DNA Methylation in Ovarian Cancer Susceptibility

Abstract
Simple Summary
Abstract
1. Introduction
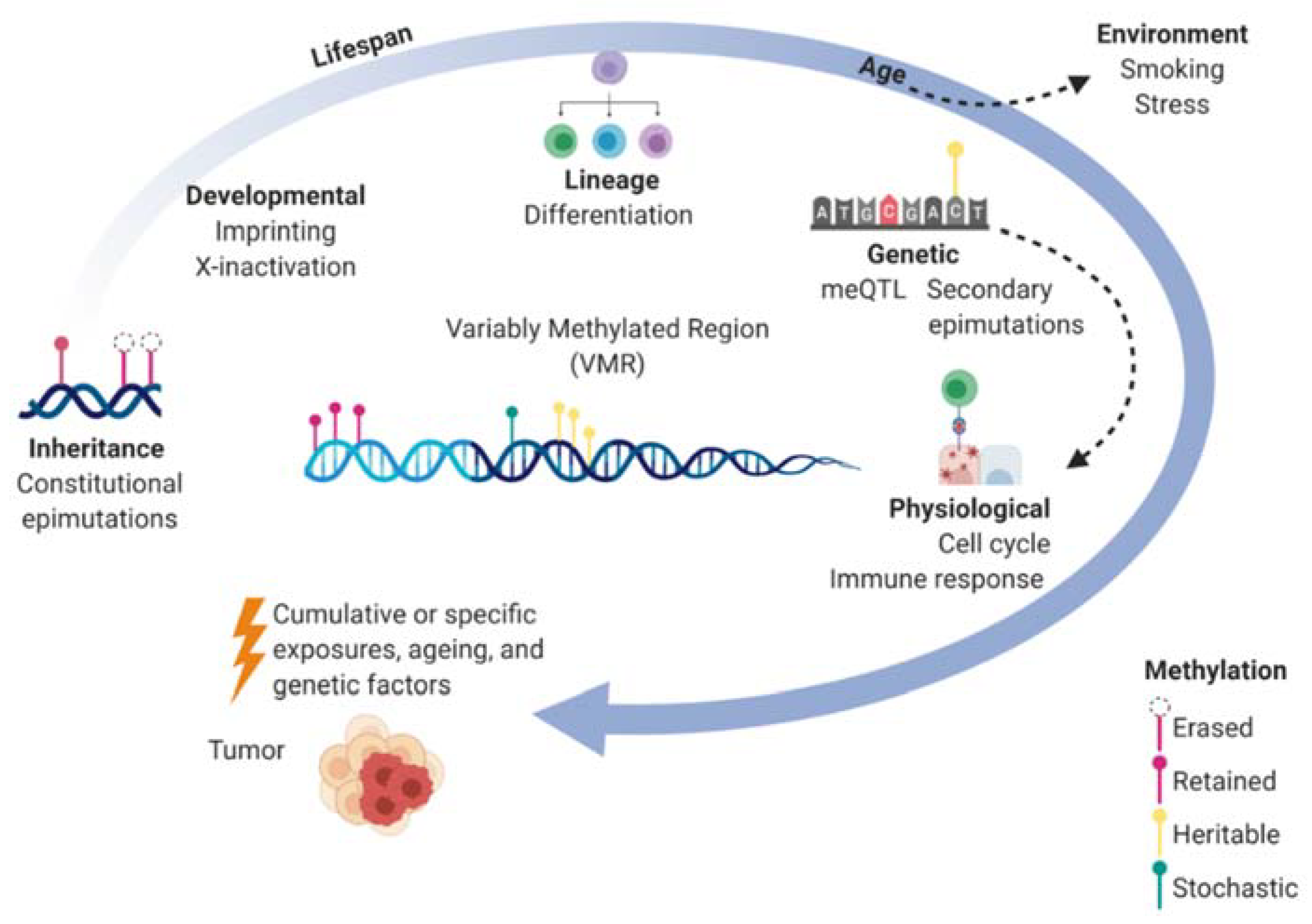
2. DNA Methylation in Epigenetic Control and Heritability
3. DNA Methylation in Ovarian Cancer Susceptibility Genes
4. Epigenome-Wide Association Studies of DNA Methylation
5. Genetic Susceptibility Mediated by DNA Methylation
6. Environmental Risk Mediated by DNA Methylation
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wray, N.R.; Yang, J.; Goddard, M.E.; Visscher, P.M. The genetic interpretation of area under the ROC curve in genomic profiling. PLoS Genet. 2010, 6, e1000864. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.Q.; Yu, H.Y.; Kanerva, A.; Forsti, A.; Sundquist, K.; Hemminki, K. Familial risks of ovarian cancer by age at diagnosis, proband type and histology. PLoS ONE 2018, 13, e0205000. [Google Scholar] [CrossRef]
- Jones, M.R.; Kamara, D.; Karlan, B.Y.; Pharoah, P.D.P.; Gayther, S.A. Genetic epidemiology of ovarian cancer and prospects for polygenic risk prediction. Gynecol. Oncol. 2017, 147, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Banta, J.A.; Richards, C.L. Quantitative epigenetics and evolution. Heredity 2018, 121, 210–224. [Google Scholar] [CrossRef] [PubMed]
- Waddington, C.H. The Epigenotype. Endeavour 1942, 1, 18–20. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Meissner, A.; Lander, E.S. The mammalian epigenome. Cell 2007, 128, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Skvortsova, K.; Iovino, N.; Bogdanovic, O. Functions and mechanisms of epigenetic inheritance in animals. Nat. Rev. Mol. Cell Biol. 2018, 19, 774–790. [Google Scholar] [CrossRef]
- Frias-Lasserre, D.; Villagra, C.A. The Importance of ncRNAs as Epigenetic Mechanisms in Phenotypic Variation and Organic Evolution. Front. Microbiol. 2017, 8, 2483. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation Distinguishes Genes of Some Human Cancers from Their Normal Counterparts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef]
- Hitchins, M.P.; Rapkins, R.W.; Kwok, C.T.; Srivastava, S.; Wong, J.J.L.; Khachigian, L.M.; Polly, P.; Goldblatt, J.; Ward, R.L. Dominantly Inherited Constitutional Epigenetic Silencing of MLH1 in a Cancer-Affected Family Is Linked to a Single Nucleotide Variant within the 5′ UTR. Cancer Cell 2011, 20, 200–213. [Google Scholar] [CrossRef]
- Lonning, P.E.; Eikesdal, H.P.; Loes, I.M.; Knappskog, S. Constitutional Mosaic Epimutations-a hidden cause of cancer? Cell Stress 2019, 3, 118–135. [Google Scholar] [CrossRef] [PubMed]
- Lacal, I.; Ventura, R. Epigenetic Inheritance: Concepts, Mechanisms and Perspectives. Front. Mol. Neurosci. 2018, 11, 292. [Google Scholar] [CrossRef] [PubMed]
- Lesch, B.J.; Tothova, Z.; Morgan, E.A.; Liao, Z.; Bronson, R.T.; Ebert, B.L.; Page, D.C. Intergenerational epigenetic inheritance of cancer susceptibility in mammals. Elife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef]
- Kim, M.; Costello, J. DNA methylation: An epigenetic mark of cellular memory. Exp. Mol. Med. 2017, 49, e322. [Google Scholar] [CrossRef]
- Wagner, A.; Regev, A.; Yosef, N. Revealing the vectors of cellular identity with single-cell genomics. Nat. Biotechnol. 2016, 34, 1145–1160. [Google Scholar] [CrossRef]
