Perspectives on Triple-Negative Breast Cancer: Current Treatment Strategies, Unmet Needs, and Potential Targets for Future Therapies

 , , , ,
, , , , 
Abstract
{kind=link}
{kind=link}
1. Introduction
2. Triple Negative Breast Cancer (TNBC)
3. Current TNBC Treatment Paradigms
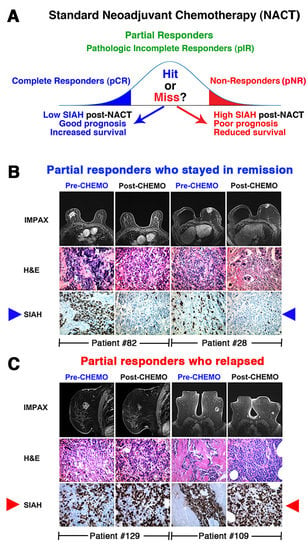
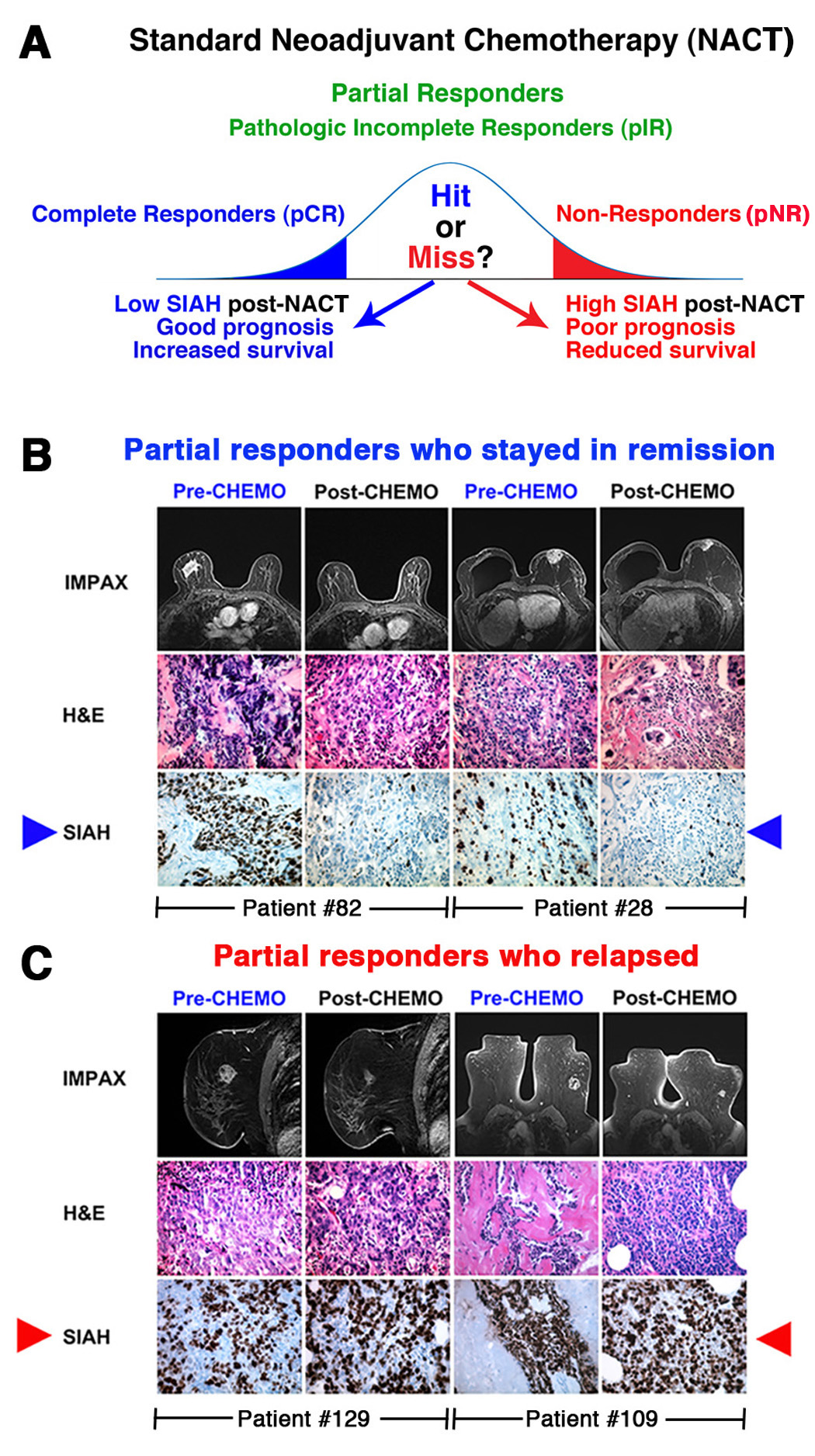
4. Prognosis and Treatment Heterogeneity in TNBC
5. Newly FDA-Approved Targeted Therapies for TNBC
5.1. Immune Checkpoint Blockade Therapies
5.2. PARP Inhibitors
5.3. Anti-Trop2 Antibody Drug Conjugate Therapy in TNBC
6. Emerging Targeted Therapies in TNBC
6.1. EGFR Targeted Therapy in TNBC
6.2. VEGF Targeted Therapy in TNBC
6.3. PI3K/AKT/mTOR Targeted Therapy in TNBC
6.4. AR Targeted Therapy in TNBC
6.5. ERβ Targeted Therapy in TNBC
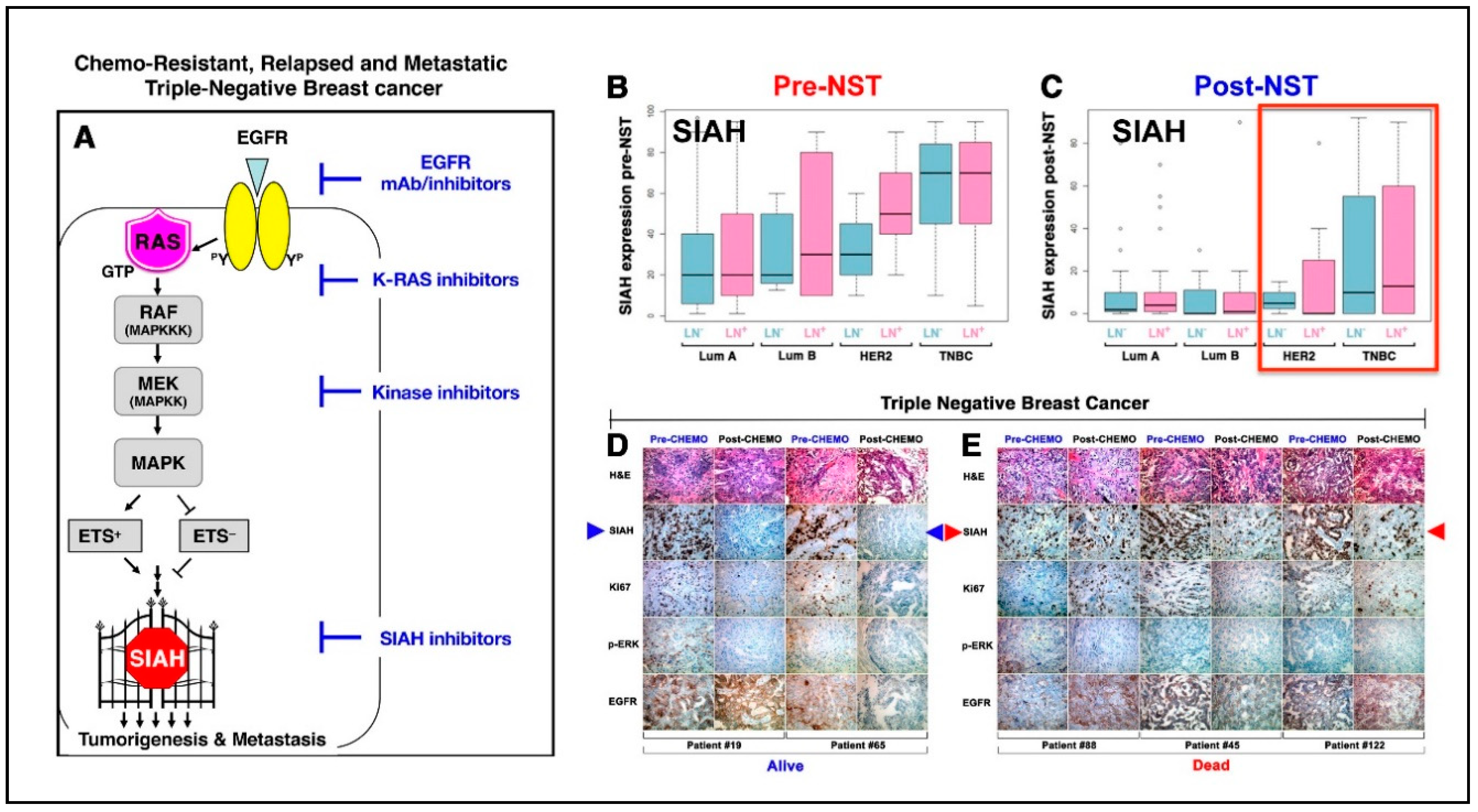
7. K-RAS/SIAH is a Major Tumor-Driving Signaling Pathway in TNBC
7.1. SIAH’s Gatekeeper Role is Indispensable for Proper K-RAS/EGFR Signal Transduction
7.2. K-RAS/SIAH/EGFR Pathway is Commonly Activated in TNBC, and SIAH is a Therapy-Responsive and Prognostic Biomarker in TNBC
7.3. SIAH as an Actionable Target Against EGFR-Driven TNBC.
8. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACT | Adriamycin: Cytoxan, and Taxotere |
| AR | Androgen receptor |
| CI | confidence interval |
| DFS | disease-free survival |
| DRFS | distant recurrence-free survival |
| EGFR | epidermal growth factor receptor |
| ER | estrogen receptor |
| HER2 | human epidermal growth factor receptor 2 |
| H&E | hematoxylin and eosin staining |
| HR | Hazard Ratio |
| IDFS | invasive disease-free survival |
| IHC | immunohistochemistry |
| LN | lymph node |
| MBC | metastatic breast cancer |
| mTNBC | metastatic TNBC |
| mTOR | the mammalian target of rapamycin |
| NACT | neoadjuvant chemotherapy |
| OS | overall survival |
| PARP | poly-ADP-ribose polymerase |
| pCR | pathological complete response |
| PD-1 | programmed cell death receptor-1 |
| PD-L1 | programmed death ligand-1 |
| PFS | progression-free survival |
| PI3K | phosphoinositide-3 kinase |
| pIR | pathological incomplete response |
| PKB | protein kinase B (AKT) |
| PR | progesterone receptor |
| RCB | Residual Cancer Burden |
| SEER | Surveillance, Epidemiology and End Results Program |
| SIAH | human homologues of Drosophila Seven-In-Absentia |
| SOC | standard of care |
| TIL | tumor infiltrating lymphocytes |
| TME | tumor microenvironment |
| TNBC | triple-negative breast cancer |
| TNM | tumor size, lymph node status, metastasis |
| Trop-2 | Trophoblast cell-surface antigen |
References
- Siegel, R.L.; Mph, K.D.M.; Jemal, A. Cancer statistics, 2020. CA A Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, C.E.; Mph, K.D.M.; Sauer, A.G.; Jemal, A.; Siegel, R.L. Cancer statistics for African Americans, 2019. CA A Cancer J. Clin. 2019, 69, 211–233. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, C.E.; Fedewa, S.A.; Kramer, J.L.; Smith, R.A.; Jemal, A.; Sauer, A.G. Breast cancer statistics, 2015: Convergence of incidence rates between black and white women. CA A Cancer J. Clin. 2015, 66, 31–42. [Google Scholar] [CrossRef]
- Miller, K.D.; Nogueira, L.; Mariotto, A.B.; Rowland, J.H.; Yabroff, K.R.; Alfano, C.M.; Jemal, A.; Kramer, J.L.; Siegel, R.L. Cancer treatment and survivorship statistics, 2019. CA A Cancer J. Clin. 2019, 69, 363–385. [Google Scholar] [CrossRef]
- Pal, S.K.; Childs, B.H.; Pegram, M. Triple negative breast cancer: Unmet medical needs. Breast Cancer Res. Treat. 2010, 125, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, M.C.; Rastogi, P.; Geyer, C.E., Jr.; Miller, L.D.; Thomas, A. Early and locally advanced metaplastic breast cancer: Presentation and survival by receptor status in surveillance, epidemiology, and end results (SEER) 2010–2014. Oncologist 2018, 23, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Gradishar, W.J.; Anderson, B.O.; Balassanian, R.; Blair, S.L.; Burstein, H.J.; Cyr, A.; Elias, A.D.; Farrar, W.B.; Forero, A.; Giordano, S.H.; et al. NCCN guidelines insights: Breast cancer, version 1.2017. J. Natl. Compr. Cancer Netw. 2017, 15, 433–451. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K. Heterogeneity in breast cancer. J. Clin. Investig. 2011, 121, 3786–3788. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Network, T.C.G.A.; Network, C.G.A.; Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer genome Landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Ciriello, G.; Gatza, M.L.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.K.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive molecular portraits of invasive lobular breast cancer. Cell 2015, 163, 506–519. [Google Scholar] [CrossRef]
