Targeting Signaling Pathways in Inflammatory Breast Cancer


 ,
,  , and
, and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. EGFR
2.1. Biological Functions of EGFR Pathway in IBC
2.2. Targeting EGFR Pathway in IBC
3. HER2
4. Inflammatory Pathways JAK/STAT, NF-ĸB, and COX-2
4.1. JAK/STAT
4.1.1. Biological Functions of JAK/STAT Pathway in IBC
4.1.2. Targeting JAK/STAT in IBC
4.2. NF-ĸB
4.3. COX-2
5. Axl
5.1. Biological Functions of Axl Pathway in IBC
5.2. Targeting Axl in IBC
6. JNK
7. VEGF
8. RhoC GTPase
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- A Jaiyesimi, I.; Buzdar, A.U.; Hortobagyi, G. Inflammatory breast cancer: A review. J. Clin. Oncol. 1992, 10, 1014–1024. [Google Scholar] [CrossRef] [PubMed]
- Levine, P.H.; Steinhorn, S.C.; Ries, L.G.; Aron, J.L. Inflammatory Breast Cancer: The Experience of the Surveillance, Epidemiology, and End Results (SEER) Program. J. Natl. Cancer Inst. 1985, 74. [Google Scholar] [CrossRef]
- Yamauchi, H.; Ueno, N.T. Targeted therapy in inflammatory breast cancer. Cancer 2010, 116, 2758–2759. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, H.; Woodward, W.A.; Valero, V.; Alvarez, R.H.; Lucci, A.; Buchholz, T.A.; Iwamoto, T.; Krishnamurthy, S.; Yang, W.; Reuben, J.M.; et al. Inflammatory Breast Cancer: What We Know and What We Need to Learn. Oncol. 2012, 17, 891–899. [Google Scholar] [CrossRef] [Green Version]
- Hance, K.W.; Anderson, W.F.; Devesa, S.S.; Young, H.A.; Levine, P.H. Trends in Inflammatory Breast Carcinoma Incidence and Survival: The Surveillance, Epidemiology, and End Results Program at the National Cancer Institute. J. Natl. Cancer Inst. 2005, 97, 966–975. [Google Scholar] [CrossRef] [PubMed]
- Dawood, S.; Ueno, N.T.; Valero, V.; Woodward, W.A.; Buchholz, T.A.; Hortobágyi, G.N.; Gonzalez-Angulo, A.M.; Cristofanilli, M. Differences in survival among women with stage III inflammatory and noninflammatory locally advanced breast cancer appear early. Cancer 2010, 117, 1819–1826. [Google Scholar] [CrossRef]
- Masuda, H.; Brewer, T.M.; Liu, D.D.; Iwamoto, T.; Shen, Y.; Hsu, L.; Willey, J.S.; Gonzalez-Angulo, A.M.; Chavez-MacGregor, M.; Fouad, T.M.; et al. Long-term treatment efficacy in primary inflammatory breast cancer by hormonal receptor- and HER2-defined subtypes. Ann. Oncol. 2014, 25, 384–391. [Google Scholar] [CrossRef]
- Pan, X.; Yang, W.; Chen, Y.; Tong, L.; Li, C.; Li, H. Nomogram for predicting the overall survival of patients with inflammatory breast cancer: A SEER-based study. Breast 2019, 47, 56–61. [Google Scholar] [CrossRef]
- Giuliano, A.E.; Edge, S.B.; Hortobagyi, G.N. Eighth Edition of the AJCC Cancer Staging Manual: Breast Cancer. Ann. Surg. Oncol. 2018, 25, 1783–1785. [Google Scholar] [CrossRef]
- Ueno, N.T.; Fernandez, J.R.E.; Cristofanilli, M.; Overmoyer, B.; Rea, D.; Berdichevski, F.; El-Shinawi, M.; Bellon, J.; Le-Petross, H.T.; Lucci, A.; et al. International Consensus on the Clinical Management of Inflammatory Breast Cancer from the Morgan Welch Inflammatory Breast Cancer Research Program 10th Anniversary Conference. J. Cancer 2018, 9, 1437–1447. [Google Scholar] [CrossRef]
- Kleer, C.G.; Zhang, Y.; Pan, Q.; Gallagher, G.; Wu, M.; Wu, Z.-F.; Merajver, S.D. WISP3 and RhoC guanosine triphosphatase cooperate in the development of inflammatory breast cancer. Breast Cancer Res. 2003, 6, R110–R115. [Google Scholar] [CrossRef] [PubMed]
- Cabioglu, N.; Gong, Y.; Islam, R.; Broglio, K.; Sneige, N.; Sahin, A.; Gonzalez-Angulo, A.; Morandi, P.; Bucana, C.; Hortobagyi, G.; et al. Expression of growth factor and chemokine receptors: New insights in the biology of inflammatory breast cancer. Ann. Oncol. 2007, 18, 1021–1029. [Google Scholar] [CrossRef]
- Zell, J.A.; Tsang, W.Y.; Taylor, T.H.; Mehta, R.S.; Anton-Culver, H. Prognostic impact of human epidermal growth factor-like receptor 2 and hormone receptor status in inflammatory breast cancer (IBC): Analysis of 2,014 IBC patient cases from the California Cancer Registry. Breast Cancer Res. 2009, 11, R9. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Saso, H.; Iwamoto, T.; Xia, W.; Gong, Y.; Pusztai, L.; Woodward, W.A.; Reuben, J.M.; Warner, S.L.; Bearss, D.J.; et al. TIG1 Promotes the Development and Progression of Inflammatory Breast Cancer through Activation of Axl Kinase. Cancer Res. 2013, 73, 6516–6525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alpaugh, M.L.; Tomlinson, J.S.; Ye, Y.; Barsky, S.H. Relationship of Sialyl-Lewisx/a Underexpression and E-Cadherin Overexpression in the Lymphovascular Embolus of Inflammatory Breast Carcinoma. Am. J. Pathol. 2002, 161, 619–628. [Google Scholar] [CrossRef] [Green Version]
- Van Golen, K.L.; Davies, S.; Wu, Z.F.; Wang, Y.; Bucana, C.D.; Root, H.; Chandrasekharappa, S.; Strawderman, M.; Ethier, S.P.; Merajver, S.D. A novel putative low-affinity insulin-like growth factor-binding protein, LIBC (lost in inflammatory breast cancer), and RhoC GTPase correlate with the inflammatory breast cancer phenotype. Clin. Cancer Res. 1999, 5, 2511–2519. [Google Scholar]
- Overmoyer, B.; Almendro, V.; Shu, S.; Peluffo, G.; Park, S.; Nakhlis, F.; Bellon, J.; Yeh, E.; Jacene, H.; Hirshfield-Bartek, J.; et al. Abstract P4-06-01: JAK2/STAT3 activity in inflammatory breast cancer supports the investigation of JAK2 therapeutic targeting. Poster Session Abstracts 2012, 72. [Google Scholar] [CrossRef]
- Marotta, L.L.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The JAK2/STAT3 signaling pathway is required for growth of CD44⁺CD24⁻ stem cell-like breast cancer cells in human tumors. J. Clin. Investig. 2011, 121, 2723–2735. [Google Scholar] [CrossRef] [PubMed]
- Lerebours, F.; Vacher, S.; Andrieu, C.; Espié, M.; Marty, M.; Lidereau, R.; Bièche, I. NF-kappa B genes have a major role in Inflammatory Breast Cancer. BMC Cancer 2008, 8, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Laere, S.J.; Van Der Auwera, I.; Eynden, G.G.V.D.; Fox, S.; Bianchi, F.; Harris, A.L.; Van Dam, P.; Van Marck, E.A.; Vermeulen, P.B.; Dirix, L.Y. Distinct Molecular Signature of Inflammatory Breast Cancer by cDNA Microarray Analysis. Breast Cancer Res. Treat. 2005, 93, 237–246. [Google Scholar] [CrossRef]
