Genetic Susceptibility to Endometrial Cancer: Risk Factors and Clinical Management


Abstract
1. Risk Factors, Biology and Genetics
1.1. Epidemiology
1.2. Biology
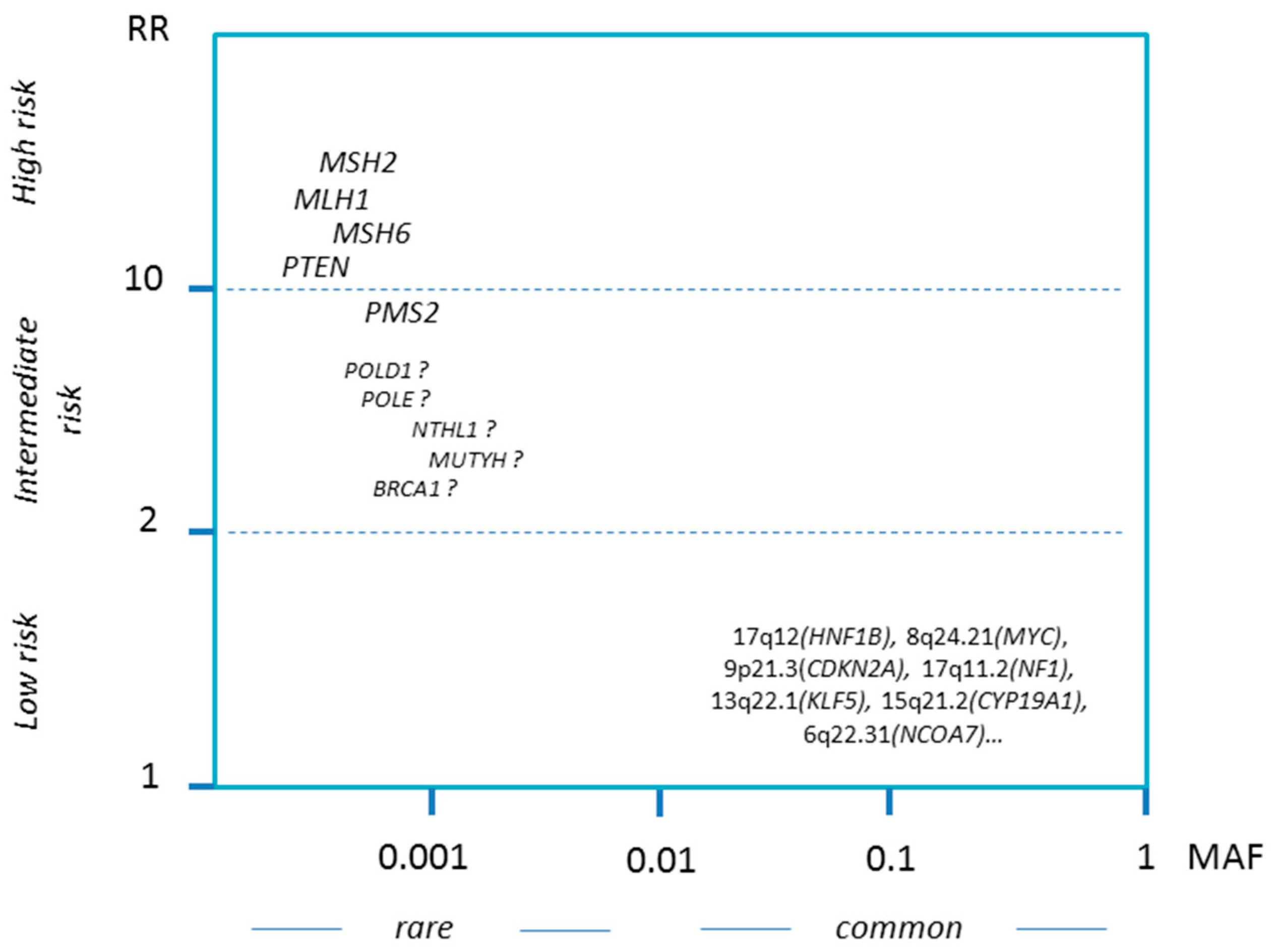
1.3. Genetic Predisposition
1.3.1. Spectrum of Hereditary Factors
1.3.2. Lynch Syndrome
1.3.3. Cowden Syndrome
1.3.4. Further Candidate Genes
POLE and POLD1
MUTYH
NTHL1
BRCA1
1.3.5. Low-Penetrance Susceptibility Loci
2. Clinical Implications and Management
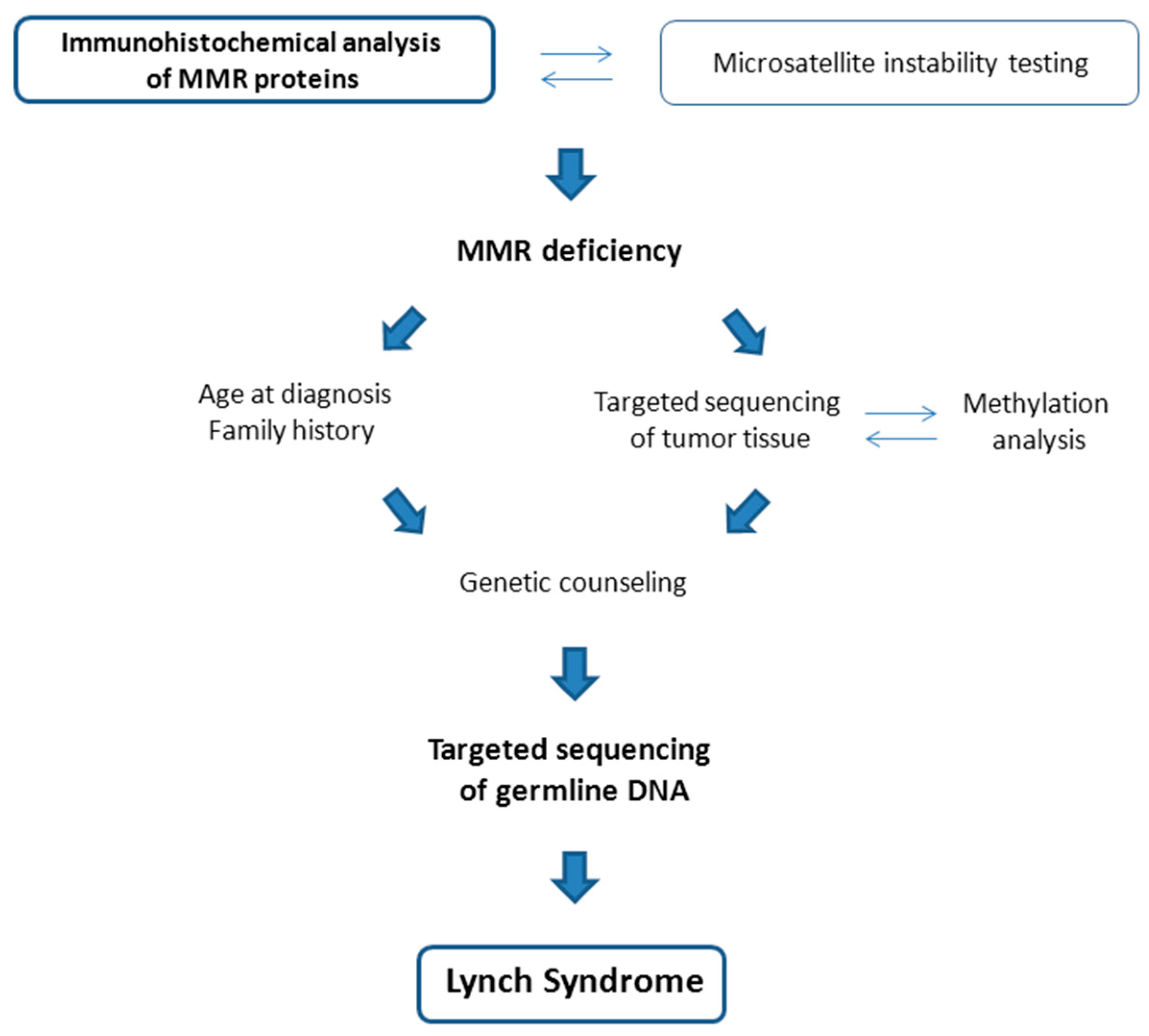
2.1. Screening for Hereditary Syndromes among Patients with Endometrial Cancer
2.2. Gynecologic Surveillance in Families with Hereditary EC
2.3. Prophylactic Surgery
2.4. MSI and Defective Mismatch Repair as Therapeutic Target in Mutation Carriers with EC
3. Conclusions
Funding
Conflicts of Interest
References
- Olson, S.H.; Jewell, E.; Rastogi, R.M. Endometrial Cancer. In Women and Health, 2nd ed.; Academic Press: Cambridge, MA, USA; Elsevier: Amsterdam, The Netherlands, 2013; pp. 1147–1158. ISBN 978-0-12-384978-6. [Google Scholar] [CrossRef]
- Amant, F.; Moerman, P.; Neven, P.; Timmerman, D.; van Limbergen, E.; Vergote, I. Endometrial cancer. Lancet 2005, 366, 491–505. [Google Scholar] [CrossRef]
- Morice, P.; Leary, A.; Creutzberg, C.; Abu-Rustum, N.; Darai, E. Endometrial cancer. Lancet 2016, 387, 1094–1108. [Google Scholar] [CrossRef]
- Shai, A.; Segev, Y.; Narod, S. Genetics of endometrial cancer. Fam. Cancer 2014, 13, 499–505. [Google Scholar] [CrossRef]
- Benedet, J.L.; Miller, D.M. Gynaecologic Cancer. In UICC International Union against Cancer Manual of Clinical Oncology, 7th ed.; Pollack, R.E., Ed.; Wiley: New York, NY, USA, 1999; pp. 537–562. [Google Scholar]
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of US adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef]
- Reeves, G.K.; Pirie, K.; Beral, V.; Green, J.; Spencer, E.; Bull, D.; Million Women Study Collaboration. Cancer incidence and mortality in relation to body mass index in the million women study: Cohort study. BMJ 2007, 335, 1134. [Google Scholar] [CrossRef]
- Kaitu’u-Lino, T.J.; Ye, L.; Gargett, C.E. Reepithelialization of the uterine surface arises from endometrial glands: Evidence from a functional mouse model of breakdown and repair. Endocrinology 2010, 151, 3386–3395. [Google Scholar] [CrossRef]
- Gargett, C.E.; Schwab, K.E.; Deane, J.A. Endometrial stem/progenitor cells: The first 10 years. Hum. Reprod. Update 2016, 22, 137–163. [Google Scholar] [CrossRef]
- Sherman, M.E.; Sturgeon, S.; Brinton, L.A.; Potischman, N.; Kurman, R.J.; Berman, M.L.; Mortel, R.; Twiggs, L.B.; Barrett, R.J.; Wilbanks, G.D. Risk factors and hormone levels in patients with serous and endometrioid uterine carcinomas. Mod. Pathol. 1997, 10, 963–968. [Google Scholar]
- Setiawan, V.W.; Yang, H.P.; Pike, M.C.; McCann, S.E.; Yu, H.; Xiang, Y.B.; Wolk, A.; Wentzensen, N.; Weiss, N.S.; Webb, P.M.; et al. Type I and II endometrial cancers: Have they different risk factors? J. Clin. Oncol. 2013, 31, 2607–2618. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network; Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef]
- Suda, K.; Nakaoka, H.; Yoshihara, K.; Ishiguro, T.; Tamura, R.; Mori, Y.; Yamawaki, K.; Adachi, S.; Takahashi, T.; Kase, H.; et al. Clonal Expansion and Diversification of Cancer-Associated Mutations in Endometriosis and Normal Endometrium. Cell Rep. 2018, 24, 1777–1789. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Dou, Y.; Kawaler, E.A.; Cui Zhou, D.; Gritsenko, M.A.; Huang, C.; Blumenberg, L.; Karpova, A.; Petyuk, V.A.; Savage, S.R.; Satpathy, S.; et al. Clinical Proteomic Tumor Analysis Consortium. Proteogenomic Characterization of Endometrial Carcinoma. Cell 2020, 180, 729–748.e26. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.; Leongamornlert, D.; Coorens, T.; Sanders, M.A.; Ellis, P.; Dentro, S.C.; Dawson, K.J.; Butler, T.; Rahbari, R.; Mitchell, T.J.; et al. The mutational landscape of normal human endometrial epithelium. Nature 2020, 580, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Bae-Jump, V.L.; Levine, D.A. A catalogue of cancer-driving mutations in healthy tissue. Nature 2020, 580, 595–596. [Google Scholar] [CrossRef]
- McKenzie, R.; Scott, R.J.; Otton, G.; Scurry, J. Early changes of endometrial neoplasia revealed by loss of mismatch repair gene protein expression in a patient diagnosed with Lynch syndrome. Pathology 2016, 48, 78–80. [Google Scholar] [CrossRef]
- Win, A.K.; Reece, J.C.; Ryan, S. Family history and risk of endometrial cancer: A systematic review and meta-analysis. Obstet. Gynecol. 2015, 125, 89–98. [Google Scholar] [CrossRef]
- Johnatty, S.E.; Tan, Y.Y.; Buchanan, D.D.; Bowman, M.; Walters, R.J.; Obermair, A.; Quinn, M.A.; Blomfield, P.B.; Brand, A.; Leung, Y.; et al. Family history of cancer predicts endometrial cancer risk independently of Lynch Syndrome: Implications for genetic counselling. Gynecol. Oncol. 2017, 147, 381–387. [Google Scholar] [CrossRef]
