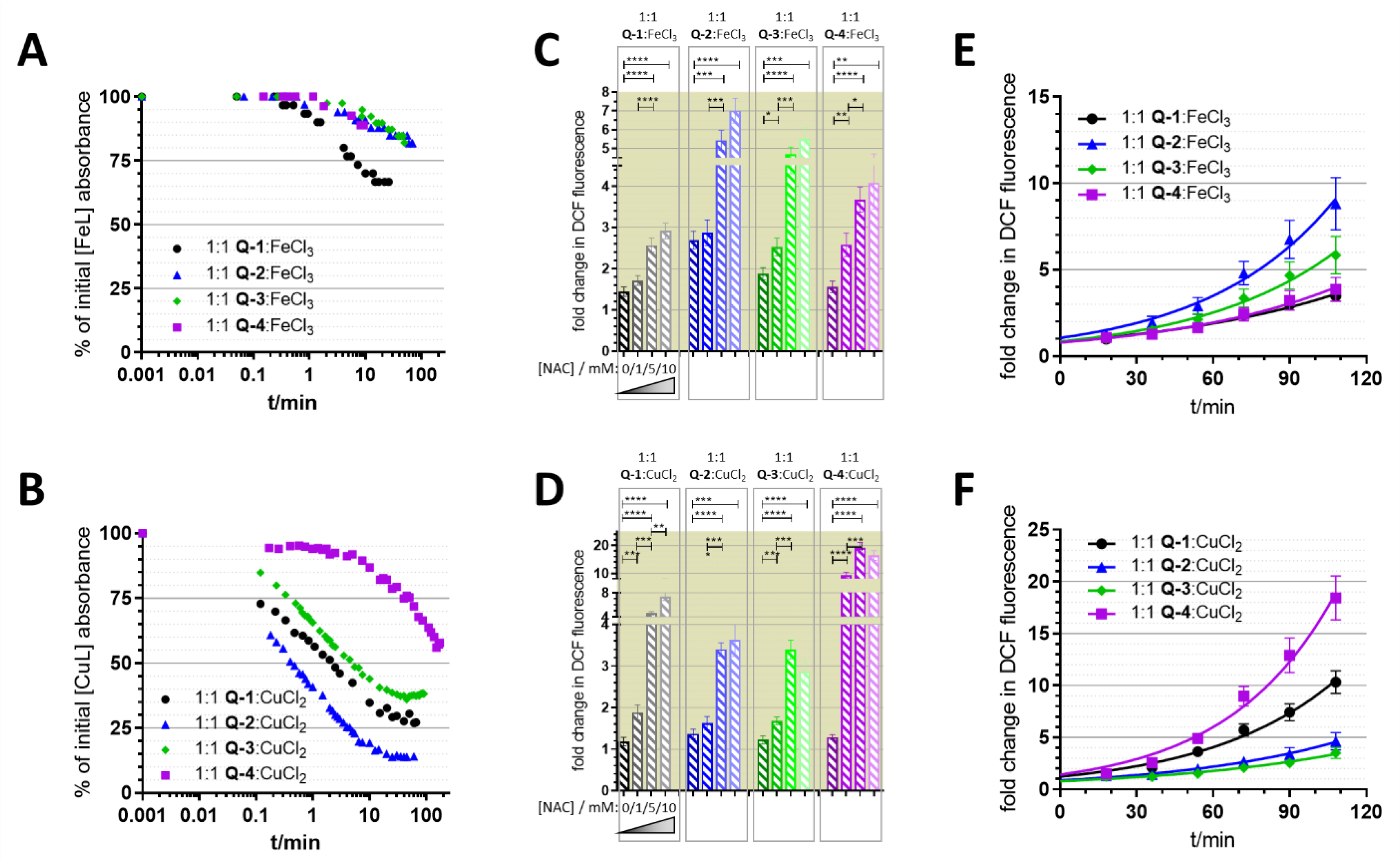
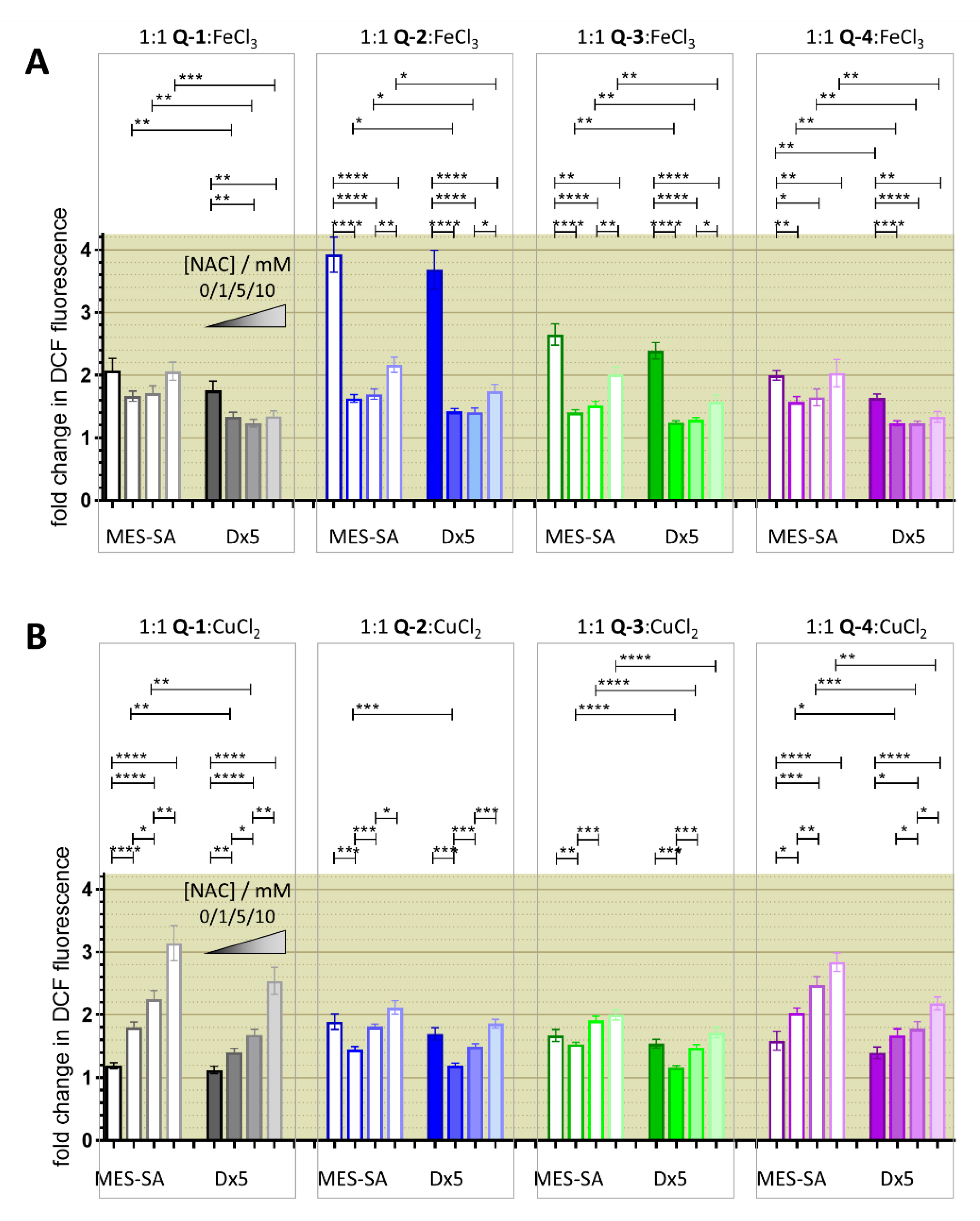
3.3. Redox Chemistry
3.4. Relevance of Redox Activity of the Complexes on Toxicity—Involvement of ROS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stewart, B.W.; Wild, C.P.; International Agency for Research on Cancer. World Cancer Report 2014, 2014 ed.; World Health Organization: Lyon, France, 2014. [Google Scholar]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of atp-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Garraway, L.A.; Jänne, P.A. Circumventing Cancer Drug Resistance in the Era of Personalized Medicine. Cancer Discov. 2012, 2, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Szakács, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef]
- Marquez, B.; Van Bambeke, F. ABC multidrug transporters: Target for modulation of drug pharmacokinetics and drug-drug interactions. Curr. Drug Targets 2011, 12, 600–620. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Ludwig, J.; Xia, D.; Szakacs, G. Defeating Drug Resistance in Cancer. Discov. Med. 2009, 6, 18–23. [Google Scholar]
- Tamaki, A.; Ierano, C.; Szakács, G.; Robey, R.W.; Bates, S.E. The controversial role of ABC transporters in clinical oncology. Essays Biochem. 2011, 50, 209–232. [Google Scholar]
- Ambudkar, S.V.; Dey, S.; Hrycyna, C.A.; Ramachandra, M.; Pastan, I.; Gottesman, M.M. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 361–398. [Google Scholar] [CrossRef]
- Polgar, O.; Bates, S.E. ABC transporters in the balance: Is there a role in multidrug resistance? Biochem. Soc. Trans. 2005, 33, 241–246. [Google Scholar] [CrossRef]
- Szakács, G.; Chen, G.K.; Gottesman, M.M. The molecular mysteries underlying P-glycoprotein-mediated multidrug resistance. Cancer Biol. Ther. 2004, 3, 382–384. [Google Scholar] [CrossRef][Green Version]
- Falasca, M.; Linton, K.J. Investigational ABC transporter inhibitors. Expert Opin. Investig. Drugs 2012, 21, 657–666. [Google Scholar] [CrossRef]
- Yu, M.; Ocana, A.; Tannock, I.F. Reversal of ATP-binding cassette drug transporter activity to modulate chemoresistance: Why has it failed to provide clinical benefit? Cancer Metastasis Rev. 2013, 32, 211–227. [Google Scholar] [CrossRef] [PubMed]
- Amiri-Kordestani, L.; Basseville, A.; Kurdziel, K.; Fojo, A.T.; Bates, S.E. Targeting MDR in breast and lung cancer: Discriminating its potential importance from the failure of drug resistance reversal studies. Drug Resist. Updates 2012, 15, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Füredi, A.; Szebényi, K.; Tóth, S.; Cserepes, M.; Hámori, L.; Nagy, V.; Karai, E.; Vajdovich, P.; Imre, T.; Szabó, P.; et al. Pegylated liposomal formulation of doxorubicin