- Van Laere, S.J.; Van Der Auwera, I.; Eynden, G.G.V.D.; Elst, H.J.; Weyler, J.; Harris, A.L.; Van Dam, P.; Van Marck, E.A.; Vermeulen, P.B.; Dirix, L.Y. Nuclear Factor- B Signature of Inflammatory Breast Cancer by cDNA Microarray Validated by Quantitative Real-time Reverse Transcription-PCR, Immunohistochemistry, and Nuclear Factor- B DNA-Binding. Clin. Cancer Res. 2006, 12, 3249–3256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Reyes, M.E.; Zhang, D.; Funakoshi, Y.; Trape, A.P.; Gong, Y.; Kogawa, T.; Eckhardt, B.L.; Masuda, H.; Pirman, D.A.; et al. EGFR signaling promotes inflammation and cancer stem-like activity in inflammatory breast cancer. Oncotarget 2017, 8, 67904–67917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirakawa, K.; Shibuya, M.; Heike, Y.; Takashima, S.; Watanabe, I.; Konishi, F.; Kasumi, F.; Goldman, C.K.; Thomas, K.A.; Bett, A.; et al. Tumor-infiltrating endothelial cells and endothelial precursor cells in inflammatory breast cancer. Int. J. Cancer 2002, 99, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Van Der Auwera, I. Increased Angiogenesis and Lymphangiogenesis in Inflammatory versus Noninflammatory Breast Cancer by Real-Time Reverse Transcriptase-PCR Gene Expression Quantification. Clin. Cancer Res. 2004, 10, 7965–7971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silvera, D.; Arju, R.; Darvishian, F.; Levine, P.H.; Zolfaghari, L.; Goldberg, J.D.; Hochman, T.; Formenti, S.C.; Schneider, R.J. Essential role for eIF4GI overexpression in the pathogenesis of inflammatory breast cancer. Nature 2009, 11, 903–908. [Google Scholar] [CrossRef]
- Charafe-Jauffret, E.; Ginestier, C.; Iovino, F.; Tarpin, C.; Diebel, M.; Esterni, B.; Houvenaeghel, G.; Extra, J.-M.; Bertucci, F.; Jacquemier, J.; et al. Aldehyde dehydrogenase 1-positive cancer stem cells mediate metastasis and poor clinical outcome in inflammatory breast cancer. Clin. Cancer Res. 2009, 16, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Allen, S.G.; Chen, Y.-C.; Madden, J.M.; Fournier, C.L.; Altemus, M.A.; Hiziroglu, A.B.; Cheng, Y.-H.; Wu, Z.F.; Bao, L.; Yates, J.; et al. Macrophages Enhance Migration in Inflammatory Breast Cancer Cells via RhoC GTPase Signaling. Sci. Rep. 2016, 6, 39190. [Google Scholar] [CrossRef] [Green Version]
- Lacerda, L.; Debeb, B.G.; Smith, D.; A Larson, R.; Solley, T.; Xu, W.; Krishnamurthy, S.; Gong, Y.; Levy, L.B.; A Buchholz, T.; et al. Mesenchymal stem cells mediate the clinical phenotype of inflammatory breast cancer in a preclinical model. Breast Cancer Res. 2015, 17, 42. [Google Scholar] [CrossRef] [Green Version]
- Reddy, S.M.; Reuben, A.; Barua, S.; Jiang, H.; Zhang, S.; Wang, L.; Gopalakrishnan, V.; Hudgens, C.; Tetzlaff, M.T.; Reuben, J.M.; et al. Poor Response to Neoadjuvant Chemotherapy Correlates with Mast Cell Infiltration in Inflammatory Breast Cancer. Cancer Immunol. Res. 2019, 7, 1025–1035. [Google Scholar] [CrossRef]
- Corkery, B.; Crown, J.; Clynes, M.; O’Donovan, N. Epidermal growth factor receptor as a potential therapeutic target in triple-negative breast cancer. Ann. Oncol. 2009, 20, 862–867. [Google Scholar] [CrossRef]
- Willmarth, N.E.; Ethier, S.P. Autocrine and Juxtacrine Effects of Amphiregulin on the Proliferative, Invasive, and Migratory Properties of Normal and Neoplastic Human Mammary Epithelial Cells. J. Boil. Chem. 2006, 281, 37728–37737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stratford, A.L.; Habibi, G.; Astanehe, A.; Jiang, H.; Hu, K.; Park, E.; Shadeo, A.; Buys, T.P.H.; Lam, W.L.; Pugh, T.J.; et al. Epidermal growth factor receptor (EGFR) is transcriptionally induced by the Y-box binding protein-1 (YB-1) and can be inhibited with Iressa in basal-like breast cancer, providing a potential target for therapy. Breast Cancer Res. 2007, 9, R61. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; LaFortune, T.A.; Krishnamurthy, S.; Esteva, F.J.; Cristofanilli, M.; Liu, P.; Lucci, A.; Singh, B.; Hung, M.-C.; Hortobagyi, G.N.; et al. Epidermal growth factor receptor tyrosine kinase inhibitor reverses mesenchymal to epithelial phenotype and inhibits metastasis in inflammatory breast cancer. Clin. Cancer Res. 2009, 15, 6639–6648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.; Ben-Batalla, I.; Schultze, A.; Loges, S. Macrophage–tumor crosstalk: Role of TAMR tyrosine kinase receptors and of their ligands. Cell. Mol. Life Sci. 2011, 69, 1391–1414. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Liu, Q.; Chen, J.; Chen, J.; Chen, F.; He, C.; Huang, D.; Wu, W.; Lin, L.; Huang, W.; et al. A Positive Feedback Loop between Mesenchymal-like Cancer Cells and Macrophages Is Essential to Breast Cancer Metastasis. Cancer Cell 2014, 25, 605–620. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Goswami, S.; Sahai, E.; Wyckoff, J.B.; Segall, J.E.; Condeelis, J.S. Tumor cells caught in the act of invading: Their strategy for enhanced cell motility. Trends Cell Boil. 2005, 15, 138–145. [Google Scholar] [CrossRef]
- Wyckoff, J.B. A Paracrine Loop between Tumor Cells and Macrophages Is Required for Tumor Cell Migration in Mammary Tumors. Cancer Res. 2004, 64, 7022–7029. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, N.; Lim, B.; Wang, X.; Ueno, N.T. Early clinical development of epidermal growth factor receptor targeted therapy in breast cancer. Expert Opin. Investig. Drugs 2017, 26, 463–479. [Google Scholar] [CrossRef]
- Matsuda, N.; Wang, X.; Lim, B.; Krishnamurthy, S.; Alvarez, R.H.; Willey, J.S.; Parker, C.A.; Song, J.; Shen, Y.; Hu, J.; et al. Safety and Efficacy of Panitumumab Plus Neoadjuvant Chemotherapy in Patients With Primary HER2-Negative Inflammatory Breast Cancer. JAMA Oncol. 2018, 4, 1207–1213. [Google Scholar] [CrossRef]
- Carboplatin and Paclitaxel With or Without Panitumumab in Treating Patients With Invasive Triple Negative Breast Cancer. Available online: https://ClinicalTrials.gov/show/NCT02876107 (accessed on 23 August 2016).