- Graham, L.J.; Shupe, M.P.; Schneble, E.J.; Flynt, F.L.; Clemenshaw, M.N.; Kirkpatrick, A.D.; Gallagher, C.; Nissan, A.; Henry, L.; Stojadinovic, A.; et al. Current approaches and challenges in monitoring treatment responses in breast cancer. J. Cancer 2014, 5, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, U.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Rakha, E.; Reis-Filho, J.S.; Ellis, I. Basal-like breast cancer: A critical review. J. Clin. Oncol. 2008, 26, 2568–2581. [Google Scholar] [CrossRef] [PubMed]
- Savard, M.F.; Khan, O.; Hunt, K.K.; Verma, S. Redrawing the lines: The next generation of treatment in metastatic breast cancer. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, e8–e21. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, C.E.; Ma, J.; Bryan, L.; Jemal, A. Breast cancer statistics, 2013. CA: A Cancer J. Clin. 2013, 64, 52–62. [Google Scholar] [CrossRef]
- Thomas, A.; Rhoads, A.; Pinkerton, E.; Schroeder, M.C.; Conway, K.M.; Hundley, W.G.; McNally, L.R.; Oleson, J.; Lynch, C.F.; Romitti, P.A. Incidence and survival among young women with stage I–III breast cancer: SEER 2000–2015. JNCI Cancer Spectr. 2019, 3. [Google Scholar] [CrossRef]
- Gupta, G.; Lee, C.D.; Guye, M.L.; Van Sciver, R.E.; Lee, M.P.; Lafever, A.C.; Pang, A.; Tang-Tan, A.M.; Winston, J.S.; Samli, B.; et al. Unmet clinical need: Developing prognostic biomarkers and precision medicine to forecast early tumor relapse, detect chemo-resistance and improve overall survival in high-risk breast cancer. Ann. Breast Cancer Ther. 2020, 4, 48–57. [Google Scholar] [CrossRef]
- Slamon, D.J.; Neven, P.; Chia, S.; Fasching, P.A.; De Laurentiis, M.; Im, S.A.; Petrakova, K.; Bianchi, G.V.; Esteva, F.J.; Martín, M.; et al. Overall survival with ribociclib plus fulvestrant in advanced breast cancer. N. Engl. J. Med. 2020, 382, 514–524. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef] [PubMed]
- Carey, L.; Winer, E.; Viale, G.; Cameron, D.; Gianni, L. Triple-negative breast cancer: Disease entity or title of convenience? Nat. Rev. Clin. Oncol. 2010, 7, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Boyle, P. Triple-negative breast cancer: Epidemiological considerations and recommendations. Ann. Oncol. 2012, 23, vi7–vi12. [Google Scholar] [CrossRef]
- Dent, R.; Hanna, W.M.; Trudeau, M.; Rawlinson, E.; Sun, P.; Narod, S.A. Pattern of metastatic spread in triple-negative breast cancer. Breast Cancer Res. Treat. 2008, 115, 423–428. [Google Scholar] [CrossRef]
- Dent, R.; Trudeau, M.E.; Pritchard, K.I.; Hanna, W.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef]
- Bauer, K.; Brown, M.; Cress, R.D.; Parise, C.A.; Caggiano, V. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype. Cancer 2007, 109, 1721–1728. [Google Scholar] [CrossRef]
- Anders, C.; Carey, L.A. Understanding and treating triple-negative breast cancer. Oncology 2008, 22, 1233–1243. [Google Scholar]
- Newman, L.A.; Kaljee, L.M. Health disparities and triple-negative breast cancer in African American women. JAMA Surg. 2017, 152, 485–493. [Google Scholar] [CrossRef]
- Huo, D.; Hu, H.; Rhie, S.K.; Gamazon, E.R.; Cherniack, A.D.; Liu, J.; Yoshimatsu, T.F.; Pitt, J.J.; Hoadley, K.A.; Troester, M.; et al. Comparison of breast cancer molecular features and survival by African and European ancestry in the cancer genome atlas. JAMA Oncol. 2017, 3, 1654–1662. [Google Scholar] [CrossRef]
- DeSantis, C.E.; Siegel, R.L.; Sauer, A.G.; Miller, K.D.; Fedewa, S.A.; Alcaraz, K.I.; Jemal, A. Cancer statistics for African Americans, 2016: Progress and opportunities in reducing racial disparities. CA A Cancer J. Clin. 2016, 66, 290–308. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Ginsburg, O.; Rochon, P.; Sun, P.; Narod, S.A. Differences in breast cancer stage at diagnosis and cancer-specific survival by race and ethnicity in the United States. JAMA 2015, 313, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Dietze, E.C.; Sistrunk, C.; Miranda-Carboni, G.; O’Regan, R.; Seewaldt, V.L. Triple-negative breast cancer in African-American women: Disparities versus biology. Nat. Rev. Cancer 2015, 15, 248–254. [Google Scholar] [CrossRef] [PubMed]