- Lichtenstein, P.; Holm, N.V.; Verkasalo, P.K.; Iliadou, A.; Kaprio, J.; Koskenvuo, M.; Pukkala, E.; Skytthe, A.; Hemminki, K. Environmental and heritable factors in the causation of cancer–analyses of cohorts of twins from Sweden, Denmark, and Finland. N. Engl. J. Med. 2000, 343, 78–85. [Google Scholar] [CrossRef]
- Lu, Y.; Ek, W.E.; Whiteman, D.; Vaughan, T.L.; Spurdle, A.B.; Easton, D.F.; Pharoah, P.D.; Thompson, D.J.; Dunning, A.M.; Hayward, N.K.; et al. Most common ‘sporadic’ cancers have a significant germline genetic component. Hum. Mol. Genet. 2014, 23, 6112–6118. [Google Scholar] [CrossRef]
- Mucci, L.A.; Hjelmborg, J.B.; Harris, J.R.; Czene, K.; Havelick, D.J.; Scheike, T.; Graff, R.E.; Holst, K.; Möller, S.; Unger, R.H.; et al. Familial risk and heritability of cancer among twins in Nordic countries. J. Am. Med. Assoc. 2016, 315, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Lynch, H.T.; Lynch, P.M.; Lanspa, S.J.; Snyder, C.L.; Lynch, J.F.; Boland, C.R. Review of the Lynch syndrome: History, molecular genetics, screening, differential diagnosis, and medicolegal ramifications. Clin. Genet. 2009, 76, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Resnick, K.E.; Hampel, H.; Fishel, R.; Cohn, D.E. Current and emerging trends in Lynch syndrome identification in women with endometrial cancer. Gynecol. Oncol. 2009, 114, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Spurdle, A.B.; Bowman, M.A.; Shamsani, J.; Kirk, J. Endometrial cancer gene panels: Clinical diagnostic vs research germline DNA testing. Mod. Pathol. 2017, 30, 1048–1068. [Google Scholar] [CrossRef] [PubMed]
- O’Mara, T.A.; Glubb, D.M.; Kho, P.F.; Thompson, D.J.; Spurdle, A.B. Genome-Wide Association Studies of Endometrial Cancer: Latest Developments and Future Directions. Cancer Epidemiol. Biomark. Prev. 2019, 28, 1095–1102. [Google Scholar] [CrossRef]
- Cerretelli, G.; Ager, A.; Arends, M.J.; Frayling, I.M. Molecular pathology of Lynch syndrome. J. Pathol. 2020, 250, 518–531. [Google Scholar] [CrossRef]
- Warthin, A.S. Heredity with reference to carcinoma. Arch. Intern. Med. 1913, 12, 546–555. [Google Scholar] [CrossRef]
- Lynch, H.T.; Shaw, M.W.; Magnuson, C.W.; Larsen, A.L.; Krush, A.J. Hereditary factors in cancer: Study of two large Midwestern kindreds. Arch. Intern. Med. 1966, 117, 206–212. [Google Scholar] [CrossRef]
- Møller, P.; Seppälä, T.T.; Bernstein, I.; Holinski-Feder, E.; Sala, P.; Gareth Evans, D.; Lindblom, A.; Macrae, F.; Blanco, I.; Sijmons, R.H.; et al. Cancer risk and survival in path_MMR carriers by gene and gender up to 75 years of age: A report from the prospective Lynch syndrome database. Gut 2018, 67, 1306–1316. [Google Scholar] [CrossRef]
- Dominguez-Valentin, M.; Sampson, J.R.; Seppälä, T.T.; Ten Broeke, S.W.; Plazzer, J.P.; Nakken, S.; Engel, C.; Aretz, S.; Jenkins, M.A.; Sunde, L.; et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: Findings from the prospective Lynch syndrome database. Genet. Med. 2020, 22, 15–25. [Google Scholar] [CrossRef]
- Joehlin-Price, A.S.; Perrino, C.M.; Stephens, J.; Backes, F.J.; Goodfellow, P.J.; Cohn, D.E.; Suarez, A.A. Mismatch repair protein expression in 1049 endometrial carcinomas, associations with body mass index, and other clinicopathologic variables. Gynecol. Oncol. 2014, 133, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Modrich, P.; Lahue, R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu. Rev. Biochem. 1996, 65, 101–133. [Google Scholar] [CrossRef] [PubMed]
- Preston, B.D.; Albertson, T.M.; Herr, A.J. DNA replication fidelity and cancer. Semin. Cancer Biol. 2010, 20, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.D.; Rathi, A.; Kamath, R.; Beardsley, D.I.; Zhan, Q.; Mannino, J.L.; Baskaran, R. The mismatch repair system is required for S-phase checkpoint activation. Nat. Genet. 2003, 33, 80–84. [Google Scholar] [CrossRef]
- Stojic, L.; Brun, R.; Jiricny, J. Mismatch repair and DNA damage signalling. DNA Repair 2004, 3, 1091–1101. [Google Scholar] [CrossRef]
- Yoshioka, K.; Yoshioka, Y.; Hsieh, P. ATR kinase activation mediated by MutSalpha and MutLalpha in response to cytotoxic O6-methylguanine adducts. Mol. Cell 2006, 22, 501–510. [Google Scholar] [CrossRef]
- Buchanan, D.D.; Tan, Y.Y.; Walsh, M.D.; Clendenning, M.; Metcalf, A.M.; Ferguson, K.; Arnold, S.T.; Thompson, B.A.; Lose, F.A.; Parsons, M.T.; et al. Tumor mismatch repair immunohistochemistry and DNA MLH1 methylation testing of patients with endometrial cancer diagnosed at age younger than 60 years optimizes triage for population-level germline mismatch repair gene mutation testing. J. Clin. Oncol. 2014, 32, 90–100. [Google Scholar] [CrossRef]
- Pasanen, A.; Loukovaara, M.; Bützow, R. Clinicopathological significance of deficient DNA mismatch repair and MLH1 promoter methylation in endometrioid endometrial carcinoma. Mod. Pathol. 2020, 33, 1443–1452. [Google Scholar] [CrossRef]
- Niessen, R.C.; Hofstra, R.M.; Westers, H.; Ligtenberg, M.J.; Kooi, K.; Jager, P.O.; de Groote, M.L.; Dijkhuizen, T.; Olderode-Berends, M.J.; Hollema, H.; et al. Germline hypermethylation of MLH1 and EPCAM deletions are a frequent cause of Lynch syndrome. Genes Chromos. Cancer 2009, 48, 737–744. [Google Scholar] [CrossRef]