- Neratinib and Paclitaxel With or Without Pertuzumab and Trastuzumab Before Combination Chemotherapy in Treating Patients With Metastatic or Locally Advanced Breast Cancer. Available online: https://ClinicalTrials.gov/show/NCT03101748 (accessed on 5 April 2017).
- Kogawa, T.; Fujii, T.; Wu, J.; Harano, K.; Fouad, T.M.; Liu, D.D.; Shen, Y.; Masuda, H.; Krishnamurthy, S.; Chavez-MacGregor, M.; et al. Prognostic Value of HER2 to CEP17 Ratio on Fluorescence In Situ Hybridization Ratio in Patients with Nonmetastatic HER2-Positive Inflammatory and Noninflammatory Breast Cancer Treated with Neoadjuvant Chemotherapy with or without Trastuzumab. Oncologist 2020, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baselga, J. Mechanism of action of trastuzumab and scientific update. Semin. Oncol. 2001, 28, 4–11. [Google Scholar] [CrossRef]
- Dawood, S.; Gong, Y.; Broglio, K.; Buchholz, T.A.; Woodward, W.A.; Lucci, A.; Valero, V.; Gonzalez-Angulo, A.M.; Hortobagyi, G.N.; Cristofanilli, M. Trastuzumab in Primary Inflammatory Breast Cancer (IBC): High Pathological Response Rates and Improved Outcome. Breast J. 2010, 16, 529–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gianni, L.; Eiermann, W.; Semiglazov, V.F.; Manikhas, A.; Lluch, A.; Tjulandin, S.; Zambetti, M.; Vazquez, F.; Byakhow, M.; Lichinitser, M.; et al. Neoadjuvant chemotherapy with trastuzumab followed by adjuvant trastuzumab versus neoadjuvant chemotherapy alone, in patients with HER2-positive locally advanced breast cancer (the NOAH trial): A randomised controlled superiority trial with a parallel HER2-negative cohort. Lancet 2010, 375, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Gianni, L.; Pienkowski, T.; Im, Y.-H.; Roman, L.; Tseng, L.-M.; Liu, M.-C.; Lluch, A.; Staroslawska, E.; De La Haba-Rodriguez, J.; Im, S.-A.; et al. Efficacy and safety of neoadjuvant pertuzumab and trastuzumab in women with locally advanced, inflammatory, or early HER2-positive breast cancer (NeoSphere): A randomised multicentre, open-label, phase 2 trial. Lancet Oncol. 2012, 13, 25–32. [Google Scholar] [CrossRef]
- Konecny, G.E.; Pegram, M.D.; Venkatesan, N.; Finn, R.; Yang, G.; Rahmeh, M.; Untch, M.; Rusnak, D.W.; Spehar, G.; Mullin, R.J.; et al. Activity of the Dual Kinase Inhibitor Lapatinib (GW572016) against HER-2-Overexpressing and Trastuzumab-Treated Breast Cancer Cells. Cancer Res. 2006, 66, 1630–1639. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, B.; Trudeau, M.; Awada, A.; Blackwell, K.; Bachelot, T.; Salazar, V.; DeSilvio, M.; Westlund, R.; Zaks, T.; Spector, N.; et al. Lapatinib monotherapy in patients with HER2-overexpressing relapsed or refractory inflammatory breast cancer: Final results and survival of the expanded HER2+ cohort in EGF103009, a phase II study. Lancet Oncol. 2009, 10, 581–588. [Google Scholar] [CrossRef]
- Eisenhauer, E.; Therasse, P.; Bogaerts, J.; Schwartz, L.; Sargent, D.J.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Boussen, H.; Cristofanilli, M.; Zaks, T.; DeSilvio, M.; Salazar, V.; Spector, N. Phase II Study to Evaluate the Efficacy and Safety of Neoadjuvant Lapatinib Plus Paclitaxel in Patients With Inflammatory Breast Cancer. J. Clin. Oncol. 2010, 28, 3248–3255. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Bartholomeusz, C.; Mansour, O.; Humphries, J.; Hortobagyi, G.N.; Ordentlich, P.; Ueno, N.T. A class I histone deacetylase inhibitor, entinostat, enhances lapatinib efficacy in HER2-overexpressing breast cancer cells through FOXO3-mediated Bim1 expression. Breast Cancer Res. Treat. 2014, 146, 259–272. [Google Scholar] [CrossRef]
- Lim, B.; Murthy, R.K.; Lee, J.; Jackson, S.A.; Iwase, T.; Davis, D.W.; Willey, J.S.; Wu, J.; Shen, Y.; Tripathy, D.; et al. A phase Ib study of entinostat plus lapatinib with or without trastuzumab in patients with HER2-positive metastatic breast cancer that progressed during trastuzumab treatment. Br. J. Cancer 2019, 120, 1105–1112. [Google Scholar] [CrossRef]
- Chmielecki, J.; Ross, J.S.; Wang, K.; Frampton, G.M.; Palmer, G.A.; Ali, S.M.; Palma, N.; Morosini, D.; Miller, V.A.; Yelensky, R.; et al. Oncogenic Alterations in ERBB2/HER2 Represent Potential Therapeutic Targets Across Tumors From Diverse Anatomic Sites of Origin. Oncologist 2014, 20, 7–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bose, R.; Kavuri, S.M.; Searleman, A.; Shen, W.; Shen, D.; Koboldt, D.C.; Monsey, J.; Goel, N.; Aronson, A.B.; Li, S.; et al. Activating HER2 Mutations in HER2 Gene Amplification Negative Breast Cancer. Cancer Discov. 2012, 3, 224–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, J.S.; Ali, S.M.; Wang, K.; Khaira, D.; Palma, N.A.; Chmielecki, J.; Palmer, G.A.; Morosini, D.; Elvin, J.A.; Fernandez, S.V.; et al. Comprehensive genomic profiling of inflammatory breast cancer cases reveals a high frequency of clinically relevant genomic alterations. Breast Cancer Res. Treat. 2015, 154, 155–162. [Google Scholar] [CrossRef]
- Neratinib +/- Fulvestrant in Metastatic HER2 Non-amplified But HER2 Mutant Breast Cancer. Available online: https://ClinicalTrials.gov/show/NCT01670877 (accessed on 22 August 2019).
