Tumors Responsive to Autophagy-Inhibition: Identification and Biomarkers

Simple Summary
Abstract
1. Introduction
2. Head and Neck Cancer
2.1. Hypoxia
2.2. PTOV1 Expression
3. Brain Cancer
3.1. EGFRvIII Expression
3.2. BRAFv600E
3.3. Neurotrophins
3.4. WNT-Signaling
4. Pancreatic Cancer
4.1. KRAS
4.2. EI24 Expression
4.3. MAPK/ERK Inhibitors
4.4. Glutamine Addicted Cancer
5. Melanoma
BRAFv600E
6. Colorectal Cancer
6.1. KRAS
6.2. JNK-Signaling
6.3. TFEB
7. Breast Cancer
7.1. JAK/STAT Signaling
7.2. HER2 Signaling
7.3. Estrogen Receptor Signaling
7.4. CDK4 and-6 Inhibition
7.5. IKBKE and JMJD6
8. Lung Cancer
8.1. TKI
8.2. KRAS and LKB1 Signaling
9. Liver Cancer
HGF Signaling
10. Prostate Cancer
KDMB4 Expression
11. Bone Cancer
HSP90AA1
12. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
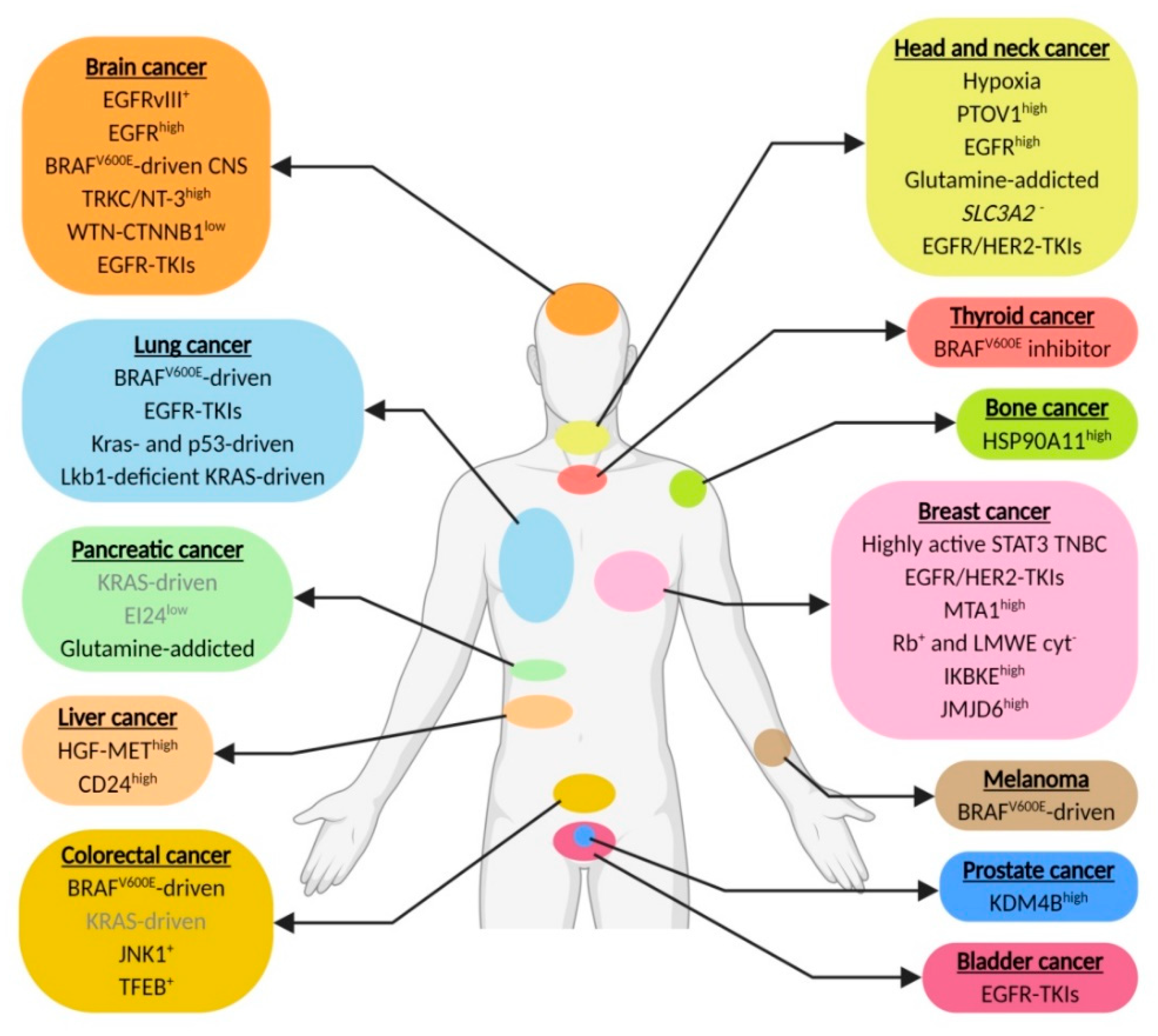
| Cancer Type | Tumor Type | Biomarker/Factor | Mechanism of Autophagy Dependency (When Known) | References |
|---|---|---|---|---|
| Head and neck cancer | HNSCC | Hypoxia | Nutrient suppletion, reduction ROS exposure | [6,10,33] |
| Laryngeal cancer cisplatin or 5-FU resistant | PTOV1high | Cisplatin resistance | [37] | |
| HNSCC | EGFRhigh | - | ||
| HNSCC | Glutamine | Glutamine suppletion | NP* | |
| HNSCC | SLC3A2- | - | [93] | |
| Esophageal HER2 amplification | TKIs against EGFR and HER2 | - | [133] | |
| Brain cancer | Hypoxic glioblastoma | EGFRvIII+ | Nutrient suppletion | [34] |
| Glioblastoma | EGFRhigh | - | [172] | |
| Central nervous system | BRAFV600E | - | [47,48] | |
| Gliomas | TRKC/NT-3high | Hypoxia tolerance | [55] | |
| Glioblastoma | WNT-CTNNB1low or mutated | - | [57] | |
| Neuroblastoma | TKIs against EGFR or multiple TKIs or Axl | - | ||
| Pancreatic cancer | Kras-driven | Kras | - | [16,64] |
| - | [67] | |||
| - | [84] | |||
| PDAC | EI24low | - | [79] | |
| KRAS-driven | Glutamine | Glutamine suppletion | [19] | |
| Thyroid cancer | BRAFV600E-driven | BRAFV600E inhibitor | - | [85] |
| Skin cancer | Melanoma | BRAFV600E | Removal of mutated protein aggregates | [104] |
| Colorectal cancer | CRC | BRAFV600E | - | [103] |
| KRAS-driven | KRAS | - | [110] | |
| CRC, metastatic CRC or adenocarcinoma | JNK1+ | - | [113] | |
| Doxorubicin resistant | TFEB+ | - | [116] | |
| Breast cancer | TNBC | STAT3 | - | [125] |
| HER2+ | TKIs against HER2 | TKI resistance | [131] | |
| TKIs against EGFR and HER2 | TKI resistance | [132] | ||
| ER+ breast cancer tamoxifen resistant | MTA1high | Tamoxifen resistance | [145] | |
| Breast cancer with Rb+ and LMWE cyt- or Letrozole or Anastrozole resistant | Rb+ and LMWE cyt- | - | [146] | |
| TNBC and HER2- | IKBKEhigh | - | [147] | |
| TNBC | JMJD6high | JMJD6 phosphorylates H2A.XY39 and regulates ATG genes transcription | [148] | |
| Lung cancer | BrafV600E-driven | BrafV600E | - | [101] |
| NSCLC with EGFR WT and mutant | TKIs against EGFR | - | [151] | |
| - | [152] | |||
| Kras- and p53-driven | Kras | - | [157,158] | |
| Lkb1-deficient Kras-driven NSCLC | Lkb1 (STK11) | TCA and fatty acid synthesis support | [163] | |
| Bladder cancer | EGFR mutant | TKIs against EGFR and HER2 | - | [153] |
| Liver cancer | HCC | HGF-METhigh | - | [164] |
| HCC sorafenib resistant | CD24high | Sorafenib resistance | [165] | |
| Prostate cancer | Castration-resistant | KDM4Bhigh | - | [170] |
| Bone cancer | Osteosarcoma doxorubicin or cisplatin or methotrexane resistant | HSP90A11high | Doxorubicin resistance | [171] |
References
- Kimmelman, A.C.; White, E. Autophagy and Tumor Metabolism. Cell Metab. 2017, 25, 1037–1043. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, J.D.; White, E. Autophagy and Metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef] [PubMed]
- Kuma, A.; Hatano, M.; Matsui, M.; Yamamoto, A.; Nakaya, H.; Yoshimori, T.; Ohsumi, Y.; Tokuhisa, T.; Mizushima, N. The role of autophagy during the early neonatal starvation period. Nature 2004, 432, 1032–1036. [Google Scholar] [CrossRef] [PubMed]
- Marx, J. Autophagy: Is it cancer’s friend or foe? Science 2006, 312, 1160–1161. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; White, E. Role of Autophagy in Cancer. Autophagy 2007, 3, 28–31. [Google Scholar] [CrossRef] [PubMed]
- Rouschop, K.; Beucken, T.V.D.; Dubois, L.; Niessen, H.; Bussink, J.; Savelkouls, K.; Keulers, T.; Mujcic, H.; Landuyt, W.; Voncken, J.W.; et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J. Clin. Investig. 2009, 120, 127–141. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef]
- Mizushima, N. The pleiotropic role of autophagy: From protein metabolism to bactericide. Cell Death Differ. 2005, 12, 1535–1541. [Google Scholar] [CrossRef]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial Autophagy Is an HIF-1-dependent Adaptive Metabolic Response to Hypoxia. J. Boil. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef]
