pH-Channeling in Cancer: How pH-Dependence of Cation Channels Shapes Cancer Pathophysiology

 ,
,  , and
, and
Abstract
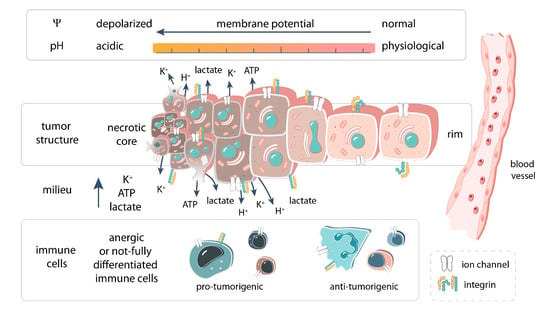
1. Introduction
2. pH Homeostasis Is Tightly Coupled to Changes in Membrane Potential
- (I).
- The plasma membrane is hyperpolarized during DNA synthesis, in the S phase, and depolarized in the G2/M phase [32,33]. Interestingly, pHi has an alkaline peak of ΔpHi ≈ 0.2 in lymphocyte populations in the S phase of the cell cycle [34,35], whereas an acidification in G2/M has not been shown to date. Since pH changes alter microtubule stability, it has been proposed that pHi may be a “clock” for mitosis [36].
- (II).
- (III).
- Glycolysis acidifies the intracellular milieu, whereas oxidative phosphorylation induces an alkalinization [26,27]. Aerobic glycolysis is prominent in the lamellipodia of multiple cancer cell types [39], which can act as a local source of metabolically produced acid during migration and further depolarize the plasma membrane. Additionally, the mitochondrial membrane potential depolarizes during glycolysis, and hyperpolarizes during oxidative phosphorylation [40,41]. It is conceivable that the plasma membrane potential is altered concordantly, as the mitochondrial membrane potential is in the phase with the plasma membrane potential [28]. Therefore, the enhanced aerobic glycolysis of cancer cells can contribute to their depolarized membrane potential.
3. Ion Channels Are Modulated Directly and Indirectly by Changes in pH
3.1. Direct Interaction between Protons and Ion Channels
3.2. pH-Dependent Interaction of Cell Adhesion Proteins with Ion Channels
4. pH-Dependent Regulation of Ion Channels in Cancer Cells
4.1. Modulation of K+ Channels in Cancer by pH
4.1.1. KV Channels
4.1.2. KCa Channels
4.1.3. Kir Channels
4.1.4. K2P Channels
4.2. TRP Channel in Tumors: Multimodal pH-Depencence
4.3. Voltage-Gated Proton Channel HV1
4.4. Other Channels
4.4.1. NaV Channels
4.4.2. Piezo1
4.4.3. Acid-Sensing Ion Channels (ASICs)
4.4.4. P2X
5. pH Dependence of Ion Channels in Tumor Immunity
5.1. Innate Immunity
5.2. Acquired Immunity
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Swietach, P.; Vaughan-Jones, R.D.; Harris, A.L.; Hulikova, A. The chemistry, physiology and pathology of pH in cancer. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369. [Google Scholar] [CrossRef] [PubMed]
- Swietach, P. What is pH regulation, and why do cancer cells need it? Cancer Metastasis Rev. 2019, 38, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, S.F.; Novak, I.; Alves, F.; Schwab, A.; Pardo, L.A. Alternating pH landscapes shape epithelial cancer initiation and progression: Focus on pancreatic cancer. Bioessays 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.C.; Singh, R.; Asters, M.; Liu, J.; Zhang, X.; Pabbidi, M.R.; Watabe, K.; Mo, Y.-Y. Regulation of breast tumorigenesis through acid sensors. Oncogene 2016, 35, 4102–4111. [Google Scholar] [CrossRef] [PubMed]
- Harguindey, S.; Alfarouk, K.; Polo Orozco, J.; Hardonnière, K.; Stanciu, D.; Fais, S.; Devesa, J. A New and Integral Approach to the Etiopathogenesis and Treatment of Breast Cancer Based upon Its Hydrogen Ion Dynamics. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Böhme, I.; Schönherr, R.; Eberle, J.; Bosserhoff, A.K. Membrane Transporters and Channels in Melanoma. Rev. Physiol. Biochem. Pharm. 2020. [Google Scholar] [CrossRef]
- Avagliano, A.; Fiume, G.; Pelagalli, A.; Sanità, G.; Ruocco, M.R.; Montagnani, S.; Arcucci, A. Metabolic Plasticity of Melanoma Cells and Their Crosstalk With Tumor Microenvironment. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Koch, A.; Schwab, A. Cutaneous pH landscape as a facilitator of melanoma initiation and progression. Acta Physiol. 2019, 225, e13105. [Google Scholar] [CrossRef]
- Stopa, K.B.; Kusiak, A.A.; Szopa, M.D.; Ferdek, P.E.; Jakubowska, M.A. Pancreatic Cancer and Its Microenvironment—Recent Advances and Current Controversies. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Harguindey, S.; Stanciu, D.; Devesa, J.; Alfarouk, K.; Cardone, R.A.; Polo Orozco, J.D.; Devesa, P.; Rauch, C.; Orive, G.; Anitua, E.; et al. Cellular acidification as a new approach to cancer treatment and to the understanding and therapeutics of neurodegenerative diseases. Semin. Cancer Biol. 2017, 43, 157–179. [Google Scholar] [CrossRef]
- Holzer, P. Acid-sensitive ion channels and receptors. Handb. Exp. Pharm. 2009, 283–332. [Google Scholar] [CrossRef]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion channels and the hallmarks of cancer. Trends Mol. Med. 2010, 16, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion Channels in Cancer: Are Cancer Hallmarks Oncochannelopathies? Physiol. Rev. 2018, 98, 559–621. [Google Scholar] [CrossRef] [PubMed]
- Litan, A.; Langhans, S.A. Cancer as a channelopathy: Ion channels and pumps in tumor development and progression. Front. Cell. Neurosci. 2015, 9. [Google Scholar] [CrossRef] [PubMed]
- Andersen, A.P.; Moreira, J.M.A.; Pedersen, S.F. Interactions of ion transporters and channels with cancer cell metabolism and the tumour microenvironment. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369. [Google Scholar] [CrossRef] [PubMed]
- Cone, C.D., Jr. Unified theory on the basic mechanism of normal mitotic control and oncogenesis. J. Theor. Biol. 1971, 30, 151–181. [Google Scholar] [CrossRef]
- Payne, S.L.; Levin, M.; Oudin, M.J. Bioelectric Control of Metastasis in Solid Tumors. Bioelectricity 2019, 1, 114–130. [Google Scholar] [CrossRef]
- Chernet, B.T.; Adams, D.S.; Lobikin, M.; Levin, M. Use of genetically encoded, light-gated ion translocators to control tumorigenesis. Oncotarget 2016, 7, 19575–19588. [Google Scholar] [CrossRef]
- Lyall, V.; Biber, T.U. Potential-induced changes in intracellular pH. Am. J. Physiol. 1994, 266, F685–F696. [Google Scholar] [CrossRef]
- Cunderlikova, B.; Moan, J.; Sjaastad, I. pH dependent uptake of porphyrin-type photosensitizers by solid tumor cells in vitro is not induced by modification of transmembrane potential. Cancer Lett. 2005, 222, 39–47. [Google Scholar] [CrossRef]
- Honasoge, A.; Shelton, K.A.; Sontheimer, H. Autocrine regulation of glioma cell proliferation via pHe-sensitive K(+) channels. Am. J. Physiol. Cell Physiol. 2014, 306, C493–C505. [Google Scholar] [CrossRef] [PubMed]
- Bear, C.E.; Davison, J.S.; Shaffer, E.A. Intracellular pH influences the resting membrane potential of isolated rat hepatocytes. Biochim. Biophys. Acta 1988, 944, 113–120. [Google Scholar] [CrossRef]
- Deitmer, J.W.; Szatkowski, M. Membrane potential dependence of intracellular pH regulation by identified glial cells in the leech central nervous system. J. Physiol. 1990, 421, 617–631. [Google Scholar] [CrossRef] [PubMed]
- Berzingi, S.; Newman, M.; Yu, H.G. Altering bioelectricity on inhibition of human breast cancer cells. Cancer Cell Int. 2016, 16, 72. [Google Scholar] [CrossRef]
- Raimondo, J.V.; Tomes, H.; Irkle, A.; Kay, L.; Kellaway, L.; Markram, H.; Millar, R.P.; Akerman, C.J. Tight Coupling of Astrocyte pH Dynamics to Epileptiform Activity Revealed by Genetically Encoded pH Sensors. J. Neurosci. 2016, 36, 7002–7013. [Google Scholar] [CrossRef]
- Javadov, S.; Huang, C.; Kirshenbaum, L.; Karmazyn, M. NHE-1 inhibition improves impaired mitochondrial permeability transition and respiratory function during postinfarction remodelling in the rat. J. Mol. Cell Cardiol. 2005, 38, 135–143. [Google Scholar] [CrossRef]
- Bonnet, S.; Archer, S.L.; Allalunis-Turner, J.; Haromy, A.; Beaulieu, C.; Thompson, R.; Lee, C.T.; Lopaschuk, G.D.; Puttagunta, L.; Harry, G.; et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007, 11, 37–51. [Google Scholar] [CrossRef]
- Azarias, G.; Perreten, H.; Lengacher, S.; Poburko, D.; Demaurex, N.; Magistretti, P.J.; Chatton, J.Y. Glutamate transport decreases mitochondrial pH and modulates oxidative metabolism in astrocytes. J. Neurosci. 2011, 31, 3550–3559. [Google Scholar] [CrossRef]
- Boron, W.F. Regulation of intracellular pH. Adv. Physiol. Educ. 2004, 28, 160–179. [Google Scholar] [CrossRef]
- Abdul Kadir, L.; Stacey, M.; Barrett-Jolley, R. Emerging Roles of the Membrane Potential: Action Beyond the Action Potential. Front. Physiol. 2018, 9, 1661. [Google Scholar] [CrossRef]
- McLaughlin, K.A.; Levin, M. Bioelectric signaling in regeneration: Mechanisms of ionic controls of growth and form. Dev. Biol. 2018, 433, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Urrego, D.; Tomczak, A.P.; Zahed, F.; Stuhmer, W.; Pardo, L.A. Potassium channels in cell cycle and cell proliferation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130094. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Brackenbury, W.J. Membrane potential and cancer progression. Front. Physiol. 2013, 4, 185. [Google Scholar] [CrossRef] [PubMed]
- Flinck, M.; Kramer, S.H.; Pedersen, S.F. Roles of pH in control of cell proliferation. Acta Physiol. 2018, 223, e13068. [Google Scholar] [CrossRef] [PubMed]
- Gerson, D.F.; Kiefer, H. High intracellular pH accompanies mitotic activity in murine lymphocytes. J. Cell Physiol. 1982, 112, 1–4. [Google Scholar] [CrossRef]
- Gagliardi, L.J.; Shain, D.H. Is intracellular pH a clock for mitosis? Theor. Biol. Med. Model 2013, 10, 8. [Google Scholar] [CrossRef]
- Martin, C.; Pedersen, S.F.; Schwab, A.; Stock, C. Intracellular pH gradients in migrating cells. Am. J. Physiol. Cell Physiol. 2011, 300, C490–C495. [Google Scholar] [CrossRef]
- Paradise, R.K.; Whitfield, M.J.; Lauffenburger, D.A.; van Vliet, K.J. Directional cell migration in an extracellular pH gradient: A model study with an engineered cell line and primary microvascular endothelial cells. Exp. Cell Res. 2013, 319, 487–497. [Google Scholar] [CrossRef]
- Shiraishi, T.; Verdone, J.E.; Huang, J.; Kahlert, U.D.; Hernandez, J.R.; Torga, G.; Zarif, J.C.; Epstein, T.; Gatenby, R.; McCartney, A.; et al. Glycolysis is the primary bioenergetic pathway for cell motility and cytoskeletal remodeling in human prostate and breast cancer cells. Oncotarget 2015, 6, 130–143. [Google Scholar] [CrossRef]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Steidl, J.V.; Yool, A.J. Differential sensitivity of voltage-gated potassium channels Kv1.5 and Kv1.2 to acidic pH and molecular identification of pH sensor. Mol. Pharmacol. 1999, 55, 812–820. [Google Scholar] [PubMed]
- Immke, D.C.; McCleskey, E.W. Protons open acid-sensing ion channels by catalyzing relief of Ca2+ blockade. Neuron 2003, 37, 75–84. [Google Scholar] [CrossRef]
- Sherwood, T.W.; Frey, E.N.; Askwith, C.C. Structure and activity of the acid-sensing ion channels. Am. J. Physiol. Cell Physiol. 2012, 303, C699–C710. [Google Scholar] [CrossRef]
- Kazmierczak, M.; Zhang, X.; Chen, B.; Mulkey, D.K.; Shi, Y.; Wagner, P.G.; Pivaroff-Ward, K.; Sassic, J.K.; Bayliss, D.A.; Jegla, T. External pH modulates EAG superfamily K+ channels through EAG-specific acidic residues in the voltage sensor. J. Gen. Physiol. 2013, 141, 721–735. [Google Scholar] [CrossRef]
- Chaves, G.; Bungert-Plumke, S.; Franzen, A.; Mahorivska, I.; Musset, B. Zinc modulation of proton currents in a new voltage-gated proton channel suggests a mechanism of inhibition. FEBS J. 2020. [Google Scholar] [CrossRef]
- Frankenhaeuser, B.; Hodgkin, A.L. The action of calcium on the electrical properties of squid axons. J. Physiol. 1957, 137, 218–244. [Google Scholar] [CrossRef]