- Ali, S.M.; Alpaugh, R.K.; Downing, S.R.; Stephens, P.J.; Yu, J.Q.; Wu, H.; Buell, J.K.; Miller, V.A.; Lipson, D.; Palmer, G.A.; et al. Response of an ERBB2-Mutated Inflammatory Breast Carcinoma to Human Epidermal Growth Factor Receptor 2–Targeted Therapy. J. Clin. Oncol. 2014, 32, e88–e91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jhaveri, K.; Teplinsky, E.; Silvera, D.; Valeta-Magara, A.; Arju, R.; Giashuddin, S.; Sarfraz, Y.; Alexander, M.; Darvishian, F.; Levine, P.H.; et al. Hyperactivated mTOR and JAK2/STAT3 Pathways: Molecular Drivers and Potential Therapeutic Targets of Inflammatory and Invasive Ductal Breast Cancers After Neoadjuvant Chemotherapy. Clin. Breast Cancer 2016, 16, 113–122.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Laere, S.J.; Van Der Auwera, I.; Eynden, G.V.D.; Van Hummelen, P.; Van Dam, P.; Van Marck, E.; Vermeulen, P.; Dirix, L. Distinct molecular phenotype of inflammatory breast cancer compared to non-inflammatory breast cancer using Affymetrix-based genome-wide gene-expression analysis. Br. J. Cancer 2007, 97, 1165–1174. [Google Scholar] [CrossRef]
- Niwa, Y.; Kanda, H.; Shikauchi, Y.; Saiura, A.; Matsubara, K.; Kitagawa, T.; Yamamoto, J.; Kubo, T.; Yoshikawa, H. Methylation silencing of SOCS-3 promotes cell growth and migration by enhancing JAK/STAT and FAK signalings in human hepatocellular carcinoma. Oncogene 2005, 24, 6406–6417. [Google Scholar] [CrossRef] [Green Version]
- Teplinsky, E.; Valeta, A.; Arju, R.; Giashuddin, S.; Sarfraz, Y.; Alexander, M.; Darvishian, F.; Silvera, D.; Levine, P.H.; Hashmi, S.; et al. Hyperactivated mTOR and JAK2/STAT3 pathways: Crucial molecular drivers and potential therapeutic targets of inflammatory breast cancer (IBC). J. Clin. Oncol. 2013, 31, 60. [Google Scholar] [CrossRef]
- Rahal, O.M.; Wolfe, A.R.; Mandal, P.; Larson, R.; Tin, S.; Jimenez, C.; Zhang, D.; Horton, J.; Reuben, J.M.; McMurray, J.S.; et al. Blocking Interleukin (IL)4- and IL13-Mediated Phosphorylation of STAT6 (Tyr641) Decreases M2 Polarization of Macrophages and Protects Against Macrophage-Mediated Radioresistance of Inflammatory Breast Cancer. Int. J. Radiat. Oncol. 2018, 100, 1034–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bièche, I.; Lerebours, F.; Tozlu, S.; Espié, M.; Marty, M.; Lidereau, R. Molecular Profiling of Inflammatory Breast Cancer: Identification of a Poor-Prognosis Gene Expression Signature. Clin. Cancer Res. 2004, 10, 6789–6795. [Google Scholar] [CrossRef] [Green Version]
- Ruxolitinib W/ Preop Chemo For Triple Negative Inflammatory Brca. Available online: https://ClinicalTrials.gov/show/NCT02041429 (accessed on 22 January 2014).
- Study Of Ruxolitinib (INCB018424) With Preoperative Chemotherapy For Triple Negative Inflammatory Breast Cancer. Available online: https://ClinicalTrials.gov/show/NCT02876302 (accessed on 23 August 2016).
- Pan, Q.; Kleer, C.G.; Van Golen, K.L.; Irani, J.; Bottema, K.M.; Bias, C.; De Carvalho, M.; A Mesri, E.; Robins, D.M.; Dick, R.D.; et al. Copper deficiency induced by tetrathiomolybdate suppresses tumor growth and angiogenesis. Cancer Res. 2002, 62, 4854–4859. [Google Scholar] [PubMed]
- Pan, Q.; Bao, L.W.; Merajver, S.D. Tetrathiomolybdate inhibits angiogenesis and metastasis through suppression of the NFkappaB signaling cascade. Mol. Cancer Res. 2003, 1, 701–706. [Google Scholar] [PubMed]
- Wang, D.; Dubois, R.N. Immunosuppression associated with chronic inflammation in the tumor microenvironment. Carcinog. 2015, 36, 1085–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, F.M.; Simeone, A.-M.; Mazumdar, A.; Shah, A.H.; McMurray, J.S.; Ghosh, S.; Cristofanilli, M. Molecular and pharmacological blockade of the EP4 receptor selectively inhibits both proliferation and invasion of human inflammatory breast cancer cells. J. Exp. Ther. Oncol. 2008, 7. [Google Scholar]
- Wang, D.; Fu, L.; Ning, W.; Guo, L.; Sun, X.; Dey, S.K.; Chaturvedi, R.; Wilson, K.T.; Dubois, R.N. Peroxisome proliferator-activated receptor promotes colonic inflammation and tumor growth. Proc. Natl. Acad. Sci. USA 2014, 111, 7084–7089. [Google Scholar] [CrossRef] [Green Version]
- Katoh, H.; Wang, D.; Daikoku, T.; Sun, H.; Dey, S.K.; Dubois, R.N. CXCR2-Expressing Myeloid-Derived Suppressor Cells Are Essential to Promote Colitis-Associated Tumorigenesis. Cancer Cell 2013, 24, 631–644. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Dubois, R.N. An Inflammatory Mediator, Prostaglandin E2, in Colorectal Cancer. Cancer J. 2013, 19, 502–510. [Google Scholar] [CrossRef] [Green Version]
- Zelenay, S.; Van Der Veen, A.G.; Böttcher, J.P.; Snelgrove, K.J.; Rogers, N.; Acton, S.E.; Chakravarty, P.; Girotti, M.R.; Marais, R.; Quezada, S.A.; et al. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 2015, 162, 1257–1270. [Google Scholar] [CrossRef] [Green Version]
- Gay, C.M.; Balaji, K.; Byers, L.A. Giving AXL the axe: Targeting AXL in human malignancy. Br. J. Cancer 2017, 116, 415–423. [Google Scholar] [CrossRef]
- Zhao, Y.; Sun, X.; Jiang, L.; Yang, F.; Zhang, Z.; Jia, L. Differential Expression of Axl and Correlation with Invasion and Multidrug Resistance in Cancer Cells. Cancer Investig. 2012, 30, 287–294. [Google Scholar] [CrossRef]
- Ben-Batalla, I.; Schultze, A.; Wroblewski, M.; Erdmann, R.; Heuser, M.; Waizenegger, J.S.; Riecken, K.; Binder, M.; Schewe, D.; Sawall, S.; et al. Axl, a prognostic and therapeutic target in acute myeloid leukemia mediates paracrine crosstalk of leukemia cells with bone marrow stroma. Blood 2013, 122, 2443–2452. [Google Scholar] [CrossRef]