- Rouschop, K.; Ramaekers, C.H.; Schaaf, M.B.; Keulers, T.G.; Savelkouls, K.G.; Lambin, P.; Koritzinsky, M.; Wouters, B. Autophagy is required during cycling hypoxia to lower production of reactive oxygen species. Radiother. Oncol. 2009, 92, 411–416. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Emr, S.D. Autophagy as a Regulated Pathway of Cellular Degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of Poly(ADP-Ribose) Polymerase in Tumors fromBRCAMutation Carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Chan, N.; Pires, I.M.; Bencokova, Z.; Coackley, C.; Luoto, K.R.; Bhogal, N.; Lakshman, M.; Gottipati, P.; Oliver, F.J.; Helleday, T.; et al. Contextual synthetic lethality of cancer cell kill based on the tumor microenvironment. Cancer Res. 2010, 70, 8045–8054. [Google Scholar] [CrossRef]
- Guo, J.Y.; Chen, H.-Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’Antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef]
- Guo, J.Y.; Teng, X.; Laddha, S.V.; Ma, S.; Van Nostrand, S.C.; Yang, Y.; Khor, S.; Chan, C.S.; Rabinowitz, J.D.; White, E. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev. 2016, 30, 1704–1717. [Google Scholar] [CrossRef]
- Zhang, N.; Yang, X.; Yuan, F.; Zhu, W.-G.; Zhao, Y. Autophagy-deficient tumor cells rely on extracellular amino acids to survive upon glutamine deprivation. Autophagy 2018, 14, 1652–1653. [Google Scholar] [CrossRef]
- Seo, J.-W.; Choi, J.; Lee, S.-Y.; Sung, S.; Yoo, H.J.; Kang, M.-J.; Cheong, H.; Son, J. Autophagy is required for PDAC glutamine metabolism. Sci. Rep. 2016, 6, 37594. [Google Scholar] [CrossRef]
- Chen, S.; Rehman, S.K.; Zhang, W.; Wen, A.-D.; Yao, L.; Zhang, J. Autophagy is a therapeutic target in anticancer drug resistance. Biochim. Et Biophys. Acta (BBA) Rev. Cancer 2010, 1806, 220–229. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef] [PubMed]
- Onorati, A.V.; Dyczynski, M.; Ojha, R.; Amaravadi, R.K. Targeting autophagy in cancer. Cancer 2018, 124, 3307–3318. [Google Scholar] [CrossRef] [PubMed]
- Buccarelli, M.; Marconi, M.; Pacioni, S.; De Pascalis, I.; D’Alessandris, Q.G.; Martini, M.; Ascione, B.; Malorni, W.; LaRocca, L.M.; Pallini, R.; et al. Inhibition of autophagy increases susceptibility of glioblastoma stem cells to temozolomide by igniting ferroptosis. Cell Death Dis. 2018, 9, 841. [Google Scholar] [CrossRef] [PubMed]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Amaravadi, R.; Kimmelman, A.C.; White, E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016, 30, 1913–1930. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.M.M.; Thorburn, A. Autophagy in cancer: Moving from understanding mechanism to improving therapy responses in patients. Cell Death Differ. 2019, 27, 843–857. [Google Scholar] [CrossRef]
- Wouters, B. Targeting hypoxia tolerance in cancer. Drug Resist. Updat. 2004, 7, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Wang, M.; Yu, M.; Zhang, J.; Bristow, R.; Hill, R.; Tannock, I.F. Role of Autophagy as a Survival Mechanism for Hypoxic Cells in Tumors. Neoplasia 2016, 18, 347–355. [Google Scholar] [CrossRef]
- Harris, A.L. Hypoxia—A key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef]
- Nordsmark, M.; Bentzen, S.M.; Rudat, V.; Brizel, D.; Lartigau, E.; Stadler, P.; Becker, A.; Adam, M.; Molls, M.; Dunst, J.; et al. Prognostic value of tumor oxygenation in 397 head and neck tumors after primary radiation therapy. An international multi-center study. Radiother. Oncol. 2005, 77, 18–24. [Google Scholar] [CrossRef]
- Overgaard, J. Hypoxic modification of radiotherapy in squamous cell carcinoma of the head and neck-A systematic review and meta-analysis. Radiother. Oncol. 2011, 100, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouyssegur, J.; Mazure, N.M. Hypoxia-Induced Autophagy Is Mediated through Hypoxia-Inducible Factor Induction of BNIP3 and BNIP3L via Their BH3 Domains. Mol. Cell. Boil. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Schaaf, M.B.; Cojocari, D.; Keulers, T.G.; Jutten, B.; Starmans, M.H.; De Jong, M.C.; Begg, A.C.; Savelkouls, K.G.; Bussink, J.; Vooijs, M.; et al. The autophagy associated gene, ULK1, promotes tolerance to chronic and acute hypoxia. Radiother. Oncol. 2013, 108, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Jutten, B.; Keulers, T.G.; Peeters, H.J.M.; Schaaf, M.B.E.; Savelkouls, K.G.M.; Compter, I.; Clarijs, R.; Schijns, O.E.M.G.; Ackermans, L.; Teernstra, O.P.M.; et al. EGFRvIII expression triggers a metabolic dependency and therapeutic vulnerability sensitive to autophagy inhibition. Autophagy 2018, 14, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Lin, H.; Wu, S.; Lei, F.; Zhu, X.; Song, L.; Hong, M.; Guo, L. Prostate Tumor Overexpressed 1 (PTOV1) Is a Novel Prognostic Marker for Nasopharyngeal Carcinoma Progression and Poor Survival Outcomes. PLoS ONE 2015, 10, e0136448. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, H.; Wang, Y.; He, Z.; Chen, H.; Liang, S.; He, S.; Wu, S.; Song, L.; Chen, Y. Prostate tumor overexpressed-1, in conjunction with human papillomavirus status, predicts outcome in early-stage human laryngeal squamous cell carcinoma. Oncotarget 2016, 7, 31878–31891. [Google Scholar] [CrossRef]
- Mayea, Y.G.; Mir, C.; Muñoz, L.; Benavente, S.; Castellví, J.; Temprana, J.; Maggio, V.; Lorente, J.; Paciucci, R.; E Lleonart, M. Autophagy inhibition as a promising therapeutic target for laryngeal cancer. Carcinogenesis 2019, 40, 1525–1534. [Google Scholar] [CrossRef]
