Inhibition of DNA Repair Pathways and Induction of ROS Are Potential Mechanisms of Action of the Small Molecule Inhibitor BOLD-100 in Breast Cancer

 ,
,
Simple Summary
Abstract
1. Introduction
2. Results
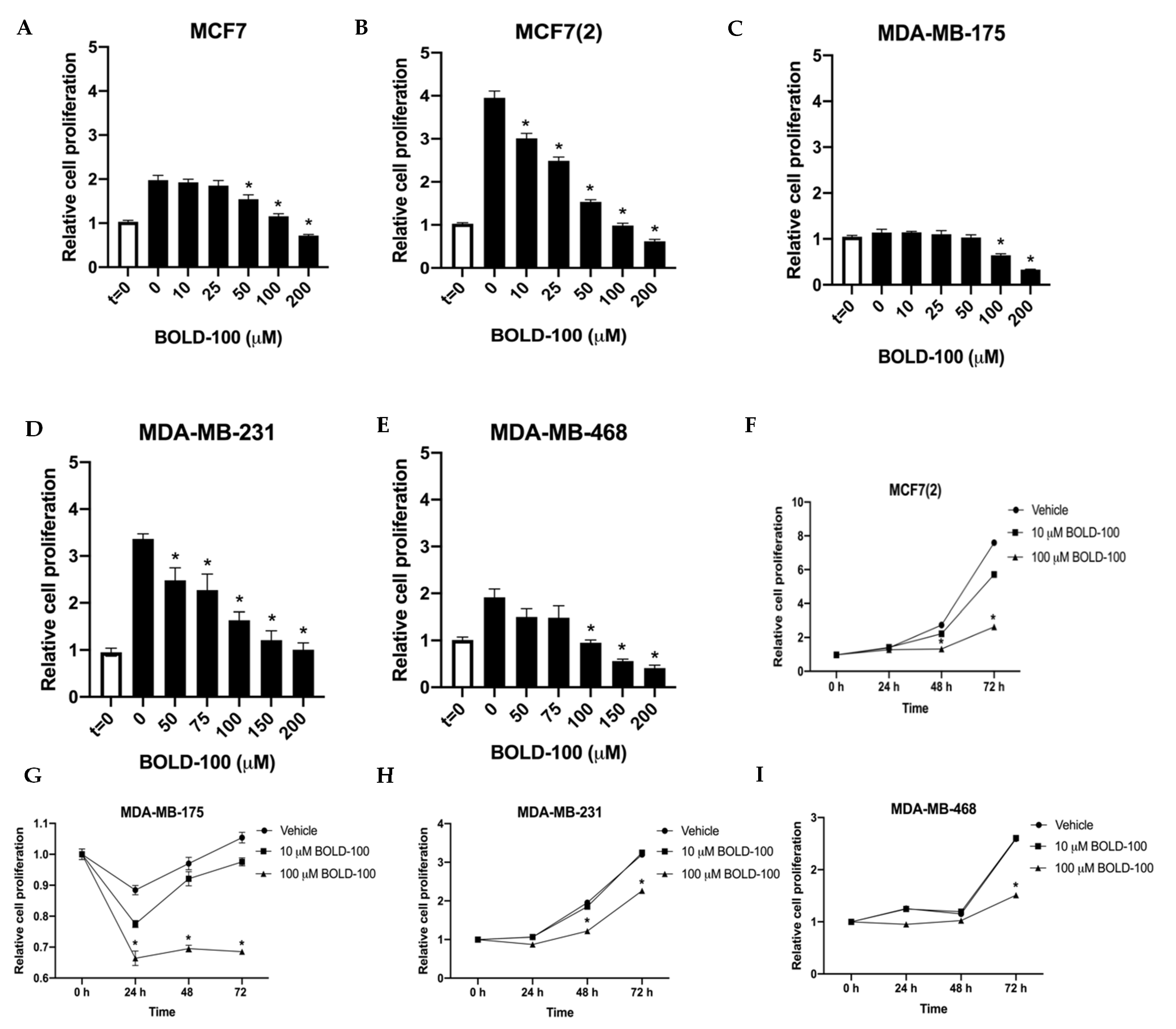
2.1. BOLD-100 Inhibits Growth of Breast Cancer Cells
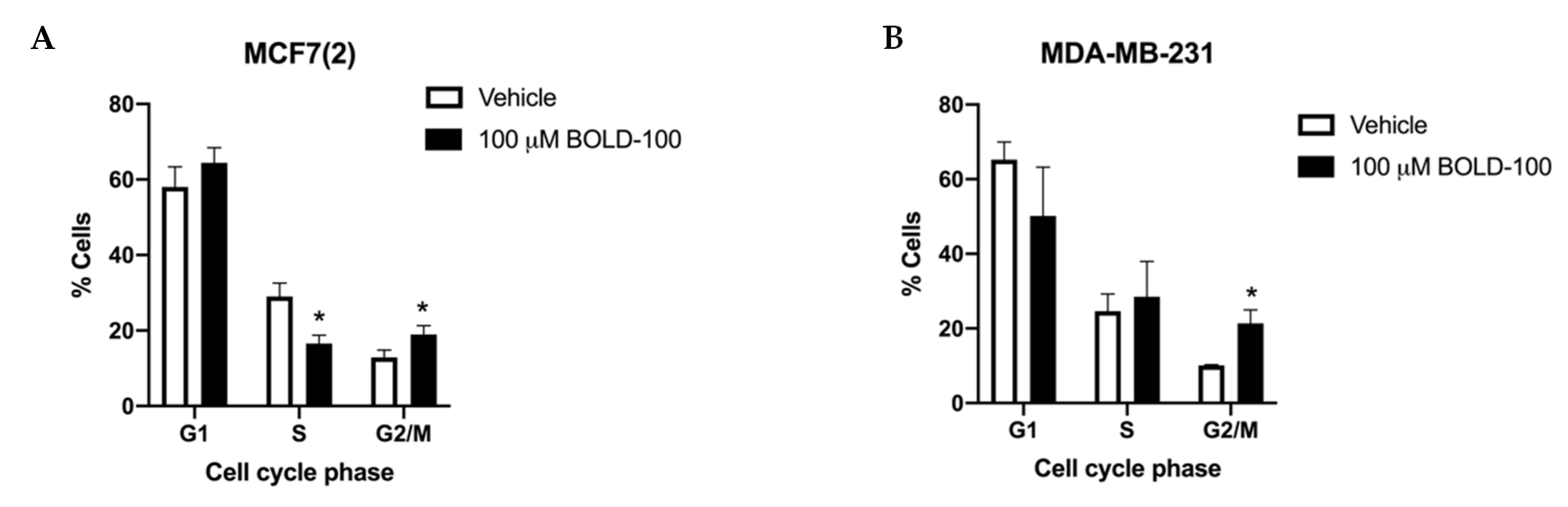
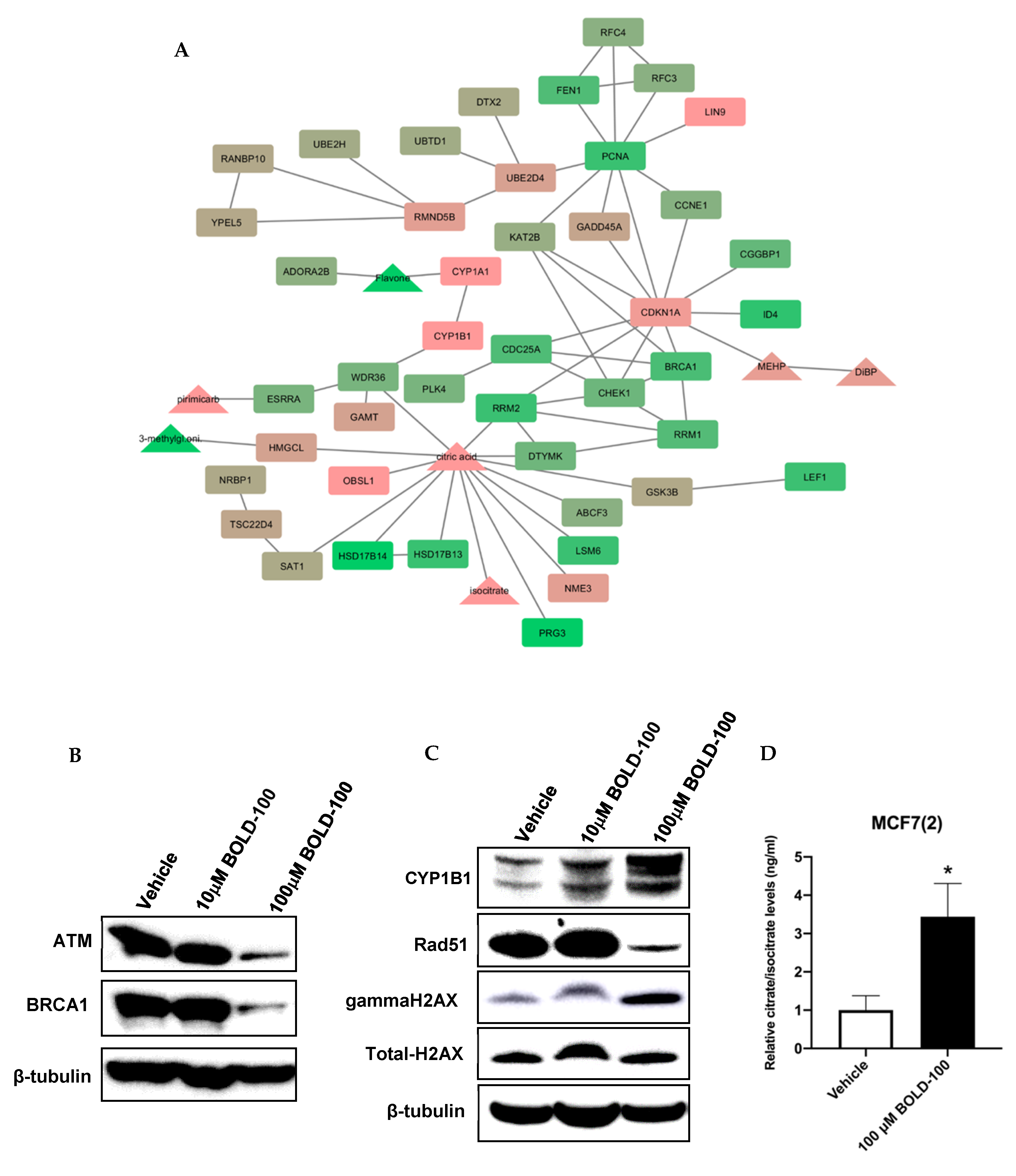
2.2. Significantly Altered Genes, Metabolites and Proteins in BOLD-100 Treated Cells Show Changes in DNA Repair Pathways
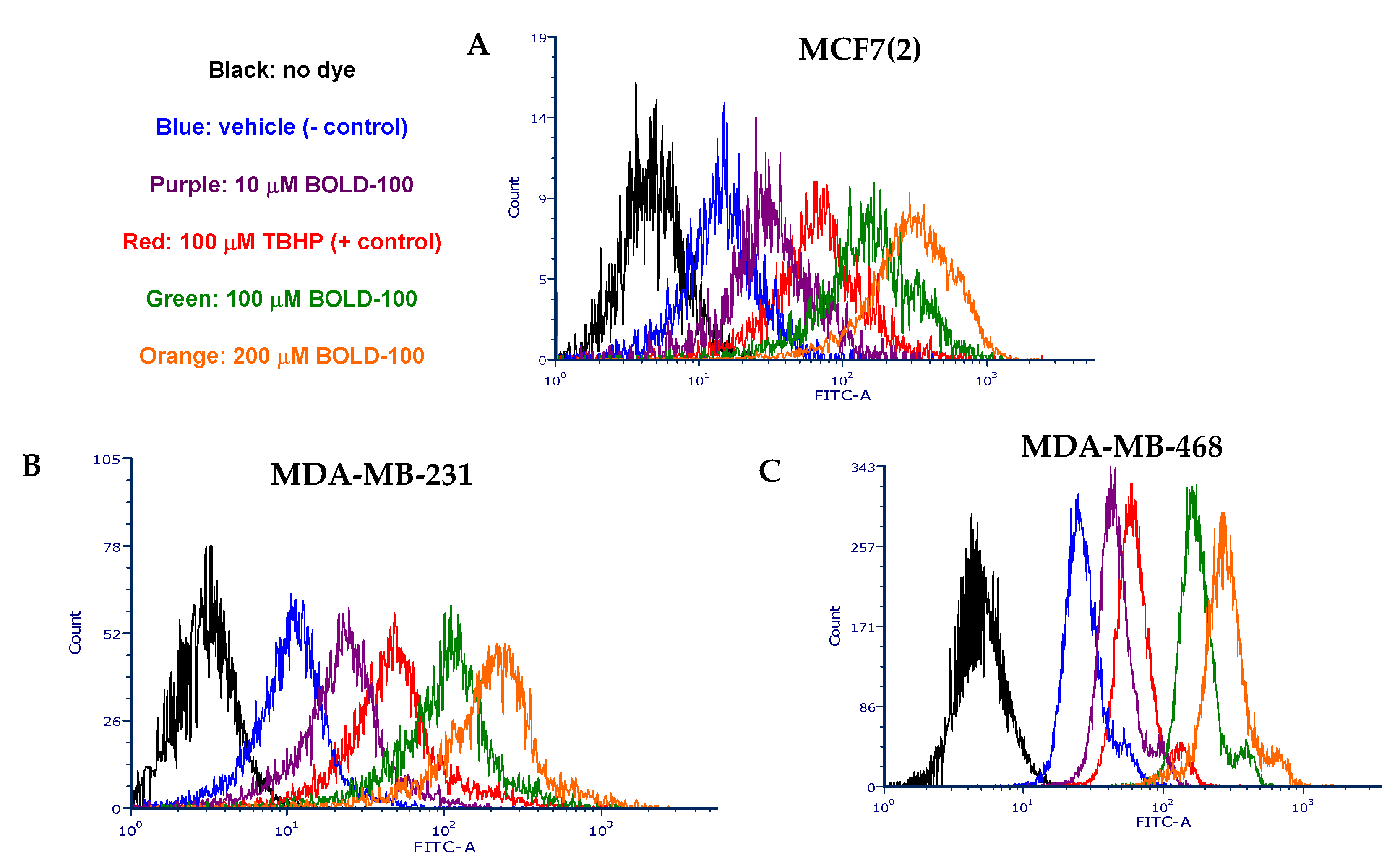
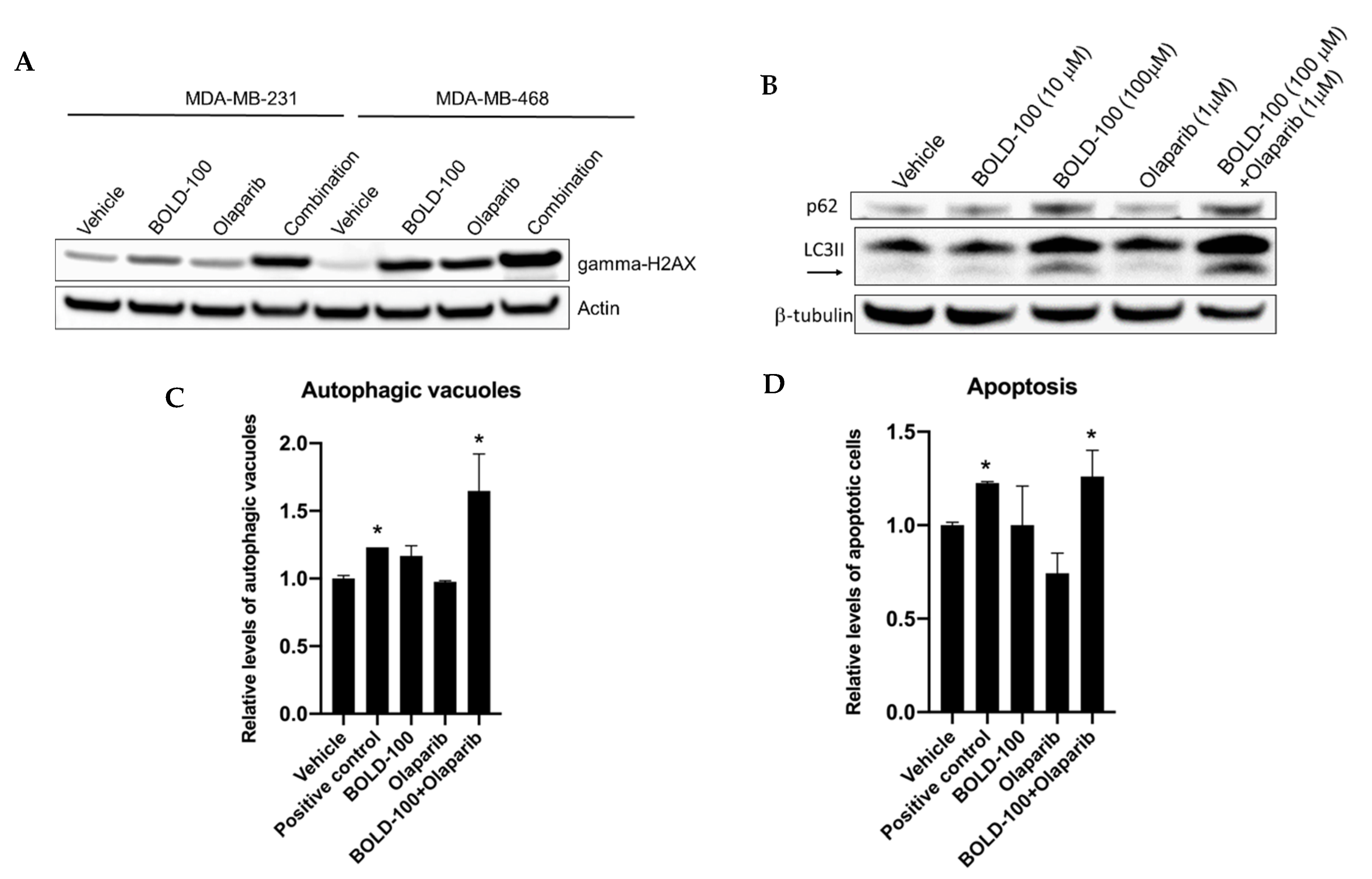
2.3. BOLD-100 Induces Reactive Oxygen Species (ROS)
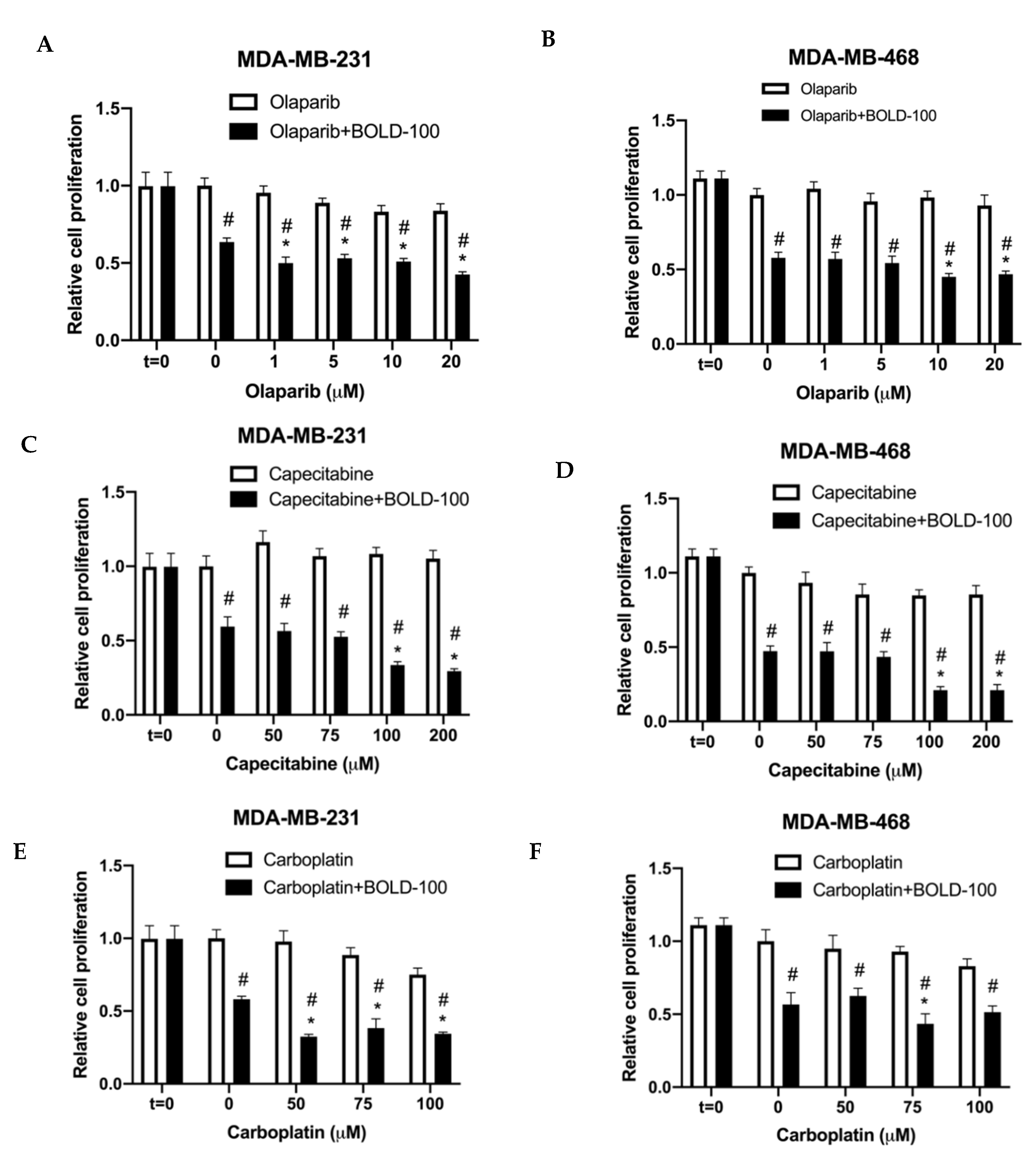
2.4. BOLD-100 Synergizes with Anticancer Agents that Target DNA in TNBC Cells
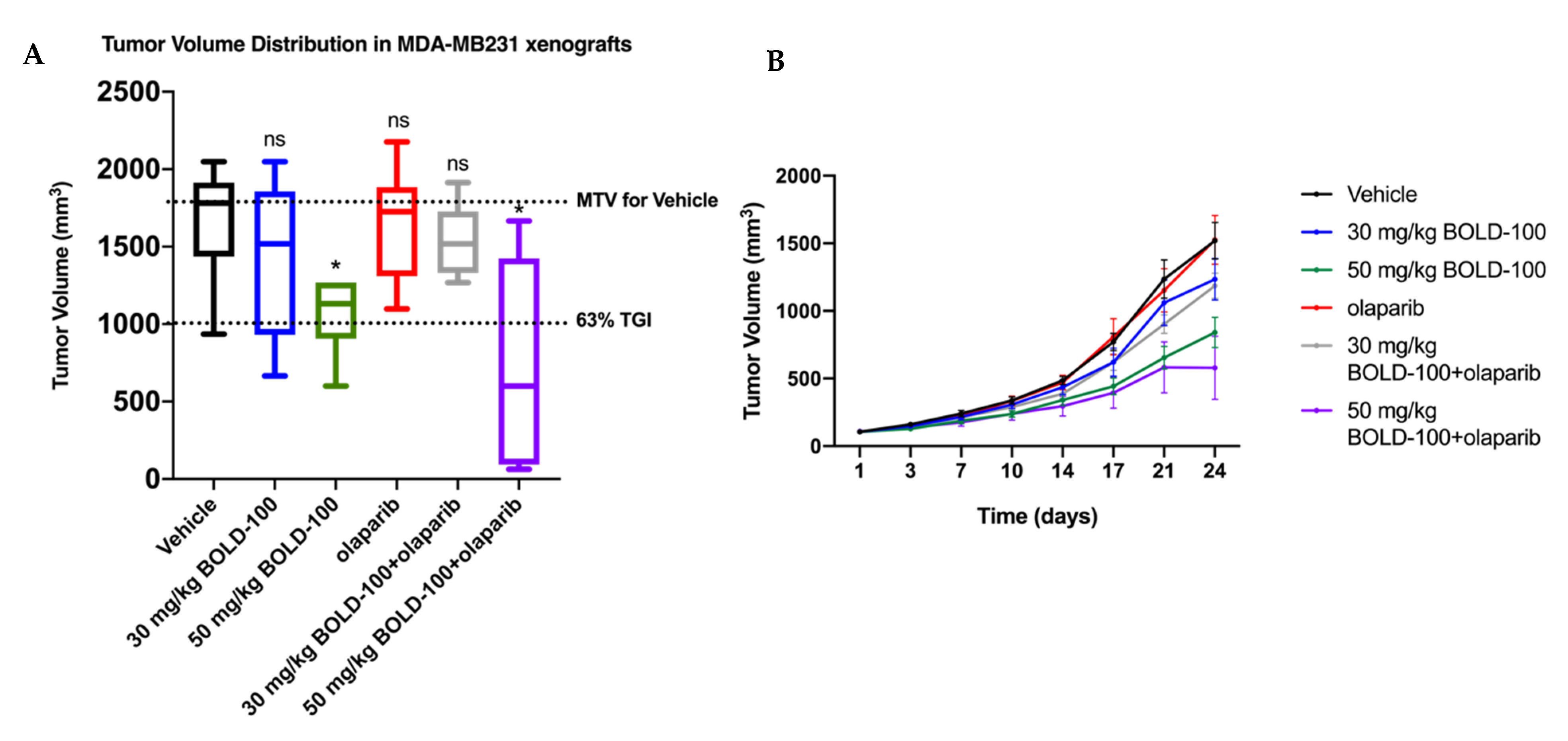
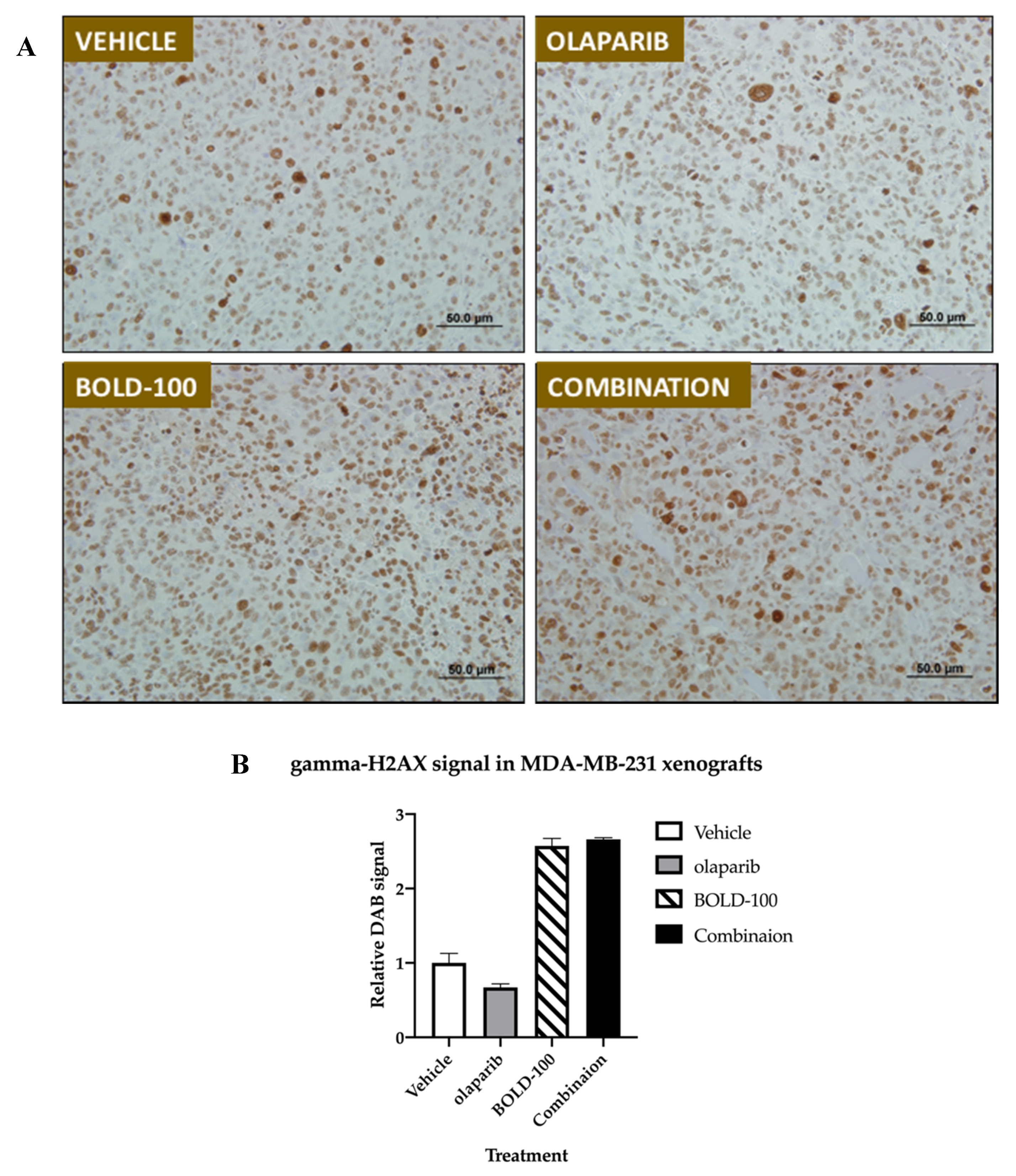
2.5. Combination of BOLD-100 and Olaparib Suppressed Growth of TNBC Tumors
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Cell Proliferation and Viability
4.3. Cell Cycle Analysis
4.4. Analysis of Cellular ROS Assay
4.5. Western Blotting
4.6. Generation, Analysis and Integration of Transcriptomics and Metabolomics Data from MCF7(2) Cells
4.7. Quantification of Citrate/Isocitrate
4.8. TNBC Xenografts and In Vivo Studies
4.9. Immunohistochemistry (IHC)
4.10. Reverse Phase Protein Array (RPPA)
4.11. Statistical Analysis and Drug Interaction for Cell Proliferation Experiments
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Clarke, R.; Skaar, T.; Baumann, K.; Leonessa, F.; James, M.; Lippman, J.; Thompson, E.W.; Freter, C.; Brunner, N. Hormonal carcinogenesis in breast cancer: Cellular and molecular studies of malignant progression. Breast Cancer Res. Treat. 