- Garg, P.; Joshi, R.S.; Watson, C.; Sharp, A.J. A survey of inter-individual variation in DNA methylation identifies environmentally responsive co-regulated networks of epigenetic variation in the human genome. PLoS Genet. 2018, 14, e1007707. [Google Scholar] [CrossRef]
- Shen, H.; Laird, P.W. Interplay between the Cancer Genome and Epigenome. Cell 2013, 153, 38–55. [Google Scholar] [CrossRef]
- Holliday, R. The inheritance of epigenetic defects. Science 1987, 238, 163–170. [Google Scholar] [CrossRef]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef]
- Earp, M.A.; Cunningham, J.M. DNA methylation changes in epithelial ovarian cancer histotypes. Genomics 2015, 106, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Natanzon, Y.; Goode, E.L.; Cunningham, J.M. Epigenetics in ovarian cancer. Semin. Cancer Biol. 2018, 51, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research, N. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Kolbe, D.L.; DeLoia, J.A.; Porter-Gill, P.; Strange, M.; Petrykowska, H.M.; Guirguis, A.; Krivak, T.C.; Brody, L.C.; Elnitski, L. Differential analysis of ovarian and endometrial cancers identifies a methylator phenotype. PLoS ONE 2012, 7, e32941. [Google Scholar] [CrossRef]
- Engqvist, H.; Parris, T.Z.; Biermann, J.; Ronnerman, E.W.; Larsson, P.; Sundfeldt, K.; Kovacs, A.; Karlsson, P.; Helou, K. Integrative genomics approach identifies molecular features associated with early-stage ovarian carcinoma histotypes. Sci. Rep. 2020, 10, 7946. [Google Scholar] [CrossRef]
- Mikkelsen, T.S.; Hanna, J.; Zhang, X.L.; Ku, M.C.; Wernig, M.; Schorderet, P.; Bernstein, B.E.; Jaenisch, R.; Lander, E.S.; Meissner, A. Dissecting direct reprogramming through integrative genomic analysis. Nature 2008, 454, 49–55. [Google Scholar] [CrossRef]
- Bjornsson, H.T.; Sigurdsson, M.I.; Fallin, M.D.; Irizarry, R.A.; Aspelund, T.; Cui, H.; Yu, W.; Rongione, M.A.; Ekstrom, T.J.; Harris, T.B.; et al. Intra-individual change over time in DNA methylation with familial clustering. JAMA 2008, 299, 2877–2883. [Google Scholar] [CrossRef]
- Kwabi-Addo, B.; Chung, W.; Shen, L.; Ittmann, M.; Wheeler, T.; Jelinek, J.; Issa, J.P. Age-related DNA methylation changes in normal human prostate tissues. Clin. Cancer Res. 2007, 13, 3796–3802. [Google Scholar] [CrossRef]
- Hitchins, M.P. Constitutional epimutation as a mechanism for cancer causality and heritability? Nat. Rev. Cancer 2015, 15. [Google Scholar] [CrossRef]
- Greenberg, M.V.C.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef]
- Rodgers, A.B.; Morgan, C.P.; Leu, N.A.; Bale, T.L. Transgenerational epigenetic programming via sperm microRNA recapitulates effects of paternal stress. Proc. Natl. Acad. Sci. USA 2015, 112, 13699–13704. [Google Scholar] [CrossRef] [PubMed]
- Van Dongen, J.; Nivard, M.G.; Willemsen, G.; Hottenga, J.J.; Helmer, Q.; Dolan, C.V.; Ehli, E.A.; Davies, G.E.; Van Iterson, M.; Breeze, C.E.; et al. Genetic and environmental influences interact with age and sex in shaping the human methylome. Nat. Commun. 2016, 7, 11115. [Google Scholar] [CrossRef] [PubMed]
- Husquin, L.T.; Rotival, M.; Fagny, M.; Quach, H.; Zidane, N.; McEwen, L.M.; MacIsaac, J.L.; Kobor, M.S.; Aschard, H.; Patin, E.; et al. Exploring the genetic basis of human population differences in DNA methylation and their causal impact on immune gene regulation. Genome. Biol. 2018, 19, 1–17. [Google Scholar] [CrossRef]