- Wang, L.; Shang, Z.; Zhou, Y.; Hu, X.; Chen, Y.; Fan, Y.; Wei, X.; Wu, L.; Liang, Q.; Zhang, J.; et al. Autophagy mediates glucose starvation-induced glioblastoma cell quiescence and chemoresistance through coordinating cell metabolism, cell cycle, and survival. Cell Death Dis. 2018, 9, 213. [Google Scholar] [CrossRef]
- Theys, J.; Jutten, B.; Dubois, L.; Rouschop, K.; Chiu, R.K.; Li, Y.; Paesmans, K.; Lambin, P.; Lammering, G.; Wouters, B. The deletion mutant EGFRvIII significantly contributes to stress resistance typical for the tumour microenvironment. Radiother. Oncol. 2009, 92, 399–404. [Google Scholar] [CrossRef]
- Sotelo, J.; Bricenño, E.; Loópez-Gonzaález, M.A. Adding Chloroquine to Conventional Treatment for Glioblastoma Multiforme. Ann. Intern. Med. 2006, 144, 337–343. [Google Scholar] [CrossRef]
- Compter, I.; Eekers, D.B.P.; Hoeben, A.; Rouschop, K.M.A.; Reymen, B.; Ackermans, L.; Beckervordersantforth, J.; Bauer, N.J.C.; Anten, M.M.; Wesseling, P.; et al. Chloroquine combined with concurrent radiotherapy and temozolomide for newly diagnosed glioblastoma: A phase IB trial. Autophagy 2020, in press. [Google Scholar]
- Rosenfeld, M.R.; Ye, X.; Supko, J.G.; Desideri, S.; Grossman, S.A.; Brem, S.; Mikkelson, T.; Wang, D.; Chang, Y.C.; Hu, J.; et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2014, 10, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, P.; Strambi, A.; Zipoli, C.; Hägg-Olofsson, M.; Buoncervello, M.; Linder, S.; De Milito, A. Acidic extracellular pH neutralizes the autophagy-inhibiting activity of chloroquine. Autophagy 2014, 10, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Nazio, F.; Strappazzon, F.; Antonioli, M.; Bielli, P.; Cianfanelli, V.; Bordi, M.; Gretzmeier, C.; Dengjel, J.; Piacentini, M.; Fimia, G.M.; et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nature 2013, 15, 406–416. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Montalto, G.; Cervello, M.; Nicoletti, F.; Fagone, P.; Malaponte, G.; Mazzarino, M.C.; et al. Mutations and Deregulation of Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR Cascades Which Alter Therapy Response. Oncotarget 2012, 3, 954–987. [Google Scholar] [CrossRef]
- Dahiya, S.; Emnett, R.J.; Haydon, D.H.; Leonard, J.R.; Phillips, J.J.; Perry, A.; Gutmann, D.H. BRAF-V600E mutation in pediatric and adult glioblastoma. Neuro Oncol. 2013, 16, 318–319. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Thompson, J.C.; Griesinger, A.M.; Amani, V.; Donson, A.M.; Birks, D.K.; Morgan, M.J.; Mirsky, D.M.; Handler, M.H.; Foreman, N.K.; et al. Autophagy Inhibition Improves Chemosensitivity in BRAFV600E Brain Tumors. Cancer Discov. 2014, 4, 773–780. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Foreman, N.K.; Thorburn, A. Using BRAF(V600E) as a marker of autophagy dependence in pediatric brain tumors. Autophagy 2014, 10, 2077–2078. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Zahedi, S.; Griesinger, A.M.; Morin, A.; Davies, K.D.; Aisner, D.L.; Kleinschmidt-DeMasters, B.; Fitzwalter, B.E.; Goodall, M.L.; Thorburn, J.; et al. Autophagy inhibition overcomes multiple mechanisms of resistance to BRAF inhibition in brain tumors. eLife 2017, 6, 358. [Google Scholar] [CrossRef]
- Zahedi, S.; Fitzwalter, B.E.; Morin, A.; Grob, S.; Desmarais, M.; Mellan, A.; Green, A.L.; Vibhakar, R.; Hankinson, T.C.; Foreman, N.K.; et al. Effect of early-stage autophagy inhibition in BRAF(V600E) autophagy-dependent brain tumor cells. Cell Death Dis. 2019, 10, 679. [Google Scholar] [CrossRef] [PubMed]
- Hamel, W.; Westphal, M. Growth factors in gliomas revisited. Acta Neurochir. 2000, 142, 113–137. [Google Scholar] [CrossRef] [PubMed]
- Arévalo, J.C.; Wu, S.H. Neurotrophin signaling: Many exciting surprises! Cell. Mol. Life Sci. 2006, 63, 1523–1537. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Kim, G.M.; Choi, Y.-J.; Kim, H.J.; Kim, Y.-J.; Jin, W. TrkC Promotes Survival and Growth of Leukemia Cells Through Akt-mTOR-Dependent Up-Regulation of PLK-1 and Twist-1. Mol. Cells 2013, 36, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Jawhari, S.; Bessette, B.; Hombourger, S.; Durand, K.; Lacroix, A.; Labrousse, F.; Jauberteau, M.-O.; Ratinaud, M.H.; Verdier, M. Autophagy and TrkC/NT-3 signaling joined forces boost the hypoxic glioblastoma cell survival. Carcinogenesis 2017, 38, 592–603. [Google Scholar] [CrossRef]
- Lee, Y.; Lee, J.-K.; Ahn, S.H.; Lee, J.; Nam, D.-H. WNT signaling in glioblastoma and therapeutic opportunities. Lab. Investig. 2015, 96, 137–150. [Google Scholar] [CrossRef]
- Nager, M.; Sallán, M.C.; Visa, A.; Pushparaj, C.; Santacana, M.; Macià, A.; Yeramian, A.; Cantí, C.; Herreros, J. Inhibition of WNT-CTNNB1 signaling upregulates SQSTM1 and sensitizes glioblastoma cells to autophagy blockers. Autophagy 2018, 14, 619–636. [Google Scholar] [CrossRef]
- Study of Pre-surgery Gemcitabine + Hydroxychloroquine (GcHc) in Stage IIb or III Adenocarcinoma of the Pancreas. 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT01128296?term=NCT01128296&draw=2&rank=1 (accessed on 17 July 2020).
