Genome-Wide Comparison of the Target Genes of the Reactive Oxygen Species and Non-Reactive Oxygen Species Constituents of Cold Atmospheric Plasma in Cancer Cells


 ,
,  and
and 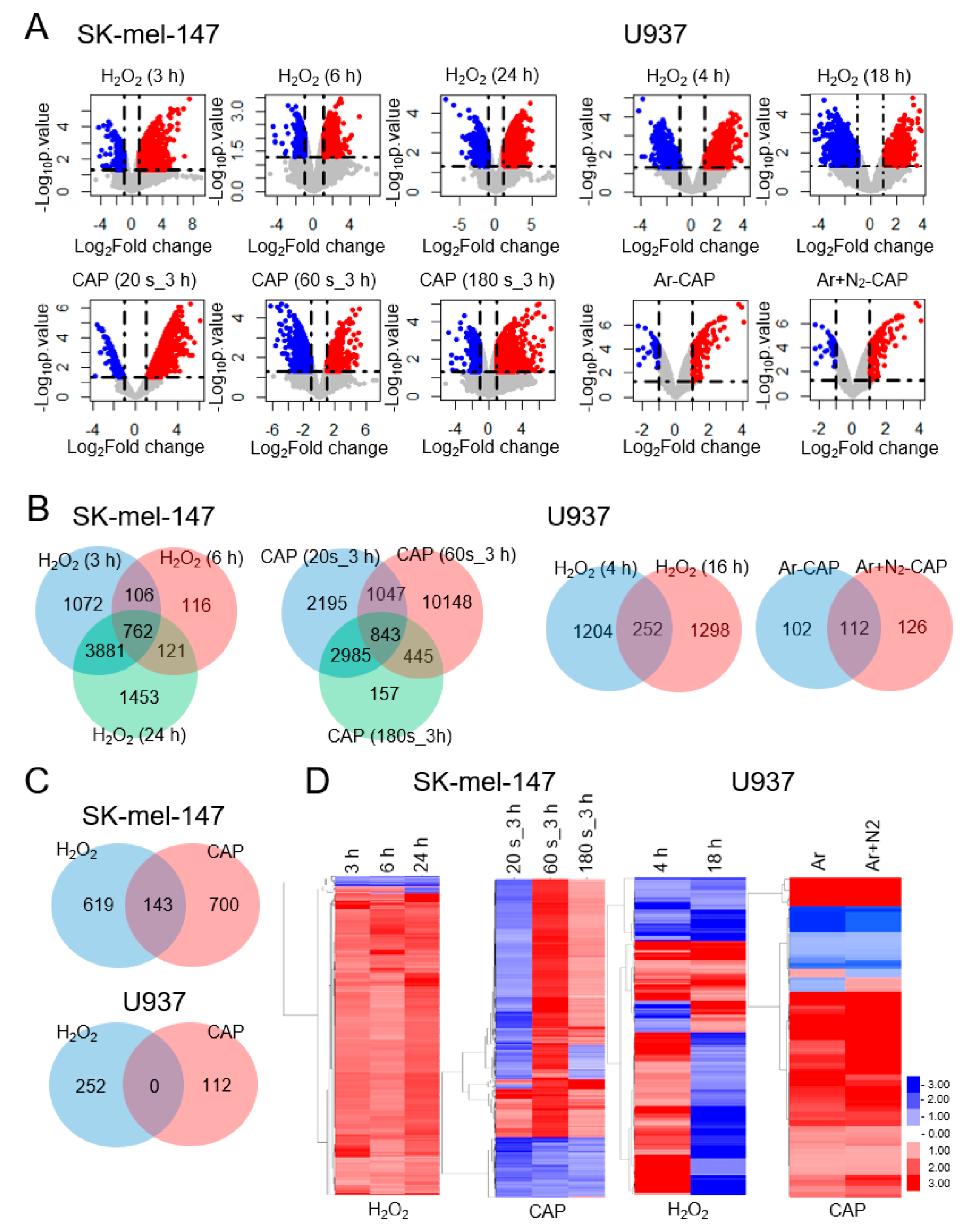{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Results
2.1. CAP and H2O2 Target a Small Number of Common Genes
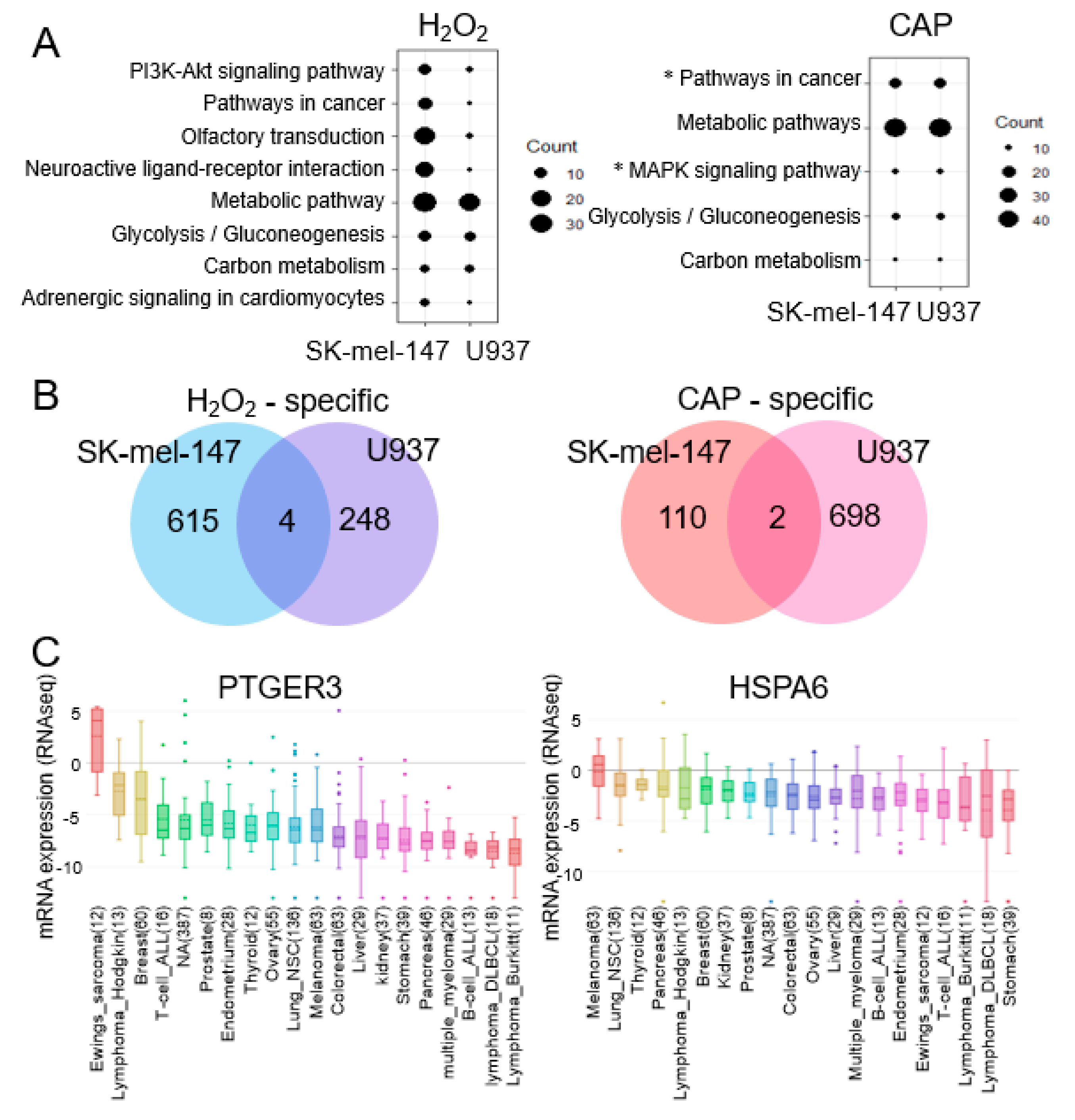
2.2. MAPK Signaling and Cancer Pathways Are Significant for CAP not H2O2
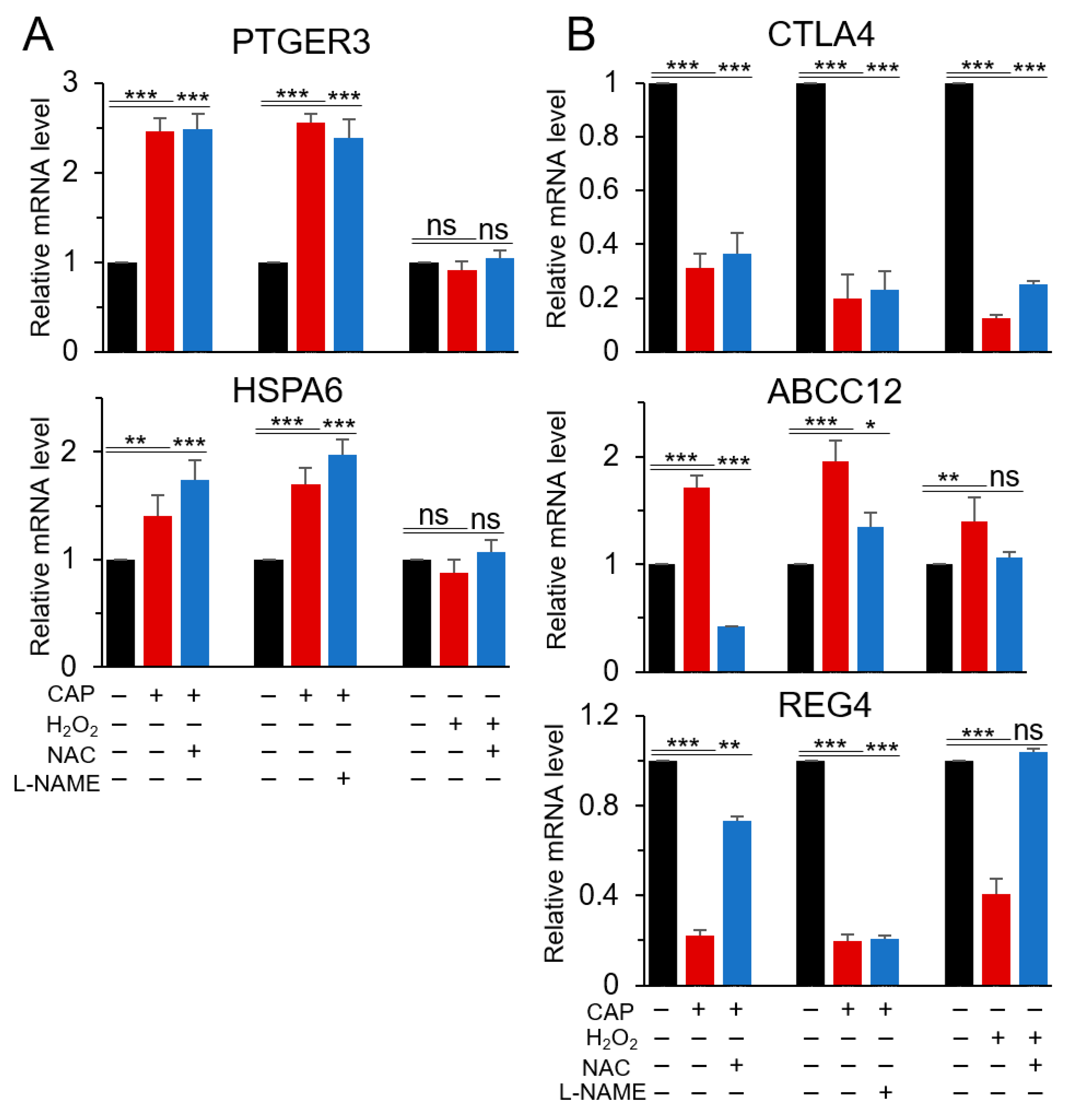
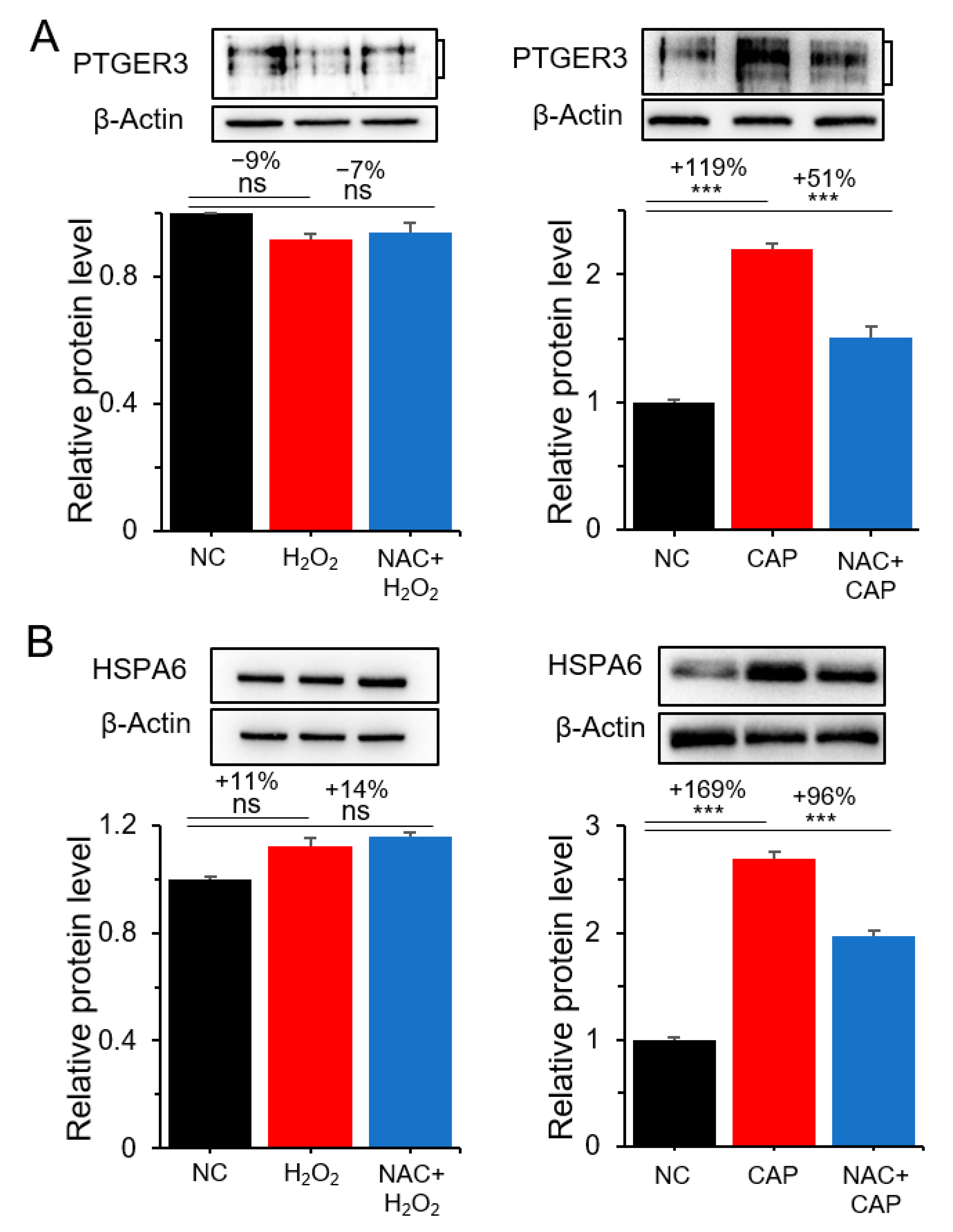
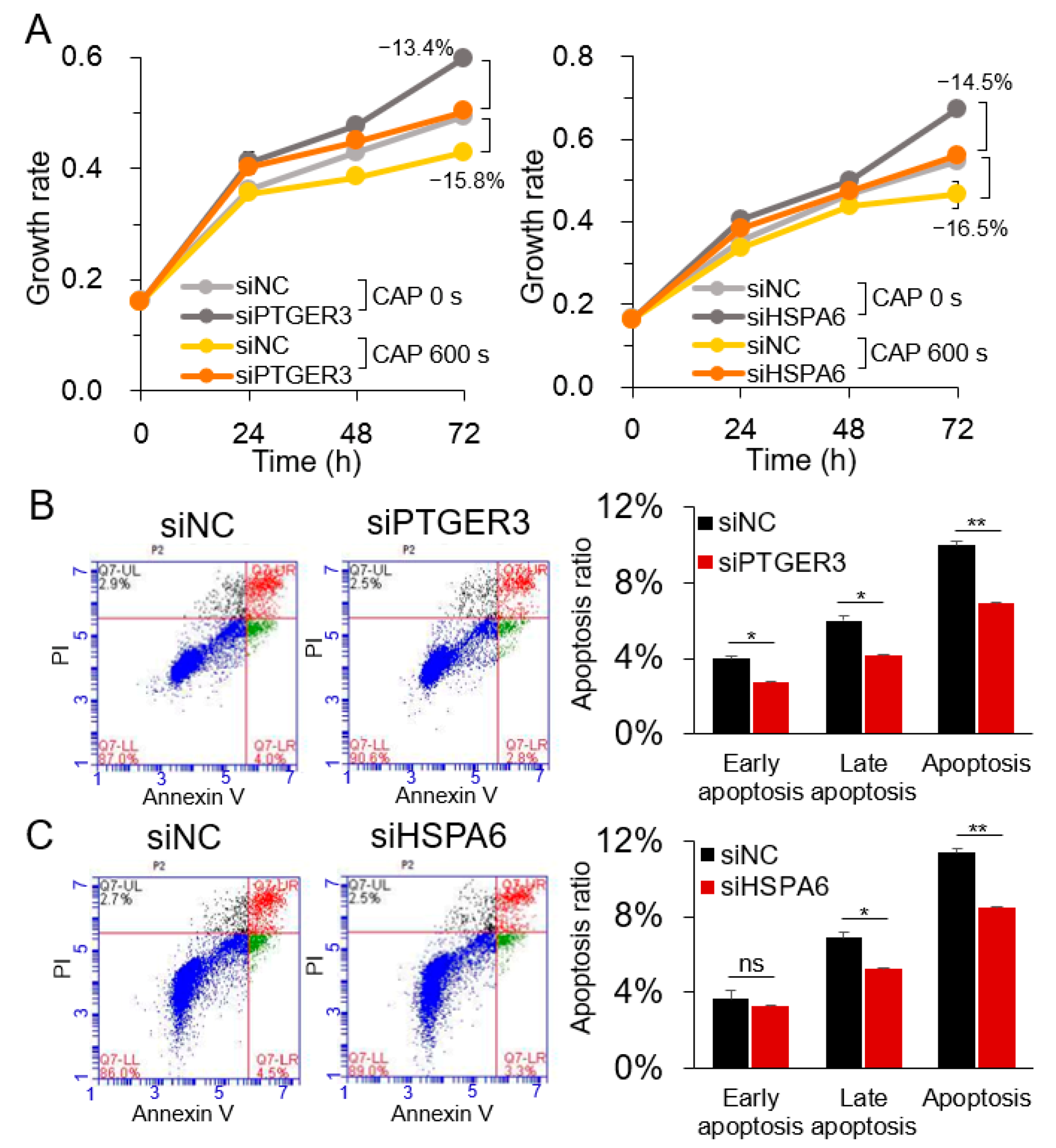
2.3. PTGER3 and HSPA6 Are Targets of CAP but not ROS or RNS
3. Discussion
4. Materials and Methods
4.1. Data Mining and Bioinformatics Analysis
4.2. Cell Culture and Treatment with CAP
4.3. Cell Transfection
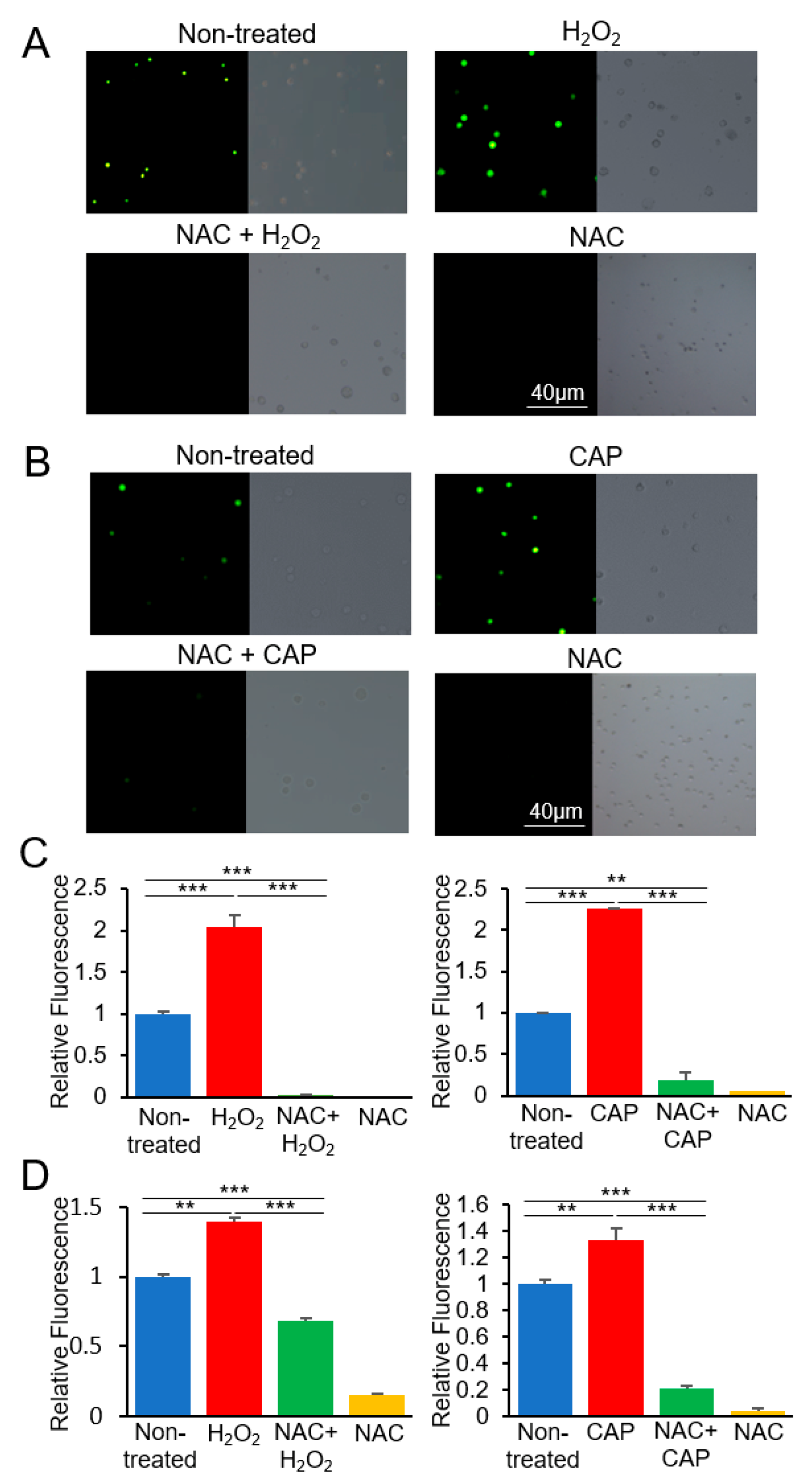
4.4. ROS Detection
4.5. Cell Proliferation Analysis
4.6. Apoptosis Assay
4.7. Quantitative Reverse Transcription Polymerase Chain Reaction (RT-qPCR)
4.8. Western Blot Analysis
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Graves, D.B. Reactive Species from Cold Atmospheric Plasma: Implications for Cancer Therapy. Plasma Process Polym. 2014, 11, 1120–1127. [Google Scholar] [CrossRef]
- Babington, P.; Rajjoub, K.; Canady, J.; Siu, A.; Keidar, M.; Sherman, J.H. Use of cold atmospheric plasma in the treatment of cancer. Biointerphases 2015, 10, 029403. [Google Scholar] [CrossRef] [PubMed]