1994, 31, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Shajahan, A.N.; Riggins, R.B.; Cho, Y.; Crawford, A.; Xuan, J.; Wang, Y.; Zwart, A.; Nehra, R.; Liu, M.C. Gene network signaling in hormone responsiveness modifies apoptosis and autophagy in breast cancer cells. J. Steroid Biochem. Mol. Biol. 2009, 114, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Iorfida, M.; Mazza, M.; Munzone, E. Fulvestrant in Combination with CDK4/6 Inhibitors for HER2- Metastatic Breast Cancers: Current Perspectives. Breast Cancer 2020, 12, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Lynce, F.; Shajahan-Haq, A.N.; Swain, S.M. CDK4/6 inhibitors in breast cancer therapy: Current practice and future opportunities. Pharmacol. Ther. 2018, 191, 65–73. [Google Scholar] [CrossRef]
- Spring, L.M.; Wander, S.A.; Andre, F.; Moy, B.; Turner, N.C.; Bardia, A. Cyclin-dependent kinase 4 and 6 inhibitors for hormone receptor-positive breast cancer: Past, present, and future. Lancet 2020, 395, 817–827. [Google Scholar] [CrossRef]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef]
- Hurvitz, S.; Mead, M. Triple-negative breast cancer: Advancements in characterization and treatment approach. Curr. Opin. Obstet. Gynecol. 2016, 28, 59–69. [Google Scholar] [CrossRef]
- Griguolo, G.; Dieci, M.V.; Miglietta, F.; Guarneri, V.; Conte, P. Olaparib for advanced breast cancer. Future Oncol. 2020. [Google Scholar] [CrossRef]
- Augusto, T.V.; Correia-da-Silva, G.; Rodrigues, C.M.P.; Teixeira, N.; Amaral, C. Acquired resistance to aromatase inhibitors: Where we stand! Endocr. Relat. Cancer 2018, 25, R283–R301. [Google Scholar] [CrossRef]
- Droog, M.; Beelen, K.; Linn, S.; Zwart, W. Tamoxifen resistance: From bench to bedside. Eur. J. Pharmacol. 2013, 717, 47–57. [Google Scholar] [CrossRef]