- Linger, R.M.; Cohen, R.A.; Cummings, C.T.; Sather, S.; Migdall-Wilson, J.; Middleton, D.H.; Lu, X.; Barón, A.E.; Franklin, W.A.; Merrick, D.T.; et al. Mer or Axl Receptor Tyrosine Kinase Inhibition Promotes Apoptosis, Blocks Growth, and Enhances Chemosensitivity of Human Non-Small Cell Lung Cancer. Oncogene 2012, 32, 3420–3431. [Google Scholar] [CrossRef] [Green Version]
- Lay, J.-D.; Hong, C.-C.; Huang, J.-S.; Yang, Y.-Y.; Pao, C.-Y.; Liu, C.-H.; Lai, Y.-P.; Lai, G.-M.; Cheng, A.-L.; Su, I.-J.; et al. Sulfasalazine Suppresses Drug Resistance and Invasiveness of Lung Adenocarcinoma Cells Expressing AXL. Cancer Res. 2007, 67, 3878–3887. [Google Scholar] [CrossRef] [Green Version]
- Hong, J.; Peng, D.; Chen, Z.; Sehdev, V.; Belkhiri, A. ABL regulation by AXL promotes cisplatin resistance in esophageal cancer. Cancer Res. 2012, 73, 331–340. [Google Scholar] [CrossRef] [Green Version]
- Dunne, P.D.; McArt, D.G.; Blayney, J.K.; Kalimutho, M.; Greer, S.; Wang, T.; Srivastava, S.; Ong, C.W.; Arthur, K.; Loughrey, M.; et al. AXL is a key regulator of inherent and chemotherapy-induced invasion and predicts a poor clinical outcome in early-stage colon cancer. Clin. Cancer Res. 2013, 20, 164–175. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Greger, J.; Shi, H.; Liu, Y.; Greshock, J.; Annan, R.; Halsey, W.; Sathe, G.M.; Martin, A.-M.; Gilmer, T.M. Novel Mechanism of Lapatinib Resistance in HER2-Positive Breast Tumor Cells: Activation of AXL. Cancer Res. 2009, 69, 6871–6878. [Google Scholar] [CrossRef] [Green Version]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Jin, G.; Wang, Z.; Wang, J.; Zhang, L.; Chen, Y.; Yuan, P.; Liu, D. Expression of Axl and its prognostic significance in human breast cancer. Oncol. Lett. 2016, 13, 621–628. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Zahari, M.S.; Ma, B.; Liu, R.; Renuse, S.; Sahasrabuddhe, N.A.; Chen, L.; Chaerkady, R.; Kim, M.-S.; Zhong, J.; et al. Global phosphotyrosine survey in triple-negative breast cancer reveals activation of multiple tyrosine kinase signaling pathways. Oncotarget 2015, 6, 29143–29160. [Google Scholar] [CrossRef] [Green Version]
- Aguilera, T.A.; Rafat, M.; Castellini, L.; Shehade, H.; Kariolis, M.S.; Hui, A.B.-Y.; Stehr, H.; Von Eyben, R.; Jiang, D.; Ellies, L.G.; et al. Reprogramming the immunological microenvironment through radiation and targeting Axl. Nat. Commun. 2016, 7, 13898. [Google Scholar] [CrossRef] [Green Version]
- Wnuk-Lipinska, K.; Davidsen, K.; Blø, M.; Engelsen, A.; Kang, J.; Hodneland, L.; Lie, M.; Bougnaud, S.; Aguilera, K.; Ahmed, L.; et al. Abstract 626: BGB324, a selective small molecule inhibitor of receptor tyrosine kinase AXL, abrogates tumor intrinsic and microenvironmental immune suppression and enhances immune checkpoint inhibitor efficacy in lung and mammary adenocarcinoma models. Immunology 2017, 77, 626. [Google Scholar] [CrossRef]
- Yoshizawa, T.; Tanaka, K.; Yasuhiro, T.; Fujikawa, R.; Ri, S.; Kawabata, K. Abstract LB-218: Development of Axl/Mer inhibitor, ONO-9330547: Preclinical evidence supporting the combination with immunotherapeutics. Experimental and Molecular Therapeutics 2016, 76. [Google Scholar] [CrossRef]
- Soh, K.K.; Kim, W.; Lee, Y.S.; Peterson, P.; Siddiqui-Jain, A.; Warner, S.L.; Bearss, D.J.; Whatcott, C.J. Abstract 235: AXL inhibition leads to a reversal of a mesenchymal phenotype sensitizing cancer cells to targeted agents and immuno-oncology therapies. Experimental and Molecular Therapeutics 2016, 76, 235. [Google Scholar] [CrossRef]
- Park, I.-K.; Giovenzana, C.; Hughes, T.L.; Yu, J.; Trotta, R.; Caligiuri, M.A. The Axl/Gas6 pathway is required for optimal cytokine signaling during human natural killer cell development. Blood 2009, 113, 2470–2477. [Google Scholar] [CrossRef] [Green Version]
- Paolino, M.; Choidas, A.; Wallner, S.; Pranjic, B.; Uribesalgo, I.; Loeser, S.; Jamieson, A.M.; Langdon, W.Y.; Ikeda, F.; Fededa, J.P.; et al. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nat. 2014, 507, 508–512. [Google Scholar] [CrossRef]
- Lu, Q.; Gore, M.; Zhang, Q.; Camenisch, T.; Boast, S.; Casagranda, F.; Lai, C.; Skinner, M.K.; Klein, R.; Matsushima, G.K.; et al. Tyro-3 family receptors are essential regulators of mammalian spermatogenesis. Nature 1999, 398, 723–728. [Google Scholar] [CrossRef]
- Lu, Q. Homeostatic Regulation of the Immune System by Receptor Tyrosine Kinases of the Tyro 3 Family. Science 2001, 293, 306–311. [Google Scholar] [CrossRef]
- Waterborg, C.E.J.; Broeren, M.G.A.; Davidson, E.B.; Koenders, M.I.; Lent, P.L.E.M.V.; Berg, W.B.V.D.; Van Der Kraan, P.M.; Loo, F.A.J.V.D. The level of synovial AXL expression determines the outcome of inflammatory arthritis, possibly depending on the upstream role of TGF-β1. Rheumatology 2018, 58, 536–546. [Google Scholar] [CrossRef]
- Bottai, G.; Raschioni, C.; Székely, B.; Di Tommaso, L.; Szász, A.M.; Losurdo, A.; Győrffy, B.; Ács, B.; Torrisi, R.; Karachaliou, N.; et al. AXL-associated tumor inflammation as a poor prognostic signature in chemotherapy-treated triple-negative breast cancer patients. Breast Cancer 2016, 2, 16033. [Google Scholar] [CrossRef]
- Rothlin, C.V.; Ghosh, S.; Zuniga, E.I.; Oldstone, M.B.; Lemke, G. TAM Receptors Are Pleiotropic Inhibitors of the Innate Immune Response. Cell 2007, 131, 1124–1136. [Google Scholar] [CrossRef] [Green Version]
- Mangelson, R.P.P.; Foulks, J.M.; Matsumura, Y.; Mouritsen, L.; Whatcott, C.J.; Bearss, D.J.; Warner, S.L. The AXL kinase inhibitor, TP-0903, demonstrates efficacy in preclinical models of colorectal cancer independent of KRAS mutation status. Cancer Res. 2019, 79, 2197. [Google Scholar]
- First-in-human Study of Oral TP-0903 (a Novel Inhibitor of AXL Kinase) in Patients With Advanced Solid Tumors. Available online: https://ClinicalTrials.gov/show/NCT02729298 (accessed on 16 April 2016).