- Biscop, E.; Lin, A.; Van Boxem, W.; Van Loenhout, J.; De Backer, J.; Deben, C.; Dewilde, S.; Smits, E.; Bogaerts, A. The Influence of Cell Type and Culture Medium on Determining Cancer Selectivity of Cold Atmospheric Plasma Treatment. Cancers 2019, 11, 1287. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Xu, R.; Gou, L.; Liu, Z.; Zhao, Y.; Liu, D.; Zhang, L.; Chen, H.; Kong, M.G. Mechanism of Virus Inactivation by Cold Atmospheric-Pressure Plasma and Plasma-Activated Water. Appl. Environ. Microbiol. 2018, 84, e00726-18. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Om, J.-Y.; Kim, Y.-H.; Kim, K.-M.; Choi, E.-H.; Kim, K.-N. Selective Killing Effects of Cold Atmospheric Pressure Plasma with NO Induced Dysfunction of Epidermal Growth Factor Receptor in Oral Squamous Cell Carcinoma. PLoS ONE 2016, 11, e0150279. [Google Scholar] [CrossRef] [PubMed]
- Soraya, S.; Mozafar, K. Oxidative Stress and Cancer: The Role of Nrf2. Curr. Cancer Drug Targets 2018, 18, 538–557. [Google Scholar]
- Pelicano, H.; Lu, W.; Zhou, Y.; Zhang, W.; Chen, Z.; Hu, Y.; Huang, P. Mitochondrial Dysfunction and Reactive Oxygen Species Imbalance Promote Breast Cancer Cell Motility through a CXCL14-Mediated Mechanism. Cancer Res. 2009, 69, 2375–2383. [Google Scholar] [CrossRef]
- Poillet-Perez, L.; Despouy, G.; Delage-Mourroux, R.; Boyer-Guittaut, M. Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol. 2015, 4, 184–192. [Google Scholar] [CrossRef]
- Liao, Z.; Chua, D.; Tan, N.S. Reactive oxygen species: A volatile driver of field cancerization and metastasis. Mol. Cancer 2019, 18, 65. [Google Scholar] [CrossRef]