- Hille, B. Ion Channels of Excitable Membranes, 3rd ed.; Oxford University Press: Sunderland, MA, USA, 2001; ISBN 0878933212 9780878933211. [Google Scholar]
- Deutsch, C.; Lee, S.C. Modulation of K+ currents in human lymphocytes by pH. J. Physiol. 1989, 413, 399–413. [Google Scholar] [CrossRef]
- Kehl, S.J.; Eduljee, C.; Kwan, D.C.; Zhang, S.; Fedida, D. Molecular determinants of the inhibition of human Kv1.5 potassium currents by external protons and Zn2+. J. Physiol. 2002, 541, 9–24. [Google Scholar] [CrossRef]
- Hille, B. Charges and potentials at the nerve surface: Divalent ions and pH. J. Gen. Physiol. 1968, 51, 221–236. [Google Scholar] [CrossRef]
- Becchetti, A.; Pillozzi, S.; Morini, R.; Nesti, E.; Arcangeli, A. New insights into the regulation of ion channels by integrins. In International Review of Cell and Molecular Biology; Elsevier: Amsterdam, The Netherlands, 2010; pp. 135–190. ISBN 1937-6448. [Google Scholar]
- Liddington, R.C.; Ginsberg, M.H. Integrin activation takes shape. J. Cell Biol. 2002, 158, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Tadokoro, S.; Shattil, S.J.; Eto, K.; Tai, V.; Liddington, R.C.; de Pereda, J.M.; Ginsberg, M.H.; Calderwood, D.A. Talin binding to integrin ß tails: A final common step in integrin activation. Science 2003, 302, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Critchley, D.R.; Gingras, A.R. Talin at a glance. J. Cell Sci. 2008, 121, 1345–1347. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, M.H.; Partridge, A.; Shattil, S.J. Integrin regulation. Curr. Opin. Cell Biol. 2005, 17, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Krähling, H.; Mally, S.; Eble, J.A.; Noël, J.; Schwab, A.; Stock, C. The glycocalyx maintains a cell surface pH nanoenvironment crucial for integrin-mediated migration of human melanoma cells. Pflügers Arch. Eur. J. Physiol. 2009, 458, 1069–1083. [Google Scholar]
- Lehenkari, P.P.; Horton, M.A. Single integrin molecule adhesion forces in intact cells measured by atomic force microscopy. Biochem. Biophys. Res. Commun. 1999, 259, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Paradise, R.K.; Lauffenburger, D.A.; van Vliet, K.J. Acidic extracellular pH promotes activation of integrin αvβ3. PLoS ONE 2011, 6, e15746. [Google Scholar] [CrossRef]
- Srivastava, J.; Barreiro, G.; Groscurth, S.; Gingras, A.R.; Goult, B.T.; Critchley; Kelly, M.J.S.; Jacobson, M.P.; Barber, D.L. Structural model and functional significance of pH-dependent talin–actin binding for focal adhesion remodeling. Proc. Natl. Acad. Sci. USA 2008, 105, 14436–14441. [Google Scholar] [CrossRef]
- Choi, C.H.; Webb, B.A.; Chimenti, M.S.; Jacobson, M.P.; Barber, D.L. pH sensing by FAK-His58 regulates focal adhesion remodeling. J. Cell Biol. 2013, 202, 849–859. [Google Scholar] [CrossRef]
- Stock, C.; Schwab, A. Ion channels and transporters in metastasis. Biochim. Biophys. Acta (BBA)-Biomembr. 2015, 1848, 2638–2646. [Google Scholar] [CrossRef] [PubMed]
- Stock, C.; Gassner, B.; Hauck, C.R.; Arnold, H.; Mally, S.; Eble, J.A.; Dieterich, P.; Schwab, A. Migration of human melanoma cells depends on extracellular pH and Na+/H+ exchange. J. Physiol. 2005, 567, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Beaumont, K.G.; Mrksich, M. The mechanostability of isolated focal adhesions is strongly dependent on pH. Chem. Biol. 2012, 19, 711–720. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Arcangeli, A.; Becchetti, A. Complex functional interaction between integrin receptors and ion channels. Trends Cell Biol. 2006, 16, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Becchetti, A.; Arcangeli, A. Integrins and Ion Channels. Molecular Complexes and Signaling; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2010; ISBN 144196066X. [Google Scholar]
- Tanner, M.R.; Pennington, M.W.; Laragione, T.; Gulko, P.S.; Beeton, C. KCa1.1 channels regulate beta1-integrin function and cell adhesion in rheumatoid arthritis fibroblast-like synoviocytes. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2017, 31, 3309–3320. [Google Scholar] [CrossRef]
- Cahalan, M.D.; Chandy, K.G. The functional network of ion channels in T lymphocytes. Immunol. Rev. 2009, 231, 59–87. [Google Scholar] [CrossRef] [PubMed]
- Katsumi, A.; Orr, A.W.; Tzima, E.; Schwartz, M.A. Integrins in mechanotransduction. J. Biol. Chem. 2004, 279, 12001–12004. [Google Scholar] [CrossRef]
- Arcangeli, A.; Becchetti, A.; Mannini, A.; Mugnai, G.; de Filippi, P.; Tarone, G.; Del Bene, M.R.; Barletta, E.; Wanke, E.; Olivotto, M. Integrin-mediated neurite outgrowth in neuroblastoma cells depends on the activation of potassium channels. J. Cell Biol. 1993, 122, 1131–1143. [Google Scholar] [CrossRef]
- Becchetti, A.; Arcangeli, A.; Del Bene, M.R.; Olivotto, M.; Wanke, E. Response to fibronectin–integrin interaction in leukaemia cells: Delayed enhancing of a K+ current. Proc. R. Soc. Lond. Ser. B Biol. Sci. 1992, 248, 235–240. [Google Scholar]
- Hofmann, G.; Bernabei, P.A.; Crociani, O.; Cherubini, A.; Guasti, L.; Pillozzi, S.; Lastraioli, E.; Polvani, S.; Bartolozzi, B.; Solazzo, V. HERG K+ channels activation during b1 integrin-mediated adhesion to fibronectin induces an up regulation of avß3 integrin in the preosteoclastic leukemia cell line FLG 29.1. J. Biol. Chem. 2000, 276, 4923–4931. [Google Scholar] [CrossRef]
- Rezzonico, R.; Cayatte, C.; Bourget-Ponzio, I.; Romey, G.; Belhacene, N.; Loubat, A.; Rocchi, S.; van Obberghen, E.; Girault, J.-A.; Rossi, B. Focal adhesion kinase pp125FAK interacts with the large conductance calcium-activated hSlo potassium channel in human osteoblasts: Potential role in mechanotransduction. J. Bone Miner. Res. 2003, 18, 1863–1871. [Google Scholar] [CrossRef]
- Wei, J.-F.; Wei, L.; Zhou, X.; Lu, Z.-Y.; Francis, K.; Hu, X.-Y.; Liu, Y.; Xiong, W.-C.; Zhang, X.; Banik, N.L. Formation of Kv2.1-FAK complex as a mechanism of FAK activation, cell polarization and enhanced motility. J. Cell. Physiol. 2008, 217, 544–557. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Chen, L.; McClafferty, H.; Sailer, C.A.; Ruth, P.; Knaus, H.-G.; Shipston, M.J. A noncanonical SH3 domain binding motif links BK channels to the actin cytoskeleton via the SH3 adapter cortactin. Faseb J. 2006, 20, 2588–2590. [Google Scholar] [CrossRef] [PubMed]
- Schwab, A.; Nechyporuk-Zloy, V.; Fabian, A.; Stock, C. Cells move when ions and water flow. Pflügers Arch. Eur. J. Physiol. 2007, 453, 421–432. [Google Scholar] [CrossRef]
- Nechyporuk-Zloy, V.; Dieterich, P.; Oberleithner, H.; Stock, C.; Schwab, A. Dynamics of single potassium channel proteins in the plasma membrane of migrating cells. Am. J. Physiol. Cell Physiol. 2008, 294, C1096–C1102. [Google Scholar] [CrossRef][Green Version]
- Du, C.; Zheng, Z.; Li, D.; Chen, L.; Li, N.; Yi, X.; Yang, Y.; Guo, F.; Liu, W.; Xie, X. BKCa promotes growth and metastasis of prostate cancer through facilitating the coupling between αvβ3 integrin and FAK. Oncotarget 2016, 7, 40174. [Google Scholar] [CrossRef] [PubMed]
- Cherubini, A.; Pillozzi, S.; Hofmann, G.; Crociani, O.; Guasti, L.; Lastraioli, E.; Polvani, S.; Masi, A.; Becchetti, A.; Wanke, E. HERG K+ channels and β1 integrins interact through the assembly of a macromolecular complex. Ann. N. Y. Acad. Sci. 2002, 973, 559–561. [Google Scholar] [CrossRef]
- Cherubini, A.; Hofmann, G.; Pillozzi, S.; Guasti, L.; Crociani, O.; Cilia, E.; Di Stefano, P.; Degani, S.; Balzi, M.; Olivotto, M. Human ether-a-go-go-related gene 1 channels are physically linked to β1 integrins and modulate adhesion-dependent signaling. Mol. Biol. Cell 2005, 16, 2972–2983. [Google Scholar] [CrossRef]
- Pillozzi, S.; Masselli, M.; de Lorenzo, E.; Accordi, B.; Cilia, E.; Crociani, O.; Amedei, A.; Veltroni, M.; D’Amico, M.; Basso, G. Chemotherapy resistance in acute lymphoblastic leukemia requires hERG1 channels and is overcome by hERG1 blockers. Blood J. Am. Soc. Hematol. 2011, 117, 902–914. [Google Scholar] [CrossRef]
- Artym, V.V.; Petty, H.R. Molecular proximity of Kv1.3 voltage-gated potassium channels and β1-integrins on the plasma membrane of melanoma cells: Effects of cell adherence and channel blockers. J. Gen. Physiol. 2002, 120, 29–37. [Google Scholar] [CrossRef]
- Levite, M.; Cahalon, L.; Peretz, A.; Hershkoviz, R.; Sobko, A.; Ariel, A.; Desai, R.; Attali, B.; Lider, O. Extracellular K+ and opening of voltage-gated potassium channels activate T cell integrin function: Physical and functional association between Kv1.3 channels and β1 integrins. J. Exp. Med. 2000, 191, 1167–1176. [Google Scholar] [CrossRef]
- Lastraioli, E.; Guasti, L.; Crociani, O.; Polvani, S.; Hofmann, G.; Witchel, H.; Bencini, L.; Calistri, M.; Messerini, L.; Scatizzi, M. HERG1 gene and HERG1 protein are overexpressed in colorectal cancers and regulate cell invasion of tumor cells. Cancer Res. 2004, 64, 606–611. [Google Scholar] [CrossRef] [PubMed]
- Crociani, O.; Zanieri, F.; Pillozzi, S.; Lastraioli, E.; Stefanini, M.; Fiore, A.; Fortunato, A.; D’Amico, M.; Masselli, M.; de Lorenzo, E. hERG1 channels modulate integrin signaling to trigger angiogenesis and tumor progression in colorectal cancer. Sci. Rep. 2013, 3, 3308. [Google Scholar] [CrossRef] [PubMed]
- Masi, A.; Becchetti, A.; Restano-Cassulini, R.; Polvani, S.; Hofmann, G.; Buccoliero, A.M.; Paglierani, M.; Pollo, B.; Taddei, G.L.; Gallina, P. hERG1 channels are overexpressed in glioblastoma multiforme and modulate VEGF secretion in glioblastoma cell lines. Br. J. Cancer 2005, 93, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Doherty, P.; Ashton, S.V.; Moore, S.E.; Walsh, F.S. Morphoregulatory activities of NCAM and N-cadherin can be accounted for by G protein-dependent activation of L-and N-type neuronal Ca2+ channels. Cell 1991, 67, 21–33. [Google Scholar] [CrossRef]
- Aissaoui, D.; Mlayah-Bellalouna, S.; Jebali, J.; Abdelkafi-Koubaa, Z.; Souid, S.; Moslah, W.; Othman, H.; Luis, J.; ElAyeb, M.; Marrakchi, N.; et al. Functional role of Kv1.1 and Kv1.3 channels in the neoplastic progression steps of three cancer cell lines, elucidated by scorpion peptides. Int. J. Biol. Macromol. 2018, 111, 1146–1155. [Google Scholar] [CrossRef] [PubMed]
- Somodi, S.; Varga, Z.; Hajdu, P.; Starkus, J.G.; Levy, D.I.; Gaspar, R.; Panyi, G. pH-dependent modulation of Kv1.3 inactivation: Role of His399. Am. J. Physiol. Cell Physiol. 2004, 287, C1067–C1076. [Google Scholar] [CrossRef]
- Somodi, S.; Hajdu, P.; Gaspar, R.; Panyi, G.; Varga, Z. Effects of changes in extracellular pH and potassium concentration on Kv1.3 inactivation. Eur. Biophys. J. 2008, 37, 1145–1156. [Google Scholar] [CrossRef]
- Jang, S.H.; Kang, K.-S.; Ryu, P.D.; Lee, S.Y. Kv1.3 voltage-gated K+ channel subunit as a potential diagnostic marker and therapeutic target for breast cancer. BMB Rep. 2009, 42, 535–539. [Google Scholar] [CrossRef]
- Eil, R.; Vodnala, S.K.; Clever, D.; Klebanoff, C.A.; Sukumar, M.; Pan, J.H.; Palmer, D.C.; Gros, A.; Yamamoto, T.N.; Patel, S.J.; et al. Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nature 2016, 537, 539–543. [Google Scholar] [CrossRef]
- Chimote, A.A.; Hajdu, P.; Sfyris, A.M.; Gleich, B.N.; Wise-Draper, T.; Casper, K.A.; Conforti, L. Kv1.3 Channels Mark Functionally Competent CD8+ Tumor-Infiltrating Lymphocytes in Head and Neck Cancer. Cancer Res. 2017, 77, 53–61. [Google Scholar] [CrossRef]