- Boone, B.A.; Bahary, N.; Zureikat, A.H.; Moser, A.J.; Normolle, D.P.; Wu, W.-C.; Singhi, A.D.; Bao, P.; Bartlett, D.L.; A Liotta, L.; et al. Safety and Biologic Response of Pre-operative Autophagy Inhibition in Combination with Gemcitabine in Patients with Pancreatic Adenocarcinoma. Ann. Surg. Oncol. 2015, 22, 4402–4410. [Google Scholar] [CrossRef]
- Randomized Phase II Trial of Pre-Operative Gemcitabine and Nab Paclitacel With or With Out Hydroxychloroquine. 2020. Available online: https://clinicaltrials.gov/ct2/show/study/NCT01978184?term=NCT01978184&draw=2&rank=1 (accessed on 17 July 2020).
- Zeh, H.J.; Bahary, N.; Boone, B.A.; Singhi, A.D.; Miller-Ocuin, J.L.; Normolle, D.P.; Zureikat, A.H.; Hogg, M.E.; Bartlett, D.L.; Lee, K.K.; et al. A Randomized Phase II Preoperative Study of Autophagy Inhibition with High-Dose Hydroxychloroquine and Gemcitabine/Nab-Paclitaxel in Pancreatic Cancer Patients. Clin. Cancer Res. 2020, 26, 3126–3134. [Google Scholar] [CrossRef]
- New, M.; Van Acker, T.; Long, J.S.; Sakamaki, J.-I.; Ryan, K.M.; Tooze, S.A. Molecular Pathways Controlling Autophagy in Pancreatic Cancer. Front. Oncol. 2017, 7, 28. [Google Scholar] [CrossRef]
- Kang, R.; Tang, D. Autophagy in pancreatic cancer pathogenesis and treatment. Am. J. Cancer Res. 2012, 2, 383–396. [Google Scholar] [PubMed]
- Yang, S.; Kimmelman, A.C. A critical role for autophagy in pancreatic cancer. Autophagy 2011, 7, 912–913. [Google Scholar] [CrossRef] [PubMed]
- Lock, R.; Roy, S.; Kenific, C.M.; Su, J.S.; Salas, E.; Ronen, S.M.; Debnath, J. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol. Boil. Cell 2011, 22, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Perera, R.M.; Stoykova, S.; Nicolay, B.N.; Ross, K.N.; Fitamant, J.; Boukhali, M.; Lengrand, J.; Deshpande, V.; Selig, M.K.; Ferrone, C.R.; et al. Transcriptional control of autophagy–lysosome function drives pancreatic cancer metabolism. Nature 2015, 524, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; RajeshKumar, N.V.; Wang, X.; Yabuuchi, S.; Alexander, B.M.; Chu, G.C.; Von Hoff, D.D.; Maitra, A.; Kimmelman, A.C. Autophagy Is Critical for Pancreatic Tumor Growth and Progression in Tumors with p53 Alterations. Cancer Discov. 2014, 4, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeldt, M.; O’Prey, J.; Morton, J.P.; Nixon, C.; Mackay, G.; Mrowinska, A.; Au, A.; Rai, T.S.; Zheng, L.; Ridgway, R.; et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature 2013, 504, 296–300. [Google Scholar] [CrossRef]
- Iacobuzio-Donahue, C.; Herman, J.M. Autophagy, p53, and Pancreatic Cancer. N. Engl. J. Med. 2014, 370, 1352–1353. [Google Scholar] [CrossRef]
- Levine, B.; Abrams, J.M. p53: The Janus of autophagy? Nature 2008, 10, 637–639. [Google Scholar] [CrossRef]
- Tang, J.; Di, J.; Cao, H.; Bai, J.; Zheng, J. p53-mediated autophagic regulation: A prospective strategy for cancer therapy. Cancer Lett. 2015, 363, 101–107. [Google Scholar] [CrossRef]
- Dall’Armi, C.; Hurtado-Lorenzo, A.; Tian, H.; Morel, E.; Nezu, A.; Chan, R.B.; Yu, W.H.; Robinson, K.S.; Yeku, O.; Small, S.A.; et al. The phospholipase D1 pathway modulates macroautophagy. Nat. Commun. 2010, 1, 1–11. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Saqcena, M.; Chatterjee, A.; Garcia, A.; Frías, M.A.; Foster, D.A. Reciprocal Regulation of AMP-activated Protein Kinase and Phospholipase D*. J. Boil. Chem. 2015, 290, 6986–6993. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.H.; Wang, Z.; Tkach, D.; Toral-Barza, L.; Ugwonali, S.; Liu, S.; Fitzgerald, S.L.; George, E.; Frias, E.; Cochran, N.; et al. Macroautophagy is dispensable for growth of KRAS mutant tumors and chloroquine efficacy. Proc. Natl. Acad. Sci. USA 2015, 113, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Nyfeler, B.; Eng, C.H. Revisiting autophagy addiction of tumor cells. Autophagy 2016, 12, 1206–1207. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Gamez, G.; Menke, C.; Hernandez, A.; Thorburn, J.; Gidan, F.; Staskiewicz, L.; Morgan, S.; Cummings, C.; Maycotte, P.; et al. Regulation of autophagy and chloroquine sensitivity by oncogenic RAS in vitro is context-dependent. Autophagy 2014, 10, 1814–1826. [Google Scholar] [CrossRef]
- Zhao, Y.G.; Zhao, H.; Miao, L.; Wang, L.; Sun, F.; Zhang, H. The p53-induced GeneEi24Is an Essential Component of the Basal Autophagy Pathway. J. Boil. Chem. 2012, 287, 42053–42063. [Google Scholar] [CrossRef]
- Galluzzi, L.; Pietrocola, F.; Pedro, J.M.B.-S.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; A Gewirtz, D.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef]
- Hwang, M.; Jun, D.W.; Kang, E.H.; Yoon, K.-A.; Cheong, H.; Kim, Y.-H.; Lee, C.-H.; Kim, S. EI24, as a Component of Autophagy, Is Involved in Pancreatic Cell Proliferation. Front. Oncol. 2019, 9, 652. [Google Scholar] [CrossRef]
- Binimetinib and Hydroxychloroquine in Treating Patients With KRAS Mutant Metastatic Pancreatic Cancer. 2019. Available online: https://clinicaltrials.gov/ct2/show/NCT04132505?term=NCT04132505&draw=2&rank=1 (accessed on 17 July 2020).