- Hansmann, T.; Pliushch, G.; Leubner, M.; Kroll, P.; Endt, D.; Gehrig, A.; Preisler-Adams, S.; Wieacker, P.; Haaf, T. Constitutive promoter methylation of BRCA1 and RAD51C in patients with familial ovarian cancer and early-onset sporadic breast cancer. Hum. Mol. Genet. 2012, 21, 4669–4679. [Google Scholar] [CrossRef] [PubMed]
- Lonning, P.E.; Berge, E.O.; Bjornslett, M.; Minsaas, L.; Chrisanthar, R.; Hoberg-Vetti, H.; Dulary, C.; Busato, F.; Bjorneklett, S.; Eriksen, C.; et al. White Blood Cell BRCA1 Promoter Methylation Status and Ovarian Cancer Risk. Ann. Intern. Med. 2018, 168, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Tabano, S.; Azzollini, J.; Pesenti, C.; Lovati, S.; Costanza, J.; Fontana, L.; Peissel, B.; Miozzo, M.; Manoukian, S. Analysis of BRCA1 and RAD51C Promoter Methylation in Italian Families at High-Risk of Breast and Ovarian Cancer. Cancers 2020, 12, 910. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.G.R.; Van Veen, E.M.; Byers, H.J.; Wallace, A.J.; Ellingford, J.M.; Beaman, G.; Santoyo-Lopez, J.; Aitman, T.J.; Eccles, D.M.; Lalloo, F.I.; et al. A Dominantly Inherited 5’ UTR Variant Causing Methylation-Associated Silencing of BRCA1 as a Cause of Breast and Ovarian Cancer. Am. J. Hum. Genet. 2018, 103, 213–220. [Google Scholar] [CrossRef]
- Sloane, M.A.; Ward, R.L.; Hesson, L.B. Defining the criteria for identifying constitutional epimutations. Clin. Epigenetics 2016, 8, 1–2. [Google Scholar] [CrossRef]
- Al-Moghrabi, N.; Al-Showimi, M.; Al-Yousef, N.; Al-Shahrani, B.; Karakas, B.; Alghofaili, L.; Almubarak, H.; Madkhali, S.; Al Humaidan, H. Methylation of BRCA1 and MGMT genes in white blood cells are transmitted from mothers to daughters. Clin. Epigenetics 2018, 10, 99. [Google Scholar] [CrossRef]
- Lonning, P.E.; Knappskog, S. BRCA1 methylation in newborns: Genetic disposition, maternal transfer, environmental influence, or by chance only? Clin. Epigenetics 2018, 10, 128. [Google Scholar] [CrossRef]
- Bibikova, M.; Barnes, B.; Tsan, C.; Ho, V.; Klotzle, B.; Le, J.M.; Delano, D.; Zhang, L.; Schroth, G.P.; Gunderson, K.L.; et al. High density DNA methylation array with single CpG site resolution. Genomics 2011, 98, 288–295. [Google Scholar] [CrossRef]
- Teschendorff, A.E.; Menon, U.; Gentry-Maharaj, A.; Ramus, S.J.; Gayther, S.A.; Apostolidou, S.; Jones, A.; Lechner, M.; Beck, S.; Jacobs, I.J.; et al. An epigenetic signature in peripheral blood predicts active ovarian cancer. PLoS ONE 2009, 4, e8274. [Google Scholar] [CrossRef] [PubMed]
- Koestler, D.C.; Marsit, C.J.; Christensen, B.C.; Accomando, W.; Langevin, S.M.; Houseman, E.A.; Nelson, H.H.; Karagas, M.R.; Wiencke, J.K.; Kelsey, K.T. Peripheral blood immune cell methylation profiles are associated with nonhematopoietic cancers. Cancer Epidemiol. Biomark. Prev. 2012, 21, 1293–1302. [Google Scholar] [CrossRef]
- Langevin, S.M.; Houseman, E.A.; Accomando, W.P.; Koestler, D.C.; Christensen, B.C.; Nelson, H.H.; Karagas, M.R.; Marsit, C.J.; Wiencke, J.K.; Kelsey, K.T. Leukocyte-adjusted epigenome-wide association studies of blood from solid tumor patients. Epigenetics 2014, 9, 884–895. [Google Scholar] [CrossRef] [PubMed]