- Zhao, Y.; Butler, E.B.; Tan, M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013, 4, e532. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A.; Bakewell, S.; Bendell, J.C.; Infante, J.; Jones, S.F.; Spigel, D.R.; Weiss, G.J.; Ramanathan, R.K.; Ogden, A.; Von Hoff, D. Safety and activity of IT-139, a ruthenium-based compound, in patients with advanced solid tumours: A first-in-human, open-label, dose-escalation phase I study with expansion cohort. ESMO Open 2016, 1, e000154. [Google Scholar] [CrossRef] [PubMed]
- Dömötör, O.; Hartinger, C.G.; Bytzek, A.K.; Kiss, T.; Keppler, B.K.; Enyedy, E.A. Characterization of the binding sites of the anticancer ruthenium(III) complexes KP1019 and KP1339 on human serum albumin via competition studies. J. Biol. Inorg. Chem. 2013, 18, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Alessio, E.; Messori, L. NAMI-A and KP1019/1339, Two Iconic Ruthenium Anticancer Drug Candidates Face-to-Face: A Case Story in Medicinal Inorganic Chemistry. Molecules 2019, 24, 1995. [Google Scholar] [CrossRef]
- Bakewell, S.J.; Rangel, D.F.; Ha, D.P.; Sethuraman, J.; Crouse, R.; Hadley, E.; Costich, T.L.; Zhou, X.; Nichols, P.; Lee, A.S. Suppression of stress induction of the 78-kilodalton glucose regulated protein (GRP78) in cancer by IT-139, an anti-tumor ruthenium small molecule inhibitor. Oncotarget 2018, 9, 29698–29714. [Google Scholar] [CrossRef]
- Gifford, J.B.; Huang, W.; Zeleniak, A.E.; Hindoyan, A.; Wu, H.; Donahue, T.R.; Hill, R. Expression of GRP78, Master Regulator of the Unfolded Protein Response, Increases Chemoresistance in Pancreatic Ductal Adenocarcinoma. Mol. Cancer Ther. 2016, 15, 1043–1052. [Google Scholar] [CrossRef]