- O’Keefe, E.B.; Meltzer, J.P.; Bethea, T.N. Health disparities and cancer: Racial disparities in cancer mortality in the United States, 2000–2010. Front. Public Health 2015, 3, 51. [Google Scholar] [CrossRef]
- Brewster, A.M.; Chavez-MacGregor, M.; Brown, P. Epidemiology, biology, and treatment of triple-negative breast cancer in women of African ancestry. Lancet Oncol. 2014, 15, e625–e634. [Google Scholar] [CrossRef]
- Howlader, N.; Cronin, K.A.; Kurian, A.W.; Andridge, R. Differences in breast cancer survival by molecular subtypes in the United States. Cancer Epidemiol. Biomark. Prev. 2018, 27, 619–626. [Google Scholar] [CrossRef]
- Lin, N.U.; Ms, A.V.; Hughes, M.E.; Theriault, R.L.; Edge, S.B.; Wong, Y.N.; Blayney, D.W.; Niland, J.C.; Winer, E.P.; Weeks, J.C. Clinicopathologic features, patterns of recurrence, and survival among women with triple-negative breast cancer in the National Comprehensive Cancer Network. Cancer 2012, 118, 5463–5472. [Google Scholar] [CrossRef]
- Andreopoulou, E.; Schweber, S.J.; Sparano, J.A.; McDaid, H.M. Therapies for triple negative breast cancer. Expert Opin. Pharmacother. 2015, 16, 983–998. [Google Scholar] [CrossRef]
- Vagia, E.; Mahalingam, D.; Cristofanilli, M. The landscape of targeted therapies in TNBC. Cancers 2020, 12, 916. [Google Scholar] [CrossRef]
- Hutchinson, L. TNBC: Can we treat the untargetable? Nat. Rev. Clin. Oncol. 2014, 11, 379. [Google Scholar] [CrossRef]
- Sharma, P. Biology and management of patients with triple-negative breast cancer. Oncologist 2016, 21, 1050–1062. [Google Scholar] [CrossRef] [PubMed]
- Isakoff, S.J.; Mayer, E.L.; He, L.; Traina, T.A.; Carey, L.A.; Krag, K.J.; Rugo, H.S.; Liu, M.C.; Stearns, V.; Come, S.E.; et al. TBCRC009: A multicenter phase II clinical trial of platinum monotherapy with biomarker assessment in metastatic triple-negative breast cancer. J. Clin. Oncol. 2015, 33, 1902–1909. [Google Scholar] [CrossRef]
- Isakoff, S.J. Triple-negative breast cancer. Cancer J. 2010, 16, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Santonja, A.; Sanchez-Muñoz, A.; Lluch, A.; Chica-Parrado, M.R.; Albanell, J.; Chacon, J.I.; Antolín, S.; Jerez, J.M.; De La Haba, J.; De Luque, V.; et al. Triple negative breast cancer subtypes and pathologic complete response rate to neoadjuvant chemotherapy. Oncotarget 2018, 9, 26406–26416. [Google Scholar] [CrossRef] [PubMed]
- Esserman, L.J.; Berry, D.A.; DeMichele, A.; Carey, L.; Davis, S.E.; Buxton, M.; Hudis, C.; Gray, J.W.; Perou, C.M.; Yau, C.; et al. Pathologic complete response predicts recurrence-free survival more effectively by cancer subset: Results from the I-SPY 1 TRIAL—CALGB 150007/150012, ACRIN 6657. J. Clin. Oncol. 2012, 30, 3242–3249. [Google Scholar] [CrossRef] [PubMed]
- Cortazar, P.; Zhang, L.; Untch, M.; Mehta, K.; Costantino, J.P.; Wolmark, N.; Bonnefoi, H.; Cameron, D.; Gianni, L.; Valagussa, P.; et al. Pathological complete response and long-term clinical benefit in breast cancer: The CTNeoBC pooled analysis. Lancet 2014, 384, 164–172. [Google Scholar] [CrossRef]
- Wu, K.-P.; Yang, Q.; Liu, Y.; Wu, A.; Yang, Z. Meta-analysis on the association between pathologic complete response and triple-negative breast cancer after neoadjuvant chemotherapy. World J. Surg. Oncol. 2014, 12, 95. [Google Scholar] [CrossRef]
- Biswas, T.; Efird, J.T.; Prasad, S.; Jindal, C.; Walker, P.R. The survival benefit of neoadjuvant chemotherapy and pCR among patients with advanced stage triple negative breast cancer. Oncotarget 2017, 8, 112712–112719. [Google Scholar] [CrossRef]
- Levasseur, N.; Sun, J.; Gondara, L.; Diocee, R.; Speers, C.; Lohrisch, C.; Chia, S. Impact of pathologic complete response on survival after neoadjuvant chemotherapy in early-stage breast cancer: A population-based analysis. J. Cancer Res. Clin. Oncol. 2019, 146, 529–536. [Google Scholar] [CrossRef]
- Carbognin, L.; Furlanetto, J.; Vicentini, C.; Nortilli, R.; Pilotto, S.; Brunelli, M.; Pellini, F.; Pollini, G.P.; Bria, E.; Tortora, G. Neoadjuvant strategies for triple negative breast cancer: ‘state-of-the-art’ and future perspectives. Anti-Cancer Agents Med. Chem. 2014, 15, 15–25. [Google Scholar] [CrossRef]
- Mougalian, S.S.; Soulos, P.R.; Killelea, B.K.; Lannin, D.R.; Abu-Khalaf, M.M.; DiGiovanna, M.P.; Sanft, T.B.; Pusztai, L.; Gross, C.P.; Chagpar, A.B. Use of neoadjuvant chemotherapy for patients with stage I to III breast cancer in the United States. Cancer 2015, 121, 2544–2552. [Google Scholar] [CrossRef]