- Kempers, M.J.; Kuiper, R.P.; Ockeloen, C.W.; Chappuis, P.O.; Hutter, P.; Rahner, N.; Schackert, H.K.; Steinke, V.; Holinski-Feder, E.; Morak, M.; et al. Risk of colorectal and endometrial cancers in EPCAM deletion-positive Lynch syndrome: A cohort study. Lancet Oncol. 2011, 12, 49–55. [Google Scholar] [CrossRef]
- Crépin, M.; Dieu, M.C.; Lejeune, S.; Escande, F.; Boidin, D.; Porchet, N.; Morin, G.; Manouvrier, S.; Mathieu, M.; Buisine, M.P. Evidence of constitutional MLH1 epimutation associated to transgenerational inheritance of cancer susceptibility. Hum. Mutat. 2012, 33, 180–188. [Google Scholar] [CrossRef]
- Morak, M.; Ibisler, A.; Keller, G.; Jessen, E.; Laner, A.; Gonzales-Fassrainer, D.; Locher, M.; Massdorf, T.; Nissen, A.M.; Benet-Pagès, A.; et al. Comprehensive analysis of the MLH1 promoter region in 480 patients with colorectal cancer and 1150 controls reveals new variants including one with a heritable constitutional MLH1 epimutation. J. Med. Genet. 2018, 55, 240–248. [Google Scholar] [CrossRef]
- Pinheiro, M.; Pinto, C.; Peixoto, A.; Veiga, I.; Mesquita, B.; Henrique, R.; Baptista, M.; Fragoso, M.; Sousa, O.; Pereira, H.; et al. A novel exonic rearrangement affecting MLH1 and the contiguous LRRFIP2 is a founder mutation in Portuguese Lynch syndrome families. Genet. Med. 2011, 13, 895–902. [Google Scholar] [CrossRef]
- Morak, M.; Koehler, U.; Schackert, H.K.; Steinke, V.; Royer-Pokora, B.; Schulmann, K.; Kloor, M.; Höchter, W.; Weingart, J.; Keiling, C.; et al. Biallelic MLH1 SNP cDNA expression or constitutional promoter methylation can hide genomic rearrangements causing Lynch syndrome. J. Med. Genet. 2011, 48, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Brieger, A.; Plotz, G.; Weber, N.; Passmann, S.; Dingermann, T.; Zeuzem, S.; Trojan, J.; Marschalek, R. An interstitial deletion at 3p21.3 results in the genetic fusion of MLH1 and ITGA9 in a Lynch syndrome family. Clin. Cancer Res. 2009, 15, 762–769. [Google Scholar] [CrossRef]
- Sjursen, W.; Bjørnevoll, I.; Engebretsen, L.F.; Fjelland, K.; Halvorsen, T.; Myrvold, H.E. A homozygote splice site PMS2 mutation as cause of Turcot syndrome gives rise to two different abnormal transcripts. Fam. Cancer 2009, 8, 179–186. [Google Scholar] [CrossRef]
- Vasen, H.F.; Ghorbanoghli, Z.; Bourdeaut, F.; Cabaret, O.; Caron, O.; Duval, A.; Entz-Werle, N.; Goldberg, Y.; Ilencikova, D.; Kratz, C.P.; et al. Guidelines for surveillance of individuals with constitutional mismatch repair-deficiency proposed by the European Consortium “Care for CMMR-D” (C4CMMR-D). J. Med. Genet. 2014, 51, 283–293. [Google Scholar] [CrossRef]
- Lavoine, N.; Colas, C.; Muleris, M.; Bodo, S.; Duval, A.; Entz-Werle, N.; Coulet, F.; Cabaret, O.; Andreiuolo, F.; Charpy, C.; et al. Constitutional mismatch repair deficiency syndrome: Clinical description in a French cohort. J. Med. Genet. 2015, 52, 770–778. [Google Scholar] [CrossRef]
- ten Broeke, S.W.; Brohet, R.M.; Tops, C.M.; van der Klift, H.M.; Velthuizen, M.E.; Bernstein, I.; Capellá Munar, G.; Gomez Garcia, E.; Hoogerbrugge, N.; Letteboer, T.G.; et al. Lynch syndrome caused by germline PMS2 mutations: Delineating the cancer risk. J. Clin. Oncol. 2015, 33, 319–325. [Google Scholar] [CrossRef]
- Blount, J.; Prakash, A. The changing landscape of Lynch syndrome due to PMS2 mutations. Clin. Genet. 2018, 94, 61–69. [Google Scholar] [CrossRef]
- Crosbie, E.J.; Ryan, N.; Arends, M.J.; Bosse, T.; Burn, J.; Cornes, J.M.; Crawford, R.; Eccles, D.; Frayling, I.M.; Ghaem-Maghami, S.; et al. The Manchester International Consensus Group recommendations for the management of gynecological cancers in Lynch syndrome. Genet. Med. 2019, 21, 2390–2400. [Google Scholar] [CrossRef]
- Modica, I.; Soslow, R.A.; Black, D.; Tornos, C.; Kauff, N.; Shia, J. Utility of immunohistochemistry in predicting microsatellite instability in endometrial carcinoma. Am. J. Surg. Pathol. 2007, 31, 744–751. [Google Scholar] [CrossRef]
- Lloyd, K.M., 2nd; Dennis, M. Cowden’s disease. A possible new symptom complex with multiple system involvement. Ann. Intern. Med. 1963, 58, 136–142. [Google Scholar] [CrossRef]
- Nelen, M.R.; Padberg, G.W.; Peeters, E.A.; Lin, A.Y.; van den Helm, B.; Frants, R.R.; Coulon, V.; Goldstein, A.M.; van Reen, M.M.; Easton, D.F.; et al. Localization of the gene for Cowden disease to chromosome 10q22-23. Nat. Genet. 1996, 13, 114–116. [Google Scholar] [CrossRef]
- Liaw, D.; Marsh, D.J.; Li, J.; Dahia, P.L.; Wang, S.I.; Zheng, Z.; Bose, S.; Call, K.M.; Tsou, H.C.; Peacocke, M.; et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat. Genet. 1997, 16, 64–67. [Google Scholar] [CrossRef]
- Pilarski, R. PTEN Hamartoma Tumor Syndrome: A Clinical Overview. Cancers 2019, 11, 844. [Google Scholar] [CrossRef]
- Mester, J.; Eng, C. Cowden syndrome: Recognizing and managing a not so-rare hereditary cancer syndrome. J. Surg. Oncol. 2015, 111, 125–130. [Google Scholar] [CrossRef]
- Tan, M.H.; Mester, J.L.; Ngeow, J.; Rybicki, L.A.; Orloff, M.S.; Eng, C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin. Cancer Res. 2012, 18, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Bubien, V.; Bonnet, F.; Brouste, V.; Hoppe, S.; Barouk-Simonet, E.; David, A.; Edery, P.; Bottani, A.; Layet, V.; Caron, O.; et al. French Cowden Disease Network. High cumulative risks of cancer in patients with PTEN hamartoma tumour syndrome. J. Med. Genet. 2013, 50, 255–263. [Google Scholar] [CrossRef]