- Schoenhacker-Alte, B.; Mohr, T.; Pirker, C.; Kryeziu, K.; Kuhn, P.S.; Buck, A.; Hofmann, T.; Gerner, C.; Hermann, G.; Koellensperger, G.; et al. Sensitivity towards the GRP78 inhibitor KP1339/IT-139 is characterized by apoptosis induction via caspase 8 upon disruption of ER homeostasis. Cancer Lett. 2017, 404, 79–88. [Google Scholar] [CrossRef]
- James, M.R.; Skaar, T.C.; Lee, R.Y.; MacPherson, A.; Zwiebel, J.A.; Ahluwalia, B.S.; Ampy, F.; Clarke, R. Constitutive expression of the steroid sulfatase gene supports the growth of MCF-7 human breast cancer cells in vitro and in vivo. Endocrinology 2001, 142, 1497–1505. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Lopes, C.T.; Franz, M.; Kazi, F.; Donaldson, S.L.; Morris, Q.; Bader, G.D. Cytoscape Web: An interactive web-based network browser. Bioinformatics 2010, 26, 2347–2348. [Google Scholar] [CrossRef]
- Abdul Aziz, A.A.; Md Salleh, M.S.; Mohamad, I.; Krishna Bhavaraju, V.M.; Mazuwin Yahya, M.; Zakaria, A.D.; Hua Gan, S.; Ankathil, R. Single-nucleotide polymorphisms and mRNA expression of CYP1B1 influence treatment response in triple negative breast cancer patients undergoing chemotherapy. J. Genet. 2018, 97, 1185–1194. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, B.T.; Lu, Y.; Gonzalez-Angulo, A.M.; Carey, M.S.; Myhre, S.; Ju, Z.; Davies, M.A.; Liu, W.; Coombes, K.; Meric-Bernstam, F.; et al. A Technical Assessment of the Utility of Reverse Phase Protein Arrays for the Study of the Functional Proteome in Non-microdissected Human Breast Cancers. Clin. Proteom. 2010, 6, 129–151. [Google Scholar] [CrossRef] [PubMed]