- Bagati, A.; Moparthy, S.; Fink, E.E.; Bianchi-Smiraglia, A.; Yun, D.H.; Kolesnikova, M.; Udartseva, O.O.; Wolff, D.W.; Roll, M.V.; Lipchick, B.C.; et al. KLF9-dependent ROS regulate melanoma progression in stage-specific manner. Oncogene 2019, 38, 3585–3597. [Google Scholar] [CrossRef]
- Ushio-Fukai, M.; Nakamura, Y. Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy. Cancer Lett. 2008, 266, 37–52. [Google Scholar] [CrossRef]
- Hofseth, L.J.; Saito, S.; Hussain, S.P.; Espey, M.G.; Miranda, K.M.; Araki, Y.; Jhappan, C.; Higashimoto, Y.; He, P.; Linke, S.P.; et al. Nitric oxide-induced cellular stress and p53 activation in chronic inflammation. Proc. Natl. Acad. Sci. USA 2003, 100, 143–148. [Google Scholar] [CrossRef]
- Li, C.-Q.; Pang, B.; Kiziltepe, T.; Trudel, L.J.; Engelward, B.P.; Dedon, P.C.; Wogan, G.N. Threshold Effects of Nitric Oxide-Induced Toxicity and Cellular Responses in Wild-Type and p53-Null Human Lymphoblastoid Cells. Chem. Res. Toxicol. 2006, 19, 399–406. [Google Scholar] [CrossRef]
- Jadeski, L.C.; Chakraborty, C.; Lala, P.K. Nitric oxide-mediated promotion of mammary tumour cell migration requires sequential activation of nitric oxide synthase, guanylate cyclase and mitogen-activated protein kinase. Int. J. Cancer 2003, 106, 496–504. [Google Scholar] [CrossRef]
- Meini, A.; Sticozzi, C.; Massai, L.; Palmi, M. A nitric oxide/Ca2+/calmodulin/ERK1/2 mitogen-activated protein kinase pathway is involved in the mitogenic effect of IL-1β in human astrocytoma cells. Br. J. Pharmacol. 2008, 153, 1706–1717. [Google Scholar] [CrossRef]