- Wolpin, B.M.; Rubinson, D.A.; Wang, X.; Chan, J.A.; Cleary, J.M.; Enzinger, P.C.; Fuchs, C.S.; McCleary, N.J.; Meyerhardt, J.A.; Ng, K.; et al. Phase II and Pharmacodynamic Study of Autophagy Inhibition Using Hydroxychloroquine in Patients With Metastatic Pancreatic Adenocarcinoma. Oncologist 2014, 19, 637–638. [Google Scholar] [CrossRef]
- Jutten, B.; Rouschop, K. EGFR signaling and autophagy dependence for growth, survival, and therapy resistance. Cell Cycle 2013, 13, 42–51. [Google Scholar] [CrossRef]
- Bryant, K.L.; Stalnecker, C.A.; Zeitouni, D.; Klomp, J.E.; Peng, S.; Tikunov, A.P.; Gunda, V.; Pierobon, M.; Waters, A.M.; George, S.D.; et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med. 2019, 25, 628–640. [Google Scholar] [CrossRef]
- Kinsey, C.G.; Camolotto, S.A.; Boespflug, A.; Guillen, K.P.; Foth, M.; Truong, A.; Schuman, S.S.; Shea, J.E.; Seipp, M.T.; Yap, J.T.; et al. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med. 2019, 25, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Kang, H.; Zhao, Y.; Min, I.; Wyrwas, B.; Moore, M.; Teng, L.; Zarnegar, R.; Jiang, X.; Fahey, T.J. Targeting Autophagy Sensitizes BRAF-Mutant Thyroid Cancer to Vemurafenib. J. Clin. Endocrinol. Metab. 2016, 102, 634–643. [Google Scholar] [CrossRef] [PubMed]
- Trametinib and Hydroxychloroquine in Treating Patients With Pancreatic Cancer (THREAD). 2019. Available online: https://clinicaltrials.gov/ct2/show/NCT03825289?term=NCT03825289&draw=2&rank=1 (accessed on 17 July 2020).
- Halbrook, C.J.; Lyssiotis, C.A. Employing Metabolism to Improve the Diagnosis and Treatment of Pancreatic Cancer. Cancer Cell 2017, 31, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Kamphorst, J.J.; Mathew, R.; Chung, M.K.; White, E.; Shlomi, T.; Rabinowitz, J.D. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol. Syst. Boil. 2013, 9, 712. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [PubMed]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2011, 481, 380–384. [Google Scholar] [CrossRef]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef]
- Digomann, D.; Kurth, I.; Tyutyunnykova, A.; Chen, O.; Löck, S.; Gorodetska, I.V.; Peitzsch, C.; Skvortsova, I.-I.; Negro, G.; Aschenbrenner, B.; et al. The CD98 Heavy Chain Is a Marker and Regulator of Head and Neck Squamous Cell Carcinoma Radiosensitivity. Clin. Cancer Res. 2019, 25, 3152–3163. [Google Scholar] [CrossRef]
- Lin, T.-C.; Chen, Y.-R.; Kensicki, E.; Li, A.Y.-J.; Kong, M.; Li, Y.; Mohney, R.P.; Shen, H.-M.; Stiles, B.; Mizushima, N.; et al. Autophagy: Resetting glutamine-dependent metabolism and oxygen consumption. Autophagy 2012, 8, 1477–1493. [Google Scholar] [CrossRef]
- Zhang, N.; Yang, X.; Yuan, F.; Zhang, L.; Wang, Y.; Wang, L.; Mao, Z.; Luo, J.; Zhang, H.; Zhu, W.-G.; et al. Increased Amino Acid Uptake Supports Autophagy-Deficient Cell Survival upon Glutamine Deprivation. Cell Rep. 2018, 23, 3006–3020. [Google Scholar] [CrossRef] [PubMed]
- Halama, A.; Kulinski, M.; Dib, S.S.; Zaghlool, S.B.; Siveen, K.S.; Iskandarani, A.; Zierer, J.; Prabhu, K.S.; Satheesh, N.J.; Bhagwat, A.M.; et al. Accelerated lipid catabolism and autophagy are cancer survival mechanisms under inhibited glutaminolysis. Cancer Lett. 2018, 430, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Zhang, L.-Q.; Huang, J.-F.; Liu, K.; Chuai, Z.-R.; Yang, Z.; Wang, Y.-X.; Shi, D.-C.; Liu, Q.; Huang, Q.; et al. BRAF Mutations in Patients with Non-Small Cell Lung Cancer: A Systematic Review and Meta-Analysis. PLoS ONE 2014, 9, e101354. [Google Scholar] [CrossRef] [PubMed]
- Thorburn, A.; Morgan, M.J. Targeting Autophagy in BRAF-Mutant Tumors. Cancer Discov. 2015, 5, 353–354. [Google Scholar] [CrossRef]
- Mancias, J.D.; Kimmelman, A.C. Targeting Autophagy Addiction in Cancer. Oncotarget 2011, 2, 1302–1306. [Google Scholar] [CrossRef]
- Strohecker, A.M.; Guo, J.Y.; Karsli-Uzunbas, G.; Price, S.M.; Chen, G.J.; Mathew, R.; McMahon, M.; White, E. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov. 2013, 3, 1272–1285. [Google Scholar] [CrossRef]
- Karsli-Uzunbas, G.; Guo, J.Y.; Price, S.; Teng, X.; Laddha, S.V.; Khor, S.; Kalaany, N.Y.; Jacks, T.; Chan, C.S.; Rabinowitz, J.D.; et al. Autophagy Is Required for Glucose Homeostasis and Lung Tumor Maintenance. Cancer Discov. 2014, 4, 914–927. [Google Scholar] [CrossRef]
- Goulielmaki, M.; Koustas, E.; Moysidou, E.; Vlassi, M.; Sasazuki, T.; Shirasawa, S.; Zografos, G.; Oikonomou, E.; Pintzas, A. BRAF associated autophagy exploitation: BRAF and autophagy inhibitors synergise to efficiently overcome resistance of BRAF mutant colorectal cancer cells. Oncotarget 2016, 7, 9188–9221. [Google Scholar] [CrossRef]
- Xie, X.; Koh, J.Y.; Price, S.; White, E.; Mehnert, J.M. Atg7 Overcomes Senescence and Promotes Growth of BrafV600E-Driven Melanoma. Cancer Discov. 2015, 5, 410–423. [Google Scholar] [CrossRef]
- Ma, X.-H.; Piao, S.-F.; Dey, S.; McAfee, Q.; Karakousis, G.; Villanueva, J.; Hart, L.S.; Levi, S.M.; Hu, J.; Zhang, G.; et al. Targeting ER stress–induced autophagy overcomes BRAF inhibitor resistance in melanoma. J. Clin. Investig. 2014, 124, 1406–1417. [Google Scholar] [CrossRef] [PubMed]
- A Phase I Trial of Vemurafenib and Hydroxychloroquine in Patients With Advanced BRAF Mutant Melanoma. 2013. Available online: https://clinicaltrials.gov/ct2/show/NCT01897116?term=NCT01897116&draw=2&rank=1 (accessed on 17 July 2020).
- Dabrafenib, Trametinib and Hydroxychloroquine in Patients With Advanced BRAF Mutant Melanoma (BAMM). 2014. Available online: https://clinicaltrials.gov/ct2/show/NCT02257424?term=NCT02257424&draw=2&rank=1 (accessed on 17 July 2020).