- Fridley, B.L.; Armasu, S.M.; Cicek, M.S.; Larson, M.C.; Wang, C.; Winham, S.J.; Kalli, K.R.; Koestler, D.C.; Rider, D.N.; Shridhar, V.; et al. Methylation of leukocyte DNA and ovarian cancer: Relationships with disease status and outcome. BMC Med. Genom. 2014, 7, 21. [Google Scholar] [CrossRef] [PubMed]
- Winham, S.J.; Armasu, S.M.; Cicek, M.S.; Larson, M.C.; Cunningham, J.M.; Kalli, K.R.; Fridley, B.L.; Goode, E.L. Genome-wide investigation of regional blood-based DNA methylation adjusted for complete blood counts implicates BNC2 in ovarian cancer. Genet. Epidemiol. 2014, 38, 457–466. [Google Scholar] [CrossRef]
- Agrawal, V.; Kim, D.Y.; Kwon, Y.G. Hhip regulates tumor-stroma-mediated upregulation of tumor angiogenesis. Exp. Mol. Med. 2017, 49, e289. [Google Scholar] [CrossRef]
- Jung, P.; Verdoodt, B.; Bailey, A.; Yates, J.R., 3rd; Menssen, A.; Hermeking, H. Induction of cullin 7 by DNA damage attenuates p53 function. Proc. Natl. Acad. Sci. USA 2007, 104, 11388–11393. [Google Scholar] [CrossRef]
- Kong, Y.J.; Wang, Z.H.; Zhou, Z.M.; Mao, X.Y.; Chen, C.S. CUL7 promotes cancer cell survival through promoting Caspase-8 ubiquitination. Cancer Res. 2019, 79. [Google Scholar] [CrossRef]
- Peterson, E.A.; Stanbery, L.; Li, C.; Kocak, H.; Makarova, O.; Petty, E.M. SEPT9_i1 and genomic instability: Mechanistic insights and relevance to tumorigenesis. Genes Chromosomes Cancer 2011, 50, 940–949. [Google Scholar] [CrossRef]
- Weber, M.; Hellmann, I.; Stadler, M.B.; Ramos, L.; Paabo, S.; Rebhan, M.; Schubeler, D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 2007, 39, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Buckley, M.A.; Woods, N.T.; Tyrer, J.P.; Mendoza-Fandino, G.; Lawrenson, K.; Hazelett, D.J.; Najafabadi, H.S.; Gjyshi, A.; Carvalho, R.S.; Lyra, P.C., Jr.; et al. Functional Analysis and Fine Mapping of the 9p22.2 Ovarian Cancer Susceptibility Locus. Cancer Res. 2019, 79, 467–481. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zhang, Y.; Jin, C.; Deng, X.; Wei, M.; Wu, Q.; Yang, T.; Zhou, Y.; Wang, Z. Impact of XRCC2 Arg188His polymorphism on cancer susceptibility: A meta-analysis. PLoS ONE 2014, 9, e91202. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zheng, H.; Huang, Y.; Huang, C.; Zhang, S.; Tian, J.; Li, P.; Sood, A.K.; Zhang, W.; Chen, K. DNA methylation signatures and coagulation factors in the peripheral blood leucocytes of epithelial ovarian cancer. Carcinogenesis 2017, 38, 797–805. [Google Scholar] [CrossRef]
- Van Der Weyden, L.; Arends, M.J.; Rust, A.G.; Poulogiannis, G.; McIntyre, R.E.; Adams, D.J. Increased tumorigenesis associated with loss of the tumor suppressor gene Cadm1. Mol. Cancer 2012, 11, 29. [Google Scholar] [CrossRef]
- Si, X.; Xu, F.; Xu, F.; Wei, M.; Ge, Y.; Chenge, S. CADM1 inhibits ovarian cancer cell proliferation and migration by potentially regulating the PI3K/Akt/mTOR pathway. Biomed. Pharm. 2020, 123, 109717. [Google Scholar] [CrossRef]
- De Strooper, L.M.; Van Zummeren, M.; Steenbergen, R.D.; Bleeker, M.C.; Hesselink, A.T.; Wisman, G.B.; Snijders, P.J.; Heideman, D.A.; Meijer, C.J. CADM1, MAL and miR124-2 methylation analysis in cervical scrapes to detect cervical and endometrial cancer. J. Clin. Pathol. 2014, 67, 1067–1071. [Google Scholar] [CrossRef]