- Klinakis, A.; Karagiannis, D.; Rampias, T. Targeting DNA repair in cancer: Current state and novel approaches. Cell Mol. Life Sci. 2020, 77, 677–703. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Feng, H.; Sun, W.; Liu, K.; Lu, J.J.; Chen, X. Tert-butyl hydroperoxide (t-BHP) induced apoptosis and necroptosis in endothelial cells: Roles of NOX4 and mitochondrion. Redox Biol. 2017, 11, 524–534. [Google Scholar] [CrossRef]
- Jhan, J.R.; Andrechek, E.R. Triple-negative breast cancer and the potential for targeted therapy. Pharmacogenomics 2017, 18, 1595–1609. [Google Scholar] [CrossRef]
- Romanelli, S.; Perego, P.; Pratesi, G.; Carenini, N.; Tortoreto, M.; Zunino, F. In vitro and in vivo interaction between cisplatin and topotecan in ovarian carcinoma systems. Cancer Chemother. Pharmacol. 1998, 41, 385–390. [Google Scholar] [CrossRef]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef]
- Bonner, W.M.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Sedelnikova, O.A.; Solier, S.; Pommier, Y. GammaH2AX and cancer. Nat. Rev. Cancer 2008, 8, 957–967. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Wernitznig, D.; Kiakos, K.; Del Favero, G.; Harrer, N.; Machat, H.; Osswald, A.; Jakupec, M.A.; Wernitznig, A.; Sommergruber, W.; Keppler, B.K. First-in-class ruthenium anticancer drug (KP1339/IT-139) induces an immunogenic cell death signature in colorectal spheroids in vitro. Metallomics 2019, 11, 1044–1048. [Google Scholar] [CrossRef] [PubMed]
- Kuo, L.J.; Yang, L.X. Gamma-H2AX—A novel biomarker for DNA double-strand breaks. In Vivo 2008, 22, 305–309. [Google Scholar] [PubMed]
- Rusz, M.; Rampler, E.; Keppler, B.K.; Jakupec, M.A.; Koellensperger, G. Single Spheroid Metabolomics: Optimizing Sample Preparation of Three-Dimensional Multicellular Tumor Spheroids. Metabolites 2019, 9, 304. [Google Scholar] [CrossRef]