- Alves, S.; Castro, L.; Fernandes, M.S.; Francisco, R.; Castro, P.; Priault, M.; Chaves, S.R.; Moyer, M.P.; Oliveira, C.; Seruca, R.; et al. Colorectal cancer-related mutant KRAS alleles function as positive regulators of autophagy. Oncotarget 2015, 6, 30787–30802. [Google Scholar] [CrossRef] [PubMed]
- Lauzier, A.; Normandeau-Guimond, J.; Vaillancourt-Lavigueur, V.; Boivin, V.; Charbonneau, M.; Rivard, N.; Scott, M.S.; Dubois, C.M.; Jean, S. Colorectal cancer cells respond differentially to autophagy inhibition in vivo. Sci. Rep. 2019, 9, 11316. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-S.; Lee, L.C.; Yuan, T.L.; Chakka, S.; Fellman, C.; Lowe, S.W.; Caplen, N.J.; McCormick, F.; Luo, J.-C. MAP kinase and autophagy pathways cooperate to maintain RAS mutant cancer cell survival. Proc. Natl. Acad. Sci. USA 2019, 116, 4508–4517. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.L.; Lin, H.H.; Kim, K.J.; Lin, A.; Forman, H.J.; Ann, D.K. Novel roles for protein kinase Cdelta-dependent signaling pathways in acute hypoxic stress-induced autophagy. J. Biol. Chem. 2008, 283, 34432–34444. [Google Scholar] [CrossRef]
- Vasilevskaya, I.A.; Selvakumaran, M.; Roberts, D.; O’Dwyer, P.J. JNK1 Inhibition Attenuates Hypoxia-Induced Autophagy and Sensitizes to Chemotherapy. Mol. Cancer Res. 2016, 14, 753–763. [Google Scholar] [CrossRef] [PubMed]
- FOLFOX/Bevacizumab/Hydroxychloroquine (HCQ) in Colorectal Cancer. 2019. Available online: https://clinicaltrials.gov/ct2/show/study/NCT01206530?term=hydroxychloroquine&cond=colorectal+cancer&draw=2&rank=4 (accessed on 17 July 2020).
- Sardiello, M.; Palmieri, M.; Di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A Gene Network Regulating Lysosomal Biogenesis and Function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef]
- Giatromanolaki, A.; Kalamida, D.; Sivridis, E.; Karagounis, I.V.; Gatter, K.C.; Harris, A.L.; Koukourakis, M.I. Increased expression of transcription factor EB (TFEB) is associated with autophagy, migratory phenotype and poor prognosis in non-small cell lung cancer. Lung Cancer 2015, 90, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.-M.; Li, B.; Guan, J.-J.; Xu, H.-D.; Shen, G.-H.; Gao, Q.-G.; Qin, Z.-H. Transcription factor EB is involved in autophagy-mediated chemoresistance to doxorubicin in human cancer cells. Acta Pharmacol. Sin. 2017, 38, 1305–1316. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Wei, S.; Gan, B.; Peng, X.; Zou, W.; Guan, J.-L. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev. 2011, 25, 1510–1527. [Google Scholar] [CrossRef]
- Huo, Y.; Cai, H.; Teplova, I.; Bowman-Colin, C.; Chen, G.; Price, S.; Barnard, N.; Ganesan, S.; Karantza, V.; White, E.; et al. Autophagy opposes p53-mediated tumor barrier to facilitate tumorigenesis in a model of PALB2-associated hereditary breast cancer. Cancer Discov. 2013, 3, 894–907. [Google Scholar] [CrossRef] [PubMed]
- Maycotte, P.; Thorburn, A. Targeting autophagy in breast cancer. World J. Clin. Oncol. 2014, 5, 224–240. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.S.; Giehl, N.; Wu, Y.; Vadgama, J.V. STAT3 activation in HER2-overexpressing breast cancer promotes epithelial-mesenchymal transition and cancer stem cell traits. Int. J. Oncol. 2013, 44, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Marotta, L.L.C.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(-) stem cell-like breast cancer cells in human tumors. J. Clin. Investig. 2011, 121, 2723–2735. [Google Scholar] [CrossRef] [PubMed]
- Keulers, T.G.; Schaaf, M.B.E.; Rouschop, K. Autophagy-Dependent Secretion: Contribution to Tumor Progression. Front. Oncol. 2016, 6, 251. [Google Scholar] [CrossRef] [PubMed]
- Maycotte, P.; Jones, K.L.; Goodall, M.L.; Thorburn, J.; Thorburn, A. Autophagy Supports Breast Cancer Stem Cell Maintenance by Regulating IL6 Secretion. Mol. Cancer Res. 2015, 13, 651–658. [Google Scholar] [CrossRef]
- Lieblein, J.C.; Ball, S.; Hutzen, B.J.; Kate, S.A.; Lin, H.-J.L.; Huang, T.H.-M.; Hall, B.M.; Lin, L. STAT3 can be activated through paracrine signaling in breast epithelial cells. BMC Cancer 2008, 8, 302. [Google Scholar] [CrossRef]
- Maycotte, P.; Gearheart, C.M.; Barnard, R.; Aryal, S.; Levy, J.M.M.; Fosmire, S.P.; Hansen, R.J.; Morgan, M.J.; Porter, C.C.; Gustafson, D.L.; et al. STAT3-mediated autophagy dependence identifies subtypes of breast cancer where autophagy inhibition can be efficacious. Cancer Res. 2014, 74, 2579–2590. [Google Scholar] [CrossRef]
- Janser, F.A.; Tschan, M.P.; Langer, R.; A Janser, F. The role of autophagy in HER2-targeted therapy. Swiss Med. Wkly. 2019, 149, w20138. [Google Scholar] [CrossRef]
- Chen, S.; Li, X.; Feng, J.; Chang, Y.; Wang, Z.; Wen, A.-D. Autophagy facilitates the Lapatinib resistance of HER2 positive breast cancer cells. Med. Hypotheses 2011, 77, 206–208. [Google Scholar] [CrossRef]
- Aveic, S.; Tonini, G.P. Resistance to receptor tyrosine kinase inhibitors in solid tumors: Can we improve the cancer fighting strategy by blocking autophagy? Cancer Cell Int. 2016, 16, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Cufí, S.; Vázquez-Martín, A.; Oliveras-Ferraros, C.; Corominas-Faja, B.; Urruticoechea, A.; Martin-Castilló, B.; Menendez, J.A. Autophagy-related gene 12 (ATG12) is a novel determinant of primary resistance to HER2-targeted therapies: Utility of transcriptome analysis of the autophagy interactome to guide breast cancer treatment. Oncotarget 2012, 3, 1600–1614. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, C.E.; Reidel, S.I.; Joffé, E.D.B.D.K.; Jasnis, M.A.; Fiszman, G.L. Autophagy Protects from Trastuzumab-Induced Cytotoxicity in HER2 Overexpressing Breast Tumor Spheroids. PLoS ONE 2015, 10, e0137920. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Martín, A.; Oliveras-Ferraros, C.; Menendez, J.A. Autophagy Facilitates the Development of Breast Cancer Resistance to the Anti-HER2 Monoclonal Antibody Trastuzumab. PLoS ONE 2009, 4, e6251. [Google Scholar] [CrossRef]
- Cufí, S.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Corominas-Faja, B.; Cuyàs, E.; Lopez-Bonet, E.; Martin-Castilló, B.; Joven, J.; Menendez, J.A. The anti-malarial chloroquine overcomes Primary resistance and restores sensitivity to Trastuzumab in HER2-positive breast cancer. Sci. Rep. 2013, 3, 2469. [Google Scholar] [CrossRef]
- Janser, F.A.; Adams, O.; Bütler, V.; Schläfli, A.M.; Dislich, B.; Seiler, C.A.; Kröll, D.; Langer, R.; Tschan, M.P. Her2-Targeted Therapy Induces Autophagy in Esophageal Adenocarcinoma Cells. Int. J. Mol. Sci. 2018, 19, 3069. [Google Scholar] [CrossRef]
- Negri, T.; Tarantino, E.; Orsenigo, M.; Reid, J.F.; Gariboldi, M.; Zambetti, M.; Pierotti, M.A.; Pilotti, S. Chromosome band 17q21 in breast cancer: Significant association between beclin 1 loss and HER2/NEU amplification. Genes Chromosom. Cancer 2010, 49, 901–909. [Google Scholar] [CrossRef]
- Han, J.; Hou, W.; Lu, C.; Goldstein, L.A.; Stolz, D.B.; Watkins, S.C.; Rabinowich, H. Interaction between Her2 and Beclin-1 Proteins Underlies a New Mechanism of Reciprocal Regulation. J. Boil. Chem. 2013, 288, 20315–20325. [Google Scholar] [CrossRef]
- Autophagy Inhibition Using Hydrochloroquine in Breast Cancer Patients. 2011. Available online: https://clinicaltrials.gov/ct2/show/NCT01292408?term=NCT01292408&draw=2&rank=1 (accessed on 17 July 2020).