- Bong, I.P.; Ng, C.C.; Fakiruddin, S.K.; Lim, M.N.; Zakaria, Z. Small interfering RNA-mediated silencing of nicotinamide phosphoribosyltransferase (NAMPT) and lysosomal trafficking regulator (LYST) induce growth inhibition and apoptosis in human multiple myeloma cells: A preliminary study. Bosn J. Basic Med. Sci. 2016, 16, 268–275. [Google Scholar] [CrossRef]
- Tarpey, P.S.; Behjati, S.; Young, M.D.; Martincorena, I.; Alexandrov, L.B.; Farndon, S.J.; Guzzo, C.; Hardy, C.; Latimer, C.; Butler, A.P.; et al. The driver landscape of sporadic chordoma. Nat. Commun. 2017, 8, 890. [Google Scholar] [CrossRef]
- Heyn, H.; Sayols, S.; Moutinho, C.; Vidal, E.; Sanchez-Mut, J.V.; Stefansson, O.A.; Nadal, E.; Moran, S.; Eyfjord, J.E.; Gonzalez-Suarez, E.; et al. Linkage of DNA methylation quantitative trait loci to human cancer risk. Cell Rep. 2014, 7, 331–338. [Google Scholar] [CrossRef]
- Heyn, H.; Moran, S.; Hernando-Herraez, I.; Sayols, S.; Gomez, A.; Sandoval, J.; Monk, D.; Hata, K.; Marques-Bonet, T.; Wang, L.; et al. DNA methylation contributes to natural human variation. Genome Res. 2013, 23, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Fridley, B.L.; Song, H.L.; Lawrenson, K.; Cunningham, J.M.; Ramus, S.J.; Cicek, M.S.; Tyrer, J.; Stram, D.; Larson, M.C.; et al. Epigenetic analysis leads to identification of HNF1B as a subtype-specific susceptibility gene for ovarian cancer. Nat. Commun. 2013, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Koestler, D.C.; Chalise, P.; Cicek, M.S.; Cunningham, J.M.; Armasu, S.; Larson, M.C.; Chien, J.; Block, M.; Kalli, K.R.; Sellers, T.A.; et al. Integrative genomic analysis identifies epigenetic marks that mediate genetic risk for epithelial ovarian cancer. BMC Med. Genom. 2014, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wu, L.; Shu, X.; Lu, Y.; Shu, X.O.; Cai, Q.; Beeghly-Fadiel, A.; Li, B.; Ye, F.; Berchuck, A.; et al. Genetic Data from Nearly 63,000 Women of European Descent Predicts DNA Methylation Biomarkers and Epithelial Ovarian Cancer Risk. Cancer Res. 2019, 79, 505–517. [Google Scholar] [CrossRef]
- Millstein, J.; Zhang, B.; Zhu, J.; Schadt, E.E. Disentangling molecular relationships with a causal inference test. BMC Genet. 2009, 10, 23. [Google Scholar] [CrossRef]
- Lugrin, J.; Martinon, F. The AIM2 inflammasome: Sensor of pathogens and cellular perturbations. Immunol. Rev. 2018, 281, 99–114. [Google Scholar] [CrossRef]
- Chang, C.M.; Wang, M.L.; Lu, K.H.; Yang, Y.P.; Juang, C.M.; Wang, P.H.; Hsu, R.J.; Yu, M.H.; Chang, C.C. Integrating the dysregulated inflammasome-based molecular functionome in the malignant transformation of endometriosis-associated ovarian carcinoma. Oncotarget 2018, 9, 3704–3726. [Google Scholar] [CrossRef]
- Luborsky, J.; Barua, A.; Edassery, S.; Bahr, J.M.; Edassery, S.L. Inflammasome expression is higher in ovarian tumors than in normal ovary. PLoS ONE 2020, 15, e0227081. [Google Scholar] [CrossRef]
- Hollmen, M.; Figueiredo, C.R.; Jalkanen, S. New tools to prevent cancer growth and spread: A ‘Clever’ approach. Brit. J. Cancer 2020, 123, 501–509. [Google Scholar] [CrossRef]