- Stultz, L.K.; Hunsucker, A.; Middleton, S.; Grovenstein, E.; O’Leary, J.; Blatt, E.; Miller, M.; Mobley, J.; Hanson, P.K. Proteomic analysis of the S. cerevisiae response to the anticancer ruthenium complex KP1019. Metallomics 2020, 12, 876–890. [Google Scholar] [CrossRef]
- Hung, K.C.; Wang, S.G.; Lin, M.L.; Chen, S.S. Citrate-Induced p85α–PTEN Complex Formation Causes G. Int. J. Mol. Sci. 2019, 20, 2105. [Google Scholar] [CrossRef]
- Dutour, R.; Poirier, D. Inhibitors of cytochrome P450 (CYP) 1B1. Eur. J. Med. Chem. 2017, 135, 296–306. [Google Scholar] [CrossRef]
- Go, R.E.; Hwang, K.A.; Choi, K.C. Cytochrome P450 1 family and cancers. J. Steroid Biochem. Mol. Biol. 2015, 147, 24–30. [Google Scholar] [CrossRef]
- Hertz, D.L.; Kidwell, K.M.; Seewald, N.J.; Gersch, C.L.; Desta, Z.; Flockhart, D.A.; Storniolo, A.M.; Stearns, V.; Skaar, T.C.; Hayes, D.F.; et al. Polymorphisms in drug-metabolizing enzymes and steady-state exemestane concentration in postmenopausal patients with breast cancer. Pharm. J. 2017, 17, 521–527. [Google Scholar] [CrossRef]
- Vasile, E.; Tibaldi, C.; Leon, G.L.; D’Incecco, A.; Giovannetti, E. Cytochrome P450 1B1 (CYP1B1) polymorphisms are associated with clinical outcome of docetaxel in non-small cell lung cancer (NSCLC) patients. J. Cancer Res. Clin. Oncol. 2015, 141, 1189–1194. [Google Scholar] [CrossRef]
- Shajahan-Haq, A.N.; Cook, K.L.; Schwartz-Roberts, J.L.; Eltayeb, A.E.; Demas, D.M.; Warri, A.M.; Facey, C.O.; Hilakivi-Clarke, L.A.; Clarke, R. MYC regulates the unfolded protein response and glucose and glutamine uptake in endocrine resistant breast cancer. Mol. Cancer 2014, 13, 239. [Google Scholar] [CrossRef]