overcomes drug resistance in a genetically engineered mouse model of breast cancer. J. Control. Release 2017, 261, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Türk, D.; Hall, M.D.; Chu, B.F.; Ludwig, J.A.; Fales, H.M.; Gottesman, M.M.; Szakács, G. Identification of Compounds Selectively Killing Multidrug-Resistant Cancer Cells. Cancer Res. 2009, 69, 8293–8301. [Google Scholar] [CrossRef] [PubMed]
- Pluchino, K.M.; Hall, M.D.; Goldsborough, A.S.; Callaghan, R.; Gottesman, M.M. Collateral sensitivity as a strategy against cancer multidrug resistance. Drug Resist. Updates 2012, 15, 98–105. [Google Scholar] [CrossRef]
- Hall, M.D.; Handley, M.D.; Gottesman, M.M. Is resistance useless? Multidrug resistance and collateral sensitivity. Trends Pharmacol. Sci. 2009, 30, 546–556. [Google Scholar] [CrossRef]
- Szakács, G.; Hall, M.D.; Gottesman, M.M.; Boumendjel, A.; Kachadourian, R.; Day, B.J.; Baubichon-Cortay, H.; Di Pietro, A. Targeting the Achilles Heel of Multidrug-Resistant Cancer by Exploiting the Fitness Cost of Resistance. Chem. Rev. 2014, 114, 5753–5774. [Google Scholar] [CrossRef]
- Füredi, A.; Tóth, S.; Szebényi, K.; Pape, V.F.S.; Türk, D.; Kucsma, N.; Cervenak, L.; Tóvári, J.; Szakács, G. Identification and Validation of Compounds Selectively Killing Resistant Cancer: Delineating Cell Line–Specific Effects from P-Glycoprotein–Induced Toxicity. Mol. Cancer Ther. 2017, 16, 45–56. [Google Scholar] [CrossRef]
- Szakács, G.; Annereau, J.-P.; Lababidi, S.; Shankavaram, U.; Arciello, A.; Bussey, K.J.; Reinhold, W.; Guo, Y.; Kruh, G.D.; Reimers, M.; et al. Predicting drug sensitivity and resistance: Profiling ABC transporter genes in cancer cells. Cancer Cell 2004, 6, 129–137. [Google Scholar] [CrossRef]
- Shoemaker, R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef]
- Ludwig, J.A.; Szakács, G.; Martin, S.E.; Chu, B.F.; Cardarelli, C.; Sauna, Z.E.; Caplen, N.J.; Fales, H.M.; Ambudkar, S.V.; Weinstein, J.N.; et al. Selective Toxicity of NSC73306 in MDR1-Positive Cells as a New Strategy to Circumvent Multidrug Resistance in Cancer. Cancer Res. 2006, 66, 4808–4815. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.D.; Salam, N.K.; Hellawell, J.L.; Fales, H.M.; Kensler, C.B.; Ludwig, J.A.; Szakács, G.; Hibbs, D.E.; Gottesman, M.M. Synthesis, Activity, and Pharmacophore Development for Isatin-β-thiosemicarbazones with Selective Activity toward Multidrug-Resistant Cells. J. Med. Chem. 2009, 52, 3191–3204. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.D.; Brimacombe, K.R.; Varonka, M.S.; Pluchino, K.M.; Monda, J.K.; Li, J.; Walsh, M.J.; Boxer, M.B.; Warren, T.H.; Fales, H.M.; et al. Synthesis and Structure–Activity Evaluation of Isatin-β-thiosemicarbazones with Improved Selective Activity toward Multidrug-Resistant Cells Expressing P-Glycoprotein. J. Med. Chem. 2011, 54, 5878–5889. [Google Scholar] [CrossRef] [PubMed]