- Huan, T.X.; Joehanes, R.; Song, C.; Peng, F.; Guo, Y.C.; Mendelson, M.; Yao, C.; Liu, C.Y.; Ma, J.T.; Richard, M.; et al. Genome-wide identification of DNA methylation QTLs in whole blood highlights pathways for cardiovascular disease. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Kannel, W.B.; Feinleib, M.; Mcnamara, P.M.; Garrison, R.J.; Castelli, W.P. An Investigation of Coronary Heart Disease in Families: The Framingham Offspring Study. Am. J. Epidemiol. 2017, 185, 1093–1102. [Google Scholar] [CrossRef]
- Shi, Y.F.; Li, Y.P.; Yan, C.; Su, H.; Ying, K.J. Identification of key genes and evaluation of clinical outcomes in lung squamous cell carcinoma using integrated bioinformatics analysis. Oncol. Lett. 2019, 18, 5859–5870. [Google Scholar] [CrossRef] [PubMed]
- Sherva, R.; Miller, M.B.; Lynch, A.I.; Devereux, R.B.; Rao, D.C.; Oberman, A.; Hopkins, P.N.; Kitzman, D.W.; Atwood, L.D.; Arnett, D.K. A whole genome scan for pulse pressure/stroke volume ratio in African Americans: The HyperGEN study. Am. J. Hypertens. 2007, 20, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Yang, H.; Winham, S.J.; Natanzon, Y.; Koestler, D.C.; Luo, T.; Fridley, B.L.; Goode, E.L.; Zhang, Y.; Cui, Y. Mediation analysis of alcohol consumption, DNA methylation, and epithelial ovarian cancer. J. Hum. Genet. 2018, 63, 339–348. [Google Scholar] [CrossRef]
- Liu, C.; Marioni, R.E.; Hedman, A.K.; Pfeiffer, L.; Tsai, P.C.; Reynolds, L.M.; Just, A.C.; Duan, Q.; Boer, C.G.; Tanaka, T.; et al. A DNA methylation biomarker of alcohol consumption. Mol. Psychiatry 2018, 23, 422–433. [Google Scholar] [CrossRef]
- Boyne, D.J.; Friedenreich, C.M.; McIntyre, J.B.; Stanczyk, F.Z.; Courneya, K.S.; King, W.D. Endogenous sex hormone exposure and repetitive element DNA methylation in healthy postmenopausal women. Cancer Causes Control. 2017, 28, 1369–1379. [Google Scholar] [CrossRef]
- Crujeiras, A.B.; Casanueva, F.F. Obesity and the reproductive system disorders: Epigenetics as a potential bridge. Hum. Reprod Update 2015, 21, 249–261. [Google Scholar] [CrossRef]
- He, J.; Chang, W.; Feng, C.; Cui, M.; Xu, T. Endometriosis Malignant Transformation: Epigenetics as a Probable Mechanism in Ovarian Tumorigenesis. Int. J. Genom. 2018, 2018, 1465348. [Google Scholar] [CrossRef]
- Jiao, J.; Sagnelli, M.; Shi, B.; Fang, Y.Y.; Shen, Z.Q.; Tang, T.Y.; Dong, B.Y.; Li, D.; Wang, X.X. Genetic and epigenetic characteristics in ovarian tissues from polycystic ovary syndrome patients with irregular menstruation resemble those of ovarian cancer. BMC Endocr. Disord. 2019, 19, 1–10. [Google Scholar] [CrossRef]
- Lam, L.L.; Emberly, E.; Fraser, H.B.; Neumann, S.M.; Chen, E.; Miller, G.E.; Kobor, M.S. Factors underlying variable DNA methylation in a human community cohort. Proc. Natl. Acad. Sci. USA 2012, 109, 17253–17260. [Google Scholar] [CrossRef]
- Ladd-Acosta, C.; Fallin, M.D. The role of epigenetics in genetic and environmental epidemiology. Epigenomics 2016, 8, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Liao, W.; Wos, F.; Johnston, A.D.; DeGrazia, J.; Ishii, J.; Bloom, T.; Zody, M.C.; Germer, S.; Greally, J.M. Whole-genome bisulfite sequencing with improved accuracy and cost. Genome Res. 2018, 28, 1364–1371. [Google Scholar] [CrossRef] [PubMed]
- Won, K.J.; Zhang, X.; Wang, T.; Ding, B.; Raha, D.; Snyder, M.; Ren, B.; Wang, W. Comparative annotation of functional regions in the human genome using epigenomic data. Nucleic Acids Res. 2013, 41, 4423–4432. [Google Scholar] [CrossRef] [PubMed]