- Kurita, H.; Haruta, N.; Uchihashi, Y.; Seto, T.; Takashima, K. Strand breaks and chemical modification of intracellular DNA induced by cold atmospheric pressure plasma irradiation. PLoS ONE 2020, 15, e0232724. [Google Scholar] [CrossRef]
- Han, I.; Ha Choi, E. The role of non-thermal atmospheric pressure biocompatible plasma in the differentiation of osteoblastic precursor cells, MC3T3-E1. Oncotarget 2017, 8, 36399–36409. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.-Y.; Hong, Y.J.; Lim, J.; Choi, J.S.; Choi, E.H.; Kang, S.; Rhim, H. Cold atmospheric plasma (CAP), a novel physicochemical source, induces neural differentiation through cross-talk between the specific RONS cascade and Trk/Ras/ERK signaling pathway. Biomaterials 2018, 156, 258–273. [Google Scholar] [CrossRef]
- Xia, J.; Zeng, W.; Xia, Y.; Wang, B.; Xu, D.; Liu, D.; Kong, M.G.; Dong, Y. Cold atmospheric plasma induces apoptosis of melanoma cells via Sestrin2-mediated nitric oxide synthase signaling. J. Biophotonics. 2019, 12, e201800046. [Google Scholar] [CrossRef]
- Tabuchi, Y.; Uchiyama, H.; Zhao, Q.-L.; Yunoki, T.; Andocs, G.; Nojima, N.; Takeda, K.; Ishikawa, K.; Hori, M.; Kondo, T. Effects of nitrogen on the apoptosis of and changes in gene expression in human lymphoma U937 cells exposed to argon-based cold atmospheric pressure plasma. Int. J. Mol. Med. 2016, 37, 1706–1714. [Google Scholar] [CrossRef]