- Pape, V.F.S.; Tóth, S.; Füredi, A.; Szebényi, K.; Lovrics, A.; Szabó, P. Design, synthesis and biological evaluation of thiosemicarbazones, hydrazinobenzothiazoles and arylhydrazones as anticancer agents with a potential to overcome multidrug resistance. Eur. J. Med. Chem. 2016, 117, 335–354. [Google Scholar] [CrossRef] [PubMed]
- Heffeter, P.; Jakupec, M.A.; Körner, W.; Chiba, P.; Pirker, C.; Dornetshuber, R.; Elbling, L.; Sutterlüty, H.; Micksche, M.; Keppler, B.K.; et al. Multidrug-resistant cancer cells are preferential targets of the new antineoplastic lanthanum compound KP772 (FFC24). Biochem. Pharmacol. 2007, 73, 1873–1886. [Google Scholar] [CrossRef]
- Alakhova, D.Y.; Rapoport, N.Y.; Batrakova, E.V.; Timoshin, A.A.; Li, S.; Nicholls, D.; Alakhov, V.Y.; Kabanov, A.V. Differential metabolic responses to pluronic in MDR and non-MDR cells: A novel pathway for chemosensitization of drug resistant cancers. J. Control. Release. 2010, 142, 89–100. [Google Scholar] [CrossRef]
- Bentley, J.; Quinn, D.M.; Pitman, R.S.; Warr, J.R.; Kellett, G.L. The human KB multidrug-resistant cell line KB-C1 is hypersensitive to inhibitors of glycosylation. Cancer Lett. 1997, 115, 221–227. [Google Scholar] [CrossRef]
- Bell, S.E.; Quinn, D.M.; Kellett, G.L.; Warr, J.R. 2-Deoxy-D-glucose preferentially kills multidrug-resistant human KB carcinoma cell lines by apoptosis. Br. J. Cancer. 1998, 78, 1464–1470. [Google Scholar] [CrossRef]
- Broxterman, H.J.; Pinedo, H.M.; Kuiper, C.M.; Schuurhuis, G.J.; Lankelma, J. Glycolysis in P-glycoprotein-overexpressing human tumor cell lines Effects of resistance-modifying agents. FEBS Lett. 1989, 247, 405–410. [Google Scholar] [CrossRef]
- Landwojtowicz, E.; Nervi, P.; Seelig, A. Real-Time Monitoring of P-Glycoprotein Activation in Living Cells. Biochemistry 2002, 41, 8050–8057. [Google Scholar] [CrossRef]
- Gatlik-Landwojtowicz, E.; Äänismaa, P.; Seelig, A. The Rate of P-Glycoprotein Activation Depends on the Metabolic State of the Cell. Biochemistry 2004, 43, 14840–14851. [Google Scholar] [CrossRef] [PubMed]
- Karwatsky, J.; Lincoln, M.C.; Georges, E. A mechanism for P-glycoprotein-mediated apoptosis as revealed by verapamil hypersensitivity. Biochemistry 2003, 42, 12163–12173. [Google Scholar] [CrossRef] [PubMed]
- Jansson, P.J.; Yamagishi, T.; Arvind, A.; Seebacher, N.; Gutierrez, E.; Stacy, A.; Maleki, S.; Sharp, D.; Sahni, S.; Richardson, D.R. Di-2-pyridylketone 4,4-Dimethyl-3-thiosemicarbazone (Dp44mT) Overcomes Multidrug-Resistance by a Novel Mechanism Involving the Hijacking of Lysosomal P-Glycoprotein (Pgp). J. Biol. Chem. 2015, 290, 9588–9603. [Google Scholar] [CrossRef] [PubMed]