- Kaplow, I.M.; MacIsaac, J.L.; Mah, S.M.; McEwen, L.M.; Kobor, M.S.; Fraser, H.B. A pooling-based approach to mapping genetic variants associated with DNA methylation. Genome Res. 2015, 25, 907–917. [Google Scholar] [CrossRef]


{kind=link}
| First Author, Year, Country | Study Design | Sample Size a | Case Criterion | Platform | Methylation Type, Thresholds | Prevalence | Comments | |
|---|---|---|---|---|---|---|---|---|
| Gene(s) | N (%) | |||||||
| Hansmann, 2012, Germany | Case-only | 641 Cases (613 BC, 39 EOC) | Familial or early- onseta BC or EOC Chemotherapy naïve BRCA1/2 negative | Bisulfite pyrosequencing (4-7 CpG) Bisulfite plasmid sequencing (27 CpG) | Hypermethylation as 75th percentile + 3xIQR, >6% for all genes | ATM BRCA1 BRCA2 PTEN RAD51C TP53 | 0 All: 9 (1.4%) EOC: 3 (8%) BC: 6 (1%) 0 0 All: 3 (0.5%) EOC: 1 (2.5%) BC: 2 (0.3%) 0 | 10 age-matched controls were tested for BRCA1 and none were hypermethylated. Formal statistical comparison was not included. Urine, saliva also profiled |
| Lonning, 2018, Norway | Two-stage case-control | Phase I: 934 Cases, 1698 Controls (332 HGSOC, 298 LGSOC, 295 Nonserous) Phase II: 607 Cases, 1984 Controls (286 HGSOC, 151 LGSOC, 170 Nonserous) | Invasive EOC Chemotherapy naïve BRCA1/2 negative | qPCR Bisulfite pyrosequencing (NR) | Manually scored positive/negative methylation status >Median | BRCA1 | Controls: 70 (4.2%) All EOC: 59 (6.4%) OR=1.8 (1.27-2.63) HGSOC: 32 (9.6%) OR=2.9 (1.85-4.56) LGSOC: 12 (4.0%) OR=0.98 (0.54-1.79) Nonserous: 15 (5.1%) OR=1.23 (0.71-2.13) | 932 post- and 784 pre-surgery 333 not tested for BRCA1/2 in phase II Age-matched controls Age-adjusted logistic regression Tumors also profiled |
| Evans, 2018, | Segregation | 49 probands | Familial BC or EOC Manchester score>34 BRCA1/2 negative | Bisulfite pyrosequencing (10 CpG) Bisulfite plasmid sequencing | Mean promoter methylation (%) | BRCA1 | Proband: 2 (4%) ~50% methylated Dominant inheritance; segregates with c.−107A>T in the BRCA1 5′ UTR | BRCA1 promoter hypermethylation was 71.4% informative Buccal mucosa, tumor, hair samples also profiled |
| Tabano, 2020, Italy | Case-control | 108 Cases, 60 Controls | Invasive or DCIS BC or high-grade non-mucinous EOC BRCA1/2 negative | MassARRAY® EpiTYPER (9 CpG BRCA1, 11 CpG RAD51C) | Hypermethylation as >UCL 95% CI b, >13.6% BRCA1 >12.1% RAD51C | BRCA1 RAD51C | 0 Case mean: 4.4% BC: 4.3% EOC: 3.9% Control mean: 4.3% 0 Case mean: 3.7% BC: 3.7% EOC: 3.5% Control mean: 4.3% | |
| First Author, Year, Region | Study Design, Population, Ancestry | Cases/ Controls | Histology, N (%) a | Platform (No. Cpg Tested) | Results | Probe/Gene Replicated | |
|---|---|---|---|---|---|---|---|
| DMP | AUC (95% CI) b | ||||||
| Teschendorff, 2009, UK | Retrospective, Population-based, European | 235/148 | Serous, 152 (57%) Endometrioid, 37, (14%) Mucinous, 30 (11%) Clear Cell, 28 (11%) Other, 19 (7%) | HumanMethylation27 (25,642) | 2714 | 0.80 (0.74–0.87) c 0.76 (0.72–0.81) d | 73/71 |
| Fridley, 2014, USA | Retrospective, Hospital-based, European | 336/338 | Serous, 243 (72%) Endometrioid, 63 (19%) Mucinous, 8 (2%) Clear Cell, 16 (5%) Other, 6 (2%) | HumanMethylation27 HumanMethylation450 (13,816) | 30 | NE | 6/6 |