- Bekeschus, S.; Lippert, M.; Diepold, K.; Chiosis, G.; Seufferlein, T.; Azoitei, N. Physical plasma-triggered ROS induces tumor cell death upon cleavage of HSP90 chaperone. Sci. Rep. 2019, 9, 4112. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Lee, S.-J.; Castro, N.J.; Yan, D.; Keidar, M.; Zhang, L.G. Synergistic Effect of Cold Atmospheric Plasma and Drug Loaded Core-shell Nanoparticles on Inhibiting Breast Cancer Cell Growth. Sci. Rep. 2016, 6, 21974. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Bekeschus, S.; von Woedtke, T.; Hasse, S. Cell migration and adhesion of a human melanoma cell line is decreased by cold plasma treatment. Clin. Plasma Med. 2015, 3, 24–31. [Google Scholar] [CrossRef]
- Park, S.-B.; Kim, B.; Bae, H.; Lee, H.; Lee, S.; Choi, E.H.; Kim, S.J. Differential Epigenetic Effects of Atmospheric Cold Plasma on MCF-7 and MDA-MB-231 Breast Cancer Cells. PLoS ONE 2015, 10, e0129931. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, H.; Bae, H.; Choi, E.H.; Kim, S.J. Epigenetic silencing of miR-19a-3p by cold atmospheric plasma contributes to proliferation inhibition of the MCF-7 breast cancer cell. Sci. Rep. 2016, 6, 30005. [Google Scholar] [CrossRef]
- Lee, S.; Park, S.; Lee, H.; Jeong, D.; Ham, J.; Choi, E.H.; Kim, S.J. ChIP-seq analysis reveals alteration of H3K4 trimethylation occupancy in cancer-related genes by cold atmospheric plasma. Free Radic. Biol. Med. 2018, 126, 133–141. [Google Scholar] [CrossRef]