- Stacy, A.E.; Palanimuthu, D.; Bernhardt, P.V.; Kalinowski, D.S.; Jansson, P.J.; Richardson, D.R. Structure–Activity Relationships of Di-2-pyridylketone, 2-Benzoylpyridine, and 2-Acetylpyridine Thiosemicarbazones for Overcoming Pgp-Mediated Drug Resistance. J. Med. Chem. 2016, 59, 8601–8620. [Google Scholar] [CrossRef]
- Lovejoy, D.B.; Jansson, P.J.; Brunk, U.T.; Wong, J.; Ponka, P.; Richardson, D.R. Antitumor Activity of Metal-Chelating Compound Dp44mT Is Mediated by Formation of a Redox-Active Copper Complex That Accumulates in Lysosomes. Cancer Res. 2011, 71, 5871–5880. [Google Scholar] [CrossRef]
- Pape, V.F.S.; Türk, D.; Szabó, P.; Wiese, M.; Enyedy, E.A.; Szakács, G. Synthesis and characterization of the anticancer and metal binding properties of novel pyrimidinylhydrazone derivatives. J. Inorg Biochem. 2015, 144, 18–30. [Google Scholar] [CrossRef]
- Dömötör, O.; Pape, V.F.S.; May, N.V.; Szakács, G.; Enyedy, É.A. Comparative solution equilibrium studies of antitumor ruthenium(η6-p-cymene) and rhodium(η5-C5Me5) complexes of 8-hydroxyquinolines. Dalton Trans. 2017, 46, 4382–4396. [Google Scholar] [CrossRef]
- Heffeter, P.; Pape, V.F.S.; Enyedy, É.A.; Keppler, B.K.; Szakacs, G.; Kowol, C.R. Anticancer Thiosemicarbazones: Chemical Properties, Interaction with Iron Metabolism, and Resistance Development. Antioxid. Redox Signal. 2019, 30, 1062–1082. [Google Scholar] [CrossRef]
- Hager, S.; Pape, V.F.S.; Pósa, V.; Montsch, B.; Uhlik, L.; Szakács, G.; T’oth, S.; Jabronka, N.; Keppler, B.K.; Kowol, C.R.; et al. High Copper Complex Stability and Slow Reduction Kinetics as Key Parameters for Improved Activity, Paraptosis Induction, and Impact on Drug-Resistant Cells of Anticancer Thiosemicarbazones. Antioxid. Redox Signal. 2020, 33, 395–414. [Google Scholar] [CrossRef]
- Enyedy, É.A.; May, N.V.; Pape, V.F.S.; Heffeter, P.; Szakács, G.; Keppler, B.K.; Kowol, C.R. Complex formation and cytotoxicity of Triapine derivatives: A comparative solution study on the effect of the chalcogen atom and NH-methylation. Dalton Trans. 2020. Available online: https://pubs.rsc.org/en/content/articlelanding/2020/dt/d0dt03465g (accessed on 6 November 2020).
- Cserepes, M.; Türk, D.; Tóth, S.; Pape, V.F.S.; Gaál, A.; Gera, M.; Szabo, J.; Kucsma, N.; Varady, G.; Vertessy, B.G.; et al. Unshielding multidrug resistant cancer through selective iron depletion of P-glycoprotein expressing cells. Cancer Res. 2019. Available online: https://cancerres.aacrjournals.org/content/early/2019/12/28/0008-5472.CAN-19-1407 (accessed on 6 January 2020).