- Echeverria, G.V.; Ge, Z.; Seth, S.; Zhang, X.; Jeter-Jones, S.; Zhou, X.; Cai, S.; Tu, Y.; McCoy, A.; Peoples, M.D.; et al. Resistance to neoadjuvant chemotherapy in triple-negative breast cancer mediated by a reversible drug-tolerant state. Sci. Transl. Med. 2019, 11, eaav0936. [Google Scholar] [CrossRef]
- Kennedy, W.R.; Tricarico, C.; Gabani, P.; Weiner, A.A.; Altman, M.B.; Ochoa, L.L.; Thomas, M.A.; Margenthaler, J.A.; Sanati, S.; Peterson, L.L.; et al. Predictors of distant metastases in triple-negative breast cancer without pathologic complete response after neoadjuvant chemotherapy. J. Natl. Compr. Cancer Netw. 2020, 18, 288–296. [Google Scholar] [CrossRef]
- Symmans, W.F.; Peintinger, F.; Hatzis, C.; Rajan, R.; Kuerer, H.; Valero, V.; Assad, L.; Poniecka, A.; Hennessy, B.; Green, M.; et al. Measurement of residual breast cancer burden to predict survival after neoadjuvant chemotherapy. J. Clin. Oncol. 2007, 25, 4414–4422. [Google Scholar] [CrossRef]
- Symmans, W.F.; Wei, C.; Gould, R.; Yu, X.; Zhang, Y.; Liu, M.; Walls, A.; Bousamra, A.; Ramineni, M.; Sinn, B.; et al. Long-term prognostic risk after neoadjuvant chemotherapy associated with residual cancer burden and breast cancer subtype. J. Clin. Oncol. 2017, 35, 1049–1060. [Google Scholar] [CrossRef]
- Masuda, N.; Lee, S.J.; Ohtani, S.; Im, Y.H.; Lee, E.S.; Yokota, I.; Kuroi, K.; Im, S.A.; Park, B.W.; Kim, S.B.; et al. Adjuvant capecitabine for breast cancer after preoperative chemotherapy. N. Engl. J. Med. 2017, 376, 2147–2159. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, Y.; Mao, F.; Zhang, X.; Shen, S.; Sun, Q. Adjuvant addition of capecitabine to early-stage triple-negative breast cancer patients receiving standard chemotherapy: A meta-analysis. Breast Cancer Res. Treat. 2019, 179, 533–542. [Google Scholar] [CrossRef]
- Garrido-Castro, A.C.; Lin, N.U.; Polyak, K. Insights into molecular classifications of triple-negative breast cancer: Improving patient selection for treatment. Cancer Discov. 2019, 9, 176–198. [Google Scholar] [CrossRef]
- Wang, D.-Y.; Jiang, Z.; Ben-David, Y.; Woodgett, J.R.; Zacksenhaus, E. Molecular stratification within triple-negative breast cancer subtypes. Sci. Rep. 2019, 9, 19107. [Google Scholar] [CrossRef]
- Masuda, H.; Baggerly, K.A.; Wang, Y.; Zhang, Y.; Gonzalez-Angulo, A.M.; Meric-Bernstam, F.; Valero, V.; Lehmann, B.D.; Pietenpol, J.A.; Hortobagyi, G.N.; et al. Differential response to neoadjuvant chemotherapy among 7 triple-negative breast cancer molecular subtypes. Clin. Cancer Res. 2013, 19, 5533–5540. [Google Scholar] [CrossRef]
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M.; Covington, K.R.; Contreras, A.; Fuqua, S.A.; Savage, M.I.; Osborne, C.K.; Hilsenbeck, S.G.; Chang, J.C.; et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin. Cancer Res. 2015, 21, 1688–1698. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Jovanović, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of triple-negative breast cancer molecular subtypes: Implications for neoadjuvant chemotherapy selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Pietenpol, J.A.; Tan, A.R. Triple-negative breast cancer: Molecular subtypes and new targets for therapy. Am. Soc. Clin. Oncol. Educ. Book 2015, e31–e39. [Google Scholar] [CrossRef] [PubMed]
- Prado-Vázquez, G.; Gamez-Pozo, A.; Trilla-Fuertes, L.; Arevalillo, J.M.; Zapater-Moros, A.; Ferrer-Gómez, M.; Díaz-Almirón, M.; López-Vacas, R.; Navarro, H.; Main, P.; et al. A novel approach to triple-negative breast cancer molecular classification reveals a luminal immune-positive subgroup with good prognoses. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Jézéquel, P.; Loussouarn, D.; Guerin-Charbonnel, C.; Campion, L.; Vanier, A.; Gouraud, W.; Lasla, H.; Guette, C.; Valo, I.; Verriele, V.; et al. Gene-expression molecular subtyping of triple-negative breast cancer tumours: Importance of immune response. Breast Cancer Res. 2015, 17, 43. [Google Scholar] [CrossRef] [PubMed]
- Sporikova, Z.; Koudelakova, V.; Trojanec, R.; Hajduch, M. Genetic markers in triple-negative breast cancer. Clin. Breast Cancer 2018, 18, e841–e850. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef]
- Jovanović, B.; Mayer, I.A.; Mayer, E.L.; Abramson, V.G.; Bardia, A.; Sanders, M.E.; Kuba, M.G.; Estrada, M.V.; Beeler, J.S.; Shaver, T.M.; et al. A randomized phase II neoadjuvant study of cisplatin, paclitaxel with or without everolimus in patients with stage II/III triple-negative breast cancer (TNBC): Responses and long-term outcome correlated with increased frequency of DNA damage response gene mutations, TNBC Subtype, AR Status, and Ki67. Clin. Cancer Res. 2017, 23, 4035–4045. [Google Scholar] [CrossRef]