- Fraser, C. Estimating Individual and Household Reproduction Numbers in an Emerging Epidemic. PLoS ONE 2007, 2, e758. [Google Scholar] [CrossRef]
- Huang, B.; Warner, M.; Gustafsson, J.-Å. Estrogen receptors in breast carcinogenesis and endocrine therapy. Mol. Cellula Endocrinol. 2015, 418, 240–244. [Google Scholar] [CrossRef]
- Calderwood, S.K.; Khaleque, M.A.; Sawyer, D.B.; Ciocca, D.R. Heat shock proteins in cancer: Chaperones of tumorigenesis. Trends Biochem. Sci. 2006, 31, 164–172. [Google Scholar] [CrossRef]
- Zhou, X.; Sun, H.; Chen, H.; Zavadil, J.; Kluz, T.; Arita, A.; Costa, M. Hypoxia Induces Trimethylated H3 Lysine 4 by Inhibition of JARID1A Demethylase. Cancer Res. 2010, 70, 4214–4221. [Google Scholar] [CrossRef]
- Sato, T.; Yokoyama, M.; Johkura, K. A key inactivation factor of HeLa cell viability by a plasma flow. J. Phys. D Appl. Phys. 2011, 44, 372001. [Google Scholar] [CrossRef]
- Liu, Y.; Tan, S.; Zhang, H.; Kong, X.; Ding, L.; Shen, J.; Lan, Y.; Cheng, C.; Zhu, T.; Xia, W. Selective effects of non-thermal atmospheric plasma on triple-negative breast normal and carcinoma cells through different cell signaling pathways. Sci. Rep. 2017, 7, 7980. [Google Scholar] [CrossRef] [PubMed]
- Ishaq, M.; Kumar, S.; Varinli, H.; Han, Z.J.; Rider, A.E.; Evans, M.D.M.; Murphy, A.B.; Ostrikov, K. Atmospheric gas plasma–induced ROS production activates TNF-ASK1 pathway for the induction of melanoma cancer cell apoptosis. Mol. Biol. Cell 2014, 25, 1523–1531. [Google Scholar] [CrossRef] [PubMed]