- Phase 1/2 Study of TP-0903 (an Inhibitor of AXL Kinase) in Patients With Previously Treated CLL. Available online: https://ClinicalTrials.gov/show/NCT03572634 (accessed on 28 June 2018).
- Xie, X.; Kaoud, T.S.; Edupuganti, R.; Zhang, T.; Kogawa, T.; Zhao, Y.; Chauhan, G.B.; Giannoukos, D.N.; Qi, Y.; Tripathy, D.; et al. c-Jun N-terminal kinase promotes stem cell phenotype in triple-negative breast cancer through upregulation of Notch1 via activation of c-Jun. Oncogene 2016, 36, 2599–2608. [Google Scholar] [CrossRef]
- Hui, L.; Zatloukal, K.; Scheuch, H.; Stepniak, E.; Wagner, E.F. Proliferation of human HCC cells and chemically induced mouse liver cancers requires JNK1-dependent p21 downregulation. J. Clin. Investig. 2008, 118, 3943–3953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, K.-I.; Sato, A.; Okada, M.; Shibuya, K.; Seino, S.; Suzuki, K.; Watanabe, E.; Narita, Y.; Shibui, S.; Kayama, T.; et al. Targeting JNK for therapeutic depletion of stem-like glioblastoma cells. Sci. Rep. 2012, 2, 516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Chao, L.; Li, X.; Ma, G.; Chen, L.; Zang, Y.; Zhou, G. Elevated expression of phosphorylated c-Jun NH2-terminal kinase in basal-like and “triple-negative” breast cancers. Hum. Pathol. 2010, 41, 401–406. [Google Scholar] [CrossRef]
- Yeh, Y.-T.; Hou, M.-F.; Chung, Y.-F.; Chen, Y.-J.; Yang, S.-F.; Chen, D.-C.; Su, J.-H.; Yuan, S.-S.F. Decreased expression of phosphorylated JNK in breast infiltrating ductal carcinoma is associated with a better overall survival. Int. J. Cancer 2006, 118, 2678–2684. [Google Scholar] [CrossRef]
- Deng, L.; Gao, X.; Liu, B.; He, X.; Xu, J.; Qiang, J.; Wu, Q.; Liu, S. NMT1 inhibition modulates breast cancer progression through stress-triggered JNK pathway. Cell Death Dis. 2018, 9, 1143. [Google Scholar] [CrossRef] [Green Version]
- Dvorak, H.F. Angiogenesis: Update 2005. J. Thromb. Haemost. 2005, 3, 1835–1842. [Google Scholar] [CrossRef]
- Weidner, N.; Semple, J.P.; Welch, W.R.; Folkman, J. Tumor Angiogenesis and Metastasis—Correlation in Invasive Breast Carcinoma. New Engl. J. Med. 1991, 324, 1–8. [Google Scholar] [CrossRef]
- McCarthy, N.J.; Yang, X.; Linnoila, I.R.; Merino, M.J.; Hewitt, S.M.; Parr, A.L.; Paik, S.; Steinberg, S.M.; Hartmann, D.P.; Mourali, N.; et al. Microvessel density, expression of estrogen receptor alpha, MIB-1, p53, and c-erbB-2 in inflammatory breast cancer. Clin. Cancer Res. 2002, 8, 3857–3862. [Google Scholar]
- Arias-Pulido, H.; Chaher, N.; Gong, Y.; Qualls, C.; Vargas, J.; Royce, M. Tumor stromal vascular endothelial growth factor A is predictive of poor outcome in inflammatory breast cancer. BMC Cancer 2012, 12, 298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierga, J.-Y.; Petit, T.; DeLozier, T.; Ferrero, J.-M.; Campone, M.; Gligorov, J.; Lerebours, F.; Roché, H.; Bachelot, T.; Charafe-Jauffret, E.; et al. Neoadjuvant bevacizumab, trastuzumab, and chemotherapy for primary inflammatory HER2-positive breast cancer (BEVERLY-2): An open-label, single-arm phase 2 study. Lancet Oncol. 2012, 13, 375–384. [Google Scholar] [CrossRef]
- Tabouret, E.; Bertucci, F.; Pierga, J.-Y.; Petit, T.; Levy, C.; Ferrero, J.-M.; Campone, M.; Gligorov, J.; Lerebours, F.; Roche, H.; et al. MMP2 and MMP9 serum levels are associated with favorable outcome in patients with inflammatory breast cancer treated with bevacizumab-based neoadjuvant chemotherapy in the BEVERLY-2 study. Oncotarget 2016, 7, 18531–18540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertucci, F.; Fekih, M.; Autret, A.; Petit, T.; Dalenc, F.; Lévy, C.; Romieu, G.; Bonneterre, J.; Ferrero, J.-M.; Kerbrat, P.; et al. Bevacizumab plus neoadjuvant chemotherapy in patients with HER2-negative inflammatory breast cancer (BEVERLY-1): A multicentre, single-arm, phase 2 study. Lancet Oncol. 2016, 17, 600–611. [Google Scholar] [CrossRef]
- A Phase 1b/2 Study of Rebastinib (DCC-2036) in Combination With Paclitaxel in Patients With Advanced or Metastatic Solid Tumors. Available online: https://ClinicalTrials.gov/show/NCT03601897 (accessed on 26 July 2018).