- Tobin, N.P.; Lundberg, A.; Lindström, L.S.; Harrell, J.C.; Foukakis, T.; Carlsson, L.; Einbeigi, Z.; Linderholm, B.K.; Loman, N.; Malmberg, M.; et al. PAM50 provides prognostic information when applied to the lymph node metastases of advanced breast cancer patients. Clin. Cancer Res. 2017, 23, 7225–7231. [Google Scholar] [CrossRef]
- Prat, A.; Parker, J.; Karginova, O.; Fan, C.; Livasy, C.; Herschkowitz, J.I.; He, X.; Perou, C.M. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010, 12, R68. [Google Scholar] [CrossRef]
- Prat, A.; Fan, C.; Fernández-Martínez, A.; Hoadley, K.A.; Martinello, R.; Vidal, M.; Viladot, M.; Pineda, E.; Arance, A.; Muñoz, M.; et al. Response and survival of breast cancer intrinsic subtypes following multi-agent neoadjuvant chemotherapy. BMC Med. 2015, 13, 303. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.M.; Tang, G.; Lucas, P.C.; Robidoux, A.; Goerlitz, D.; Harris, B.T.; Bandos, H.; Geyer, C.E.; Rastogi, P.; Mamounas, E.P.; et al. Pathologic complete response and outcomes by intrinsic subtypes in NSABP B-41, a randomized neoadjuvant trial of chemotherapy with trastuzumab, lapatinib, or the combination. Breast Cancer Res. Treat. 2019, 178, 389–399. [Google Scholar] [CrossRef]
- Swain, S.M.; Tang, G.; Brauer, H.A.; Goerlitz, D.S.; Lucas, P.C.; Robidoux, A.; Harris, B.T.; Bandos, H.; Ren, Y.; Geyer, C.E., Jr.; et al. NSABP B-41, a randomized neoadjuvant trial: Genes and signatures associated with pathologic complete response. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Esserman, L.J.; Berry, D.A.; Cheang, M.C.U.; Yau, C.; Perou, C.M.; Carey, L.; DeMichele, A.; Gray, J.W.; Conway-Dorsey, K.; Lenburg, M.; et al. Chemotherapy response and recurrence-free survival in neoadjuvant breast cancer depends on biomarker profiles: Results from the I-SPY 1 TRIAL (CALGB 150007/150012; ACRIN 6657). Breast Cancer Res. Treat. 2011, 132, 1049–1062. [Google Scholar] [CrossRef]
- Amin, M.B.; Greene, F.L.; Edge, S.B.; Compton, C.C.; Gershenwald, J.E.; Brookland, R.K.; Meyer, L.; Gress, D.M.; Byrd, D.R.; Winchester, D.P. The Eighth Edition AJCC Cancer Staging Manual: Continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA A Cancer J. Clin. 2017, 67, 93–99. [Google Scholar] [CrossRef]
- Li, J.P.; Zhang, X.M.; Zhang, Y.S.; Zheng, L.H.; Liu, Y.J. The prognostic value of the 8th edition of the American Joint Committee on Cancer (AJCC) staging system in triple-negative breast cancer. Neoplasma 2019, 66, 810–817. [Google Scholar] [CrossRef]
- He, J.; Tsang, J.Y.; Xu, X.; Li, J.; Li, M.; Chao, X.; Xu, Y.; Luo, R.; Tse, G.M.; Sun, P. AJCC 8th edition prognostic staging provides no better discriminatory ability in prognosis than anatomical staging in triple negative breast cancer. BMC Cancer 2020, 20, 1–9. [Google Scholar] [CrossRef]
- Eastman, A.; Tammaro, Y.; Moldrem, A.; Andrews, V.; Huth, J.; Euhus, D.; Leitch, M.; Rao, R. Outcomes of delays in time to treatment in triple negative breast cancer. Ann. Surg. Oncol. 2013, 20, 1880–1885. [Google Scholar] [CrossRef]
- Marra, A.; Viale, G.; Curigliano, G. Recent advances in triple negative breast cancer: The immunotherapy era. BMC Med. 2019, 17, 90. [Google Scholar] [CrossRef]
- Pandy, J.G.P.; Balolong-Garcia, J.C.; Cruz-Ordinario, M.V.B.; Que, F.V.F. Triple negative breast cancer and platinum-based systemic treatment: A meta-analysis and systematic review. BMC Cancer 2019, 19, 1065–1069. [Google Scholar] [CrossRef]
- Vaz-Luis, I.; Ottesen, R.A.; Hughes, M.E.; Mamet, R.; Burstein, H.J.; Edge, S.B.; Gonzalez-Angulo, A.M.; Moy, B.; Rugo, H.S.; Theriault, R.L.; et al. Outcomes by tumor subtype and treatment pattern in women with small, node-negative breast cancer: A multi-institutional study. J. Clin. Oncol. 2014, 32, 2142–2150. [Google Scholar] [CrossRef] [PubMed]
- Theriault, R.L.; Litton, J.; Mittendorf, E.A.; Chen, H.; Meric-Bernstam, F.; Chavez-MacGregor, M.; Morrow, P.K.; Woodward, W.A.; Sahin, A.; Hortobagyi, G.N.; et al. Age and survival estimates in patients who have node-negative T1ab breast cancer by breast cancer subtype. Clin. Breast Cancer 2011, 11, 325–331. [Google Scholar] [CrossRef]
- Leon-Ferre, R.A.; Polley, M.-Y.; Liu, H.; Gilbert, J.A.; Cafourek, V.; Hillman, D.W.; Elkhanany, A.; Akinhanmi, M.; Lilyquist, J.; Thomas, A.; et al. Impact of histopathology, tumor-infiltrating lymphocytes, and adjuvant chemotherapy on prognosis of triple-negative breast cancer. Breast Cancer Res. Treat. 2017, 167, 89–99. [Google Scholar] [CrossRef]