- Trapani, J.G.; Korn, S.J. Effect of external pH on activation of the Kv1.5 potassium channel. Biophys. J. 2003, 84, 195–204. [Google Scholar] [CrossRef]
- Xie, J.; Wang, B.S.; Yu, D.H.; Lu, Q.; Ma, J.; Qi, H.; Fang, C.; Chen, H.Z. Dichloroacetate shifts the metabolism from glycolysis to glucose oxidation and exhibits synergistic growth inhibition with cisplatin in HeLa cells. Int. J. Oncol. 2011, 38, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Lan, M.; Shi, Y.; Han, Z.; Hao, Z.; Pan, Y.; Liu, N.; Guo, C.; Hong, L.; Wang, J.; Qiao, T.; et al. Expression of delayed rectifier potassium channels and their possible roles in proliferation of human gastric cancer cells. Cancer Biol. Ther. 2005, 4, 1342–1347. [Google Scholar] [CrossRef]
- Suzuki, T.; Takimoto, K. Selective expression of HERG and Kv2 channels influences proliferation of uterine cancer cells. Int. J. Oncol. 2004, 25, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Fujii, T.; Takahashi, Y.; Takahashi, Y.; Suzuki, T.; Ukai, M.; Tauchi, K.; Horikawa, N.; Tsukada, K.; Sakai, H. Up-regulation of Kv7.1 channels in thromboxane A2-induced colonic cancer cell proliferation. Pflug. Arch. Eur. J. Physiol. 2014, 466, 541–548. [Google Scholar] [CrossRef]
- Yamane, T.; Furukawa, T.; Horikawa, S.; Hiraoka, M. External pH regulates the slowly activating potassium current IsK expressed in Xenopus oocytes. FEBS Lett. 1993, 319, 229–232. [Google Scholar] [CrossRef]
- Unsöld, B.; Kerst, G.; Brousos, H.; Hübner, M.; Schreiber, R.; Nitschke, R.; Greger, R.; Bleich, M. KCNE1 reverses the response of the human K+ channel KCNQ1 to cytosolic pH changes and alters its pharmacology and sensitivity to temperature. Pflügers Arch. Eur. J. Physiol. 2000, 441, 368–378. [Google Scholar] [CrossRef]
- Martínez, R.; Stühmer, W.; Martin, S.; Schell, J.; Reichmann, A.; Rohde, V.; Pardo, L. Analysis of the expression of Kv10.1 potassium channel in patients with brain metastases and glioblastoma multiforme: Impact on survival. BMC Cancer 2015, 15. [Google Scholar] [CrossRef]
- Terlau, H.; Ludwig, J.; Steffan, R.; Pongs, O.; Stühmer, W.; Heinemann, S.H. Extracellular Mg2+ regulates activation of rat eag potassium channel. Pflügers Arch. Eur. J. Physiol. 1996, 432, 301–312. [Google Scholar] [CrossRef]
- Spitzner, M.; Ousingsawat, J.; Scheidt, K.; Kunzelmann, K.; Schreiber, R. Voltage-gated K+ channels support proliferation of colonic carcinoma cells. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2007, 21, 35–44. [Google Scholar] [CrossRef]
- Ding, X.-W.; Luo, H.-s.; Jin, X.; Yan, J.-j.; Ai, Y.-w. Aberrant expression of Eag1 potassium channels in gastric cancer patients and cell lines. Med. Oncol. 2007, 24, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Lai, Q.; Wang, T.; Guo, Q.; Zhang, Y.; Wang, Y.; Yuan, L.; Ling, R.; He, Y.; Wang, W. Positive correlation between the expression of hEag1 and HIF-1α in breast cancers: An observational study. BMJ Open 2014, 4, e005049. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhong, D.; Fu, X.; Liu, Q.; Kang, L.; Ding, Z. Silencing of Ether à go-go 1 by shRNA inhibits osteosarcoma growth and cell cycle progression. Int. J. Mol. Sci. 2014, 15, 5570–5581. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wu, X.; Zhong, D.; Zhai, W.; Ding, Z.; Zhou, Y. Short Hairpin RNA (shRNA) Ether à go-go 1 (Eag1) Inhibition of Human Osteosarcoma Angiogenesis via VEGF/PI3K/AKT Signaling. Int. J. Mol. Sci. 2012, 13, 12573–12583. [Google Scholar] [CrossRef]
- van Slyke, A.C.; Cheng, Y.M.; Mafi, P.; Allard, C.R.; Hull, C.M.; Shi, Y.P.; Claydon, T.W. Proton block of the pore underlies the inhibition of hERG cardiac K+ channels during acidosis. Am. J. Physiol. Cell Physiol. 2012, 302, C1797–C1806. [Google Scholar] [CrossRef]
- Wang, Y.; Guo, J.; Perissinotti, L.L.; Lees-Miller, J.; Teng, G.; Durdagi, S.; Duff, H.J.; Noskov, S.Y. Role of the pH in state-dependent blockade of hERG currents. Sci. Rep. 2016, 6, 32536. [Google Scholar] [CrossRef]
- Lastraioli, E.; Iorio, J.; Arcangeli, A. Ion channel expression as promising cancer biomarker. Biochim. Biophys. Acta 2015, 1848, 2685–2702. [Google Scholar] [CrossRef]
- Klumpp, L.; Sezgin, E.C.; Skardelly, M.; Eckert, F.; Huber, S.M. KCa3.1 Channels and Glioblastoma: In Vitro Studies. Curr. Neuropharmacol. 2018, 16, 627–635. [Google Scholar] [CrossRef]
- Doroszewicz, J.; Waldegger, P.; Jeck, N.; Seyberth, H.; Waldegger, S. pH dependence of extracellular calcium sensing receptor activity determined by a novel technique. Kidney Int. 2005, 67, 187–192. [Google Scholar] [CrossRef]
- Strupp, M.; Staub, F.; Grafe, P. A Ca2+- and pH-dependent K+ channel of rat C6 glioma cells and its possible role in acidosis-induced cell swelling. Glia 1993, 9, 136–145. [Google Scholar] [CrossRef]
- Faouzi, M.; Hague, F.; Geerts, D.; Ay, A.-S.; Potier-Cartereau, M.; Ahidouch, A.; Ouadid-Ahidouch, H. Functional cooperation between KCa3.1 and TRPC1 channels in human breast cancer: Role in cell proliferation and patient prognosis. Oncotarget 2016, 7, 36419–36435. [Google Scholar] [CrossRef] [PubMed]
- Ouadid-Ahidouch, H.; Roudbaraki, M.; Delcourt, P.; Ahidouch, A.; Joury, N.; Prevarskaya, N. Functional and molecular identification of intermediate-conductance Ca2+-activated K+ channels in breast cancer cells: Association with cell cycle progression. Am. J. Physiol. Cell Physiol. 2004, 287, C125–C134. [Google Scholar] [CrossRef] [PubMed]
- Bonito, B.; Sauter, D.R.P.; Schwab, A.; Djamgoz, M.B.A.; Novak, I. KCa3.1 (IK) modulates pancreatic cancer cell migration, invasion and proliferation: Anomalous effects on TRAM-34. Pflug. Arch. Eur. J. Physiol. 2016, 468, 1865–1875. [Google Scholar] [CrossRef] [PubMed]
- Storck, H.; Hild, B.; Schimmelpfennig, S.; Sargin, S.; Nielsen, N.; Zaccagnino, A.; Budde, T.; Novak, I.; Kalthoff, H.; Schwab, A. Ion channels in control of pancreatic stellate cell migration. Oncotarget 2017, 8, 769–784. [Google Scholar] [CrossRef] [PubMed]
- Vodnala, S.K.; Eil, R.; Kishton, R.J.; Sukumar, M.; Yamamoto, T.N.; Ha, N.H.; Lee, P.H.; Shin, M.; Patel, S.J.; Yu, Z.; et al. T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science 2019, 363. [Google Scholar] [CrossRef]
- Chimote, A.A.; Balajthy, A.; Arnold, M.J.; Newton, H.S.; Hajdu, P.; Qualtieri, J.; Wise-Draper, T.; Conforti, L. A defect in KCa3.1 channel activity limits the ability of CD8+ T cells from cancer patients to infiltrate an adenosine-rich microenvironment. Sci. Signal 2018, 11. [Google Scholar] [CrossRef]
- Chimote, A.A.; Gawali, V.S.; Newton, H.S.; Wise-Draper, T.M.; Conforti, L. A Compartmentalized Reduction in Membrane-Proximal Calmodulin Reduces the Immune Surveillance Capabilities of CD8+ T Cells in Head and Neck Cancer. Front. Pharm. 2020, 11, 143. [Google Scholar] [CrossRef]
- Guo, Z.; Liu, J.; Zhang, L.; Su, B.; Xing, Y.; He, Q.; Ci, W.; Li, X.; Zhou, L. KCNJ1 inhibits tumor proliferation and metastasis and is a prognostic factor in clear cell renal cell carcinoma. Tumour Biol. 2015, 36, 1251–1259. [Google Scholar] [CrossRef]
- Schulte, U.; Hahn, H.; Konrad, M.; Jeck, N.; Derst, C.; Wild, K.; Weidemann, S.; Ruppersberg, J.P.; Fakler, B.; Ludwig, J. pH gating of ROMK (Kir1.1) channels: Control by an Arg-Lys-Arg triad disrupted in antenatal Bartter syndrome. Proc. Natl. Acad. Sci. USA 1999, 96, 15298–15303. [Google Scholar] [CrossRef]
- Higashimori, H.; Sontheimer, H. Role of Kir4.1 channels in growth control of glia. Glia 2007, 55, 1668–1679. [Google Scholar] [CrossRef]
- Pessia, M.; Imbrici, P.; D’Adamo, M.C.; Salvatore, L.; Tucker, S.J. Differential pH sensitivity of Kir4.1 and Kir4.2 potassium channels and their modulation by heteropolymerisation with Kir5.1. J. Physiol. 2001, 532, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.M.; Wan, F.N.; Qin, X.J.; Cao, D.L.; Zhang, H.L.; Zhu, Y.; Dai, B.; Shi, G.H.; Ye, D.W. Prognostic significance of the TREK-1 K2P potassium channels in prostate cancer. Oncotarget 2015, 6, 18460–18468. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sandoz, G.; Douguet, D.; Chatelain, F.; Lazdunski, M.; Lesage, F. Extracellular acidification exerts opposite actions on TREK1 and TREK2 potassium channels via a single conserved histidine residue. Proc. Natl. Acad. Sci. USA 2009, 106, 14628–14633. [Google Scholar] [CrossRef] [PubMed]
- Maingret, F.; Patel, A.J.; Lesage, F.; Lazdunski, M.; Honoré, E. Mechano- or acid stimulation, two interactive modes of activation of the TREK-1 potassium channel. J. Biol. Chem. 1999, 274, 26691–26696. [Google Scholar] [CrossRef] [PubMed]
- Sauter, D.R.P.; Sørensen, C.E.; Rapedius, M.; Brüggemann, A.; Novak, I. pH-sensitive K+ channel TREK-1 is a novel target in pancreatic cancer. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 1994–2003. [Google Scholar] [CrossRef] [PubMed]
- Meuth, S.G.; Herrmann, A.M.; Ip, C.W.; Kanyshkova, T.; Bittner, S.; Weishaupt, A.; Budde, T.; Wiendl, H. The two-pore domain potassium channel TASK3 functionally impacts glioma cell death. J. Neuro-Oncol. 2008, 87, 263–270. [Google Scholar] [CrossRef]
- Pei, L.; Wiser, O.; Slavin, A.; Mu, D.; Powers, S.; Lily, Y.J.; Hoey, T. Oncogenic potential of TASK3 (Kcnk9) depends on K+ channel function. Proc. Natl. Acad. Sci. USA 2003, 100, 7803–7807. [Google Scholar] [CrossRef]
- Alvarez-Baron, C.P.; Jonsson, P.; Thomas, C.; Dryer, S.E.; Williams, C. The two-pore domain potassium channel KCNK5: Induction by estrogen receptor α and role in proliferation of breast cancer cells. Mol. Endocrinol. 2011, 25, 1326–1336. [Google Scholar] [CrossRef]
- Niemeyer, M.I.; Cid, L.P.; Gonzalez, W.; Sepulveda, F.V. Gating, Regulation, and Structure in K2P K+ Channels: In Varietate Concordia? Mol. Pharmacol. 2016, 90, 309–317. [Google Scholar] [CrossRef]
- Bustos, D.; Bedoya, M.; Ramírez, D.; Concha, G.; Zúñiga, L.; Decher, N.; Hernández-rodríguez, E.W.; Sepúlveda, F.V.; Martínez, L.; González, W. Elucidating the structural basis of the intracellular ph sensing mechanism of TASK-2 K2P channels. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Ratnayake, R.; Covell, D.; Ransom, T.T.; Gustafson, K.R.; Beutler, J.A. Englerin A, a Selective Inhibitor of Renal Cancer Cell Growth, from Phyllanthus engleri. Org. Lett. 2009, 11, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Semtner, M.; Schaefer, M.; Pinkenburg, O.; Plant, T.D. Potentiation of TRPC5 by protons. J. Biol. Chem. 2007, 282, 33868–33878. [Google Scholar] [CrossRef] [PubMed]
- Chigurupati, S.; Venkataraman, R.; Barrera, D.; Naganathan, A.; Madan, M.; Paul, L.; Pattisapu, J.V.; Kyriazis, G.A.; Sugaya, K.; Bushnev, S.; et al. Receptor channel TRPC6 is a key mediator of Notch-driven glioblastoma growth and invasiveness. Cancer Res. 2010, 70, 418–427. [Google Scholar] [CrossRef]
- El Boustany, C.; Bidaux, G.; Enfissi, A.; Delcourt, P.; Prevarskaya, N.; Capiod, T. Capacitative calcium entry and transient receptor potential canonical 6 expression control human hepatoma cell proliferation. Hepatology 2008, 47, 2068–2077. [Google Scholar] [CrossRef] [PubMed]
- Steele, C.W.; Karim, S.A.; Leach, J.D.G.; Bailey, P.; Upstill-Goddard, R.; Rishi, L.; Foth, M.; Bryson, S.; McDaid, K.; Wilson, Z.; et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell 2016, 29, 832–845. [Google Scholar] [CrossRef]