- Nieuwenhuis, M.H.; Kets, C.M.; Murphy-Ryan, M.; Yntema, H.G.; Evans, D.G.; Colas, C.; Møller, P.; Hes, F.J.; Hodgson, S.V.; Olderode-Berends, M.J.; et al. Cancer risk and genotype-phenotype correlations in PTEN hamartoma tumor syndrome. Fam. Cancer 2013, 13, 57–63. [Google Scholar] [CrossRef]
- Elnaggar, A.C.; Spunt, S.L.; Smith, W.; Depas, M.; Santoso, J.T. Endometrial cancer in a 15-year-old girl: A complication of Cowden syndrome. Gynecol. Oncol. Case Rep. 2012, 3, 18–19. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Baker, W.D.; Soisson, A.P.; Dodson, M.K. Endometrial cancer in a 14-year old girl with Cowden syndrome: A case report. J. Obstet. Gynaecol. Res. 2013, 39, 876–878. [Google Scholar] [CrossRef]
- Mutter, G.L.; Lin, M.C.; Fitzgerald, J.T.; Kum, J.B.; Baak, J.P.; Lees, J.A.; Weng, L.P.; Eng, C. Altered PTEN expression as a diagnostic marker for the earliest endometrial precancers. J. Natl. Cancer Inst. 2000, 92, 924–930. [Google Scholar] [CrossRef]
- Djordjevic, B.; Barkoh, B.A.; Luthra, R.; Broaddus, R.R. Relationship between PTEN, DNA mismatch repair, and tumor histotype in endometrial carcinoma: Retained positive expression of PTEN preferentially identifies sporadic non-endometrioid carcinomas. Mod. Pathol. 2013, 26, 1401–1412. [Google Scholar] [CrossRef]
- Black, D.; Bogomolniy, F.; Robson, M.E.; Offit, K.; Barakat, R.R.; Boyd, J. Evaluation of germline PTEN mutations in endometrial cancer patients. Gynecol. Oncol. 2005, 96, 21–24. [Google Scholar] [CrossRef]
- Orloff, M.S.; He, X.; Peterson, C.; Chen, F.; Chen, J.L.; Mester, J.L.; Eng, C. Germline PIK3CA and AKT1 Mutations in Cowden and Cowden-like Syndromes. Am. J. Hum. Genet. 2013, 92, 76–80. [Google Scholar] [CrossRef]
- Palles, C.; Cazier, J.B.; Howarth, K.M.; Domingo, E.; Jones, A.M.; Broderick, P.; Kemp, Z.; Spain, S.L.; Guarino, E.; Salguero, I.; et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet. 2013, 45, 136–144. [Google Scholar] [CrossRef]
- Elsayed, F.A.; Kets, C.M.; Ruano, D.; van den Akker, B.; Mensenkamp, A.R.; Schrumpf, M.; Nielsen, M.; Wijnen, J.T.; Tops, C.M.; Ligtenberg, M.J.; et al. Germline variants in POLE are associated with early onset mismatch repair deficient colorectal cancer. Eur. J. Hum. Genet. 2014, 23, 1080–1084. [Google Scholar] [CrossRef]
- Rohlin, A.; Zagoras, T.; Nilsson, S.; Lundstam, U.; Wahlström, J.; Hultén, L.; Martinsson, T.; Karlsson, G.B.; Nordling, M. A mutation in POLE predisposing to a multi-tumour phenotype. Int. J. Oncol. 2014, 45, 77–81. [Google Scholar] [CrossRef]
- Bellido, F.; Pineda, M.; Aiza, G.; Valdés-Mas, R.; Navarro, M.; Puente, D.A.; Pons, T.; González, S.; Iglesias, S.; Darder, E.; et al. POLE and POLD1 mutations in 529 kindreds with familial colorectal cancer and/or polyposis: Review of reported cases and recommendations for genetic testing and surveillance. Genet. Med. 2016, 18, 325–332. [Google Scholar] [CrossRef]
- Siraj, A.K.; Parvathareddy, S.K.; Bu, R.; Iqbal, K.; Siraj, S.; Masoodi, T.; Concepcion, R.M.; Ghazwani, L.O.; AlBadawi, I.; Al-Dayel, F.; et al. Germline POLE and POLD1 proofreading domain mutations in endometrial carcinoma from Middle Eastern region. Cancer Cell Int. 2019, 19, 334. [Google Scholar] [CrossRef]
- Logan, C.V.; Murray, J.E.; Parry, D.A.; Robertson, A.; Bellelli, R.; Tarnauskaitė, Ž; Challis, R.; Cleal, L.; Borel, V.; Fluteau, A.; et al. DNA Polymerase Epsilon Deficiency Causes IMAGe Syndrome with Variable Immunodeficiency. Am. J. Hum. Genet. 2018, 103, 1038–1044. [Google Scholar] [CrossRef] [PubMed]
- Pachlopnik Schmid, J.; Lemoine, R.; Nehme, N.; Cormier-Daire, V.; Revy, P.; Debeurme, F.; Debré, M.; Nitschke, P.; Bole-Feysot, C.; Legeai-Mallet, L.; et al. Polymerase ε1 mutation in a human syndrome with facial dysmorphism, immunodeficiency, livedo, and short stature (“FILS syndrome”). J. Exp. Med. 2012, 209, 2323–2330. [Google Scholar] [CrossRef]
- Win, A.K.; Dowty, J.G.; Cleary, S.P.; Kim, H.; Buchanan, D.D.; Young, J.P.; Clendenning, M.; Rosty, C.; MacInnis, R.J.; Giles, G.G.; et al. Risk of colorectal cancer for carriers of mutations in MUTYH, with and without a family history of cancer. Gastroenterology 2014, 146, 1208–1211.e5. [Google Scholar] [CrossRef] [PubMed]
- Vogt, S.; Jones, N.; Christian, D.; Engel, C.; Nielsen, M.; Kaufmann, A.; Steinke, V.; Vasen, H.F.; Propping, P.; Sampson, J.R.; et al. Expanded extracolonic tumor spectrum in MUTYH-associated polyposis. Gastroenterology 2009, 137, 1976–1985.e10. [Google Scholar] [CrossRef] [PubMed]
- Win, A.K.; Reece, J.C.; Dowty, J.G.; Buchanan, D.D.; Clendenning, M.; Rosty, C.; Southey, M.C.; Young, J.P.; Cleary, S.P.; Kim, H.; et al. Risk of extracolonic cancers for people with biallelic and monoallelic mutations in MUTYH. Int. J. Cancer 2016, 139, 1557–1563. [Google Scholar] [CrossRef] [PubMed]