- Park, J.H.; Ahn, J.-H.; Kim, S.-B. How shall we treat early triple-negative breast cancer (TNBC): From the current standard to upcoming immuno-molecular strategies. ESMO Open 2018, 3, e000357. [Google Scholar] [CrossRef]
- Blum, J.L.; Flynn, P.J.; Yothers, G.; Asmar, L.; Geyer, C.E.; Jacobs, S.A.; Robert, N.J.; Hopkins, J.O.; O’Shaughnessy, J.A.; Dang, C.T.; et al. Anthracyclines in early breast cancer: The ABC Trials–USOR 06-090, NSABP B-46-I/USOR 07132, and NSABP B-49 (NRG Oncology). J. Clin. Oncol. 2017, 35, 2647–2655. [Google Scholar] [CrossRef]
- Martin, M.; Rodríguez-Lescure, A.; Ruiz, A.; Alba, E.; Calvo, L.; Ruiz-Borrego, M.; Santaballa, A.; Rodríguez, C.A.; Crespo, C.; Abad, M.; et al. Molecular predictors of efficacy of adjuvant weekly paclitaxel in early breast cancer. Breast Cancer Res. Treat. 2009, 123, 149–157. [Google Scholar] [CrossRef]
- Xia, L.Y.; Hu, Q.L.; Zhang, J.; Xu, W.Y.; Li, X.S. Survival outcomes of neoadjuvant versus adjuvant chemotherapy in triple-negative breast cancer: A meta-analysis of 36,480 cases. World J. Surg. Oncol. 2020, 18, 1–8. [Google Scholar] [CrossRef]
- Haddad, T.C.; Goetz, M.P. Landscape of neoadjuvant therapy for breast cancer. Ann. Surg. Oncol. 2015, 22, 1408–1415. [Google Scholar] [CrossRef]
- Arteaga, C.L.; Sliwkowski, M.X.; Osborne, C.K.; Perez, E.A.; Puglisi, F.; Gianni, L. Treatment of HER2-positive breast cancer: Current status and future perspectives. Nat. Rev. Clin. Oncol. 2011, 9, 16–32. [Google Scholar] [CrossRef]
- Thompson, A.M.; Moulder-Thompson, S.L. Neoadjuvant treatment of breast cancer. Ann. Oncol. 2012, 23, x231–x236. [Google Scholar] [CrossRef]
- Zardavas, D.; Baselga, J.; Piccart, M. Emerging targeted agents in metastatic breast cancer. Nat. Rev. Clin. Oncol. 2013, 10, 191–210. [Google Scholar] [CrossRef] [PubMed]
- Tevaarwerk, A.; Gray, R.J.; Schneider, B.P.; Smith, M.L.; Wagner, L.I.; Fetting, J.H.; Davidson, N.; Goldstein, L.J.; Miller, K.D.; Sparano, J.A. Survival in patients with metastatic recurrent breast cancer after adjuvant chemotherapy: Little evidence of improvement over the past 30 years. Cancer 2012, 119, 1140–1148. [Google Scholar] [CrossRef] [PubMed]
- Nagayama, A.; Hayashida, T.; Jinno, H.; Takahashi, M.; Seki, T.; Matsumoto, A.; Murata, T.; Ashrafian, H.; Athanasiou, T.; Okabayashi, K.; et al. Comparative effectiveness of neoadjuvant therapy for HER2–positive breast cancer: A network meta-analysis. J. Natl. Cancer Inst. 2014, 106, dju203. [Google Scholar] [CrossRef] [PubMed]
- King, T.A.; Morrow, M. Surgical issues in patients with breast cancer receiving neoadjuvant chemotherapy. Nat. Rev. Clin. Oncol. 2015, 12, 335–343. [Google Scholar] [CrossRef] [PubMed]
- DeMichele, A.; Yee, D.; Berry, N.A.; Albain, K.S.; Benz, C.C.; Boughey, J.; Buxton, M.; Chia, S.K.; Chien, A.J.; Chui, S.Y.; et al. The neoadjuvant model is still the future for drug development in breast cancer. Clin. Cancer Res. 2015, 21, 2911–2915. [Google Scholar] [CrossRef] [PubMed]
- Bevers, T.B.; Anderson, B.O.; Bonaccio, E.; Buys, S.; Buys, S.; Daly, M.B.; Dempsey, P.J.; Farrar, W.B.; Fleming, I.; Garber, J.E.; et al. NCCN clinical practice guidelines in oncology: Breast cancer screening and diagnosis. J. Natl. Compr. Cancer Netw. 2009, 7, 1060–1096. [Google Scholar] [CrossRef]
- Redden, M.H.; Fuhrman, G.M. Neoadjuvant chemotherapy in the treatment of breast cancer. Surg. Clin. North. Am. 2013, 93, 493–499. [Google Scholar] [CrossRef]
- Tolaney, S.M.; Barry, W.T.; Dang, C.T.; Yardley, D.A.; Moy, B.; Marcom, P.K.; Albain, K.S.; Rugo, H.S.; Ellis, M.; Shapira, I.; et al. Adjuvant paclitaxel and trastuzumab for node-negative, HER2-positive breast cancer. N. Engl. J. Med. 2015, 372, 134–141. [Google Scholar] [CrossRef]
- Baselga, J.; Segalla, J.G.M.; Roché, H.; Del Giglio, A.; Pinczowski, H.; Ciruelos, E.M.; Filho, S.C.; Gómez, P.; Van Eyll, B.; Bermejo, B.; et al. Sorafenib in Combination with Capecitabine: An Oral Regimen for Patients With HER2-Negative Locally Advanced or Metastatic Breast Cancer. J. Clin. Oncol. 2012, 30, 1484–1491. [Google Scholar] [CrossRef]
- Baselga, J.; Gómez, P.; Greil, R.; Braga, S.; Climent, M.A.; Wardley, A.; Kaufman, B.; Stemmer, S.M.; Pêgo, A.; Chan, A.; et al. Randomized Phase II Study of the Anti–Epidermal Growth Factor Receptor Monoclonal Antibody Cetuximab With Cisplatin Versus Cisplatin Alone in Patients with Metastatic Triple-Negative Breast Cancer. J. Clin. Oncol. 2013, 31, 2586–2592. [Google Scholar] [CrossRef]