- Lindemann, O.; Umlauf, D.; Frank, S.; Schimmelpfennig, S.; Bertrand, J.; Pap, T.; Hanley, P.J.; Fabian, A.; Dietrich, A.; Schwab, A. TRPC6 Regulates CXCR2-Mediated Chemotaxis of Murine Neutrophils. J. Immunol. 2013, 190, 5496–5505. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, N.; Kondratska, K.; Ruck, T.; Hild, B.; Kovalenko, I.; Schimmelpfennig, S.; Welzig, J.; Sargin, S.; Lindemann, O.; Christian, S.; et al. TRPC6 channels modulate the response of pancreatic stellate cells to hypoxia. Pflug. Arch. Eur. J. Physiol. 2017, 469, 1567–1577. [Google Scholar] [CrossRef]
- Gershkovitz, M.; Caspi, Y.; Fainsod-Levi, T.; Katz, B.; Michaeli, J.; Khawaled, S.; Lev, S.; Polyansky, L.; Shaul, M.E.; Sionov, R.V.; et al. TRPM2 Mediates Neutrophil Killing of Disseminated Tumor Cells. Cancer Res. 2018, 78, 2680–2690. [Google Scholar] [CrossRef]
- Gershkovitz, M.; Fainsod-Levi, T.; Zelter, T.; Sionov, R.V.; Granot, Z. TRPM2 modulates neutrophil attraction to murine tumor cells by regulating CXCL2 expression. Cancer Immunol. Immunother. 2019, 68, 33–43. [Google Scholar] [CrossRef]
- Starkus, J.G.; Fleig, A.; Penner, R. The calcium-permeable non-selective cation channel TRPM2 is modulated by cellular acidification. J. Physiol. 2010, 588, 1227–1240. [Google Scholar] [CrossRef]
- Zeng, X.; Sikka, S.C.; Huang, L.; Sun, C.; Xu, C.; Jia, D.; Abdel-Mageed, A.B.; Pottle, J.E.; Taylor, J.T.; Li, M. Novel role for the transient receptor potential channel TRPM2 in prostate cancer cell proliferation. Prostate Cancer Prostatic Dis. 2010, 13, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-P.; Luan, Y.; You, C.-X.; Chen, X.-H.; Luo, R.-C.; Li, R. TRPM7 regulates the migration of human nasopharyngeal carcinoma cell by mediating Ca2+ influx. Cell Calcium 2010, 47, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Chokshi, R.; Matsushita, M.; Kozak, J.A. Detailed examination of Mg2+ and pH sensitivity of human TRPM7 channels. Am. J. Physiol. Cell Physiol. 2012, 302, C1004–C1011. [Google Scholar] [CrossRef]
- Gao, H.; Chen, X.; Du, X.; Guan, B.; Liu, Y.; Zhang, H. EGF enhances the migration of cancer cells by up-regulation of TRPM7. Cell Calcium 2011, 50, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Rybarczyk, P.; Gautier, M.; Hague, F.; Dhennin-Duthille, I.; Chatelain, D.; Kerr-Conte, J.; Pattou, F.; Regimbeau, J.-M.; Sevestre, H.; Ouadid-Ahidouch, H. Transient receptor potential melastatin-related 7 channel is overexpressed in human pancreatic ductal adenocarcinomas and regulates human pancreatic cancer cell migration. Int. J. Cancer 2012, 131, E851–E861. [Google Scholar] [CrossRef]
- Guilbert, A.; Gautier, M.; Dhennin-Duthille, I.; Haren, N.; Sevestre, H.; Ouadid-Ahidouch, H. Evidence that TRPM7 is required for breast cancer cell proliferation. Am. J. Physiol. Cell Physiol. 2009, 297, C493–C502. [Google Scholar] [CrossRef]
- Weber, L.V.; Al-Refae, K.; Wölk, G.; Bonatz, G.; Altmüller, J.; Becker, C.; Gisselmann, G.; Hatt, H. Expression and functionality of TRPV1 in breast cancer cells. Breast Cancer Targets Ther. 2016, 8, 243–252. [Google Scholar] [CrossRef]
- Caterina, M.J.; Schumacher, M.A.; Tominaga, M.; Rosen, T.A.; Levine, J.D.; Julius, D. The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature 1997, 389, 816–824. [Google Scholar] [CrossRef]
- Hou, N.; He, X.; Yang, Y.; Fu, J.; Zhang, W.; Guo, Z.; Hu, Y.; Liang, L.; Xie, W.; Xiong, H.; et al. TRPV1 Induced Apoptosis of Colorectal Cancer Cells by Activating Calcineurin-NFAT2-p53 Signaling Pathway. Biomed Res. Int. 2019, 2019, 1–8. [Google Scholar] [CrossRef]
- Stock, K.; Kumar, J.; Synowitz, M.; Petrosino, S.; Imperatore, R.; Smith, E.S.J.; Wend, P.; Purfürst, B.; Nuber, U.A.; Gurok, U.; et al. Neural precursor cells induce cell death of high-grade astrocytomas through stimulation of TRPV1. Nat. Med. 2012, 18, 1232–1238. [Google Scholar] [CrossRef]
- Fonseca, B.M.; Correia-da-Silva, G.; Teixeira, N.A. Cannabinoid-induced cell death in endometrial cancer cells: Involvement of TRPV1 receptors in apoptosis. J. Physiol. Biochem. 2018, 74, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Thoppil, R.J.; Cappelli, H.C.; Adapala, R.K.; Kanugula, A.K.; Paruchuri, S.; Thodeti, C.K. TRPV4 channels regulate tumor angiogenesis via modulation of Rho/Rho kinase pathway. Oncotarget 2016, 7, 25849–25861. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Mizuno, A.; Kodaira, K.; Imai, M. Impaired pressure sensation in mice lacking TRPV4. J. Biol. Chem. 2003, 278, 22664–22668. [Google Scholar] [CrossRef] [PubMed]
- Fiorio Pla, A.; Ong, H.L.; Cheng, K.T.; Brossa, A.; Bussolati, B.; Lockwich, T.; Paria, B.; Munaron, L.; Ambudkar, I.S. TRPV4 mediates tumor-derived endothelial cell migration via arachidonic acid-activated actin remodeling. Oncogene 2012, 31, 200–212. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Liu, G.; Xie, C.; Qian, K.; Lei, X.; Liu, Q.; Liu, G.; Cao, Z.; Fu, J.; Du, H.; et al. Pharmacological inhibition of TRPV4 channel suppresses malignant biological behavior of hepatocellular carcinoma via modulation of ERK signaling pathway. Biomed. Pharmacother. 2018, 101, 910–919. [Google Scholar] [CrossRef]
- Xie, R.; Xu, J.; Xiao, Y.; Wu, J.; Wan, H.; Tang, B.; Liu, J.; Fan, Y.; Wang, S.; Wu, Y.; et al. Calcium Promotes Human Gastric Cancer via a Novel Coupling of Calcium-Sensing Receptor and TRPV4 Channel. Cancer Res. 2017, 77, 6499–6512. [Google Scholar] [CrossRef]
- Bare, D.J.; Cherny, V.V.; DeCoursey, T.E.; Abukhdeir, A.M.; Morgan, D. Expression and function of voltage gated proton channels (Hv1) in MDA-MB-231 cells. PLoS ONE 2020, 15, e0227522. [Google Scholar] [CrossRef]
- Wang, Y.; Li, S.J.; Wu, X.; Che, Y.; Li, Q. Clinicopathological and biological significance of human voltage-gated proton channel Hv1 protein overexpression in breast cancer. J. Biol. Chem. 2012, 287, 13877–13888. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, X.; Li, Q.; Zhang, S.; Li, S.J. Human voltage-gated proton channel hv1: A new potential biomarker for diagnosis and prognosis of colorectal cancer. PLoS ONE 2013, 8, e70550. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, S.; Li, S.J. Zn2+ induces apoptosis in human highly metastatic SHG-44 glioma cells, through inhibiting activity of the voltage-gated proton channel Hv1. Biochem. Biophys. Res. Commun. 2013, 438, 312–317. [Google Scholar] [CrossRef]
- Ribeiro-Silva, L.; Queiroz, F.O.; da Silva, A.M.; Hirata, A.E.; Arcisio-Miranda, M. Voltage-Gated Proton Channel in Human Glioblastoma Multiforme Cells. ACS Chem. Neurosci. 2016, 7, 864–869. [Google Scholar] [CrossRef] [PubMed]
- Hondares, E.; Brown, M.A.; Musset, B.; Morgan, D.; Cherny, V.V.; Taubert, C.; Bhamrah, M.K.; Coe, D.; Marelli-Berg, F.; Gribben, J.G.; et al. Enhanced activation of an amino-terminally truncated isoform of the voltage-gated proton channel HVCN1 enriched in malignant B cells. Proc. Natl. Acad. Sci. USA 2014, 111, 18078–18083. [Google Scholar] [CrossRef] [PubMed]
- Gavriliouk, D.; Scrimgeour, N.R.; Grigoryev, S.; Ma, L.; Zhou, F.H.; Barritt, G.J.; Rychkov, G.Y. Regulation of Orai1/STIM1 mediated ICRAC by intracellular pH. Sci. Rep. 2017, 7, 9829. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; Lkhagvadorj, S.; Lee, M.-R.; Hwang, K.-H.; Chung, H.C.; Jung, J.H.; Cha, S.-K.; Eom, M. Orai1 and STIM1 are critical for cell migration and proliferation of clear cell renal cell carcinoma. Biochem. Biophys. Res. Commun. 2014, 448, 76–82. [Google Scholar] [CrossRef]
- Tsujikawa, H.; Yu, A.S.; Xie, J.; Yue, Z.; Yang, W.; He, Y.; Yue, L. Identification of key amino acid residues responsible for internal and external pH sensitivity of Orai1/STIM1 channels. Sci. Rep. 2015, 5, 16747. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, J.J.; Huang, X.-Y. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell 2009, 15, 124–134. [Google Scholar] [CrossRef]
- Zhu, H.; Zhang, H.; Jin, F.; Fang, M.; Huang, M.; Yang, C.S.; Chen, T.; Fu, L.; Pan, Z. Elevated Orai1 expression mediates tumor-promoting intracellular Ca2+ oscillations in human esophageal squamous cell carcinoma. Oncotarget 2014, 5, 3455–3471. [Google Scholar] [CrossRef]
- Zhu, M.; Chen, L.; Zhao, P.; Zhou, H.; Zhang, C.; Yu, S.; Lin, Y.; Yang, X. Store-operated Ca2+ entry regulates glioma cell migration and invasion via modulation of Pyk2 phosphorylation. J. Exp. Clin. Cancer Res. 2014, 33, 98. [Google Scholar] [CrossRef]
- Motiani, R.K.; Hyzinski-García, M.C.; Zhang, X.; Henkel, M.M.; Abdullaev, I.F.; Kuo, Y.-H.; Matrougui, K.; Mongin, A.A.; Trebak, M. STIM1 and Orai1 mediate CRAC channel activity and are essential for human glioblastoma invasion. Pflug. Arch. Eur. J. Physiol. 2013, 465, 1249–1260. [Google Scholar] [CrossRef]
- Zhan, Z.-Y.; Zhong, L.-X.; Feng, M.; Wang, J.-F.; Liu, D.-B.; Xiong, J.-P. Over-expression of Orai1 mediates cell proliferation and associates with poor prognosis in human non-small cell lung carcinoma. Int. J. Clin. Exp. Pathol. 2015, 8, 5080–5088. [Google Scholar]
- Umemura, M.; Baljinnyam, E.; Feske, S.; de Lorenzo, M.S.; Xie, L.-H.; Feng, X.; Oda, K.; Makino, A.; Fujita, T.; Yokoyama, U.; et al. Store-operated Ca2+ entry (SOCE) regulates melanoma proliferation and cell migration. PLoS ONE 2014, 9, e89292. [Google Scholar] [CrossRef] [PubMed]
- Kondratska, K.; Kondratskyi, A.; Yassine, M.; Lemonnier, L.; Lepage, G.; Morabito, A.; Skryma, R.; Prevarskaya, N. Orai1 and STIM1 mediate SOCE and contribute to apoptotic resistance of pancreatic adenocarcinoma. Biochim. Biophys. Acta 2014, 1843, 2263–2269. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Wang, H.; Huang, H.; Sun, L.; Dong, S.; Huang, N.; Shi, M.; Bin, J.; Liao, Y.; Liao, W. Elevated Orai1 and STIM1 expressions upregulate MACC1 expression to promote tumor cell proliferation, metabolism, migration, and invasion in human gastric cancer. Cancer Lett. 2016, 381, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Diez-Bello, R.; Jardin, I.; Salido, G.M.; Rosado, J.A. Orai1 and Orai2 mediate store-operated calcium entry that regulates HL60 cell migration and FAK phosphorylation. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2017, 1864, 1064–1070. [Google Scholar] [CrossRef]
- Wu, Z.; Qing, J.; Xia, Y.; Wang, K.; Zhang, F. Suppression of stromal interaction molecule 1 inhibits SMMC7721 hepatocellular carcinoma cell proliferation by inducing cell cycle arrest. Biotechnol. Appl. Biochem. 2015, 62, 107–111. [Google Scholar] [CrossRef]
- Gui, L.; Wang, Z.; Han, J.; Ma, H.; Li, Z. High Expression of Orai1 Enhances Cell Proliferation and is Associated with Poor Prognosis in Human Colorectal Cancer. Clin. Lab. 2016, 62, 1689–1698. [Google Scholar] [CrossRef]
- Weidinger, C.; Shaw, P.J.; Feske, S. STIM1 and STIM2-mediated Ca2+ influx regulates antitumour immunity by CD8(+) T cells. EMBO Mol. Med. 2013, 5, 1311–1321. [Google Scholar] [CrossRef]
- Zhou, X.; Friedmann, K.S.; Lyrmann, H.; Zhou, Y.; Schoppmeyer, R.; Knorck, A.; Mang, S.; Hoxha, C.; Angenendt, A.; Backes, C.S.; et al. A calcium optimum for cytotoxic T lymphocyte and natural killer cell cytotoxicity. J. Physiol. 2018, 596, 2681–2698. [Google Scholar] [CrossRef]
- Motiani, R.K.; Abdullaev, I.F.; Trebak, M. A Novel Native Store-operated Calcium Channel Encoded by Orai3: Selective requirement of Orai3 versus Orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells. J. Biol. Chem. 2010, 285, 19173–19183. [Google Scholar] [CrossRef]