- Shajahan-Haq, A.N.; Boca, S.M.; Jin, L.; Bhuvaneshwar, K.; Gusev, Y.; Cheema, A.K.; Demas, D.D.; Raghavan, K.S.; Michalek, R.; Madhavan, S.; et al. EGR1 regulates cellular metabolism and survival in endocrine resistant breast cancer. Oncotarget 2017, 8, 96865–96884. [Google Scholar] [CrossRef] [PubMed]
- Vindeløv, L.L.; Christensen, I.J.; Nissen, N.I. A detergent-trypsin method for the preparation of nuclei for flow cytometric DNA analysis. Cytometry 1983, 3, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, K.D.; Khanna, S.; Byers, S.W.; Fornace, A., Jr.; Cheema, A.K. Small molecule metabolite extraction strategy for improving LC/MS detection of cancer cell metabolome. J. Biomol. Tech. 2011, 22, 1–4. [Google Scholar] [PubMed]
- Crowe, A.R.; Yue, W. Semi-quantitative Determination of Protein Expression using Immunohistochemistry Staining and Analysis: An Integrated Protocol. Bio-protocol 2019, 9, e3465. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PROTEIN NAME | p-Value | ≥20% Difference in 100 μM BOLD-100 Treated Cells |
|---|---|---|
| PDK1 | <0.001 | 24% increased |
| TSC1 | <0.001 | 27% increased |
| SOD2 | <0.001 | 22% increased |
| G6PD | <0.001 | 39% increased |
| HER2 | 0.002 | 31% increased |
| PDK1_pS241 | 0.004 | 35% increased |
| Notch1 | 0.006 | 21% increased |
| TFAM | 0.006 | 30% increased |
| PR (Progesterone receptor) | 0.009 | 64% decreased |
| DJ1 | 0.010 | 24% increased |
| MUC1 (EMA) | 0.014 | 65% increased |
| UGT1A | 0.015 | 36% increased |
| Collagen-VI | 0.025 | 21% increased |
| c-Jun_pS73 | 0.026 | 20% decreased |
| ATRX | 0.031 | 24% decreased |
| LC3A-B | 0.035 | 21% increased |
| Cyclin-D3 | 0.044 | 39% increased |
| Group | n | Administration of BOLD-100 | Administration of Olaparib | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Drug | Route | mg/kg | Schedule | Drug | Route | mg/kg | Schedule | ||