- Weren, R.D.; Ligtenberg, M.J.; Kets, C.M.; de Voer, R.M.; Verwiel, E.T.; Spruijt, L.; van Zelst-Stams, W.A.; Jongmans, M.C.; Gilissen, C.; Hehir-Kwa, J.Y.; et al. A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat. Genet. 2015, 47, 668–671. [Google Scholar] [CrossRef]
- Grolleman, J.E.; de Voer, R.M.; Elsayed, F.A.; Nielsen, M.; Weren, R.; Palles, C.; Ligtenberg, M.; Vos, J.R.; Ten Broeke, S.W.; de Miranda, N.; et al. Mutational Signature Analysis Reveals NTHL1 Deficiency to Cause a Multi-tumor Phenotype. Cancer Cell 2019, 35, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Thompson, D.; Easton, D.F.; Breast Cancer Linkage Consortium. Cancer Incidence in BRCA1 mutation carriers. J. Natl. Cancer Inst. 2002, 94, 1358–1365. [Google Scholar] [CrossRef]
- Beiner, M.E.; Finch, A.; Rosen, B.; Lubinski, J.; Møller, P.; Ghadirian, P.; Lynch, H.T.; Friedman, E.; Sun, P.; Narod, S.A.; et al. The risk of endometrial cancer in women with BRCA1 and BRCA2 mutations. A prospective study. Gynecol. Oncol. 2007, 104, 7–10. [Google Scholar] [CrossRef]
- Segev, Y.; Iqbal, J.; Lubinski, J.; Gronwald, J.; Lynch, H.T.; Møller, P.; Ghadirian, P.; Rosen, B.; Tung, N.; Kim-Sing, C.; et al. The incidence of endometrial cancer in women with BRCA1 and BRCA2 mutations: An international prospective cohort study. Gynecol. Oncol. 2013, 130, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Shu, C.A.; Pike, M.C.; Jotwani, A.R.; Friebel, T.M.; Soslow, R.A.; Levine, D.A.; Nathanson, K.L.; Konner, J.A.; Arnold, A.G.; Bogomolniy, F.; et al. Uterine cancer after risk-reducing salpingo-oophorectomy without hysterectomy in women with BRCA mutations. JAMA Oncol. 2016, 2, 1434–1440. [Google Scholar] [CrossRef] [PubMed]
- de Jonge, M.M.; Ritterhouse, L.L.; de Kroon, C.D.; Vreeswijk, M.; Segal, J.P.; Puranik, R.; Hollema, H.; Rookus, M.A.; van Asperen, C.J.; van Leeuwen, F.E.; et al. Germline BRCA-Associated Endometrial Carcinoma Is a Distinct Clinicopathologic Entity. Clin. Cancer Res. 2019, 25, 7517–7526. [Google Scholar] [CrossRef]
- Spurdle, A.B.; Thompson, D.J.; Ahmed, S.; Ferguson, K.; Healey, C.S.; O’Mara, T.; Walker, L.C.; Montgomery, S.B.; Dermitzakis, E.T.; Australian National Endometrial Cancer Study Group; et al. Genome-wide association study identifies a common variant associated with risk of endometrial cancer. Nat. Genet. 2011, 43, 451–454. [Google Scholar] [CrossRef]
- Chen, M.M.; O’Mara, T.A.; Thompson, D.J.; Painter, J.N.; Australian National Endometrial Cancer Study Group (ANECS); Attia, J.; Black, A.; Brinton, L.; Chanock, S.; Chen, C.; et al. GWAS meta-analysis of 16 852 women identifies new susceptibility locus for endometrial cancer. Hum. Mol. Genet. 2016, 25, 2612–2620. [Google Scholar] [CrossRef]
- Cheng, T.H.; Thompson, D.J.; O’Mara, T.A.; Painter, J.N.; Glubb, D.M.; Flach, S.; Lewis, A.; French, J.D.; Freeman-Mills, L.; Church, D.; et al. Five endometrial cancer risk loci identified through genome-wide association analysis. Nat. Genet. 2016, 48, 667–674. [Google Scholar] [CrossRef]
- O’Mara, T.A.; Glubb, D.M.; Amant, F.; Annibali, D.; Ashton, K.; Attia, J.; Auer, P.L.; Beckmann, M.W.; Black, A.; Bolla, M.K.; et al. Identification of nine new susceptibility loci for endometrial cancer. Nat. Comm. 2018, 9, 3166. [Google Scholar] [CrossRef]
- Painter, J.N.; Kaufmann, S.; O’Mara, T.A.; Hillman, K.M.; Sivakumaran, H.; Darabi, H.; Cheng, T.; Pearson, J.; Kazakoff, S.; Waddell, N.; et al. A Common Variant at the 14q32 Endometrial Cancer Risk Locus Activates AKT1 through YY1 Binding. Am. J. Hum. Genet. 2016, 98, 1159–1169. [Google Scholar] [CrossRef]
- Cheng, T.H.; Thompson, D.; Painter, J.; O’Mara, T.; Gorman, M.; Martin, L.; Palles, C.; Jones, A.; Buchanan, D.D.; Win, A.K.; et al. Meta-analysis of genome-wide association studies identifies common susceptibility polymorphisms for colorectal and endometrial cancer near SH2B3 and TSHZ1. Sci. Rep. 2015, 5, 17369. [Google Scholar] [CrossRef]
- Painter, J.N.; O’Mara, T.A.; Morris, A.P.; Cheng, T.; Gorman, M.; Martin, L.; Hodson, S.; Jones, A.; Martin, N.G.; Gordon, S.; et al. Genetic overlap between endometriosis and endometrial cancer: Evidence from cross-disease genetic correlation and GWAS meta-analyses. Cancer Med. 2018, 7, 1978–1987. [Google Scholar] [CrossRef]
- O’Mara, T.A.; Crosbie, E.J. Polygenic risk score opportunities for early detection and prevention strategies in endometrial cancer. Br. J. Cancer 2020, in press. [Google Scholar] [CrossRef]
- Nead, K.T.; Sharp, S.J.; Thompson, D.J.; Painter, J.N.; Savage, D.B.; Semple, R.K.; Barker, A.; Australian National Endometrial Cancer Study Group (ANECS); Perry, J.R.; Attia, J.; et al. Evidence of a causal association between insulinemia and endometrial cancer: A mendelian randomization analysis. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- Painter, J.N.; O’Mara, T.A.; Marquart, L.; Webb, P.M.; Attia, J.; Medland, S.E.; Cheng, T.; Dennis, J.; Holliday, E.G.; McEvoy, M.; et al. Genetic risk score Mendelian randomization shows that obesity measured as body mass index, but not waist:hip ratio, is causal for endometrial cancer. Cancer Epidemiol. Biomark. Prev. 2016, 25, 1503–1510. [Google Scholar] [CrossRef] [PubMed]