- Holzmann, C.; Kilch, T.; Kappel, S.; Armbrüster, A.; Jung, V.; Stöckle, M.; Bogeski, I.; Schwarz, E.C.; Peinelt, C. ICRAC controls the rapid androgen response in human primary prostate epithelial cells and is altered in prostate cancer. Oncotarget 2013, 4, 2096–2107. [Google Scholar] [CrossRef]
- Brackenbury, W.J.; Chioni, A.M.; Diss, J.K.; Djamgoz, M.B. The neonatal splice variant of NaV1.5 potentiates in vitro invasive behaviour of MDA-MB-231 human breast cancer cells. Breast Cancer Res. Treat. 2007, 101, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; James, A.D.; Suman, R.; Kasprowicz, R.; Nelson, M.; O’Toole, P.J.; Brackenbury, W.J. Voltage-dependent activation of Rac1 by Nav1.5 channels promotes cell migration. J. Cell Physiol. 2020, 235, 3950–3972. [Google Scholar] [CrossRef]
- Vilin, Y.Y.; Peters, C.H.; Ruben, P.C. Acidosis differentially modulates inactivation in NaV1.2, NaV1.4, and NaV1.5 channels. Front. Pharmacol. 2012, 3, 109. [Google Scholar] [CrossRef]
- Yang, X.-N.; Lu, Y.-P.; Liu, J.-J.; Huang, J.-K.; Liu, Y.-P.; Xiao, C.-X.; Jazag, A.; Ren, J.-L.; Guleng, B. Piezo1 is as a novel trefoil factor family 1 binding protein that promotes gastric cancer cell mobility in vitro. Dig. Dis. Sci. 2014, 59, 1428–1435. [Google Scholar] [CrossRef]
- Bae, C.; Sachs, F.; Gottlieb, P.A. Protonation of the human PIEZO1 ion channel stabilizes inactivation. J. Biol. Chem. 2015, 290, 5167–5173. [Google Scholar] [CrossRef] [PubMed]
- Kuntze, A.; Goetsch, O.; Fels, B.; Najder, K.; Unger, A.; Wilhelmi, M.; Sargin, S.; Schimmelpfennig, S.; Neumann, I.; Schwab, A.; et al. Protonation of Piezo1 Impairs Cell-Matrix Interactions of Pancreatic Stellate Cells. Front. Physiol. 2020, 11, 89. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Muraki, Y.; Hatano, N.; Suzuki, H.; Muraki, K. PIEZO1 Channel Is a Potential Regulator of Synovial Sarcoma Cell-Viability. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wanggou, S.; Bodalia, A.; Zhu, M.; Dong, W.; Fan, J.J.; Yin, W.C.; Min, H.-K.; Hu, M.; Draghici, D.; et al. A Feedforward Mechanism Mediated by Mechanosensitive Ion Channel PIEZO1 and Tissue Mechanics Promotes Glioma Aggression. Neuron 2018, 100, 799–815. [Google Scholar] [CrossRef]
- Li, C.; Rezania, S.; Kammerer, S.; Sokolowski, A.; Devaney, T.; Gorischek, A.; Jahn, S.; Hackl, H.; Groschner, K.; Windpassinger, C.; et al. Piezo1 forms mechanosensitive ion channels in the human MCF-7 breast cancer cell line. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef]
- Han, Y.; Liu, C.; Zhang, D.; Men, H.; Huo, L.; Geng, Q.; Wang, S.; Gao, Y.; Zhang, W.; Zhang, Y.; et al. Mechanosensitive ion channel Piezo1 promotes prostate cancer development through the activation of the Akt/mTOR pathway and acceleration of cell cycle. Int. J. Oncol. 2019, 55, 629–644. [Google Scholar] [CrossRef]
- Zhu, S.; Zhou, H.-Y.; Deng, S.-C.; Deng, S.-J.; He, C.; Li, X.; Chen, J.-Y.; Jin, Y.; Hu, Z.-L.; Wang, F.; et al. ASIC1 and ASIC3 contribute to acidity-induced EMT of pancreatic cancer through activating Ca2+/RhoA pathway. Cell Death Dis. 2017, 8, e2806. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.-h.; Song, J.-w.; Li, W.; Liu, X.; Cao, L.; Wan, L.-m.; Tan, Y.-x.; Ji, S.-p.; Liang, Y.-m.; Gong, F. The acid-sensing ion channel, ASIC2, promotes invasion and metastasis of colorectal cancer under acidosis by activating the calcineurin/NFAT1 axis. J. Exp. Clin. Cancer Res. 2017, 36. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Gao, B.; Xiong, Q.-J.; Wang, Y.-C.; Huang, D.-K.; Wu, W.-N. Acid-sensing ion channels contribute to the effect of extracellular acidosis on proliferation and migration of A549 cells. Tumor Biol. 2017, 39, 101042831770575. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, E.; Raffaghello, L.; Giuliani, A.L.; Cavazzini, L.; Capece, M.; Chiozzi, P.; Bianchi, G.; Kroemer, G.; Pistoia, V.; Di Virgilio, F. Expression of P2X7 Receptor Increases In Vivo Tumor Growth. Cancer Res. 2012, 72, 2957–2969. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ma, W.; Surprenant, A.; Jiang, L.-H. Identification of the amino acid residues in the extracellular domain of rat P2X7 receptor involved in functional inhibition by acidic pH. Br. J. Pharmacol. 2009, 156, 135–142. [Google Scholar] [CrossRef][Green Version]
- Wang, Q.; Wang, L.; Feng, Y.-H.; Li, X.; Zeng, R.; Gorodeski, G.I. P2X7 receptor-mediated apoptosis of human cervical epithelial cells. Am. J. Physiol. Cell Physiol. 2004, 287, C1349–C1358. [Google Scholar] [CrossRef]
- Giannuzzo, A.; Pedersen, S.F.; Novak, I. The P2X7 receptor regulates cell survival, migration and invasion of pancreatic ductal adenocarcinoma cells. Mol. Cancer 2015, 14, 203. [Google Scholar] [CrossRef]
- Bianchi, G.; Vuerich, M.; Pellegatti, P.; Marimpietri, D.; Emionite, L.; Marigo, I.; Bronte, V.; Di Virgilio, F.; Pistoia, V.; Raffaghello, L. ATP/P2X7 axis modulates myeloid-derived suppressor cell functions in neuroblastoma microenvironment. Cell Death Dis. 2014, 5, e1135. [Google Scholar] [CrossRef]
- Starkus, J.G.; Varga, Z.; Schonherr, R.; Heinemann, S.H. Mechanisms of the inhibition of Shaker potassium channels by protons. Pflug. Arch. Eur. J. Physiol. 2003, 447, 44–54. [Google Scholar] [CrossRef]
- Ahn, D.S.; Hume, J.R. pH regulation of voltage-dependent K+ channels in canine pulmonary arterial smooth muscle cells. Pflug. Arch. Eur. J. Physiol. 1997, 433, 758–765. [Google Scholar] [CrossRef]
- Starace, D.M.; Bezanilla, F. A proton pore in a potassium channel voltage sensor reveals a focused electric field. Nature 2004, 427, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Shorthouse, D.; Rahrmann, E.; Kosmidou, C.; Greenwood, B.; Hall, M.; Devonshire, G.; Gilbertson, R.; Fitzgerald, R.C.; Hall, B.A. KCNQ gene family members act as both tumor suppressors and oncogenes in gastrointestinal cancers. bioRxiv 2020. [Google Scholar] [CrossRef]
- Mohr, C.J.; Steudel, F.A.; Gross, D.; Ruth, P.; Lo, W.Y.; Hoppe, R.; Schroth, W.; Brauch, H.; Huber, S.M.; Lukowski, R. Cancer-Associated Intermediate Conductance Ca2+-Activated K+ Channel KCa3.1. Cancers 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.L.; Ikeuchi, M.; Fujimoto, W.Y. Lowering of pHi inhibits Ca2+-activated K+ channels in pancreatic B-cells. Nature 1984, 311, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, K.A.; Jorgensen, N.K.; Jensen, B.S.; Olesen, S.P. Inhibition of the human intermediate-conductance, Ca2+-activated K+ channel by intracellular acidification. Pflug. Arch. Eur. J. Physiol. 2000, 440, 153–156. [Google Scholar] [CrossRef]
- Kume, H.; Takagi, K.; Satake, T.; Tokuno, H.; Tomita, T. Effects of intracellular pH on calcium-activated potassium channels in rabbit tracheal smooth muscle. J. Physiol. 1990, 424, 445–457. [Google Scholar] [CrossRef]
- Christensen, O.; Zeuthen, T. Maxi K+ channels in leaky epithelia are regulated by intracellular Ca2+, pH and membrane potential. Pflug. Arch. Eur. J. Physiol. 1987, 408, 249–259. [Google Scholar] [CrossRef]
- Paynter, J.J.; Shang, L.; Bollepalli, M.K.; Baukrowitz, T.; Tucker, S.J. Random mutagenesis screening indicates the absence of a separate H+-sensor in the pH-sensitive Kir channels. Channels 2010, 4, 390–397. [Google Scholar] [CrossRef][Green Version]
- Qu, Z.; Zhu, G.; Yang, Z.; Cui, N.; Li, Y.; Chanchevalap, S.; Sulaiman, S.; Haynie, H.; Jiang, C. Identification of a critical motif responsible for gating of Kir2.3 channel by intracellular protons. J. Biol. Chem. 1999, 274, 13783–13789. [Google Scholar] [CrossRef]
- Olsen, M.L.; Sontheimer, H. Mislocalization of Kir channels in malignant glia. Glia 2004, 46, 63–73. [Google Scholar] [CrossRef]
- Lee, I.; Park, C.; Kang, W.K. Knockdown of inwardly rectifying potassium channel Kir2.2 suppresses tumorigenesis by inducing reactive oxygen species-mediated cellular senescence. Mol. Cancer Ther. 2010, 9, 2951–2959. [Google Scholar] [CrossRef] [PubMed]
- Tan, G.; Sun, S.Q.; Yuan, D.L. Expression of Kir 4.1 in human astrocytic tumors: Correlation with pathologic grade. Biochem. Biophys. Res. Commun. 2008, 367, 743–747. [Google Scholar] [CrossRef] [PubMed]
- DeHart, G.W.; Jin, T.; McCloskey, D.E.; Pegg, A.E.; Sheppard, D. The α9β1 integrin enhances cell migration by polyamine-mediated modulation of an inward-rectifier potassium channel. Proc. Natl. Acad. Sci. USA 2008, 105, 7188–7193. [Google Scholar] [CrossRef]
- Williams, S.; Bateman, A.; O’Kelly, I. Altered Expression of Two-Pore Domain Potassium (K2P) Channels in Cancer. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Ghatak, S.; Sikdar, S.K. Lactate modulates the intracellular pH sensitivity of human TREK1 channels. Pflug. Arch. Eur. J. Physiol. 2016, 468, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Honoré, E. The neuronal background K2P channels: Focus on TREK1. Nat. Rev. Neurosci. 2007, 8, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Innamaa, A.; Jackson, L.; Asher, V.; van Schalkwyk, G.; Warren, A.; Keightley, A.; Hay, D.; Bali, A.; Sowter, H.; Khan, R. Expression and effects of modulation of the K2P potassium channels TREK-1 (KCNK2) and TREK-2 (KCNK10) in the normal human ovary and epithelial ovarian cancer. Clin. Transl. Oncol. Off. Publ. Fed. Span. Oncol. Soc. Natl. Cancer Inst. Mex. 2013, 15, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Sepúlveda, F.V.; Cid, L.P.; Teulon, J.; Niemeyer, M.I. Molecular aspects of structure, gating, and physiology of pH-sensitive background K2P and Kir K+-transport channels. Physiol. Rev. 2015, 95, 179–217. [Google Scholar] [CrossRef]
- Patel, A.J.; Lazdunski, M. The 2P-domain K+ channels: Role in apoptosis and tumorigenesis. Pflug. Arch. Eur. J. Physiol. 2004, 448, 261–273. [Google Scholar] [CrossRef]
- Bittner, S.; Budde, T.; Wiendl, H.; Meuth, S.G. From the background to the spotlight: TASK channels in pathological conditions. Brain Pathol. 2010, 20, 999–1009. [Google Scholar] [CrossRef]
- Czirják, G.; Enyedi, P. TASK-3 dominates the background potassium conductance in rat adrenal glomerulosa cells. Mol. Endocrinol. 2002, 16, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, K.; Montell, C. TRP Channels. Annu. Rev. Biochem. 2007, 76, 387–417. [Google Scholar] [CrossRef] [PubMed]
- Glitsch, M. Protons and Ca2+: Ionic Allies in Tumor Progression? Physiology 2011, 26, 252–265. [Google Scholar] [CrossRef]
- Fels, B.; Bulk, E.; Petho, Z.; Schwab, A. The Role of TRP Channels in the Metastatic Cascade. Pharmaceuticals 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Ghilardi, J.R. Selective Blockade of the Capsaicin Receptor TRPV1 Attenuates Bone Cancer Pain. J. Neurosci. 2005, 25, 3126–3131. [Google Scholar] [CrossRef]
- Tominaga, M.; Caterina, M.J.; Malmberg, A.B.; Rosen, T.A.; Gilbert, H.; Skinner, K.; Raumann, B.E.; Basbaum, A.I.; Julius, D. The Cloned Capsaicin Receptor Integrates Multiple Pain-Producing Stimuli. Neuron 1998, 21, 531–543. [Google Scholar] [CrossRef]
- Jordt, S.-E.; Tominaga, M.; Julius, D. Acid potentiation of the capsaicin receptor determined by a key extracellular site. Proc. Natl. Acad. Sci. USA 2000, 97, 8134–8139. [Google Scholar] [CrossRef]
- Aneiros, E.; Cao, L.; Papakosta, M.; Stevens, E.B.; Phillips, S.; Grimm, C. The biophysical and molecular basis of TRPV1 proton gating. EMBO J. 2011, 30, 994–1002. [Google Scholar] [CrossRef]
- Hellwig, N.; Plant, T.D.; Janson, W.; Schafer, M.; Schultz, G.; Schaefer, M. TRPV1 acts as proton channel to induce acidification in nociceptive neurons. J. Biol. Chem. 2004, 279, 34553–34561. [Google Scholar] [CrossRef]