- A Phase 2 Randomized, Double-blind Trial Evaluating the Effects of Chloroquine in Breast Cancer (CUBiC). 2015. Available online: https://clinicaltrials.gov/ct2/show/NCT02333890?term=NCT02333890&draw=2&rank=1 (accessed on 17 July 2020).
- Study of the Efficacy of Chloroquine in the Treatment of Ductal Carcinoma in Situ (The PINC Trial). 2009. Available online: https://clinicaltrials.gov/ct2/show/NCT01023477?term=NCT01023477&draw=2&rank=1 (accessed on 17 July 2020).
- Chloroquine With Taxane Chemotherapy for Advanced or Metastatic Breast Cancer After Anthracycline Failure (CAT) (CAT). 2011. Available online: https://clinicaltrials.gov/ct2/show/NCT01446016?term=NCT01446016&draw=2&rank=1 (accessed on 17 July 2020).
- CLEVER Pilot Trial: A Phase II Pilot Trial of HydroxyChLoroquine, EVErolimus or the Combination for Prevention of Recurrent Breast Cancer. 2017. Available online: https://clinicaltrials.gov/ct2/show/NCT03032406?term=NCT03032406&draw=2&rank=1 (accessed on 17 July 2020).
- Gedatolisib, Hydroxychloroquine or the Combination for Prevention of Recurrent Breast Cancer (“GLACIER”) (GLACIER). 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT03400254?term=NCT03400254&draw=2&rank=1 (accessed on 17 July 2020).
- Cook, K.L.; Shajahan, A.N.; Clarke, R. Autophagy and endocrine resistance in breast cancer. Expert Rev. Anticancer. Ther. 2011, 11, 1283–1294. [Google Scholar] [CrossRef]
- Samaddar, J.S.; Gaddy, V.T.; Duplantier, J.; Thandavan, S.P.; Shah, M.; Smith, M.J.; Browning, D.D.; Rawson, J.V.; Smith, S.B.; Barrett, J.T.; et al. A role for macroautophagy in protection against 4-hydroxytamoxifen-induced cell death and the development of antiestrogen resistance. Mol. Cancer Ther. 2008, 7, 2977–2987. [Google Scholar] [CrossRef]
- Crawford, A.C.; Riggins, R.B.; Shajahan, A.N.; Zwart, A.; Clarke, R. Co-Inhibition of BCL-W and BCL2 Restores Antiestrogen Sensitivity through BECN1 and Promotes an Autophagy-Associated Necrosis. PLoS ONE 2010, 5, e8604. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-H.; Koh, D.; Na, H.; Ka, N.-L.; Kim, S.; Kim, H.-J.; Hong, S.; Shin, Y.K.; Seong, J.K.; Lee, M.-O. MTA1 is a novel regulator of autophagy that induces tamoxifen resistance in breast cancer cells. Autophagy 2018, 14, 812–824. [Google Scholar] [CrossRef] [PubMed]
- Vijayaraghavan, S.; Karakas, C.; Doostan, I.; Chen, X.; Bui, T.; Yi, M.; Raghavendra, A.S.; Zhao, Y.; Bashour, S.I.; Ibrahim, N.K.; et al. CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin E negative cancers. Nat. Commun. 2017, 8, 15916. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, M.; Perna, E.; Tronnolone, S.; Colecchia, D.; Chiariello, M. Activated kinase screening identifies the IKBKE oncogene as a positive regulator of autophagy. Autophagy 2018, 15, 312–326. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Long, Y.-H.; Wang, S.-Q.; Zhang, Y.-Y.; Li, Y.-F.; Mi, J.-S.; Yu, C.-H.; Li, D.-Y.; Zhang, J.-H.; Zhang, X.-J. JMJD6 regulates histone H2A.X phosphorylation and promotes autophagy in triple-negative breast cancer cells via a novel tyrosine kinase activity. Oncogene 2018, 38, 980–997. [Google Scholar] [CrossRef]
- Aveic, S.; Pantile, M.; Polo, P.; Sidarovich, V.; De Mariano, M.; Quattrone, A.; Longo, L.; Tonini, G.P. Autophagy inhibition improves the cytotoxic effects of receptor tyrosine kinase inhibitors. Cancer Cell Int. 2018, 18, 63. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Hino, H.; Moriya, S.; Kazama, H.; Miyazaki, M.; Takano, N.; Hiramoto, M.; Tsukahara, K.; Miyazawa, K. Comparison of autophagy inducibility in various tyrosine kinase inhibitors and their enhanced cytotoxicity via inhibition of autophagy in cancer cells in combined treatment with azithromycin. Biochem. Biophys. Rep. 2020, 22, 100750. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Pan, H.; Chen, Y.; Sun, J.; Wang, Y.; Li, J.; Ge, W.; Feng, L.; Lin, X.; Wang, X.; et al. EGFR Tyrosine Kinase Inhibitors Activate Autophagy as a Cytoprotective Response in Human Lung Cancer Cells. PLoS ONE 2011, 6, e18691. [Google Scholar] [CrossRef]
- Sugita, S.; Ito, K.; Yamashiro, Y.; Moriya, S.; Che, X.-F.; Yokoyama, T.; Hiramoto, M.; Miyazawa, K. EGFR-independent autophagy induction with gefitinib and enhancement of its cytotoxic effect by targeting autophagy with clarithromycin in non-small cell lung cancer cells. Biochem. Biophys. Res. Commun. 2015, 461, 28–34. [Google Scholar] [CrossRef]
- Kang, M.; Lee, K.-H.; Lee, H.S.; Jeong, C.W.; Kwak, C.; Kim, H.H.; Ku, J.H. Concurrent Autophagy Inhibition Overcomes the Resistance of Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Human Bladder Cancer Cells. Int. J. Mol. Sci. 2017, 18, 321. [Google Scholar] [CrossRef]
- Kwon, Y.; Kim, M.; Jung, H.S.; Kim, Y.; Jeoung, D. Targeting Autophagy for Overcoming Resistance to Anti-EGFR Treatments. Cancers 2019, 11, 1374. [Google Scholar] [CrossRef] [PubMed]
- Bokobza, S.M.; Jiang, Y.; Weber, A.M.; Devery, A.M.; Ryan, A.J. Combining AKT inhibition with chloroquine and gefitinib prevents compensatory autophagy and induces cell death in EGFR mutated NSCLC cells. Oncotarget 2014, 5, 4765–4778. [Google Scholar] [CrossRef] [PubMed]
- Erlotinib With or Without Hydroxychloroquine in Chemo-Naive Advanced NSCLC and (EGFR) Mutations. 2009. Available online: https://clinicaltrials.gov/ct2/show/NCT00977470?term=NCT00977470&draw=2&rank=1 (accessed on 17 July 2020).