| #1 | 8 | Vehicle | iv | -- | qd4 to end | -- | -- | -- | -- |
| #2 | 8 | BOLD-100 | iv | 30 | qd4 to end | -- | -- | -- | -- |
| #3 | 8 | BOLD-100 | iv | 50 | qwk to end | -- | -- | -- | -- |
| #4 | 8 | olaparib | po | 50 | qd × 32 (start on Day 2) | -- | -- | -- | -- |
| #5 | 8 | BOLD-100 | iv | 30 | q4d to end | olaparib | 50 | po | qd × 32 (start on Day 2) |
| #6 | 8 | BOLD-100 | iv | 50 | qwk to end | olaparib | 50 | po | qd × 32 (start on Day 2) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bakewell, S.; Conde, I.; Fallah, Y.; McCoy, M.; Jin, L.; Shajahan-Haq, A.N. Inhibition of DNA Repair Pathways and Induction of ROS Are Potential Mechanisms of Action of the Small Molecule Inhibitor BOLD-100 in Breast Cancer. Cancers 2020, 12, 2647. https://doi.org/10.3390/cancers12092647
Bakewell S, Conde I, Fallah Y, McCoy M, Jin L, Shajahan-Haq AN. Inhibition of DNA Repair Pathways and Induction of ROS Are Potential Mechanisms of Action of the Small Molecule Inhibitor BOLD-100 in Breast Cancer. Cancers. 2020; 12(9):2647. https://doi.org/10.3390/cancers12092647
Chicago/Turabian StyleBakewell, Suzanne, Isabel Conde, Yassi Fallah, Mathew McCoy, Lu Jin, and Ayesha N. Shajahan-Haq. 2020. "Inhibition of DNA Repair Pathways and Induction of ROS Are Potential Mechanisms of Action of the Small Molecule Inhibitor BOLD-100 in Breast Cancer" Cancers 12, no. 9: 2647. https://doi.org/10.3390/cancers12092647
APA StyleBakewell, S., Conde, I., Fallah, Y., McCoy, M., Jin, L., & Shajahan-Haq, A. N. (2020). Inhibition of DNA Repair Pathways and Induction of ROS Are Potential Mechanisms of Action of the Small Molecule Inhibitor BOLD-100 in Breast Cancer. Cancers, 12(9), 2647. https://doi.org/10.3390/cancers12092647