- Capiod, T. Cell proliferation, calcium influx and calcium channels. Biochimie 2011, 93, 2075–2079. [Google Scholar] [CrossRef]
- Stewart, T.A.; Yapa, K.T.D.S.; Monteith, G.R. Altered calcium signaling in cancer cells. Biochim. Biophys. Acta 2015, 1848, 2502–2511. [Google Scholar] [CrossRef] [PubMed]
- Prevarskaya, N.; Ouadid-Ahidouch, H.; Skryma, R.; Shuba, Y. Remodelling of Ca2+ transport in cancer: How it contributes to cancer hallmarks? Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130097. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, M.G.; Sánchez, A.M.; Collado, B.; Malagarie-Cazenave, S.; Olea, N.; Carmena, M.J.; Prieto, J.C.; Díaz-Laviada, I. Expression of the transient receptor potential vanilloid 1 (TRPV1) in LNCaP and PC-3 prostate cancer cells and in human prostate tissue. Eur. J. Pharmacol. 2005, 515, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, B.; Loblaw, D.A.; Nam, R. Capsaicin may slow PSA doubling time: Case report and literature review. Can. Urol. Assoc. J. 2013, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Montcourrier, P.; Silver, I.; Farnoud, R.; Bird, I.; Rochefort, H. Breast cancer cells have a high capacity to acidify extracellular milieu by a dual mechanism. Clin. Exp. Metastasis 1997, 15, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Granlund, K.L.; Tee, S.-S.; Vargas, H.A.; Lyashchenko, S.K.; Reznik, E.; Fine, S.; Laudone, V.; Eastham, J.A.; Touijer, K.A.; Reuter, V.E.; et al. Hyperpolarized MRI of Human Prostate Cancer Reveals Increased Lactate with Tumor Grade Driven by Monocarboxylate Transporter 1. Cell Metab. 2020, 31, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Pecze, L.; Jósvay, K.; Blum, W.; Petrovics, G.; Vizler, C.; Oláh, Z.; Schwaller, B. Activation of endogenous TRPV1 fails to induce overstimulation-based cytotoxicity in breast and prostate cancer cells but not in pain-sensing neurons. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2016, 1863, 2054–2064. [Google Scholar] [CrossRef]
- Dömötör, A.; Peidl, Z.; Vincze, Á.; Hunyady, B.; Szolcsányi, J.; Kereskay, L.; Szekeres, G.; Mózsik, G. Immunohistochemical distribution of vanilloid receptor, calcitonin-gene related peptide and substance P in gastrointestinal mucosa of patients with different gastrointestinal disorders. InflammoPharmacology 2005, 13, 161–177. [Google Scholar] [CrossRef]
- Lazzeri, M.; Vannucchi, M.G.; Spinelli, M.; Bizzoco, E.; Beneforti, P.; Turini, D.; Faussone-Pellegrini, M.-S. Transient Receptor Potential Vanilloid Type 1 (TRPV1) Expression Changes from Normal Urothelium to Transitional Cell Carcinoma of Human Bladder. Eur. Urol. 2005, 48, 691–698. [Google Scholar] [CrossRef]
- Lee, W.H.; Choong, L.Y.; Mon, N.N.; Lu, S.; Lin, Q.; Pang, B.; Yan, B.; Krishna, V.S.R.; Singh, H.; Tan, T.Z.; et al. TRPV4 Regulates Breast Cancer Cell Extravasation, Stiffness and Actin Cortex. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Gaunt, H.J.; Vasudev, N.S.; Beech, D.J. Transient receptor potential canonical 4 and 5 proteins as targets in cancer therapeutics. Eur. Biophys. J. 2016, 45, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-S.; Wen, J.; Yang, F.; Cai, X.-L.; Yang, H.; Luo, K.-J.; Liu, Q.W.; Hu, R.-G.; Xie, X.; Huang, Q.-Y.; et al. High expression of Transient potential receptor C6 correlated with poor prognosis in patients with esophageal squamous cell carcinoma. Med. Oncol. 2013, 30. [Google Scholar] [CrossRef] [PubMed]
- Starkus, J.; Beck, A.; Fleig, A.; Penner, R. Regulation of TRPM2 by Extra- and Intracellular Calcium. J. Gen. Physiol. 2007, 130, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Zou, J.; Xia, R.; Vaal, M.L.; Seymour, V.A.; Luo, J.; Beech, D.J.; Jiang, L.-H. State-dependent Inhibition of TRPM2 Channel by Acidic pH. J. Biol. Chem. 2010, 285, 30411–30418. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Xie, J.; Yue, L. Modulation of TRPM2 by acidic pH and the underlying mechanisms for pH sensitivity. J. Gen. Physiol. 2009, 134, 471–488. [Google Scholar] [CrossRef]
- Schumacker, P.T. Reactive oxygen species in cancer cells: Live by the sword, die by the sword. Cancer Cell 2006, 10, 175–176. [Google Scholar] [CrossRef]
- Miller, B.A. TRPM2 in Cancer. Cell Calcium 2019, 80, 8–17. [Google Scholar] [CrossRef]
- Li, M.; Du, J.; Jiang, J.; Ratzan, W.; Su, L.-T.; Runnels, L.W.; Yue, L. Molecular Determinants of Mg2+ and Ca2+ Permeability and pH Sensitivity in TRPM6 and TRPM7. J. Biol. Chem. 2007, 282, 25817–25830. [Google Scholar] [CrossRef]
- Jiang, J.; Li, M.; Yue, L. Potentiation of TRPM7 Inward Currents by Protons. J. Gen. Physiol. 2005, 126, 137–150. [Google Scholar] [CrossRef]
- Schmitz, C.; Perraud, A.-L.; Johnson, C.O.; Inabe, K.; Smith, M.K.; Penner, R.; Kurosaki, T.; Fleig, A.; Scharenberg, A.M. Regulation of Vertebrate Cellular Mg2+ Homeostasis by TRPM7. Cell 2003, 114, 191–200. [Google Scholar] [CrossRef]
- Zierler, S.; Yao, G.; Zhang, Z.; Kuo, W.C.; Pörzgen, P.; Penner, R.; Horgen, F.D.; Fleig, A. Waixenicin A Inhibits Cell Proliferation through Magnesium-dependent Block of Transient Receptor Potential Melastatin 7 (TRPM7) Channels. J. Biol. Chem. 2011, 286, 39328–39335. [Google Scholar] [CrossRef] [PubMed]
- Macianskiene, R.; Almanaityte, M.; Jekabsone, A.; Mubagwa, K. Modulation of Human Cardiac TRPM7 Current by Extracellular Acidic pH Depends upon Extracellular Concentrations of Divalent Cations. PLoS ONE 2017, 12, e0170923. [Google Scholar] [CrossRef] [PubMed]
- Musset, B.; Decoursey, T. Biophysical properties of the voltage gated proton channel H(V)1. Wiley Interdiscip. Rev. Membr. Transp. Signal 2012, 1, 605–620. [Google Scholar] [CrossRef]
- DeCoursey, T.E. Voltage and pH sensing by the voltage-gated proton channel, HV1. J. R. Soc. Interface 2018, 15. [Google Scholar] [CrossRef]
- DeCoursey, T.E.; Cherny, V.V. Potential, pH, and arachidonate gate hydrogen ion currents in human neutrophils. Biophys. J. 1993, 65, 1590–1598. [Google Scholar] [CrossRef]
- De-la-Rosa, V.; Suarez-Delgado, E.; Rangel-Yescas, G.E.; Islas, L.D. Currents through HV1 channels deplete protons in their vicinity. J. Gen. Physiol. 2016, 147, 127–136. [Google Scholar] [CrossRef]
- Berridge, M.J. Calcium microdomains: Organization and function. Cell Calcium 2006, 40, 405–412. [Google Scholar] [CrossRef]
- Pathak, M.M.; Tran, T.; Hong, L.; Joos, B.; Morris, C.E.; Tombola, F. The Hv1 proton channel responds to mechanical stimuli. J. Gen. Physiol. 2016, 148, 405–418. [Google Scholar] [CrossRef]
- Petho, Z.; Najder, K.; Bulk, E.; Schwab, A. Mechanosensitive ion channels push cancer progression. Cell Calcium 2019, 80, 79–90. [Google Scholar] [CrossRef]
- Stock, C.; Schwab, A. Protons make tumor cells move like clockwork. Pflug. Arch. Eur. J. Physiol. 2009, 458, 981–992. [Google Scholar] [CrossRef]
- Boedtkjer, E.; Bunch, L.; Pedersen, S.F. Physiology, pharmacology and pathophysiology of the pH regulatory transport proteins NHE1 and NBCn1: Similarities, differences, and implications for cancer therapy. Curr. Pharm. Des. 2012, 18, 1345–1371. [Google Scholar] [CrossRef] [PubMed]
- Spugnini, E.P.; Sonveaux, P.; Stock, C.; Perez-Sayans, M.; de Milito, A.; Avnet, S.; Garcia, A.G.; Harguindey, S.; Fais, S. Proton channels and exchangers in cancer. Biochim. Biophys. Acta 2015, 1848, 2715–2726. [Google Scholar] [CrossRef] [PubMed]
- Krysiak, K.; Gomez, F.; White, B.S.; Matlock, M.; Miller, C.A.; Trani, L.; Fronick, C.C.; Fulton, R.S.; Kreisel, F.; Cashen, A.F.; et al. Recurrent somatic mutations affecting B-cell receptor signaling pathway genes in follicular lymphoma. Blood 2017, 129, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Gosselin-Badaroudine, P.; Delemotte, L.; Moreau, A.; Klein, M.L.; Chahine, M. Gating pore currents and the resting state of Nav1.4 voltage sensor domains. Proc. Natl. Acad. Sci. USA 2012, 109, 19250–19255. [Google Scholar] [CrossRef]
- Jones, D.K.; Peters, C.H.; Allard, C.R.; Claydon, T.W.; Ruben, P.C. Proton sensors in the pore domain of the cardiac voltage-gated sodium channel. J. Biol. Chem. 2013, 288, 4782–4791. [Google Scholar] [CrossRef]
- Peters, C.H.; Ghovanloo, M.R.; Gershome, C.; Ruben, P.C. pH Modulation of Voltage-Gated Sodium Channels. Handb. Exp. Pharm. 2018, 246, 147–160. [Google Scholar] [CrossRef]
- Ghovanloo, M.R.; Peters, C.H.; Ruben, P.C. Effects of acidosis on neuronal voltage-gated sodium channels: Nav1.1 and Nav1.3. Channels 2018, 12, 367–377. [Google Scholar] [CrossRef]
- Khan, A.; Kyle, J.W.; Hanck, D.A.; Lipkind, G.M.; Fozzard, H.A. Isoform-dependent interaction of voltage-gated sodium channels with protons. J. Physiol. 2006, 576, 493–501. [Google Scholar] [CrossRef]
- Nonner, W.; Spalding, B.C.; Hille, B. Low intracellular pH and chemical agents slow inactivation gating in sodium channels of muscle. Nature 1980, 284, 360–363. [Google Scholar] [CrossRef]
- House, C.D.; Vaske, C.J.; Schwartz, A.M.; Obias, V.; Frank, B.; Luu, T.; Sarvazyan, N.; Irby, R.; Strausberg, R.L.; Hales, T.G.; et al. Voltage-gated Na+ channel SCN5A is a key regulator of a gene transcriptional network that controls colon cancer invasion. Cancer Res. 2010, 70, 6957–6967. [Google Scholar] [CrossRef]
- Yildirim, S.; Altun, S.; Gumushan, H.; Patel, A.; Djamgoz, M.B.A. Voltage-gated sodium channel activity promotes prostate cancer metastasis in vivo. Cancer Lett. 2012, 323, 58–61. [Google Scholar] [CrossRef]
- Fraser, S.P.; Ozerlat-Gunduz, I.; Brackenbury, W.J.; Fitzgerald, E.M.; Campbell, T.M.; Coombes, R.C.; Djamgoz, M.B. Regulation of voltage-gated sodium channel expression in cancer: Hormones, growth factors and auto-regulation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130105. [Google Scholar] [CrossRef] [PubMed]
- Gillet, L.; Roger, S.; Besson, P.; Lecaille, F.; Gore, J.; Bougnoux, P.; Lalmanach, G.; Le Guennec, J.Y. Voltage-gated Sodium Channel Activity Promotes Cysteine Cathepsin-dependent Invasiveness and Colony Growth of Human Cancer Cells. J. Biol. Chem. 2009, 284, 8680–8691. [Google Scholar] [CrossRef] [PubMed]
- Gradek, F.; Lopez-Charcas, O.; Chadet, S.; Poisson, L.; Ouldamer, L.; Goupille, C.; Jourdan, M.L.; Chevalier, S.; Moussata, D.; Besson, P.; et al. Sodium Channel Nav1.5 Controls Epithelial-to-Mesenchymal Transition and Invasiveness in Breast Cancer Cells Through its Regulation by the Salt-Inducible Kinase-1. Sci. Rep. 2019, 9, 18652. [Google Scholar] [CrossRef] [PubMed]
- Onkal, R.; Fraser, S.; Djamgoz, M. Cationic Modulation of Voltage-Gated Sodium Channel (Nav1.5): Neonatal Versus Adult Splice Variants—1. Monovalent H+ Ions. Bioelectricity 2019, 1, 139–147. [Google Scholar] [CrossRef]
- Guzel, R.M.; Ogmen, K.; Ilieva, K.M.; Fraser, S.P.; Djamgoz, M.B.A. Colorectal cancer invasiveness in vitro: Predominant contribution of neonatal Nav1.5 under normoxia and hypoxia. J. Cell Physiol. 2019, 234, 6582–6593. [Google Scholar] [CrossRef]
- Ou, S.W.; Kameyama, A.; Hao, L.Y.; Horiuchi, M.; Minobe, E.; Wang, W.Y.; Makita, N.; Kameyama, M. Tetrodotoxin-resistant Na+ channels in human neuroblastoma cells are encoded by new variants of Nav1.5/SCN5A. Eur. J. Neurosci. 2005, 22, 793–801. [Google Scholar] [CrossRef]
- Nelson, M.; Yang, M.; Dowle, A.A.; Thomas, J.R.; Brackenbury, W.J. The sodium channel-blocking antiepileptic drug phenytoin inhibits breast tumour growth and metastasis. Mol. Cancer 2015, 14, 13. [Google Scholar] [CrossRef]