- Torti, S.V.; Torti, F.M. Ironing out Cancer. Cancer Res. 2011, 71, 1511–1514. [Google Scholar] [CrossRef]
- Lui, G.Y.L.; Kovacevic, Z.; Richardson, V.; Merlot, A.M.; Kalinowski, D.S.; Richardson, D.R. Targeting cancer by binding iron: Dissecting cellular signaling pathways. Oncotarget 2015, 6, 18748–18779. [Google Scholar] [CrossRef] [PubMed]
- Gupte, A.; Mumper, R.J. Elevated copper and oxidative stress in cancer cells as a target for cancer treatment. Cancer Treat. Rev. 2009, 35, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Tapiero, H.; Townsend, D.M.; Tew, K.D. Trace elements in human physiology and pathology. Copper. Biomed. Pharmacother. 2003, 57, 386–398. [Google Scholar] [CrossRef]
- Kalinowski, D.S.; Richardson, D.R. The Evolution of Iron Chelators for the Treatment of Iron Overload Disease and Cancer. Pharmacol. Rev. 2005, 57, 547–583. [Google Scholar] [CrossRef]
- Merlot, A.M.; Kalinowski, D.S.; Richardson, D.R. Novel Chelators for Cancer Treatment: Where Are We Now? Antioxid. Redox Signal. 2013, 18, 973–1006. [Google Scholar] [CrossRef]
- Lane, D.J.; Mills, T.M.; Shafie, N.H.; Merlot, A.M.; Moussa, R.S.; Kalinowski, D.S.; Kovacevic, Z.; Richardson, D.R. Expanding horizons in iron chelation and the treatment of cancer: Role of iron in the regulation of ER stress and the epithelial–mesenchymal transition. Biochim. Biophys. Acta BBA—Rev. Cancer 2014, 1845, 166–181. [Google Scholar] [CrossRef]
- Shvartsman, M.; Cabantchik, Z.I. Intracellular iron trafficking: Role of cytosolic ligands. BioMetals 2012, 25, 711–723. [Google Scholar] [CrossRef]
- Finch, R.A.; Liu, M.C.; Cory, A.H.; Cory, J.G.; Sartorelli, A.C. Triapine (3-aminopyridine-2-carboxaldehyde thiosemicarbazone; 3-AP): An inhibitor of ribonucleotide reductase with antineoplastic activity. Adv. Enzyme Regul. 1999, 39, 3–12. [Google Scholar] [CrossRef]
- Kowol, C.R.; Berger, R.; Eichinger, R.; Roller, A.; Jakupec, M.A.; Schmidt, P.P.; Arion, V.B.; Keppler, B.K. Gallium(III) and Iron(III) Complexes of α-N-Heterocyclic Thiosemicarbazones: Synthesis, Characterization, Cytotoxicity, and Interaction with Ribonucleotide Reductase. J. Med. Chem. 2007, 50, 1254–1265. [Google Scholar] [CrossRef]
- Chitambar, C.R.; Antholine, W.E. Iron-Targeting Antitumor Activity of Gallium Compounds and Novel Insights Into Triapine® -Metal Complexes. Antioxid. Redox Signal. 2013, 18, 956–972. [Google Scholar] [CrossRef]
- Yu, Y.; Wong, J.; Lovejoy, D.B.; Kalinowski, D.S.; Richardson, D.R. Chelators at the Cancer Coalface: Desferrioxamine to Triapine and Beyond. Clin. Cancer Res. 2006, 12, 6876–6883. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Lovejoy, D.B.; Richardson, D.R. Novel Di-2-Pyridyl-Derived Iron Chelators with Marked and Selective Anti-Tumor Activity: In Vitro and In Vivo Assessment. Blood 2004, 104, 1450–1458. [Google Scholar] [CrossRef] [PubMed]
- Saletta, F.; Rahmanto, Y.S.; Siafakas, A.R.; Richardson, D.R. Cellular Iron Depletion and the Mechanisms Involved in the Iron-dependent Regulation of the Growth Arrest and DNA Damage Family of Genes. J. Biol. Chem. 2011, 286, 35396–35406. [Google Scholar] [CrossRef]
- Kalinowski, D.S.; Stefani, C.; Toyokuni, S.; Ganz, T.; Anderson, G.J.; Subramaniam, N.V.; Trinder, D.; Olynyk, J.K.; Chua, A.; Jansson, P.J.; et al. Redox cycling metals: Pedaling their roles in metabolism and their use in the development of novel therapeutics. Biochim. Biophys. Acta BBA—Mol. Cell Res. 2016, 1863, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Jungwirth, U.; Kowol, C.R.; Keppler, B.K.; Hartinger, C.G.; Berger, W.; Heffeter, P. Anticancer Activity of Metal Complexes: Involvement of Redox Processes. Antioxid. Redox Signal. 2011, 15, 1085–1127. [Google Scholar] [CrossRef] [PubMed]
- Santini, C.; Pellei, M.; Gandin, V.; Porchia, M.; Tisato, F.; Marzano, C. Advances in Copper Complexes as Anticancer Agents. Chem. Rev. 2014, 114, 815–862. [Google Scholar] [CrossRef] [PubMed]