| Winham, 2014, USA | Retrospective, Hospital-based, European | 242/181 e | Serous, 168 (70%) Endometrioid, 45 (19%) Mucinous, 7 (3%) Clear Cell, 12 (5%) Other, 10 (4%) | HumanMethylation27 HumanMethylation450 (22,278) | 62 | NE | 61/59 |
| Li, 2017, China | Retrospective, Hospital-based, Han Chinese | 230/229 | Serous, 109 (49%) Endometrioid, 59 (26%) Mucinous, 24 (11%) Other, 32 (14%) | HumanMethylation450 (450k) | Overall: | 6/5 | |
| 40 | 0.77 (0.73–0.82) | ||||||
| Serous: | |||||||
| 32 | 0.77 (0.71–0.83) | ||||||
| Endometrioid: | |||||||
| 34 | 0.80 (0.74–0.86) | ||||||
| Mucinous: | |||||||
| 11 | 0.73 (0.61–0.84) | ||||||
| TOTAL | 801/715 | Serous, 504 (52%) Endometrioid, 267 (28%) Mucinous, 69 (7%) Clear Cell, 56 (6%) Other, 67 (7%) | 2846 | 82/71 | |||
| Study | Locus | Genetic Risk | DNA Methylation | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| SNP a (Gene) | Histotype | OR (95% CI) | p-Value | CpG Site b | Correlated Expression | Histotype c | Case Status | p-Value | ||
| Shen, 2013 | 17q12 | rs7405776 (HNF1B) | Serous | 1.13 (1.09–1.17) | 3.1 × 10−10 | cg14487292 | HNF1B | Serous | Hyper | NE |
| rs11651755 (HNF1B) | Clear cell | 0.77 (0.70–0.84) | 1.6 × 10−8 | Clear cell | Hypo | NE | ||||
| Yang. 2019 | 2q31.1 | rs2072590 (HAGLR) | Serous | 1.20 (1.14–1.25) | 3.8 × 10−14 | cg25137403 | HOXD4 | Serous | Hyper | 9.1 × 10−14 |
| rs711830 (HOXD3) | Mucinous | 1.30 (1.20–1.40) | 7.5 × 10−12 | Mucinous | Hyper | 9.7 × 10−9 | ||||
| rs2072590 (HAGLR) | Endometrioid | 1.13 (1.04–1.22) | 2.4 × 10−3 | Endometrioid | Hyper | 5.8 × 10−5 | ||||
| 3q25 | rs7651446 (TIPARP) | All | 1.44 (1.35–1.53) | 1.5 × 10−28 | cg26405475 | SSR3 | HG Serous | Hypo | 1.9 × 10−26 | |
| 7p22.3 | NA | cg03634833 | ADAP1 | All | Hypo | 5.8 × 10−7 | ||||
| 8q24.21 | rs10088218 (LINC00824) | Serous | 0.76 (0.70–0.81) | 8.0 × 10−15 | cg08478672 | NA | All | Hyper | 3.8 × 10−7 | |
| 9q34.2 | rs635634 | All | 1.11 (1.07–1.16) | 4.4 × 10−9 | cg14653977 | GBGT1 ABO | All | Hyper | 2.0 × 10−9 | |
| 10p12 | rs1243180 (MLLT10) | All | 1.10 (1.06–1.13) | 1.8 × 10−8 | cg04231319 | MLLT10 | All | Hypo | 1.1 × 10−8 | |
| 17q21.31 | rs2960000 (PLEKHM1) | Serous | 1.16 (1.12–1.20) | 3.3 × 10−10 | cg07067577 | ARHGAP27MAPT | All | Hypo | 6.9 × 10−14 | |
| HG Serous | Hypo | 1.4 × 10−10 | ||||||||
| 17q21.32 | rs9303542 (SKAP1) | All | 1.12 (1.08–1.16) | 6.0 × 10−11 | cg19139618 | SKAP1 HOXB3 HOXB8 | All | Hypo | 7.1 × 10−7 | |
| 19p13.11 | rs2363956 (ANKLE1) | Serous | 1.16 (1.11–1.21) | 3.8 × 10−11 | cg21956434 | ABHD8 | HG Serous | Hyper | 5.7 × 10−20 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reid, B.M.; Fridley, B.L. DNA Methylation in Ovarian Cancer Susceptibility. Cancers 2021, 13, 108. https://doi.org/10.3390/cancers13010108
Reid BM, Fridley BL. DNA Methylation in Ovarian Cancer Susceptibility. Cancers. 2021; 13(1):108. https://doi.org/10.3390/cancers13010108
Chicago/Turabian StyleReid, Brett M., and Brooke L. Fridley. 2021. "DNA Methylation in Ovarian Cancer Susceptibility" Cancers 13, no. 1: 108. https://doi.org/10.3390/cancers13010108
APA StyleReid, B. M., & Fridley, B. L. (2021). DNA Methylation in Ovarian Cancer Susceptibility. Cancers, 13(1), 108. https://doi.org/10.3390/cancers13010108