- Djamgoz, M.B.A.; Onkal, R. Persistent current blockers of voltage-gated sodium channels: A clinical opportunity for controlling metastatic disease. Recent Pat. Anticancer. Drug Discov. 2013, 8, 66–84. [Google Scholar] [CrossRef]
- Coste, B.; Mathur, J.; Schmidt, M.; Earley, T.J.; Ranade, S.; Petrus, M.J.; Dubin, A.E.; Patapoutian, A. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science 2010, 330, 55–60. [Google Scholar] [CrossRef]
- Ridone, P.; Vassalli, M.; Martinac, B. Piezo1 mechanosensitive channels: What are they and why are they important. Biophys. Rev. 2019, 11, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Beech, D.J.; Xiao, B. Piezo channel mechanisms in health and disease. J. Physiol. 2018, 596, 965–967. [Google Scholar] [CrossRef] [PubMed]
- Boris Martinac; Charles, D. Cox. Mechanosensory Transduction: Focus on Ion Channels. In Reference Module in Life Sciences: Comprehensive Biophysics; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar] [CrossRef]
- Sun, Y.; Li, M.; Liu, G.; Zhang, X.; Zhi, L.; Zhao, J.; Wang, G. The function of Piezo1 in colon cancer metastasis and its potential regulatory mechanism. J. Cancer Res. Clin. Oncol. 2020, 146, 1139–1152. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Ho, K.K.Y.; Tong, Z.; Deng, L.; Liu, A.P. Compressive Stress Enhances Invasive Phenotype of Cancer Cells via Piezo1 Activation. bioRxiv 2019. [Google Scholar] [CrossRef]
- Kellenberger, S.; Schild, L. Epithelial sodium channel/degenerin family of ion channels: A variety of functions for a shared structure. Physiol. Rev. 2002, 82, 735–767. [Google Scholar] [CrossRef]
- Pattison, L.A.; Callejo, G.; St John Smith, E. Evolution of acid nociception: Ion channels and receptors for detecting acid. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2019, 374, 20190291. [Google Scholar] [CrossRef]
- Waldmann, R.; Champigny, G.; Bassilana, F.; Heurteaux, C.; Lazdunski, M. A proton-gated cation channel involved in acid-sensing. Nature 1997, 386, 173–177. [Google Scholar] [CrossRef]
- Vullo, S.; Kellenberger, S. A molecular view of the function and pharmacology of acid-sensing ion channels. Pharmacol. Res. 2020, 154, 104166. [Google Scholar] [CrossRef]
- Stoop, R.; Surprenant, A.; North, R.A. Different Sensitivities to pH of ATP-Induced Currents at Four Cloned P2X Receptors. J. Neurophysiol. 1997, 78, 1837–1840. [Google Scholar] [CrossRef]
- Sekar, P.; Huang, D.-Y.; Chang, S.-F.; Lin, W.-W. Coordinate effects of P2X7 and extracellular acidification in microglial cells. Oncotarget 2018, 9, 12718–12731. [Google Scholar] [CrossRef]
- Giannuzzo, A.; Saccomano, M.; Napp, J.; Ellegaard, M.; Alves, F.; Novak, I. Targeting of the P2X7 receptor in pancreatic cancer and stellate cells. Int. J. Cancer 2016, 139, 2540–2552. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, E.; Capece, M.; Amoroso, F.; Marchi, E.; Franceschini, A. Emerging Roles of P2X Receptors in Cancer. Curr. Med. Chem. 2015, 22, 878–890. [Google Scholar] [CrossRef] [PubMed]
- Engblom, C.; Pfirschke, C.; Pittet, M.J. The role of myeloid cells in cancer therapies. Nat. Rev. Cancer 2016, 16, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 Agonists Alter Tumor Stroma and Show Efficacy Against Pancreatic Carcinoma in Mice and Humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef] [PubMed]
- Vesely, M.D.; Kershaw, M.H.; Schreiber, R.D.; Smyth, M.J. Natural Innate and Adaptive Immunity to Cancer. Annu. Rev. Immunol. 2011, 29, 235–271. [Google Scholar] [CrossRef]
- Najder, K.; Rugi, M.; Lebel, M.; Schröder, J.; Oster, L.; Pethő, Z.; Bulk, E.; Schwab, A. Role of the Intracellular Sodium Homeostasis in Chemotaxis of Activated Murine Neutrophils. Front Immunol 2020. [Google Scholar] [CrossRef]
- Woo, S.-R.; Corrales, L.; Gajewski, T.F. Innate Immune Recognition of Cancer. Annu. Rev. Immunol. 2015, 33, 445–474. [Google Scholar] [CrossRef]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.-X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef]
- Wu, L.; Saxena, S.; Awaji, M.; Singh, R.K. Tumor-Associated Neutrophils in Cancer: Going Pro. Cancers 2019, 11. [Google Scholar] [CrossRef]
- Yamamoto, S.; Shimizu, S.; Kiyonaka, S.; Takahashi, N.; Wajima, T.; Hara, Y.; Negoro, T.; Hiroi, T.; Kiuchi, Y.; Okada, T.; et al. TRPM2-mediated Ca2+ influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat. Med. 2008, 14, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Cao, L.; Liu, X.; Sieracki, N.A.; Di, A.; Wen, X.; Chen, Y.; Taylor, S.; Huang, X.; Tiruppathi, C.; et al. Oxidant Sensing by TRPM2 Inhibits Neutrophil Migration and Mitigates Inflammation. Dev. Cell 2016, 38, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-h.; Rong, M.-y.; Wang, L.; Ren, Z.; Chen, L.-n.; Jia, J.-f.; Li, X.-y.; Wu, Z.-b.; Chen, Z.-n.; Zhu, P. CD147 up-regulates calcium-induced chemotaxis, adhesion ability and invasiveness of human neutrophils via a TRPM-7-mediated mechanism. Rheumatology 2014, 53, 2288–2296. [Google Scholar] [CrossRef] [PubMed]
- Nadolni, W.; Zierler, S. The Channel-Kinase TRPM7 as Novel Regulator of Immune System Homeostasis. Cells 2018, 7, 109. [Google Scholar] [CrossRef] [PubMed]
- Najder, K.; Musset, B.; Lindemann, O.; Bulk, E.; Schwab, A.; Fels, B. The function of TRP channels in neutrophil granulocytes. Pflug. Arch. Eur. J. Physiol. 2018, 470, 1017–1033. [Google Scholar] [CrossRef] [PubMed]
- Musset, B.; Cherny, V.V.; Morgan, D.; DeCoursey, T.E. The intimate and mysterious relationship between proton channels and NADPH oxidase. FEBS Lett. 2009, 583, 7–12. [Google Scholar] [CrossRef]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef]
- Schilling, T.; Miralles, F.; Eder, C. TRPM7 regulates proliferation and polarisation of macrophages. J. Cell Sci. 2014, 127, 4561–4566. [Google Scholar] [CrossRef]
- Beceiro, S.; Radin, J.N.; Chatuvedi, R.; Piazuelo, M.B.; Horvarth, D.J.; Cortado, H.; Gu, Y.; Dixon, B.; Gu, C.; Lange, I.; et al. TRPM2 ion channels regulate macrophage polarization and gastric inflammation during Helicobacter pylori infection. Mucosal Immunol. 2017, 10, 493–507. [Google Scholar] [CrossRef]
- Chauhan, A.; Sun, Y.; Sukumaran, P.; Quenum Zangbede, F.O.; Jondle, C.N.; Sharma, A.; Evans, D.L.; Chauhan, P.; Szlabick, R.E.; Aaland, M.O.; et al. M1 Macrophage Polarization Is Dependent on TRPC1-Mediated Calcium Entry. iScience 2018, 8, 85–102. [Google Scholar] [CrossRef]
- Di Virgilio, F.; Dal Ben, D.; Sarti, A.C.; Giuliani, A.L.; Falzoni, S. The P2X7 Receptor in Infection and Inflammation. Immunity 2017, 47, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Wiley, J.S.; Sluyter, R.; Gu, B.J.; Stokes, L.; Fuller, S.J. The human P2X7 receptor and its role in innate immunity. Tissue Antigens 2011, 78, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Brandau, S.; Chen, S.-H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Hegde, V.L.; Nagarkatti, P.S.; Nagarkatti, M. Role of Myeloid-Derived Suppressor Cells in Amelioration of Experimental Autoimmune Hepatitis Following Activation of TRPV1 Receptors by Cannabidiol. PLoS ONE 2011, 6, e18281. [Google Scholar] [CrossRef] [PubMed]
- Freitas, C.M.T.; Johnson, D.K.; Weber, K.S. T Cell Calcium Signaling Regulation by the Co-Receptor CD5. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Fox, C.J.; Hammerman, P.S.; Thompson, C.B. Fuel feeds function: Energy metabolism and the T-cell response. Nat. Rev. Immunol. 2005, 5, 844–852. [Google Scholar] [CrossRef]
- Menk, A.V.; Scharping, N.E.; Moreci, R.S.; Zeng, X.; Guy, C.; Salvatore, S.; Bae, H.; Xie, J.; Young, H.A.; Wendell, S.G.; et al. Early TCR Signaling Induces Rapid Aerobic Glycolysis Enabling Distinct Acute T Cell Effector Functions. Cell Rep. 2018, 22, 1509–1521. [Google Scholar] [CrossRef]
- Vaeth, M.; Maus, M.; Klein-Hessling, S.; Freinkman, E.; Yang, J.; Eckstein, M.; Cameron, S.; Turvey, S.E.; Serfling, E.; Berberich-Siebelt, F.; et al. Store-Operated Ca2+ Entry Controls Clonal Expansion of T Cells through Metabolic Reprogramming. Immunity 2017, 47, 664–679. [Google Scholar] [CrossRef]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef]
- Feske, S.; Skolnik, E.Y.; Prakriya, M. Ion channels and transporters in lymphocyte function and immunity. Nat. Rev. Immunol. 2012, 12, 532–547. [Google Scholar] [CrossRef]
- Feske, S.; Wulff, H.; Skolnik, E.Y. Ion channels in innate and adaptive immunity. Annu. Rev. Immunol. 2015, 33, 291–353. [Google Scholar] [CrossRef] [PubMed]
- Feske, S.; Prakriya, M.; Rao, A.; Lewis, R.S. A severe defect in CRAC Ca2+ channel activation and altered K+ channel gating in T cells from immunodeficient patients. J. Exp. Med. 2005, 202, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Zhang, R.; Liu, Y.; Feng, J.; Wang, W.; Wu, Y.; Ding, J. Physiological role of Kv1.3 channel in T lymphocyte cell investigated quantitatively by kinetic modeling. PLoS ONE 2014, 9, e89975. [Google Scholar] [CrossRef]
- Malayev, A.; Nelson, D.J. Extracellular pH modulates the Ca2+ current activated by depletion of intracellular Ca2+ stores in human macrophages. J. Membr. Biol. 1995, 146. [Google Scholar] [CrossRef] [PubMed]
- Demaurex, N.; Nunes, P. The role of STIM and ORAI proteins in phagocytic immune cells. Am. J. Physiol. Cell Physiol. 2016, 310, C496–C508. [Google Scholar] [CrossRef] [PubMed]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006, 441, 179–185. [Google Scholar] [CrossRef]
- Angenendt, A.; Steiner, R.; Knorck, A.; Schwar, G.; Konrad, M.; Krause, E.; Lis, A. Orai, STIM, and PMCA contribute to reduced calcium signal generation in CD8+ T cells of elderly mice. Aging 2020, 12, 3266–3286. [Google Scholar] [CrossRef]
- Schwarz, E.C.; Qu, B.; Hoth, M. Calcium, cancer and killing: The role of calcium in killing cancer cells by cytotoxic T lymphocytes and natural killer cells. Biochim. Biophys. Acta 2013, 1833, 1603–1611. [Google Scholar] [CrossRef]
- Modiano, J.F.; Kelepouris, E.; Kern, J.A.; Nowell, P.C. Requirement for extracellular calcium or magnesium in mitogen-induced activation of human peripheral blood lymphocytes. J. Cell Physiol. 1988, 135, 451–458. [Google Scholar] [CrossRef]
- Kuras, Z.; Yun, Y.H.; Chimote, A.A.; Neumeier, L.; Conforti, L. KCa3.1 and TRPM7 channels at the uropod regulate migration of activated human T cells. PLoS ONE 2012, 7, e43859. [Google Scholar] [CrossRef] [PubMed]
- Philipp, S.; Strauss, B.; Hirnet, D.; Wissenbach, U.; Mery, L.; Flockerzi, V.; Hoth, M. TRPC3 mediates T-cell receptor-dependent calcium entry in human T-lymphocytes. J. Biol. Chem. 2003, 278, 26629–26638. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.Y.; Sun, M.R.; Wu, C.L.; Li, Y.; Du, J.J.; Zeng, J.Y.; Bi, H.L.; Sun, Y.H. Activation of calcium-sensing receptor increases TRPC3/6 expression in T lymphocyte in sepsis. Mol. Immunol. 2015, 64, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Rao, G.K.; Kaminski, N.E. Induction of intracellular calcium elevation by Delta9-tetrahydrocannabinol in T cells involves TRPC1 channels. J. Leukoc. Biol. 2006, 79, 202–213. [Google Scholar] [CrossRef]
- Gamberucci, A.; Giurisato, E.; Pizzo, P.; Tassi, M.; Giunti, R.; McIntosh, D.P.; Benedetti, A. Diacylglycerol activates the influx of extracellular cations in T-lymphocytes independently of intracellular calcium-store depletion and possibly involving endogenous TRP6 gene products. Biochem. J. 2002, 364, 245–254. [Google Scholar] [CrossRef]