- Pape, V.F.S.; May, N.V.; Gál, G.T.; Szatmári, I.; Szeri, F.; Fulop, F.; Szakács, G.; Enyedy, É.A. Impact of copper and iron binding properties on the anticancer activity of 8-hydroxyquinoline derived Mannich bases. Dalton Trans. 2018, 47, 17032–17045. [Google Scholar] [CrossRef]
- Windt, T.; Tóth, S.; Patik, I.; Sessler, J.; Kucsma, N.; Szepesi, Á.; Zdrazil, B.; Özvegy-Laczka, C.; Szakács, G. Identification of anticancer OATP2B1 substrates by an in vitro triple-fluorescence-based cytotoxicity screen. Arch. Toxicol. 2019, 93, 953–964. [Google Scholar] [CrossRef]
- Bársony, O.; Szalóki, G.; Türk, D.; Tarapcsák, S.; Gutay-Tóth, Z.; Bacsó, Z.; Holb, I.J.; Szekvolgyi, L.; Szabo, G.; Csanady, L.; et al. A single active catalytic site is sufficient to promote transport in P-glycoprotein. Sci. Rep. 2016, 6, 24810. [Google Scholar] [CrossRef]
- Juvale, K.; Pape, V.F.S.; Wiese, M. Investigation of chalcones and benzochalcones as inhibitors of breast cancer resistance protein. Bioorg. Med. Chem. 2012, 20, 346–355. [Google Scholar] [CrossRef]
- Mueller, H.; Kassack, M.U.; Wiese, M. Comparison of the Usefulness of the MTT, ATP, and Calcein Assays to Predict the Potency of Cytotoxic Agents in Various Human Cancer Cell Lines. J. Biomol. Screen. 2004, 9, 506–515. [Google Scholar] [CrossRef]
- Polgári, Z.; Ajtony, Z.; Kregsamer, P.; Streli, C.; Mihucz, V.G.; Réti, A.; Budai, B.; Kralovánszky, J.; Szoboszlai, N.; Záray, G. Microanalytical method development for Fe, Cu and Zn determination in colorectal cancer cells. Talanta 2011, 85, 1959–1965. [Google Scholar] [CrossRef] [PubMed]
- Szoboszlai, N.; Polgári, Z.; Mihucz, V.G.; Záray, G. Recent trends in total reflection X-ray fluorescence spectrometry for biological applications. Anal. Chim. Acta 2009, 633, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Bakker, M.; Renes, J.; Groenhuijzen, A.; Visser, P.; Timmer-Bosscha, H.; Müller, M.; Groen, H.J.M.; Smit, E.F.; de Vries, E.G.E. Mechanisms for high methoxymorpholino doxorubicin cytotoxicity in doxorubicin-resistant tumor cell lines. Int. J. Cancer 1997, 73, 362–366. [Google Scholar] [CrossRef]
- Harker, W.G.; Sikic, B.I. Multidrug (Pleiotropic) Resistance in Doxorubicin-selected Variants of the Human Sarcoma Cell Line MES-SA. Cancer Res. 1985, 45, 4091–4096. [Google Scholar]
- Shen, D.-W.; Cardarelli, C.; Hwang, J.; Cornwell, M.; Richert, N.; Ishii, S.; Pastan, I.; Gottesman, M.M. Multiple drug-resistant human KB carcinoma cells independently selected for high-level resistance to colchicine, adriamycin, or vinblastine show changes in expression of specific proteins. J. Biol. Chem. 1986, 261, 7762–7770. [Google Scholar]
- Gaál, A.; Orgován, G.; Polgári, Z.; Réti, A.; Mihucz, V.G.; Bősze, S.; Szoboszlai, N.; Streli, C. Complex forming competition and in-vitro toxicity studies on the applicability of di-2-pyridylketone-4,4,-dimethyl-3-thiosemicarbazone (Dp44mT) as a metal chelator. J. Inorg. Biochem. 2014, 130, 52–58. [Google Scholar] [CrossRef]
- Gaál, A.; Orgován, G.; Mihucz, V.G.; Pape, I.; Ingerle, D.; Streli, C.; Szoboszlai, N. Metal transport capabilities of anticancer copper chelators. J. Trace Elem. Med. Biol. 2018, 47, 79–88. [Google Scholar] [CrossRef]
- Eruslanov, E.; Kusmartsev, S. Identification of ROS Using Oxidized DCFDA and Flow-Cytometry. In Advanced Protocols in Oxidative Stress II; Armstrong, D., Ed.; Humana Press: Totowa, NJ, USA, 2010; pp. 57–72. [Google Scholar]
- LeBel, C.P.; Ischiropoulos, H.; Bondy, S.C. Evaluation of the probe 2′, 7′-dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress. Chem. Res. Toxicol. 1992, 5, 227–231. [Google Scholar] [CrossRef]
- Kowol, C.R.; Heffeter, P.; Miklos, W.; Gille, L.; Trondl, R.; Cappellacci, L.; Berger, W.; Keppler, B.K. Mechanisms underlying reductant-induced reactive oxygen species formation by anticancer copper(II) compounds. J. Biol. Inorg. Chem. 2012, 17, 409–423. [Google Scholar] [CrossRef]