- Gass, P.; Lux, M.P.; Rauh, C.; Hein, A.; Bani, M.R.; Fiessler, C.; Hartmann, A.; Häberle, L.; Pretscher, J.; Erber, R.; et al. Prediction of pathological complete response and prognosis in patients with neoadjuvant treatment for triple-negative breast cancer. BMC Cancer 2018, 18, 1051. [Google Scholar] [CrossRef] [PubMed]
- Pelizzari, G.; Gerratana, L.; Basile, D.; Fanotto, V.; Bartoletti, M.; Liguori, A.; Fontanella, C.; Spazzapan, S.; Puglisi, F. Post-neoadjuvant strategies in breast cancer: From risk assessment to treatment escalation. Cancer Treat. Rev. 2019, 72, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Bear, H.D.; Tang, G.; Rastogi, P.; Geyer, C.E.; Liu, Q.; Robidoux, A.; Baez-Diaz, L.; Brufsky, A.M.; Mehta, R.S.; Fehrenbacher, L.; et al. Neoadjuvant plus adjuvant bevacizumab in early breast cancer (NSABP B-40 [NRG Oncology]): Secondary outcomes of a phase 3, randomised controlled trial. Lancet Oncol. 2015, 16, 1037–1048. [Google Scholar] [CrossRef]
- Liedtke, C.; Mazouni, C.; Hess, K.R.; André, F.; Tordai, A.; Mejia, J.A.; Symmans, W.F.; Gonzalez-Angulo, A.M.; Hennessy, B.; Green, M.C.; et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2008, 26, 1275–1281. [Google Scholar] [CrossRef]
- Gluz, O.; Liedtke, C.; Gottschalk, N.; Pusztai, L.; Nitz, U.; Harbeck, N. Triple-negative breast cancer—Current status and future directions. Ann. Oncol. 2009, 20, 1913–1927. [Google Scholar] [CrossRef]
- Von Minckwitz, G.; Untch, M.; Blohmer, J.U.; Costa, S.D.; Eidtmann, H.; Fasching, P.A.; Gerber, B.; Eiermann, W.; Hilfrich, J.; Huober, J.; et al. Definition and impact of pathologic complete response on prognosis after neoadjuvant chemotherapy in various intrinsic breast cancer subtypes. J. Clin. Oncol. 2012, 30, 1796–1804. [Google Scholar] [CrossRef]
- Weiss, A.; Bashour, S.I.; Hess, K.R.; Thompson, A.M.; Ibrahim, N. Effect of neoadjuvant chemotherapy regimen on relapse-free survival among patients with breast cancer achieving a pathologic complete response: An early step in the de-escalation of neoadjuvant chemotherapy. Breast Cancer Res. 2018, 20, 27. [Google Scholar] [CrossRef]
- Schott, A.F.; Hayes, D.F. Defining the benefits of neoadjuvant chemotherapy for breast cancer. J. Clin. Oncol. 2012, 30, 1747–1749. [Google Scholar] [CrossRef]
- Parinyanitikul, N.; Lei, X.; Chavez-MacGregor, M.; Liu, S.; Mittendorf, E.A.; Litton, J.K.; Woodward, W.A.; Zhang, A.H.; Hortobágyi, G.N.; Valero, V.; et al. Receptor status change from primary to residual breast cancer after neoadjuvant chemotherapy and analysis of survival outcomes. Clin. Breast Cancer 2015, 15, 153–160. [Google Scholar] [CrossRef]
- Luen, S.; Salgado, R.; Dieci, M.V.; Vingiani, A.; Curigliano, G.; Gould, R.; Castaneda, C.A.; D’Alfonso, T.; Sanchez, J.; Cheng, E.; et al. Prognostic implications of residual disease tumor-infiltrating lymphocytes and residual cancer burden in triple-negative breast cancer patients after neoadjuvant chemotherapy. Ann. Oncol. 2019, 30, 236–242. [Google Scholar] [CrossRef]
- Sheri, A.; Smith, I.; Johnston, S.R.; A’Hern, R.; Nerurkar, A.; Jones, R.L.; Hills, M.; Detre, S.; Pinder, S.E.; Symmans, W.F.; et al. Residual proliferative cancer burden to predict long-term outcome following neoadjuvant chemotherapy. Ann. Oncol. 2015, 26, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.I.; Yau, C.; Krass, P.; Moore, D.; Carey, L.A.; Au, A.; Chhieng, D.; Giri, D.; Livasy, C.; Mies, C.; et al. Comparison of residual cancer burden, American Joint Committee on Cancer staging and pathologic complete response in breast cancer after neoadjuvant chemotherapy: Results from the I-SPY 1 TRIAL (CALGB 150007/150012; ACRIN 6657). Breast Cancer Res. Treat. 2017, 165, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Cain, H.; MacPherson, I.; Beresford, M.; Pinder, S.; Pong, J.; Dixon, J. Neoadjuvant therapy in early breast cancer: Treatment considerations and common debates in practice. Clin. Oncol. 2017, 29, 642–652. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Yost, S.E.; Yuan, Y. Neoadjuvant treatment for triple negative breast cancer: Recent progresses and challenges. Cancers 2020, 12, 1404. [Google Scholar] [CrossRef] [PubMed]