- Majhi, R.K.; Sahoo, S.S.; Yadav, M.; Pratheek, B.M.; Chattopadhyay, S.; Goswami, C. Functional expression of TRPV channels in T cells and their implications in immune regulation. FEBS J. 2015, 282, 2661–2681. [Google Scholar] [CrossRef]
- Capasso, M.; Bhamrah, M.K.; Henley, T.; Boyd, R.S.; Langlais, C.; Cain, K.; Dinsdale, D.; Pulford, K.; Khan, M.; Musset, B.; et al. HVCN1 modulates BCR signal strength via regulation of BCR-dependent generation of reactive oxygen species. Nat. Immunol. 2010, 11, 265–272. [Google Scholar] [CrossRef]
- Calcinotto, A.; Filipazzi, P.; Grioni, M.; Iero, M.; de Milito, A.; Ricupito, A.; Cova, A.; Canese, R.; Jachetti, E.; Rossetti, M.; et al. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res. 2012, 72, 2746–2756. [Google Scholar] [CrossRef]
- Chimote, A.A.; Hajdu, P.; Kucher, V.; Boiko, N.; Kuras, Z.; Szilagyi, O.; Yun, Y.H.; Conforti, L. Selective inhibition of KCa3.1 channels mediates adenosine regulation of the motility of human T cells. J. Immunol. 2013, 191, 6273–6280. [Google Scholar] [CrossRef]


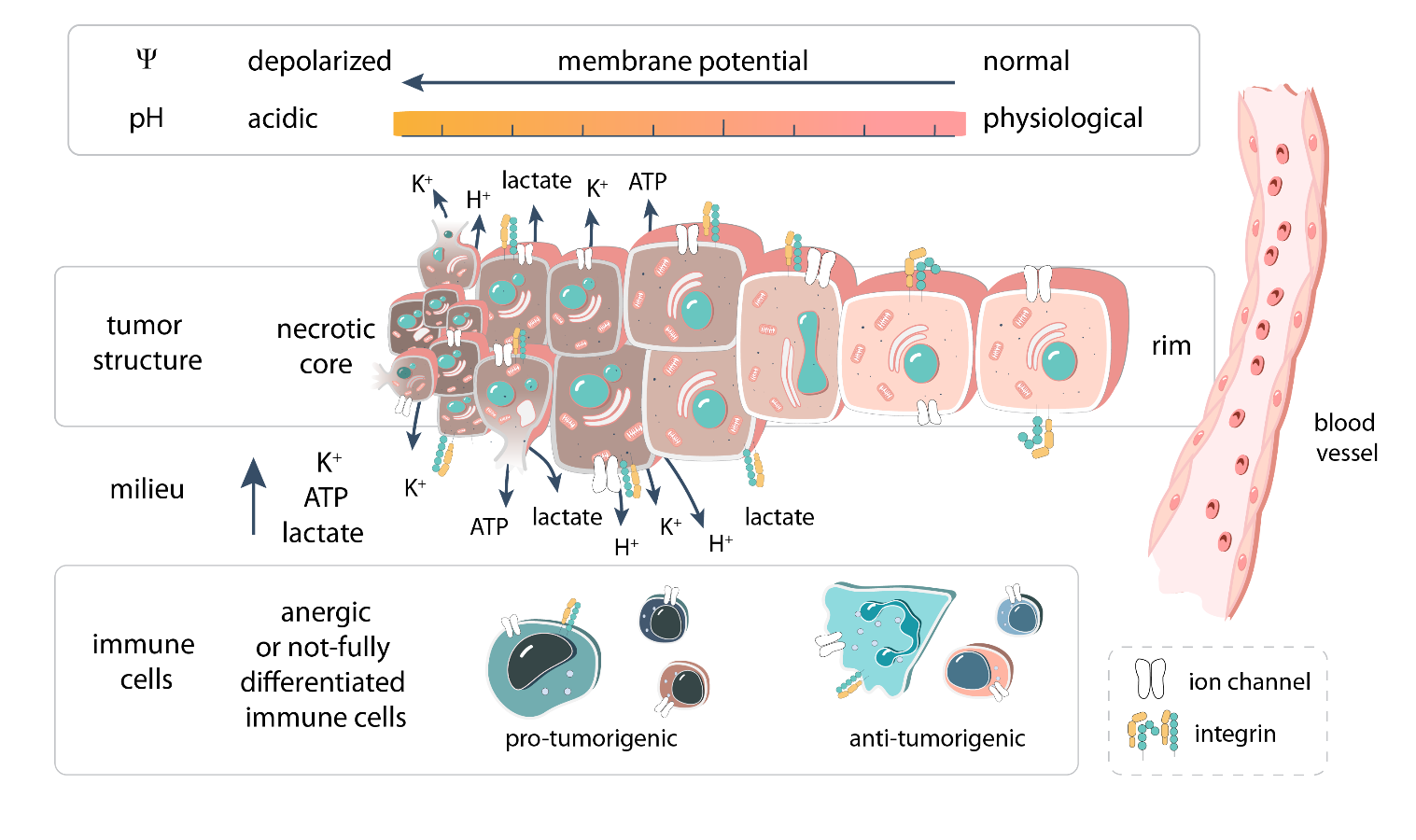{kind=link}
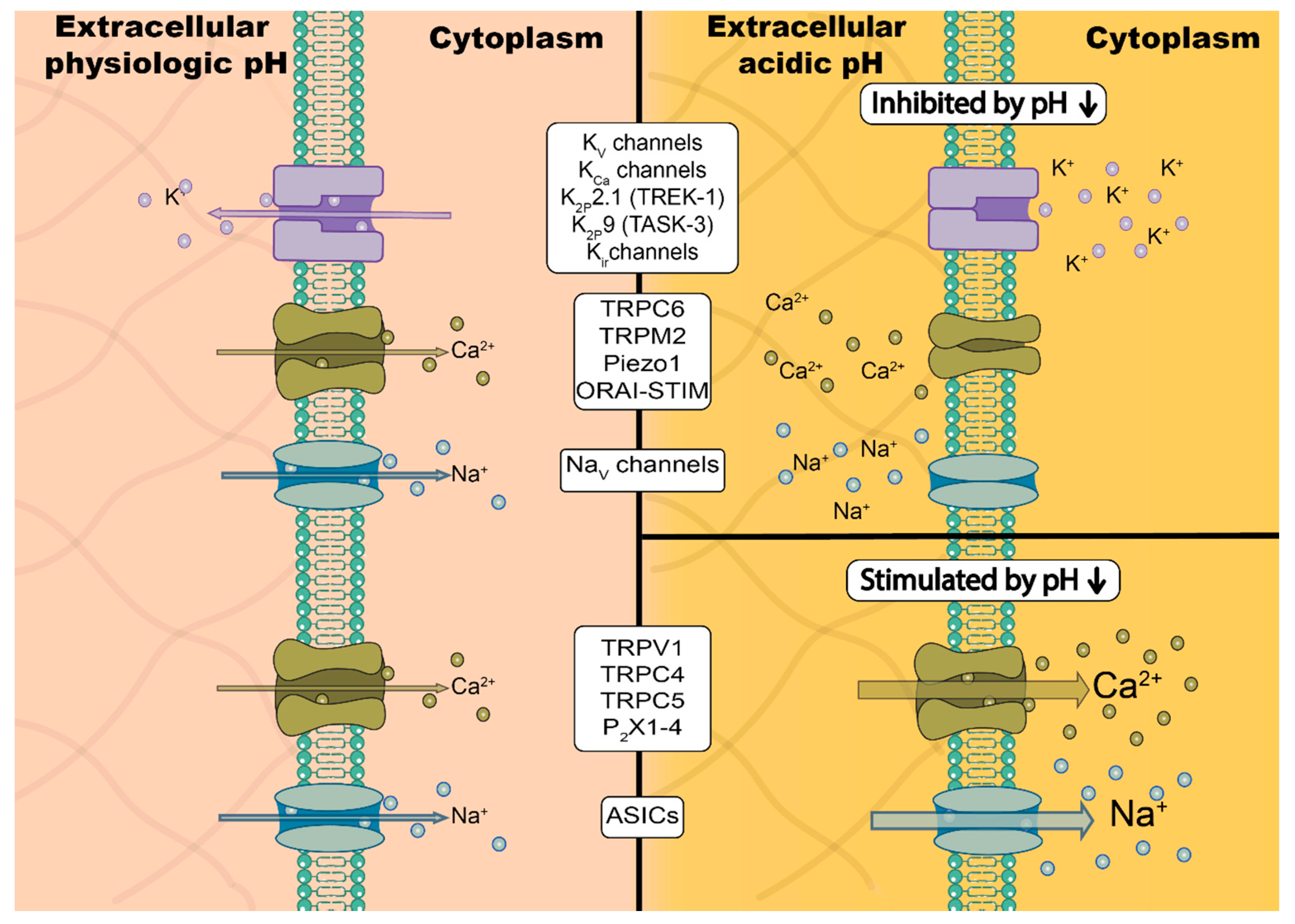{kind=link}
| Gene Name | Channel Name | Cancer Type | Functions in Cancers | pHo Acidification | pHi Acidification |
|---|---|---|---|---|---|
| KCNA3 | KV1.3 | colorectal (cell line) | ↑ migration, adhesiveness [88] | slight inhibition [89,90] | |
| breast (cell line) | ↑ migration, adhesiveness, proliferation [88,91] | ||||
| melanoma (T lymphocyte) | ↑ antitumor immunity in tumor-bearing mice [92] | ||||
| head and neck (T lymphocyte) | defects are associated with diminished cytotoxicity [93] | ||||
| KCNA5 | KV1.5 | lung (cell line, tumor) | apoptosis, H2O2 production, ↓ tumor growth [27] | strong inhibition, pKa = 7.2 [94] | inhibition [27,95] |
| cervical (cell line) | apoptosis, H2O2 production [95] | ||||
| gastric (cell line) | ↑ proliferation [96] | ||||
| KCNB1 | KV2.1 | uterine (cell line) | ↑ cell cycle progression [97] | slight inhibition [94] | |
| KCNQ1 | KV.7.1 | colorectal (cell line) | ↑ proliferation [98] | slight inhibition, pKa = 5.5 [99] | inhibition, BUT stimulation in presence of KCNE1 [100] |
| KCNH1 | KV10.1 (hEAG1) | glioblastoma (cell line, tumor) | ↑ tumor growth and metastasis [101] | inhibition, pKa ≈ 6.5 [102] | |
| colorectal (cell line) | ↑ proliferation [103] | ||||
| gastric (cell line) | ↑ proliferation [104] | ||||
| breast (cell line, tumor) | ↑ tumor progression and hypoxia signaling [105] | ||||
| osteosarcoma (cell line) | ↑ proliferation, cell cycle progression [106] | ||||
| osteosarcoma (endothelium) | knockdown inhibitis tumor angiogenesis and VEGF-signaling [107] | ||||
| KCNH2 | KV11.1 (hERG1) | pancreatic (cell line) | ↑ EGFR-signaling, proliferation, migration [85] | slight inhibition, pKa = 5.1 [45,108] | slight stimulation [109] |
| gastric (cell line, tumor) | ↑ PI3K/Akt pathway, promotes angiogenesis, proliferation, migration and tumor growth [110] | ||||
| KCNN4 | KCa3.1 | glioblastoma (cell line, tumor) | ↑ migration, swelling, invasion, radiotherapy resistance, tumor growth (reviewed in [111] | possibly insensitive between pH 6.5 and pH 8 [112] | inhibition [113] |
| breast (cell line, tumor) | ↑ cell cycle progression, proliferation and tumor growth [114,115] | ||||
| pancreatic (cell line) | ↑ migration, invasion, proliferation [116] | inhibition [113] | |||
| pancreatic (stromal cell) | ↑ migration, chemotaxis [117] | ||||
| melanoma (T lymphocyte) | ↑ antitumor immunity in tumor-bearing mice [92,118] | ||||
| head and neck cancer (T lymphocyte) | ↑ tumor infiltration [119,120] | ||||
| KCNJ1 | Kir1.1 | renal (cell line) | ↓ cancer cell growth, invasion and promotes cancer cell apoptosis [121] | strong inhibition pKa = 6.9 [122] | |
| KCNJ10 | Kir4.1 | glioblastoma (cell line) | ↓ cell cycle progression [123] | inhibition pKa = 6 [124] | |
| KCNK2 | K2P2.1 (TREK1) | prostate (cell line) | ↑ proliferation, contributes to setting the membrane potential [125] | strong inhibition pKa = 7.35 [126] | slight stimulation [127] |
| pancreatic (cell line) | ↑ proliferation, migration, contributes to setting the membrane potential [128] | ||||
| KCNK3, KCNK9 | K2P3.1(TASK1), K2P9.1(TASK3) | glioblastoma (cell line) | ↑ apoptosis and hypoxia resistance, contribute to setting the membrane potential [129] | inhibition [126] | |
| lung (cell line) | ↑ proliferation, contribute to setting the membrane potential [130] | ||||
| KCNK5 | K2P5.1(TASK2) | breast (cell line) | ↑ proliferation, contribute to setting the membrane potential [131] | inhibition [132] | inhibition [132,133] |
| TRPC4, TRPC5 | TRPC4, TRPC5 | renal (cell line) | activation leads to cancer cell death [134] | stimulation [135] | |
| TRPC6 | TRPC6 | glioblastoma (U373MG cell line) | ↑ cancer cell proliferation, invasion, angiogenesis [136] | slight inhibition [135] | |
| liver (cell line) | ↑ proliferation [137] | ||||
| pancreatic (tumor-associated immune cell) | ↑ CXCL1/CXCR2 induced neutrophil infiltration [138,139] | ||||
| pancreatic (stromal cell) | ↑ migration, hypoxia response [140] | ||||
| TRPM2 | TRPM2 | breast (cell lines and murine model) | mediates H2O2 cytotoxicity [141,142] | inhibition pKa = 6.5 [143] | complete inhibition by pH = 6 [143] |
| prostate (cell lines) | ↑ proliferation [144] | ||||
| TRPM7 | TRPM7 | nasopharyngeal (cell lines) | ↑ migration [145] | inhibition pKa = 6.32 [146] | |
| lung (A549 cell line) | ↑ migration [147] | ||||
| pancreatic (cell line) | ↑ migration [148] | ||||
| breast | ↑ proliferation and migration (overexpressed in cancer tissue) [149] | ||||
| TRPV1 | TRPV1 | breast (cell line) | apoptosis, tumor suppressor (mRNA overexpression) [150] | stimulation [151] | |
| gastric and colorectal (cell line) | apoptosis, tumor suppressor (decreased expression in cancer tissues) [152] | ||||
| astrocytoma (murine model) | apoptosis, tumor suppressor [153] | ||||
| endometrial (cell line) | apoptosis, tumor suppressor [154] | ||||
| TRPV4 | TRPV4 | lung (murine model) | ↓ endothelial tumor cell migration, normalization of tumor vascularization [155] | stimulation [156] | |
| breast (tumor-derived EC, cancer cell line) | ↑ promotes tumor-derived endothelial cell migration and metastasis [157] | ||||
| hepatocellular carcinoma (cell line and murine model) | TRPV4 inhibition leads to cell apoptosis [158] | ||||
| gastric (cell line) | ↑ cancer proliferation, migration and invasion [159] | ||||
| HVCN1 | HV1 | breast (cell line, tumor) | promotes cell migration, tumor growth, pHi regulation [160,161] | pHo/pHi relation and membrane-potential dependent, detailed in Section 4.3 | |
| colorectal (cell line) | promotes cell migration, pHi regulation [162,163] | ||||
| glioblastoma (cell line) | promotes cell migration, survival, pHi regulation [164] | ||||
| B-cell lymphoma (patient samples, cell line) | short variant of HV1 promotes cell migration and proliferation [165] | ||||
| CRAC channel (ORAI-STIM) | inhibition pKa = 8.3 [166] | iinhibition [166] | |||
| ORAI1/STIM1 | CRAC channel | renal (primary cells) | ↑ migration and proliferation [167] | inhibition pKa = 8.3 [168] | inhibition pKa = 7.5 [166] |
| breast (primary cells, cell line) | ↑ migration, invasion [169] | ||||
| esophageal (cell line) | ↑ migration and proliferation [170] | ||||
| glioblastoma (primary cells, cell line) | ↑ migration, invasion, proliferation and ↓ apoptosis [171,172] | ||||
| lung (cell line) | ↑ proliferation [173] | ||||
| melanoma (cell line) | ↑ proliferation, migration and invasion; metastasis formation [174] | ||||
| pancreatic (cell line) | apoptosis resistance [175] | ||||
| gastric (cell line) | ↑ proliferation, migration, invasion [176] | ||||
| acut myeloid leukemia (cell line) | ↑ proliferation and migration [177] | ||||
| liver (cell line) | ↑ proliferation [178] | ||||
| colorectal (primary cells, cell line) | ↑ proliferation, invasion and metastasis [179] | ||||
| colorectal (T lymphocyte) | ↑ tumor cell killing by CD8+ T cells, regulates antitumor immunity [180] | ||||
| melanoma (T lymphocyte) | ↑ tumor cell killing by CD8+ T cells, regulates antitumor immunity [180] | ||||
| leukemia (T lymphocyte and NK cell) | ↓ tumor cell killing due to verly high Ca2+ signals [181] | ||||
| ORAI2/STIM1 | CRAC channel | acut myeloid leukemia (cell line) | ↑ proliferation and migration [177] | inhibition pKa = 8.3 [168] | |
| ORAI3/STIM1 | CRAC channel | breast (cell line) | ↑ proliferation [182] | inhibition pKa = 8.5 [168] | |
| prostate (primary cells, cell line) | ↑ tumor growth, apoptosis resistance [183] | ||||
| SCN5A | NaV1.5 | breast (cell line, biopsies) | ↑ migration, invasion and cancer progression [33,184,185] | inhbition pKa = 6.1 [186] | |
| PIEZO1 | Piezo1 | gastric (cell line) | ↑ proliferation, migration, chemotherapy resistance [187] | inhibition [188,189] | inhibition [189] |
| synovial (cell line) | ↑ viability and migration [190] | ||||
| gliobastoma (cell line) | ↑ cell proliferation [191] | ||||
| breast (cell line) | ↑ invasion, cell migration [192] | ||||
| prostate (cell line) | ↑ proliferation, migration [193] | ||||
| pancreatic (stromal cell) | ↑ migration and invasiveness [189] | ||||
| ASIC1 | ASIC1 | breast (cell line and murine model) | ↑ invasion, metastasis and ROS production [4] | stimulation (reviewed in [44]) | |
| ASIC1, ASIC3 | ASIC1/3 | pancreatic (cell lines) | ↑ epithelial–mesenchymal transition [194,171] | ||
| ASIC2 | ASIC2 | colorectal (cell line and murine model) | ↑ cancer proliferation, invasion and metastasis [195] | ||
| ASIC1a | lung (cell line) | ↑ proliferation and migration [196] | |||
| P2X7 | P2X7 | melanoma (murine model) | ↑ tumor growth, reduced apoptosis, increased angiogenesis [197] | inhibition pKa = 6.7 [198] | |
| cervical cancer (cell line) | ↑ cancer cell apoptosis [199] | ||||
| pancreatic (cell line) | ↑ tumor progression [200] | ||||
| neuroblastoma (tumor-associated immune cell) | promotion of MDSCs; CCL2 release, macrophage recruitment [201] | ||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pethő, Z.; Najder, K.; Carvalho, T.; McMorrow, R.; Todesca, L.M.; Rugi, M.; Bulk, E.; Chan, A.; Löwik, C.W.G.M.; Reshkin, S.J.; et al. pH-Channeling in Cancer: How pH-Dependence of Cation Channels Shapes Cancer Pathophysiology. Cancers 2020, 12, 2484. https://doi.org/10.3390/cancers12092484
Pethő Z, Najder K, Carvalho T, McMorrow R, Todesca LM, Rugi M, Bulk E, Chan A, Löwik CWGM, Reshkin SJ, et al. pH-Channeling in Cancer: How pH-Dependence of Cation Channels Shapes Cancer Pathophysiology. Cancers. 2020; 12(9):2484. https://doi.org/10.3390/cancers12092484
Chicago/Turabian StylePethő, Zoltán, Karolina Najder, Tiago Carvalho, Roisin McMorrow, Luca Matteo Todesca, Micol Rugi, Etmar Bulk, Alan Chan, Clemens W. G. M. Löwik, Stephan J. Reshkin, and et al. 2020. "pH-Channeling in Cancer: How pH-Dependence of Cation Channels Shapes Cancer Pathophysiology" Cancers 12, no. 9: 2484. https://doi.org/10.3390/cancers12092484
APA StylePethő, Z., Najder, K., Carvalho, T., McMorrow, R., Todesca, L. M., Rugi, M., Bulk, E., Chan, A., Löwik, C. W. G. M., Reshkin, S. J., & Schwab, A. (2020). pH-Channeling in Cancer: How pH-Dependence of Cation Channels Shapes Cancer Pathophysiology. Cancers, 12(9), 2484. https://doi.org/10.3390/cancers12092484