- Guo, J.Y.; Karsli-Uzunbas, G.; Mathew, R.; Aisner, S.C.; Kamphorst, J.J.; Strohecker, A.M.; Chen, G.; Price, S.; Lu, W.; Teng, X.; et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013, 27, 1447–1461. [Google Scholar] [CrossRef]
- Guo, J.Y.; White, E. Autophagy is required for mitochondrial function, lipid metabolism, growth, and fate of KRASG12D-driven lung tumors. Autophagy 2013, 9, 1636–1638. [Google Scholar] [CrossRef] [PubMed]
- El Osta, B.; Behera, M.; Kim, S.; Berry, L.D.; Sica, G.; Pillai, R.N.; Owonikoko, T.K.; Kris, M.G.; Johnson, B.E.; Kwiatkowski, D.J.; et al. Characteristics and Outcomes of Patients With Metastatic KRAS-Mutant Lung Adenocarcinomas: The Lung Cancer Mutation Consortium Experience. J. Thorac. Oncol. 2019, 14, 876–889. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.; Iwakawa, R.; Takahashi, K.; Kohno, T.; Nakanishi, Y.; Matsuno, Y.; Suzuki, K.; Nakamoto, M.; Shimizu, E.; Minna, J.D.; et al. Prevalence and specificity of LKB1 genetic alterations in lung cancers. Oncogene 2007, 26, 5911–5918. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, I.; Lim, A.L.; Laderoute, K.; Denko, N.C. Hypoxia signals autophagy in tumor cells via AMPK activity, independent of HIF-1, BNIP3, and BNIP3L. Cell Death Differ. 2008, 15, 1572–1581. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.Y.; White, E. Autophagy, metabolism, and cancer. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: Woodbury, NY, USA, 2016; Volume 81, pp. 73–78. [Google Scholar]
- Bhatt, V.; Khayati, K.; Hu, Z.S.; Lee, A.; Kamran, W.; Su, X.; Guo, J.Y. Autophagy modulates lipid metabolism to maintain metabolic flexibility for Lkb1-deficient Kras-driven lung tumorigenesis. Genes Dev. 2019, 33, 150–165. [Google Scholar] [CrossRef]
- Huang, X.; Gan, G.; Wang, X.; Xu, T.; Xie, W. The HGF-MET axis coordinates liver cancer metabolism and autophagy for chemotherapeutic resistance. Autophagy 2019, 15, 1258–1279. [Google Scholar] [CrossRef]
- Lu, S.; Yao, Y.; Xu, G.; Zhou, C.; Zhang, Y.; Sun, J.; Jiang, R.; Shao, Q.; Chen, Y. CD24 regulates sorafenib resistance via activating autophagy in hepatocellular carcinoma. Cell Death Dis. 2018, 9, 646. [Google Scholar] [CrossRef]
- Santanam, U.; Banach-Petrosky, W.; Abate-Shen, C.; Shen, M.M.; White, E.; DiPaola, R.S. Atg7 cooperates with Pten loss to drive prostate cancer tumor growth. Genes Dev. 2016, 30, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Gaughan, L.; Stockley, J.; Coffey, K.; O’Neill, D.; Jones, D.L.; Wade, M.; Wright, J.; Moore, M.; Tse, S.; Rogerson, L.; et al. KDM4B is a Master Regulator of the Estrogen Receptor Signalling Cascade. Nucleic Acids Res. 2013, 41, 6892–6904. [Google Scholar] [CrossRef] [PubMed]
- Coffey, K.; Rogerson, L.; Ryan-Munden, C.; Alkharaif, D.; Stockley, J.; Heer, R.; Sahadevan, K.; O’Neill, D.; Jones, D.; Darby, S.; et al. The lysine demethylase, KDM4B, is a key molecule in androgen receptor signalling and turnover. Nucleic Acids Res. 2013, 41, 4433–4446. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.J.; Harris, A.L.; Davidoff, A.M. Hypoxia and Hormone-Mediated Pathways Converge at the Histone Demethylase KDM4B in Cancer. Int. J. Mol. Sci. 2018, 19, 240. [Google Scholar] [CrossRef] [PubMed]
- Sha, J.; Han, Q.; Chi, C.; Zhu, Y.; Pan, J.; Dong, B.; Huang, Y.; Xia, W.; Xue, W. Upregulated KDM4B promotes prostate cancer cell proliferation by activating autophagy. J. Cell. Physiol. 2019, 235, 2129–2138. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Wang, W.; Li, Y.; Yang, D.; Li, X.; Shen, C.; Liu, Y.; Ke, X.; Guo, S.; Guo, Z. HSP90AA1-mediated autophagy promotes drug resistance in osteosarcoma. J. Exp. Clin. Cancer Res. 2018, 37, 201. [Google Scholar] [CrossRef]
- Jutten, B.; Keulers, T.G.; Schaaf, M.B.; Savelkouls, K.; Theys, J.; Span, P.N.; Vooijs, M.; Bussink, J.; Rouschop, K. EGFR overexpressing cells and tumors are dependent on autophagy for growth and survival. Radiother. Oncol. 2013, 108, 479–483. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbeau, L.M.O.; Keulers, T.G.H.; Rouschop, K.M.A. Tumors Responsive to Autophagy-Inhibition: Identification and Biomarkers. Cancers 2020, 12, 2463. https://doi.org/10.3390/cancers12092463
Barbeau LMO, Keulers TGH, Rouschop KMA. Tumors Responsive to Autophagy-Inhibition: Identification and Biomarkers. Cancers. 2020; 12(9):2463. https://doi.org/10.3390/cancers12092463
Chicago/Turabian StyleBarbeau, Lydie M.O., Tom G.H. Keulers, and Kasper M.A. Rouschop. 2020. "Tumors Responsive to Autophagy-Inhibition: Identification and Biomarkers" Cancers 12, no. 9: 2463. https://doi.org/10.3390/cancers12092463
APA StyleBarbeau, L. M. O., Keulers, T. G. H., & Rouschop, K. M. A. (2020). Tumors Responsive to Autophagy-Inhibition: Identification and Biomarkers. Cancers, 12(9), 2463. https://doi.org/10.3390/cancers12092463