- Thompson, D.J.; O’Mara, T.A.; Glubb, D.M.; Painter, J.N.; Cheng, T.; Folkerd, E.; Doody, D.; Dennis, J.; Webb, P.M.; Australian National Endometrial Cancer Study Group (ANECS); et al. CYP19A1 fine-mapping and Mendelian randomization: Estradiol is causal for endometrial cancer. Endocr. Relat. Cancer 2016, 23, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Kho, P.-F.; Amant, F.; Annibali, D.; Ashton, K.; Attia, J.; Auer, P.L.; Beckmann, M.W.; Black, A.; Brinton, L.; Buchanan, D.D.; et al. Mendelian randomization analyses suggest a role for cholesterol in the development of endometrial cancer. Int. J. Cancer 2020, in press. [Google Scholar] [CrossRef]
- Lu, K.H.; Dinh, M.; Kohlmann, W.; Watson, P.; Green, J.; Syngal, S.; Bandipalliam, P.; Chen, L.M.; Allen, B.; Conrad, P.; et al. Gynecologic cancer as a “sentinel cancer” for women with hereditary nonpolyposis colorectal cancer syndrome. Obstet Gynecol. 2005, 105, 569–574. [Google Scholar] [CrossRef]
- Lipton, L.R.; Johnson, V.; Cummings, C.; Fisher, S.; Risby, P.; Eftekhar Sadat, A.T.; Cranston, T.; Izatt, L.; Sasieni, P.; Hodgson, S.V.; et al. Refining the Amsterdam Criteria and Bethesda Guidelines: Testing algorithms for the prediction of mismatch repair mutation status in the familial cancer clinic. J. Clin. Oncol. 2004, 22, 4934–4943, with published correction in J. Clin. Oncol. 2005, 23, 3652. [Google Scholar] [CrossRef]
- Egoavil, C.; Alenda, C.; Castillejo, A.; Paya, A.; Peiro, G.; Sánchez-Heras, A.B.; Castillejo, M.I.; Rojas, E.; Barberá, V.M.; Cigüenza, S.; et al. Prevalence of Lynch syndrome among patients with newly diagnosed endometrial cancers. PLoS ONE 2013, 8, e79737. [Google Scholar] [CrossRef]
- Ferguson, S.E.; Aronson, M.; Pollett, A.; Eiriksson, L.R.; Oza, A.M.; Gallinger, S.; Lerner-Ellis, J.; Alvandi, Z.; Bernardini, M.Q.; MacKay, H.J.; et al. Performance characteristics of screening strategies for Lynch syndrome in unselected women with newly diagnosed endometrial cancer who have undergone universal germline mutation testing. Cancer 2014, 120, 3932–3939. [Google Scholar] [CrossRef]
- Hampel, H.; Frankel, W.; Panescu, J.; Lockman, J.; Sotamaa, K.; Fix, D.; Comeras, I.; La Jeunesse, J.; Nakagawa, H.; Westman, J.A.; et al. Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res. 2006, 66, 7810–7817. [Google Scholar] [CrossRef]
- Hampel, H.; Panescu, J.; Lockman, J.; Sotamaa, K.; Fix, D.; Comeras, I.; LaJeunesse, J.; Nakagawa, H.; Westman, J.A.; Prior, T.W.; et al. Comment on: Screening for Lynch Syndrome (Hereditary Nonpolyposis Colorectal Cancer) among Endometrial Cancer Patients. Cancer Res. 2007, 67, 9603. [Google Scholar] [CrossRef] [PubMed]
- Leenen, C.H.; van Lier, M.G.; van Doorn, H.C.; van Leerdam, M.E.; Kooi, S.G.; de Waard, J.; Hoedemaeker, R.F.; van den Ouweland, A.M.; Hulspas, S.M.; Dubbink, H.J.; et al. Prospective evaluation of molecular screening for Lynch syndrome in patients with endometrial cancer ≤ 70 years. Gynecol. Oncol. 2012, 125, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Snowsill, T.; Huxley, N.; Hoyle, M.; Jones-Hughes, T.; Coelho, H.; Cooper, C.; Frayling, I.; Hyde, C. A systematic review and economic evaluation of diagnostic strategies for Lynch syndrome. Health Technol. Assess. 2014, 18, 1–406. [Google Scholar] [CrossRef]
- Backes, F.J.; Leon, M.E.; Ivanov, I.; Suarez, A.; Frankel, W.L.; Hampel, H.; Fowler, J.M.; Copeland, L.J.; O’Malley, D.M.; Cohn, D.E. Prospective evaluation of DNA mismatch repair protein expression in primary endometrial cancer. Gynecol. Oncol. 2009, 114, 486–490. [Google Scholar] [CrossRef]
- Moline, J.; Mahdi, H.; Yang, B.; Biscotti, C.; Roma, A.A.; Heald, B.; Rose, P.G.; Michener, C.; Eng, C. Implementation of tumor testing for lynch syndrome in endometrial cancers at a large academic medical center. Gynecol. Oncol. 2013, 130, 121–126. [Google Scholar] [CrossRef]
- Gausachs, M.; Mur, P.; Corral, J.; Pineda, M.; González, S.; Benito, L.; Menéndez, M.; Espinàs, J.A.; Brunet, J.; Iniesta, M.D.; et al. MLH1 promoter hypermethylation in the analytical algorithm of Lynch syndrome: A cost-effectiveness study. Eur. J. Hum. Genet. 2012, 20, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.; Bell, I.; Crawley, S.; Gum, J.; Terdiman, J.P.; Allen, B.A.; Truta, B.; Sleisenger, M.H.; Kim, Y.S. BRAF mutation is frequently present in sporadic colorectal cancer with methylated hMLH1, but not in hereditary nonpolyposis colorectal cancer. Clin. Cancer Res. 2004, 10 Pt 1, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, M.; Yanokura, M.; Banno, K.; Kobayashi, Y.; Kuwabara, Y.; Kobayashi, M.; Nomura, H.; Hirasawa, A.; Susumu, N.; Aoki, D. Analysis of a correlation between the BRAF V600E mutation and abnormal DNA mismatch repair in patients with sporadic endometrial cancer. Int. J. Oncol. 2009, 34, 1541–1547. [Google Scholar] [CrossRef] [PubMed]
- Dove-Edwin, I.; Boks, D.; Goff, S.; Kenter, G.G.; Carpenter, R.; Vasen, H.F.; Thomas, H.J. The outcome of endometrial carcinoma surveillance by ultrasound scan in women at risk of hereditary nonpolyposis colorectal carcinoma and familial colorectal carcinoma. Cancer 2002, 94, 1708–1712. [Google Scholar] [CrossRef]