- Noszál, B.; Visky, D.; Kraszni, M. Population, Acid−Base, and Redox Properties of N-Acetylcysteine Conformers. J. Med. Chem. 2000, 43, 2176–2182. [Google Scholar] [CrossRef]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Song, Y.; Xu, H.; Chen, W.; Zhan, P.; Liu, X. 8-Hydroxyquinoline: A privileged structure with a broad-ranging pharmacological potential. MedChemComm 2015, 6, 61–74. [Google Scholar] [CrossRef]
- Oliveri, V.; Vecchio, G. 8-Hydroxyquinolines in medicinal chemistry: A structural perspective. Eur. J. Med. Chem. 2016, 120, 252–274. [Google Scholar] [CrossRef] [PubMed]
- Haas, K.L.; Franz, K.J. Application of Metal Coordination Chemistry To Explore and Manipulate Cell Biology. Chem. Rev. 2009, 109, 4921–4960. [Google Scholar] [CrossRef]
- Enyedy, É.A.; Zsigó, É.; Nagy, N.V.; Kowol, C.R.; Roller, A.; Keppler, B.K.; Kiss, T. Complex-Formation Ability of Salicylaldehyde Thiosemicarbazone towards ZnII, CuII, FeII, FeIII and GaIII Ions. Eur. J. Inorg. Chem. 2012, 2012, 4036–4047. [Google Scholar] [CrossRef]
- Pierre, J.; Baret, P.; Serratrice, G. Hydroxyquinolines as iron chelators. Curr. Med. Chem. 2003, 10, 1077–1084. [Google Scholar] [CrossRef]
- Biaso, F.; Baret, P.; Pierre, J.-L.; Serratrice, G. Comparative studies on the iron chelators O-TRENSOX and TRENCAMS: Selectivity of the complexation towards other biologically relevant metal ions and Al3+. J. Inorg. Biochem. 2002, 89, 123–130. [Google Scholar] [CrossRef]
- Jansson, P.J.; Hawkins, C.L.; Lovejoy, D.B.; Richardson, D.R. The iron complex of Dp44mT is redox-active and induces hydroxyl radical formation: An EPR study. J. Inorg. Biochem. 2010, 104, 1224–1228. [Google Scholar] [CrossRef]
- Yu, Y.; Kalinowski, D.S.; Kovacevic, Z.; Siafakas, A.R.; Jansson, P.J.; Stefani, C.; Lovejoy, D.B.; Sharpe, P.C.; Bernhardt, P.V.; Richardson, D.R. Thiosemicarbazones from the Old to New: Iron Chelators That Are More Than Just Ribonucleotide Reductase Inhibitors. J. Med. Chem. 2009, 52, 5271–5294. [Google Scholar] [CrossRef]
- Barilli, A.; Atzeri, C.; Bassanetti, I.; Ingoglia, F.; Dall’Asta, V.; Bussolati, O.; Maffini, M.; Mucchino, C.; Marchiò, L. Oxidative Stress Induced by Copper and Iron Complexes with 8-Hydroxyquinoline Derivatives Causes Paraptotic Death of HeLa Cancer Cells. Mol. Pharm. 2014, 11, 1151–1163. [Google Scholar] [CrossRef] [PubMed]
- Kerksick, C.; Willoughby, D. The Antioxidant Role of Glutathione and N-Acetyl-Cysteine Supplements and Exercise-Induced Oxidative Stress. J. Int. Soc. Sports Nutr. 2005, 2, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Tardito, S.; Barilli, A.; Bassanetti, I.; Tegoni, M.; Bussolati, O.; Franchi-Gazzola, R.; Mucchino, C.; Marchiò, L. Copper-Dependent Cytotoxicity of 8-Hydroxyquinoline Derivatives Correlates with Their Hydrophobicity and Does Not Require Caspase Activation. J. Med. Chem. 2012, 55, 10448–10459. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 (µM) | Q-1 | SR | Q-2 | SR | Q-3 | SR | Q-4 | SR |
|---|---|---|---|---|---|---|---|---|
| MES-SA | 4.19 ± 0.74 (5.08 ± 0.48) | 6.97 ± 1.75 (4.65 ± 0.92) | 4.11 ± 1.20 (2.99 ± 0.91) | 2.75 ± 0.46 (2.22 ± 0.36) | ||||
| MES-SA/Dx5 | 2.46 ± 0.69 a (2.94 ± 0.66) | 1.70 ** (1.73 *) | 3.27 ± 0.39 a (5.49 ± 1.02) | 2.13 *** (0.85) | 0.86 ± 0.26 a (3.15 ± 0.93) | 4.75 **** (0.95) | 0.20 ± 0.06 a (3.28 ± 0.51) | 13.68 **** (0.68) |
| MES-SA/B1 | 3.68 ± 0.95 (4.92 ± 1.32) | 1.14 (1.03) | 3.12 ± 0.28 (3.60 ± 0.63) | 2.23 *** (1.29) | 0.83 ± 0.02 (1.50 ± 0.22) | 4.95 **** (1.99) | 0.30 ± 0.06 (1.03 ± 0.29) | 9.24 **** (2.16) |
| OVCAR-8 | 3.45 ± 0.13 (3.43 ± 0.40) | 3.16 ± 0.52 (3.28 ± 0.41) | 3.15 ± 0.36 (3.72 ± 0.55) | 1.08 ± 0.10 (1.09 ± 0.12) | ||||
| NCI-ADRres | 1.84 ± 0.30 (2.65 ± 0.8) | 1.88 (1.29) | 2.84 ± 0.81 (5.41 ± 1.55) | 1.11 (0.61) | 0.68 ± 0.09 (4.22 ± 0.86) | 4.60 ** (0.88) | 0.14 ± 0.07 (1.15 ± 1.24) | 7.84 * (0-95) |
| KB-3-1 | 8.59 ± 2.92 (10.89 ± 3.04) | 9.55 ± 2.55 (11.69 ± 3.31) | 6.19 ± 0.12 (6.69 ± 0.15) | 1.75 ± 0.62 (2.65 ± 1.57) | ||||