- Duchesne, C.; Banzet, S.; Lataillade, J.-J.; Rousseau, A.; Frescaline, N. Cold atmospheric plasma modulates endothelial nitric oxide synthase signalling and enhances burn wound neovascularisation. J. Pathol. 2019, 249, 368–380. [Google Scholar] [CrossRef] [PubMed]
- GEO. Available online: https://www.ncbi.nlm.nih.gov/geo/ (accessed on 3 September 2019).
- Meja, K.K.; Rajendrasozhan, S.; Adenuga, D.; Biswas, S.K.; Sundar, I.K.; Spooner, G.; Marwick, J.A.; Chakravarty, P.; Fletcher, D.; Whittaker, P.; et al. Curcumin restores corticosteroid function in monocytes exposed to oxidants by maintaining HDAC2. Am. J. Respir. Cell Mol. Biol. 2008, 39, 312–323. [Google Scholar] [CrossRef]
- Krysan, K.; Reckamp, K.L.; Dalwadi, H.; Sharma, S.; Rozengurt, E.; Dohadwala, M.; Dubinett, S.M. Prostaglandin E2 Activates Mitogen-Activated Protein Kinase/Erk Pathway Signaling and Cell Proliferation in Non–Small Cell Lung Cancer Cells in an Epidermal Growth Factor Receptor–Independent Manner. Cancer Res. 2005, 65, 6275–6281. [Google Scholar] [CrossRef]
- Shin, S.-S.; Song, J.-H.; Hwang, B.; Noh, D.-H.; Park, S.L.; Kim, W.T.; Park, S.-S.; Kim, W.-J.; Moon, S.-K. HSPA6 augments garlic extract-induced inhibition of proliferation, migration, and invasion of bladder cancer EJ cells; Implication for cell cycle dysregulation, signaling pathway alteration, and transcription factor-associated MMP-9 regulation. PLoS ONE 2017, 12, e0171860. [Google Scholar] [CrossRef]
- CCLE. Available online: https://portals.broadinstitute.org/ccle (accessed on 24 January 2020).