- Rijcken, F.E.; Mourits, M.J.; Kleibeuker, J.H.; Hollema, H.; van der Zee, A.G. Gynecologic screening in hereditary nonpolyposis colorectal cancer. Gynecol. Oncol. 2003, 91, 74–80. [Google Scholar] [CrossRef]
- Auranen, A.; Joutsiniemi, T. A systematic review of gynecological cancer surveillance in women belonging to hereditary nonpolyposis colorectal cancer (Lynch syndrome) families. Acta Obstet. Gynecol. Scand. 2011, 90, 437–444. [Google Scholar] [CrossRef]
- Renkonen-Sinisalo, L.; Bützow, R.; Leminen, A.; Lehtovirta, P.; Mecklin, J.P.; Järvinen, H.J. Surveillance for endometrial cancer in hereditary nonpolyposis colorectal cancer syndrome. Int. J. Cancer 2006, 120, 821–824. [Google Scholar] [CrossRef] [PubMed]
- Lécuru, F.; Le Frère Belda, M.A.; Bats, A.S.; Tulpin, L.; Metzger, U.; Olschwang, S.; Laurent-Puig, P. Performance of office hysteroscopy and endometrial biopsy for detecting endometrial disease in women at risk of human non-polyposis colon cancer: A prospective study. Int. J. Gynecol. Cancer 2008, 18, 1326–1331. [Google Scholar] [CrossRef] [PubMed]
- Manchanda, R.; Saridogan, E.; Abdelraheim, A.; Johnson, M.; Rosenthal, A.N.; Benjamin, E.; Brunell, C.; Side, L.; Gessler, S.; Jacobs, I.; et al. Annual outpatient hysteroscopy and endometrial sampling (OHES) in HNPCC/Lynch syndrome (LS). Arch. Gynecol. Obstet. 2012, 286, 1555–1562. [Google Scholar] [CrossRef]
- Helder-Woolderink, J.M.; De Bock, G.H.; Sijmons, R.H.; Hollema, H.; Mourits, M.J. The additional value of endometrial sampling in the early detection of endometrial cancer in women with Lynch syndrome. Gynecol. Oncol. 2013, 131, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Ryan, N.; Morris, J.; Green, K.; Lalloo, F.; Woodward, E.R.; Hill, J.; Crosbie, E.J.; Evans, D.G. Association of Mismatch Repair Mutation With Age at Cancer Onset in Lynch Syndrome: Implications for Stratified Surveillance Strategies. JAMA Oncol. 2017, 3, 1702–1706. [Google Scholar] [CrossRef]
- Vasen, H.F.; Blanco, I.; Aktan-Collan, K.; Gopie, J.P.; Alonso, A.; Aretz, S.; Bernstein, I.; Bertario, L.; Burn, J.; Capella, G.; et al. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): Recommendations by a group of European experts. Gut 2013, 62, 812–823. [Google Scholar] [CrossRef]
- Schmeler, K.M.; Lynch, H.T.; Chen, L.M.; Munsell, M.F.; Soliman, P.T.; Clark, M.B.; Daniels, M.S.; White, K.G.; Boyd-Rogers, S.G.; Conrad, P.G.; et al. Prophylactic surgery to reduce the risk of gynecologic cancers in the Lynch syndrome. N. Engl. J. Med. 2006, 354, 261–269. [Google Scholar] [CrossRef]
- Bartosch, C.; Pires-Luís, A.S.; Meireles, C.; Baptista, M.; Gouveia, A.; Pinto, C.; Shannon, K.M.; Jerónimo, C.; Teixeira, M.R.; Lopes, J.M.; et al. Pathologic Findings in Prophylactic and Nonprophylactic Hysterectomy Specimens of Patients With Lynch Syndrome. Am. J. Surg. Pathol. 2016, 40, 1177–1191. [Google Scholar] [CrossRef]
- Shaw, P.A.; Clarke, B.A. Prophylactic gynecologic specimens from hereditary cancer carriers. Surg. Pathol. Clin. 2016, 9, 307–328. [Google Scholar] [CrossRef]
- Ramchander, N.C.; Ryan, N.; Walker, T.; Harries, L.; Bolton, J.; Bosse, T.; Evans, D.G.; Crosbie, E.J. Distinct Immunological Landscapes Characterize Inherited and Sporadic Mismatch Repair Deficient Endometrial Cancer. Front. Immunol. 2020, 10, 3023. [Google Scholar] [CrossRef] [PubMed]
- Bohaumilitzky, L.; von Knebel Doeberitz, M.; Kloor, M.; Ahadova, A. Implications of Hereditary Origin on the Immune Phenotype of Mismatch Repair-Deficient Cancers: Systematic Literature Review. J. Clin. Med. 2020, 9, 1741. [Google Scholar] [CrossRef]
- Ott, P.A.; Bang, Y.J.; Berton-Rigaud, D.; Elez, E.; Pishvaian, M.J.; Rugo, H.S.; Puzanov, I.; Mehnert, J.M.; Aung, K.L.; Lopez, J.; et al. Safety and Antitumor Activity of Pembrolizumab in Advanced Programmed Death Ligand 1-Positive Endometrial Cancer: Results From the KEYNOTE-028 Study. J. Clin. Oncol. 2017, 35, 2535–2541. [Google Scholar] [CrossRef]
- Makker, V.; Rasco, D.; Vogelzang, N.J.; Brose, M.S.; Cohn, A.L.; Mier, J.; Di Simone, C.; Hyman, D.M.; Stepan, D.E.; Dutcus, C.E.; et al. Lenvatinib plus pembrolizumab in patients with advanced endometrial cancer: An interim analysis of a multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 711–718. [Google Scholar] [CrossRef]
- Dedes, K.J.; Wetterskog, D.; Mendes-Pereira, A.M.; Natrajan, R.; Lambros, M.B.; Geyer, F.C.; Vatcheva, R.; Savage, K.; Mackay, A.; Lord, C.J.; et al. PTEN deficiency in endometrioid endometrial adenocarcinomas predicts sensitivity to PARP inhibitors. Sci. Transl. Med. 2010, 2, 53ra75. [Google Scholar] [CrossRef] [PubMed]
- Bian, X.; Gao, J.; Luo, F.; Rui, C.; Zheng, T.; Wang, D.; Wang, Y.; Roberts, T.M.; Liu, P.; Zhao, J.J.; et al. PTEN deficiency sensitizes endometrioid endometrial cancer to compound PARP-PI3K inhibition but not PARP inhibition as monotherapy. Oncogene 2018, 37, 341–351. [Google Scholar] [CrossRef]
- U.S. National Library of Medicine, ClinicalTrials.gov Database. Available online: https://www.clinicaltrials.gov/ (accessed on 27 May 2020).