- Slamon, D.; Gomez, H.L.; Kabbinavar, F.F.; Amit, O.; Richie, M.; Pandite, L.; Goodman, V. Randomized study of pazopanib + lapatinib vs. lapatinib alone in patients with HER2-positive advanced or metastatic breast cancer. J. Clin. Oncol. 2008, 26, 1016. [Google Scholar] [CrossRef]
- Slamon, D.; Stemmer, S.; Johnston, S.; Kim, S.; Durante, M.; Pandite, L.; Roychowdhury, D.; Goodman, V. Phase 2 study of dual VEGF/HER2 blockade with pazopanib + lapatinib in patients with first-line HER2 positive advanced or metastatic (adv/met) breast cancer. Poster Session Abstracts 2009, 69, 4114. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Johnston, S.R.D.; Manikhas, A.; Gomez, H.; Gladkov, O.; Shao, Z.; Safina, S.; Blackwell, K.L.; Alvarez, R.H.; Rubin, S.D.; et al. A randomized phase II study of lapatinib + pazopanib versus lapatinib in patients with HER2+ inflammatory breast cancer. Breast Cancer Res. Treat. 2012, 137, 471–482. [Google Scholar] [CrossRef] [Green Version]
- Lehman, H.L.; Van Laere, S.J.; Van Golen, C.M.; Vermeulen, P.B.; Dirix, L.Y.; Van Golen, K.L. Regulation of Inflammatory Breast Cancer Cell Invasion through Akt1/PKB Phosphorylation of RhoC GTPase. Mol. Cancer Res. 2012, 10, 1306–1318. [Google Scholar] [CrossRef] [Green Version]
- Van Golen, K.L.; Bao, L.; DiVito, M.M.; Wu, Z.; Prendergast, G.C.; Merajver, S.D. Reversion of RhoC GTPase-induced inflammatory breast cancer phenotype by treatment with a farnesyl transferase inhibitor. Mol. Cancer Ther. 2002, 1, 575–583. [Google Scholar]
- Van Golen, K.L.; Wu, Z.-F.; Qiao, X.; Bao, L.; Merajver, S.D. RhoC GTPase Overexpression Modulates Induction of Angiogenic Factors in Breast Cells. Neoplasia 2000, 2, 418–425. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.-B.; Erickson, J.W.; Fuji, R.; Ramachandran, S.; Gao, P.; Dinavahi, R.; Wilson, K.F.; Ambrosio, A.L.; Dias, S.M.G.; Dang, C.V.; et al. Targeting Mitochondrial Glutaminase Activity Inhibits Oncogenic Transformation. Cancer Cell 2010, 18, 207–219. [Google Scholar] [CrossRef] [Green Version]
- Wynn, M.L.; Yates, J.; Evans, C.; Van Wassenhove, L.D.; Wu, Z.F.; Bridges, S.; Bao, L.; Fournier, C.; Ashrafzadeh, S.; Merrins, M.J.; et al. RhoC GTPase Is a Potent Regulator of Glutamine Metabolism andN-Acetylaspartate Production in Inflammatory Breast Cancer Cells. J. Boil. Chem. 2016, 291, 13715–13729. [Google Scholar] [CrossRef] [Green Version]
- Connie, R.; Shi, D.T.R.; Liwei, B.; Judy, C.; Pang, Z.W.; Steven, G.A.; Sofia, D.M. Effect of novel RhoC inhibitor on breast cancer progression and metastasis in vivo. In Proceedings of the 103rd Annual Meeting of the American Association for Cancer Research, Chicago, IL, USA, April 2012; p. 72. [Google Scholar]
- Andreopoulou, E.; Vigoda, I.S.; Valero, V.; Hershman, D.L.; Raptis, G.; Vahdat, L.T.; Han, H.S.; Wright, J.J.; Pellegrino, C.M.; Cristofanilli, M.; et al. Phase I-II study of the farnesyl transferase inhibitor tipifarnib plus sequential weekly paclitaxel and doxorubicin-cyclophosphamide in HER2/neu-negative inflammatory carcinoma and non-inflammatory estrogen receptor-positive breast carcinoma. Breast Cancer Res. Treat. 2013, 141, 429–435. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.S.; Dering, J.; Conklin, D.; Kalous, O.; Cohen, D.J.; Desai, A.J.; Ginther, C.; Atefi, M.; Chen, I.; Fowst, C.; et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009, 11, R77. [Google Scholar] [CrossRef] [Green Version]