- Kim, H.W.; Jeong, D.; Ham, J.; Kim, H.; Ji, H.W.; Choi, E.H.; Kim, S.J. ZNRD1 and Its Antisense Long Noncoding RNA ZNRD1-AS1 Are Oppositely Regulated by Cold Atmospheric Plasma in Breast Cancer Cells. Oxid. Med. Cell. Longev. 2020, 2020, 9490567. [Google Scholar] [CrossRef]
- Vaquero, J.; Judée, F.; Vallette, M.; Decauchy, H.; Arbelaiz, A.; Aoudjehane, L.; Scatton, O.; Gonzalez-Sanchez, E.; Merabtene, F.; Augustin, J.; et al. Cold-Atmospheric Plasma Induces Tumor Cell Death in Preclinical In Vivo and In Vitro Models of Human Cholangiocarcinoma. Cancers 2020, 12, 1280. [Google Scholar] [CrossRef]
- Jalili, A.; Irani, S.; Mirfakhraie, R. Combination of cold atmospheric plasma and iron nanoparticles in breast cancer: Gene expression and apoptosis study. Onco Targets Ther. 2016, 9, 5911–5917. [Google Scholar]
- Niethammer, P.; Grabher, C.; Look, A.T.; Mitchison, T.J. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature 2009, 459, 996–999. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Braný, D.; Dvorská, D.; Halašová, E.; Škovierová, H. Cold Atmospheric Plasma: A Powerful Tool for Modern Medicine. Int. J. Mol. Sci. 2020, 21, 2932. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Shan, H.; Miao, M.Z.; Jiang, Z.; Meng, Y.; Chen, R.; Zhang, L.; Liu, Y. Identification of the key genes and pathways involved in the tumorigenesis and prognosis of kidney renal clear cell carcinoma. Sci. Rep. 2020, 10, 4271. [Google Scholar] [CrossRef] [PubMed]
- Rohde, M.; Daugaard, M.; Jensen, M.H.; Helin, K.; Nylandsted, J.; Jäättelä, M. Members of the heat-shock protein 70 family promote cancer cell growth by distinct mechanisms. Genes Dev. 2005, 19, 570–582. [Google Scholar] [CrossRef] [PubMed]
- Cui, N.; Xu, Y.; Cao, Z.; Xu, F.; Zhang, P.; Jin, L. Effects of heat stress on the level of heat shock protein 70 on the surface of hepatocellular carcinoma Hep G2 cells: Implications for the treatment of tumors. Tumour Biol. 2013, 34, 743–748. [Google Scholar] [CrossRef]
- An, S.; Yang, J.; So, S.W.; Zeng, L.; Goetzl, E.J. Isoforms of the EP3 Subtype of Human Prostaglandin E2 Receptor Transduce Both Intracellular Calcium and cAMP Signals. Biochemistry 1994, 33, 14496–14502. [Google Scholar] [CrossRef]
- Erfani, N.; Razmkhah, M.; Ghaderi, A. Circulating Soluble CTLA4 (sCTLA4) Is Elevated in Patients with Breast Cancer. Cancer Invest. 2010, 28, 828–832. [Google Scholar] [CrossRef]
- Kadowaki, Y.; Ishihara, S.; Miyaoka, Y.; Rumi, M.A.K.; Sato, H.; Kazumori, H.; Adachi, K.; Takasawa, S.; Okamoto, H.; Chiba, T.; et al. Reg protein is overexpressed in gastric cancer cells, where it activates a signal transduction pathway that converges on ERK1/2 to stimulate growth. FEBS Lett. 2002, 530, 59–64. [Google Scholar] [CrossRef]
- Bera, T.K.; Iavarone, C.; Kumar, V.; Lee, S.; Lee, B.; Pastan, I. MRP9, an unusual truncated member of the ABC transporter superfamily, is highly expressed in breast cancer. Proc. Natl. Acad. Sci. USA 2002, 99, 6997–7002. [Google Scholar] [CrossRef]
- NCBI. Available online: https://www.ncbi.nlm.nih.gov/gds (accessed on 21 August 2019).
- GEO2R. Available online: https://www.ncbi.nlm.nih.gov/geo/geo2r/ (accessed on 30 August 2019).
- DAVID. Available online: https://david.ncifcrf.gov/ (accessed on 16 November 2019).
- KEGG. Available online: http://www.genome.jp/kegg (accessed on 17 November 2019).
- Clustering 3.0. Available online: http://bonsai.hgc.jp/~mdehoon/software/cluster/software.htm#ctv (accessed on 7 December 2019).
- TreeView 3.0. Available online: https://bitbucket.org/TreeView3Dev/treeview3/src/master/ (accessed on 15 December 2019).
- Lee, S.; Lee, H.; Jeong, D.; Ham, J.; Park, S.; Choi, E.H.; Kim, S.J. Cold atmospheric plasma restores tamoxifen sensitivity in resistant MCF-7 breast cancer cell. Free Radic. Biol. Med. 2017, 110, 280–290. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, H.W.; Kim, H.; Kim, H.W.; Yun, S.H.; Park, J.E.; Choi, E.H.; Kim, S.J. Genome-Wide Comparison of the Target Genes of the Reactive Oxygen Species and Non-Reactive Oxygen Species Constituents of Cold Atmospheric Plasma in Cancer Cells. Cancers 2020, 12, 2640. https://doi.org/10.3390/cancers12092640
Ji HW, Kim H, Kim HW, Yun SH, Park JE, Choi EH, Kim SJ. Genome-Wide Comparison of the Target Genes of the Reactive Oxygen Species and Non-Reactive Oxygen Species Constituents of Cold Atmospheric Plasma in Cancer Cells. Cancers. 2020; 12(9):2640. https://doi.org/10.3390/cancers12092640
Chicago/Turabian StyleJi, Hwee Won, Heejoo Kim, Hyeon Woo Kim, Sung Hwan Yun, Jae Eun Park, Eun Ha Choi, and Sun Jung Kim. 2020. "Genome-Wide Comparison of the Target Genes of the Reactive Oxygen Species and Non-Reactive Oxygen Species Constituents of Cold Atmospheric Plasma in Cancer Cells" Cancers 12, no. 9: 2640. https://doi.org/10.3390/cancers12092640
APA StyleJi, H. W., Kim, H., Kim, H. W., Yun, S. H., Park, J. E., Choi, E. H., & Kim, S. J. (2020). Genome-Wide Comparison of the Target Genes of the Reactive Oxygen Species and Non-Reactive Oxygen Species Constituents of Cold Atmospheric Plasma in Cancer Cells. Cancers, 12(9), 2640. https://doi.org/10.3390/cancers12092640