- de Jonge, M.M.; Auguste, A.; van Wijk, L.M.; Schouten, P.C.; Meijers, M.; Ter Haar, N.T.; Smit, V.; Nout, R.A.; Glaire, M.A.; Church, D.N.; et al. Frequent Homologous Recombination Deficiency in High-grade Endometrial Carcinomas. Clin. Cancer Res. 2019, 25, 1087–1097. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Gene | Location | Frequency * | Associated Syndrome | Lifetime Risk EC | Other Cancers | |
|---|---|---|---|---|---|---|
| Biallelic | Monoallelic | |||||
| Established | ||||||
| MLH1 | 3p22.2 | 1/1500 | CMMRD | Lynch Syndrome | ~40–45% | Colon, Ovary, Stomach, Pancreas, Brain **, HS ** |
| MSH2 | 2p21-16 | 1/10,000 | CMMRD | Lynch Syndrome | ~50% | Colon, Ovary, Skin, Brain **, HS ** |
| MSH6 | 2p16.3 | 1/2500 | CMMRD | Lynch Syndrome | ~40–45% | Colon, Ovary, Stomach, Pancreas, Breast?, Brain **, HS ** |
| PMS2 | 7p22.1 | 1/600 | CMMRD | Lynch Syndrome | ~15–20% | Colon, Ovary, Breast?, Brain **, HS ** |
| PTEN | 10q23.31 | 1/10,000 | unknown | Cowden Syndrome | ~25% | Breast, Thyroid, Kidney, Colon, Skin |
| Candidates | ||||||
| POLD1 | 19q13.33 | not defined * | unknown | Lynch Syndrome-like | likely increased | Colon |
| POLE | 12q24.33 | not defined * | (FILS or IMAGEI Syndrome) | Lynch Syndrome-like | likely increased | Colon |
| NTHL1 | 16p13.3 | 1/250 | NTHL1 multicancer syndrome | – | likely increased for homozygotes | Colon, Breast |
| MUTYH | 1p34.1 | 1/200 | MUTYH-associated polyposis | MUTYH-associated polyposis | possibly increased for homozygotes | Colon |
| BRCA1 | 17q21.31 | 1/600 | Fanconi Anemia S | Hereditary Breast and Ovarian Cancer | possibly increased for serous EC | Breast, Ovary, Skin, Pancreas |
| Study | Number | Design | Patients | Strategy | Results |
|---|---|---|---|---|---|
| Backes et al., 2009 [94] | n = 140 | Prospective | Unselected EC patients | IHC MMR proteins → invitation for genetic counseling when suggestive for LS. | 30 patients (21%) with loss of one or more MMR proteins, 15/30 invited to genetic counseling, 2/15 accepted both negative for LS. |
| Buchanan et al., 2014 [35] | n = 702 | Prospective (multicentric) | Unselected EC patients | IHC MMR proteins + DNA MLH1 methylation status for all tumors exhibiting MLH1/PMS2 loss → genetic testing in IHC MRD patients. | 170 (24%) of 702 patients showed MMR loss. 158/170 available for genetic testing. 22/158 truncating gene variants. Overall carrier frequency 3%. Testing MMR loss by IHC in women <60 years at diagnosis was optimal regarding sensitivity and cost-effectiveness. |
| Egoavil et al., 2013 [88] | n = 173 | Prospective (monocentric) | Unselected EC patients | MMR-IHC and MSI testing MMR mutation testing in positive cases. If MMR gene mutation was detected or MLH1 methylation in the blood test was positive, patients were classified as LS positive. | 61/173 patients had abnormal IHC or MSI results. 8/61 patients tested positive for LS (prevalence 4.6% (8/173)). |
| Ferguson et al., 2014 [89] | n = 117 | Prospective (monocentric) | Unselected EC patients | Family history assessment, IHC screening for MMR, MSI testing, tumor morphology followed by germline testing for MMR gene mutations. | 34/117 had MMR deficiency in IHC. 27/117 had MSI, 7/27 LS (5.9%). IHC < 60 had sensitivity of 100%, specificity of 86.1%, with PPV of 58.3% and NPV of 100%, family history and tumor morphology had poorest performance with a specificity of 42.1%. |
| Gausachs et al., 2012 [96] | n.a. | n.a. | n = 122 CRC patients with MMRD, 57 LS, 48 MSS cancers and positive family history for CRC, 73 sporadic CRC. | BRAF mutation and MLH1 promoter hypermethylation were assessed and a decision model was developed to estimate incremental costs of alternative case finding methods for detecting MLH1 mutation carriers. | Sensitivity of the absence of BRAF mutations for depiction of LS patients was 96% (23/24) and specificity was 28% (13/47). Specificity of MLH1 promoter hypermethylation for depiction of sporadic tumors was 66% (31/47) and sensitivity of 96% (23/24). The cost per additional mutation detected by hypermethylation analysis lower when compared with BRAF and germinal MLH1 mutation study. |
| Hampel et al., 2006 [90]; Hampel et al., 2007 [91] | n = 562 | Prospective (multicentric) | Unselected EC patients | MSI testing, if positive germline mutations in MMR genes were tested. | 119/562 were MSI positive, 11 germline mutations in at least one MMR gene, one patient not MSI positive but germline mutation in MSH6, one patient’s MSI test failed. 8/13 patients w/o criteria for HNPCC syndrome, 8/13 diagnosed >50 years. |
| Leenen et al., 2012 [92] | n = 179 | Prospective (multicentric) | Unselected EC patients | MSI/IHC for MMR proteins. MLH1 promoter hypermethylation if MSI high and MLH1 absent. Tumors classified as: (1) likely to be caused by LS, (MSI high and MMR protein deficiency) (2) sporadic MSI-H (MSI high, MLH1 absent, and MLH1 promoter hyper-methylated), or (3) MSS. | Eleven EC patients found likely to have LS (6%) Germline analyses revealed 7 MMR mutations. Ten patients likely to have LS (92%) were >50 years. 31 sporadic MSI-H tumors with MLH1 promoter hypermethylation (17%; 95% CI 13–24%) identified. |
| Moline et al., 2013 [95] | n = 245 | Prospective | EC patients <50 years or suspicious personal history or histo-pathologic features. EC <69 years or at any age with suspicious features | MSI and IHC, later IHC for two proteins, and MLH1 promoter methylation analysis when indicated. Genetic counselor contacted patients to offer counseling appointments. | 245 EC screened. 62 (25%) abnormal results, 42 patients referred for genetic counseling. 34/42 patients underwent genetic counseling, 28 pursued genetic testing, 11 LS. Age and pathology overlooked 27 eligible cases, 3 cases of LS were only found by clinician request. |
| Study | Number | Design | Patients | Intervention | Results |
|---|---|---|---|---|---|
| Dove-Edwin et al., 2002 [100] | n = 269 | Prospective (multicentric) | Unselected women from HNPCC or HNPCC-like families. | Annual or biannual TVU. | During surveillance two EC, none detected by screening. |
| Helder-Woolderink et al., 2013 [106] | n = 75 | Prospective (monocentric) | Women >30 years with LS or first-degree relatives with LS. | Period 1 TVU and CA 125; Period 2 TVU, CA 125 and EB to detect EC or precancerous lesions. | Six pre-malignancies and one EC detected. 0/6 would have been missed without EB, annual TVU seems to detect pre-malignancies in women with LS or first-degree relatives with LS. |
| Lécuru et al., 2008 [104] | n = 62 | Prospective (monocentric) | Unselected women with LS/meeting Amsterdam II Criteria. | Women with least one hysteroscopy and EB during standard screening. | Three possibly malignant lesions detected, none of them missed w/o hysteroscopy due to abnormal uterine bleeding. |
| Manchanda et al., 2012 [105] | n = 41 | Prospective observational | Unselected women with LS. | Annual OHES vs. annual TVS. | OHES detected 4/4 EC/AEH, TVS 2/4; OHES has similar specificity, higher PLR and lower NLR. |
| Renkonen-Sinisalo et al., 2007 [103] | n = 175 | Prospective Cohort Study | Unselected women with LS. | TVU and EB. | 14/175 patients diagnosed with EC. 11/14 diagnosed by surveillance. 4/11 diagnosed by TVUS only. EB detected 14 cases of potentially premalignant hyperplasia. Cases detected by surveillance at more favorable disease stage. 0/14 detected patients but 6/83 symptomatic LS patients died of EC (n.s., p = 0.4). |
| Rijcken et al., 2003 [101] | n = 41 | Prospective | Women with LS. | Annual TVU and serum level CA 125. | 17/179 TVUs suggested biopsy. 3/17 AEH. One EC as an interval carcinoma, no OC. |
| Ryan et al., 2017 [107] | n = 162 | Retrospective | Unselected women with LS diagnosed with EC. | Comparison of mutated MMR genes and type of mutation. | Patients with MSH6 variants and those with truncating MLH1 variants diagnosed with EC at later age (median difference 6.6 years; 95% CI 2.7–10.4; p = 0.002 for truncating MLH1 variants). |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dörk, T.; Hillemanns, P.; Tempfer, C.; Breu, J.; Fleisch, M.C. Genetic Susceptibility to Endometrial Cancer: Risk Factors and Clinical Management. Cancers 2020, 12, 2407. https://doi.org/10.3390/cancers12092407
Dörk T, Hillemanns P, Tempfer C, Breu J, Fleisch MC. Genetic Susceptibility to Endometrial Cancer: Risk Factors and Clinical Management. Cancers. 2020; 12(9):2407. https://doi.org/10.3390/cancers12092407
Chicago/Turabian StyleDörk, Thilo, Peter Hillemanns, Clemens Tempfer, Julius Breu, and Markus C. Fleisch. 2020. "Genetic Susceptibility to Endometrial Cancer: Risk Factors and Clinical Management" Cancers 12, no. 9: 2407. https://doi.org/10.3390/cancers12092407
APA StyleDörk, T., Hillemanns, P., Tempfer, C., Breu, J., & Fleisch, M. C. (2020). Genetic Susceptibility to Endometrial Cancer: Risk Factors and Clinical Management. Cancers, 12(9), 2407. https://doi.org/10.3390/cancers12092407