- Van Laere, S.J.; Ueno, N.T.; Finetti, P.; Vermeulen, P.; Lucci, A.; Robertson, F.M.; Marsan, M.; Iwamoto, T.; Krishnamurthy, S.; Masuda, H.; et al. Uncovering the molecular secrets of inflammatory breast cancer biology: An integrated analysis of three distinct affymetrix gene expression datasets. Clin. Cancer Res. 2013, 19, 4685–4696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
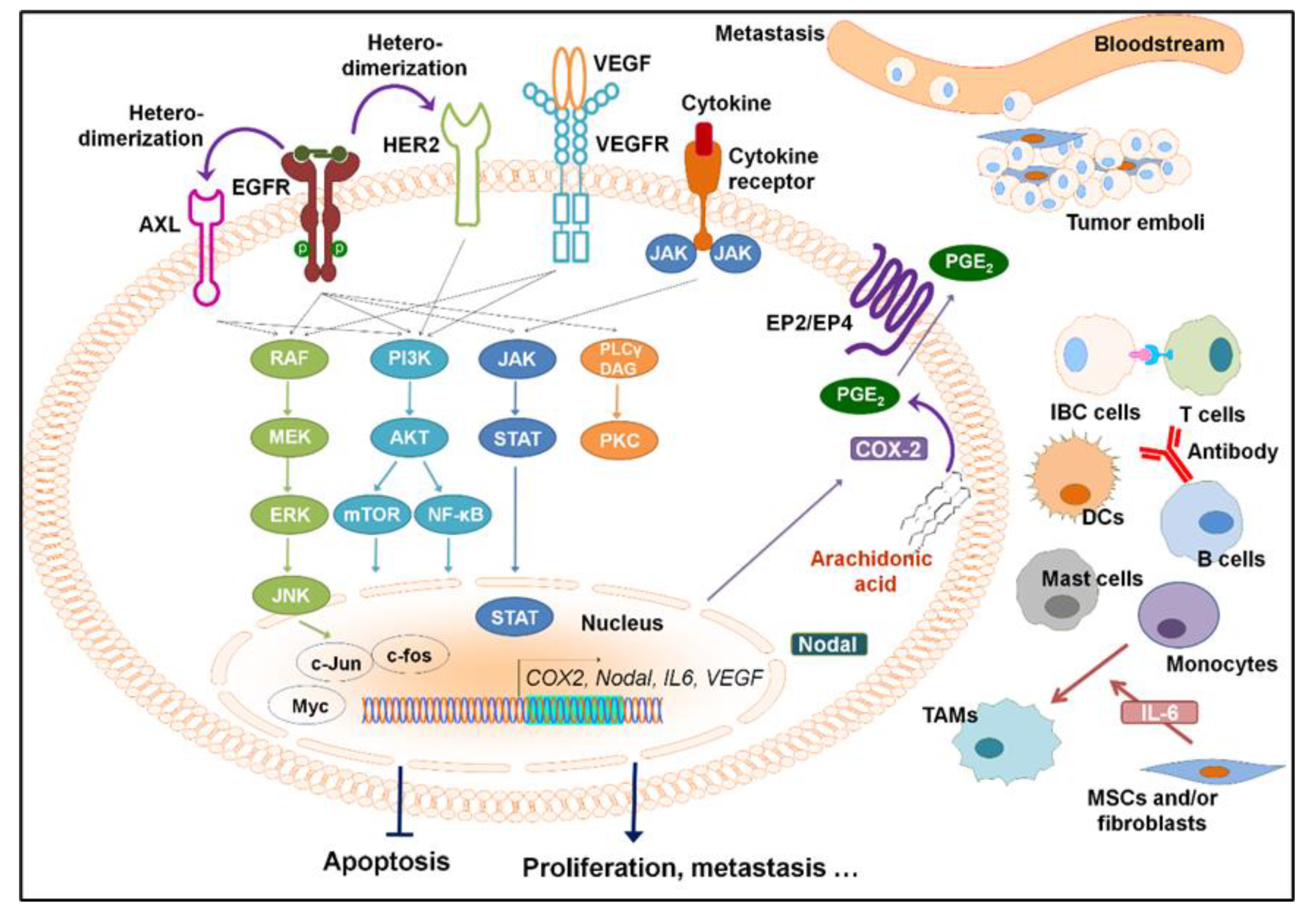

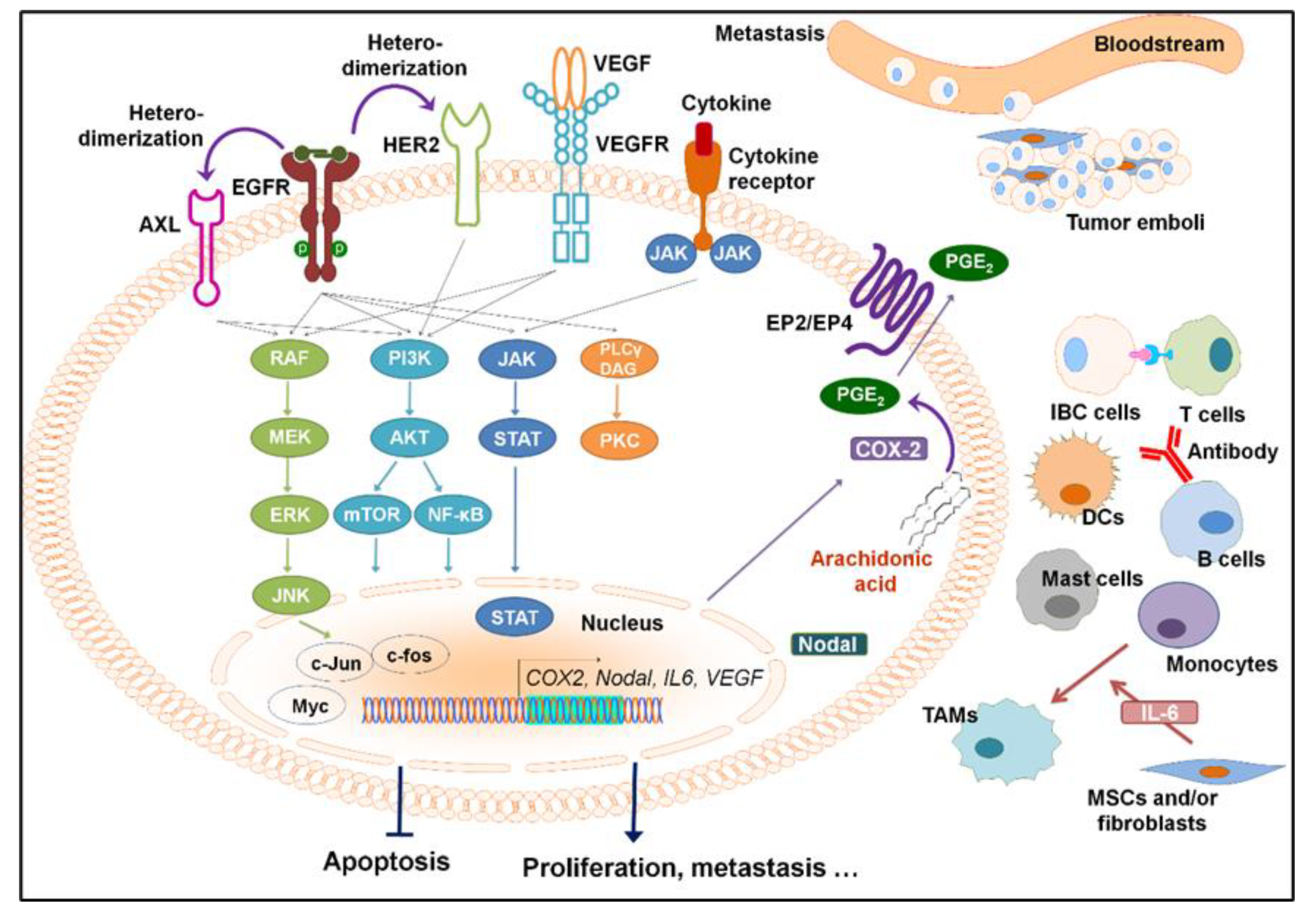{kind=link}
| Drug Studied | Target(s) | Combined Agents | Type of Study | Patient Population | Trial Identification No. |
|---|---|---|---|---|---|
| Panitumumab | EGFR | Carboplatin, nab-paclitaxel, 5-fluorouracil, epirubicin, and cyclophosphamide | Phase 2, single-arm | Primary HER2− newly diagnosed IBC | NCT01036087 |
| Panitumumab | EGFR | Carboplatin, nab-paclitaxel, 5-fluorouracil, epirubicin, and cyclophosphamide | Phase 2, randomized | TN-IBC | NCT02876107 |
| Neratinib | EGFR, HER2, and HER4 | Neratinib, pertuzumab, and trastuzumab with paclitaxel | Phase 2 | HER2+ IBC; HR+/HER2− IBC | NCT03101748 |
| Trastuzumab | HER2 | Trastuzumab, doxorubicin, paclitaxel, cyclophosphamide, methotrexate, 5-fluorouracil, and tamoxifen | Phase 3 | Newly diagnosed HER2+ locally advanced breast cancer or IBC | ISRCTN86043495 |
| Pertuzumab and trastuzumab | HER2 | Pertuzumab, trastuzumab, and docetaxel | Phase 2 | Locally advanced, inflammatory or early stage HER2+ breast cancer | NCT00545688 |
| Lapatinib | HER2 | Lapatinib | Phase 2 | HER2+ IBC | NCT00105950 |
| Lapatinib | HER2 | Lapatinib and paclitaxel | Phase 2 | Newly diagnosed IBC | EGF102580 |
| Lapatinib | HER2 | Lapatinib, entinostat, with or without trastuzumab | Phase 1b | HER2+ breast cancer | NCT01434303 |
| Ruxolitinib | JAK1/2 | Ruxolitinib with preoperative chemotherapy | Phase 1/2 | TN-IBC | NCT02041429 |
| Ruxolitinib | JAK1/2 | Ruxolitinib, paclitaxel, doxorubicin, and cyclophosphamide | Phase 2, randomized | TN-IBC | NCT02876302 |
| Bevacizumab | VEGF | Bevacizumab, doxorubicin, and docetaxel | Interventional | Inflammatory and locally advanced breast cancer | NCT00717405 |
| Bevacizumab | VEGF | Bevacizumab, fluorouracil, epirubicin, cyclophosphamide, and docetaxel | Phase 2 | Non-metastatic HER2− IBC | NCT00820547 |
| Rebastinib | Tie2 | Rebastinib and paclitaxel | Phase 1b/2 | Advanced or metastatic solid tumors including IBC | NCT03601897 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Semba, T.; Phi, L.T.H.; Chainitikun, S.; Iwase, T.; Lim, B.; Ueno, N.T. Targeting Signaling Pathways in Inflammatory Breast Cancer. Cancers 2020, 12, 2479. https://doi.org/10.3390/cancers12092479
Wang X, Semba T, Phi LTH, Chainitikun S, Iwase T, Lim B, Ueno NT. Targeting Signaling Pathways in Inflammatory Breast Cancer. Cancers. 2020; 12(9):2479. https://doi.org/10.3390/cancers12092479
Chicago/Turabian StyleWang, Xiaoping, Takashi Semba, Lan Thi Hanh Phi, Sudpreeda Chainitikun, Toshiaki Iwase, Bora Lim, and Naoto T. Ueno. 2020. "Targeting Signaling Pathways in Inflammatory Breast Cancer" Cancers 12, no. 9: 2479. https://doi.org/10.3390/cancers12092479
APA StyleWang, X., Semba, T., Phi, L. T. H., Chainitikun, S., Iwase, T., Lim, B., & Ueno, N. T. (2020). Targeting Signaling Pathways in Inflammatory Breast Cancer. Cancers, 12(9), 2479. https://doi.org/10.3390/cancers12092479