| KB-v1 | 8.71 ± 4.43 (10.85 ± 2.52) | 0.99 (1.00) | 6.42 ± 2.19 (11.34 ± 3.11) | 1.49 (1.03) | 1.56 ± 0.57 (7.47 ± 0.84) | 3.96 * (0.89) | 0.27 ± 0.12 (2.27 ± 0.43) | 6.51 * (1.17) |
| A431 | 2.35 ± 0.92 (2.90 ± 1.37) | 3.31 ± 1.61 (3.18 ± 1.45) | 3.91 ± 0.83 (4.09 ± 0.88) | 5.82 ± 1.15 (4.54 ± 1.42) | ||||
| A431-B1 | 8.21 ± 4.04 (7.06 ± 4.75) | 0.31 (0.41) | 4.05 ± 1.86 (7.64 ± 5.32) | 0.82 (0.42) | 1.14 ± 0.16 (5.58 ± 1.67) | 3.96 *** (0.73) | 2.57 ± 0.56 (7.95 ± 2.62) | 2.26 *** 0.57*) |
| MDCK | 3.27 ± 1.24 (3.08 ± 1.41) | 9.15 ± 3.51 (11.10 ± 4.87) | 7.61 ± 3.06 (10.18 ± 3.12) | 4.55 ± 1.53 (5.92 ± 2.01) | ||||
| MDCK-B1 | 4.58 ± 1.75 (15.17 ± 10.11) | 0.71 (0.20) | 11.98 ± 7.84 (12.08 ± 5.07) | 0.76 (0.92) | 3.22 ± 1.02 (12.41 ± 6.27) | 2.36 * (0.82) | 0.89 ± 0.35 (6.95 ± 3.49) | 5.13 **** (0.85) |
| MDCK-MM | 3.34 ± 0.60 (3.59 ± 1.01) | 0.97 (0.86) | 9.74 ± 4.04 (9.01 ± 3.80) | 0.82 (1.23) | 6.67 ± 2.00 (8.45 ± 2.30) | 1.14(1.20) | 5.23 ± 2.11 (6.27 ± 2.29) | 0.87 (0.94) |
| Compound | Structure | MES-SA | MES-SA/Dx5 | SRMES-SA/Dx5 | pKa (OH) | pKa (NquinoliniumH+) | pKa (additional) |
|---|---|---|---|---|---|---|---|
| Q-1 |  | 4.19 ± 0.74 (5.08 ± 0.48) | 2.46 ± 0.69 a (2.94 ± 0.66) | 1.70 ** (1.73 *) | 9.51 a | 4.99 a | |
| 5-Cl-Q-1 |  | 10.44 ± 1.71 (12.10 ± 0.42) | 9.66 ± 2.48 (8.32 ± 0.53) | 1.08 (1.46 *) | 8.37 b | 4.01 b | |
| Q-2 |  | 6.97 ± 1.75 (4.65 ± 0.92) | 3.27 ± 0.39 a (5.49 ± 1.02) | 2.13 *** (0.85) | 10.37 a | 2.59 a | 6.25 a (NmorpholiniumH+) |
| 5-Cl-Q-2 |  | 1.40 ± 0.24 (1.57 ± 0.17) | 0.64 ± 0.03 (1.13 ± 0.28) | 2.19 * (1.39) | 9.68 ± 0.03 | <2 | 5.83 ± 0.02 (NmorpholiniumH+) |
| NC-2 |  | 421.68 ± 34.90 | 561.61 ± 36.8 | 0.75 | 9.21 ± 0.02 c | ‒ | 6.61 ± 0.02 c (NmorpholiniumH+) |
| Q-3 |  | 4.11 ± 1.20 (2.99 ± 0.91) | 0.86 ± 0.26 a (3.15 ± 0.93) | 4.75 **** (0.95) | 6.99 a | 2.69 a | >11.5 a |
| 5-Cl-Q-3 |  | 1.30 ± 0.21 (1.46 ± 0.15) | 0.15 ± 0.01 (0.90 ± 0.12) | 8.40 *** (1.61 *) | 4.85 ± 0.02 | 1.61 ± 0.04 | 7.16 ± 0.03 (NpiperidiniumH+) |
| NC-3 |  | 139.53 ± 47.89 | 162.78 ± 16.63 | 0.86 | 6.06 ± 0.01 c | ‒ | 10.12 ± 0.01 c (NpiperidiniumH+) |
| De-Cl-Q-4 |  | 1.78 ± 0.08 (2.25 ± 0.54) | 0.34 ± 0.01 (1.31 ± 0.04) | 5.22 ** (1.72) | 6.42 ± 0.04 | 3.21 ± 0.04 | 10.69 ± 0.04 (NbenzylamineH2+) |
| Q-4 |  | 2.75 ± 0.46 (2.22 ± 0.36) | 0.20 ± 0.06 a (3.28 ± 0.51) | 13.68 **** (0.68) | 5.16 a | < 2 a | 8.54 a (NbenzylamineH2+) |
| NC-4 |  | 44.69 ± 4.52 | 31.09 ± 2.17 | 1.44 | 5.50 ± 0.02 | ‒ | 9.72 ± 0.02 (NbenzylamineH2+) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pape, V.F.S.; Gaál, A.; Szatmári, I.; Kucsma, N.; Szoboszlai, N.; Streli, C.; Fülöp, F.; Enyedy, É.A.; Szakács, G. Relation of Metal-Binding Property and Selective Toxicity of 8-Hydroxyquinoline Derived Mannich Bases Targeting Multidrug Resistant Cancer Cells. Cancers 2021, 13, 154. https://doi.org/10.3390/cancers13010154
Pape VFS, Gaál A, Szatmári I, Kucsma N, Szoboszlai N, Streli C, Fülöp F, Enyedy ÉA, Szakács G. Relation of Metal-Binding Property and Selective Toxicity of 8-Hydroxyquinoline Derived Mannich Bases Targeting Multidrug Resistant Cancer Cells. Cancers. 2021; 13(1):154. https://doi.org/10.3390/cancers13010154
Chicago/Turabian StylePape, Veronika F.S., Anikó Gaál, István Szatmári, Nóra Kucsma, Norbert Szoboszlai, Christina Streli, Ferenc Fülöp, Éva A. Enyedy, and Gergely Szakács. 2021. "Relation of Metal-Binding Property and Selective Toxicity of 8-Hydroxyquinoline Derived Mannich Bases Targeting Multidrug Resistant Cancer Cells" Cancers 13, no. 1: 154. https://doi.org/10.3390/cancers13010154
APA StylePape, V. F. S., Gaál, A., Szatmári, I., Kucsma, N., Szoboszlai, N., Streli, C., Fülöp, F., Enyedy, É. A., & Szakács, G. (2021). Relation of Metal-Binding Property and Selective Toxicity of 8-Hydroxyquinoline Derived Mannich Bases Targeting Multidrug Resistant Cancer Cells. Cancers, 13(1), 154. https://doi.org/10.3390/cancers13010154