- García-Martínez, E.; Luengo-Gil, G.; Benito, A.C.; González-Billalabeitia, E.; Conesa, M.A.V.; García, T.G.; García-Garre, E.; Vicente, V.; De La Peña, F.A. Tumor-infiltrating immune cell profiles and their change after neoadjuvant chemotherapy predict response and prognosis of breast cancer. Breast Cancer Res. 2014, 16, 488. [Google Scholar] [CrossRef]
- Ladoire, S.; Mignot, G.; Dabakuyo, S.; Arnould, L.; Apetoh, L.; Rébé, C.; Coudert, B.; Martin, F.; Bizollon, M.H.; Vanoli, A.; et al. In situ immune response after neoadjuvant chemotherapy for breast cancer predicts survival. J. Pathol. 2011, 224, 389–400. [Google Scholar] [CrossRef]
- Asano, Y.; Kashiwagi, S.; Goto, W.; Takada, K.; Takahashi, K.; Hatano, T.; Noda, S.; Takashima, T.; Onoda, N.; Tomita, S.; et al. Prediction of survival after neoadjuvant chemotherapy for breast cancer by evaluation of tumor-infiltrating lymphocytes and residual cancer burden. BMC Cancer 2017, 17, 888. [Google Scholar] [CrossRef]
- Asano, Y.; Kashiwagi, S.; Goto, W.; Takada, K.; Takahashi, K.; Morisaki, T.; Fujita, H.; Takashima, T.; Tomita, S.; Ohsawa, M.; et al. Prediction of treatment responses to neoadjuvant chemotherapy in triple-negative breast cancer by analysis of immune checkpoint protein expression. J. Transl. Med. 2018, 16, 87. [Google Scholar] [CrossRef]
- Pinard, C.; Debled, M.; Ben Rejeb, H.; Velasco, V.; De Lara, C.T.; Hoppe, S.; Richard, E.; Brouste, V.; Bonnefoi, H.; MacGrogan, G. Residual cancer burden index and tumor-infiltrating lymphocyte subtypes in triple-negative breast cancer after neoadjuvant chemotherapy. Breast Cancer Res. Treat. 2019, 179, 11–23. [Google Scholar] [CrossRef]
- Dieci, M.V.; Radosevic-Robin, N.; Fineberg, S.; Eynden, G.V.D.; Ternes, N.; Penault-Llorca, F.; Pruneri, G.; D’Alfonso, T.M.; DeMaria, S.; Castaneda, C.A.; et al. Update on tumor-infiltrating lymphocytes (TILs) in breast cancer, including recommendations to assess TILs in residual disease after neoadjuvant therapy and in carcinoma in situ: A report of the International Immuno-Oncology Biomarker Working Group on Breast Cancer. Semin. Cancer Biol. 2018, 52, 16–25. [Google Scholar] [CrossRef]
- Dieci, M.V.; Criscitiello, C.; Goubar, A.; Viale, G.; Conte, P.; Guarneri, V.; Ficarra, G.; Mathieu, M.C.; Delaloge, S.; Curigliano, G.; et al. Prognostic value of tumor-infiltrating lymphocytes on residual disease after primary chemotherapy for triple-negative breast cancer: A retrospective multicenter study. Ann. Oncol. 2015, 26, 1518. [Google Scholar] [CrossRef] [PubMed]
- Byrne, A.; Savas, P.; Sant, S.; Li, R.; Virassamy, B.; Luen, S.J.; Beavis, P.A.; Mackay, L.K.; Neeson, P.J.; Loi, S. Tissue-resident memory T cells in breast cancer control and immunotherapy responses. Nat. Rev. Clin. Oncol. 2020, 17, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Von Minckwitz, G.; Schneeweiss, A.; Loibl, S.; Salat, C.; Denkert, C.; Rezai, M.; Blohmer, J.U.; Jackisch, C.; Paepke, S.; Gerber, B.; et al. Neoadjuvant carboplatin in patients with triple-negative and HER2-positive early breast cancer (GeparSixto; GBG 66): A randomised phase 2 trial. Lancet Oncol. 2014, 15, 747–756. [Google Scholar] [CrossRef]
- Sikov, W.M.; Berry, D.A.; Perou, C.M.; Singh, B.; Cirrincione, C.T.; Tolaney, S.M.; Kuzma, C.S.; Pluard, T.J.; Somlo, G.; Port, E.R.; et al. Impact of the addition of carboplatin and/or bevacizumab to neoadjuvant once-per-week paclitaxel followed by dose-dense doxorubicin and cyclophosphamide on pathologic complete response rates in Stage II to III triple-negative breast cancer: CALGB 40603 (Alliance). J. Clin. Oncol. 2015, 33, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Loibl, S.; O’Shaughnessy, J.A.; Untch, M.; Sikov, W.M.; Rugo, H.S.; McKee, M.D.; Huober, J.; Golshan, M.; Von Minckwitz, G.; Maag, D.; et al. Addition of the PARP inhibitor veliparib plus carboplatin or carboplatin alone to standard neoadjuvant chemotherapy in triple-negative breast cancer (BrighTNess): A randomised, phase 3 trial. Lancet Oncol. 2018, 19, 497–509. [Google Scholar] [CrossRef]
- Denkert, C.; Von Minckwitz, G.; Brase, J.C.; Sinn, B.V.; Gade, S.; Kronenwett, R.; Pfitzner, B.M.; Salat, C.; Loi, S.; Schmitt, W.D.; et al. Tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy with or without carboplatin in human epidermal growth factor receptor 2–Positive and triple-negative primary breast cancers. J. Clin. Oncol. 2015, 33, 983–991. [Google Scholar] [CrossRef]
- Denkert, C.; Von Minckwitz, G.; Darb-Esfahani, S.; Lederer, B.; Heppner, B.I.; Weber, K.E.; Budczies, J.; Huober, J.; Klauschen, F.; Furlanetto, J.; et al. Tumour-infiltrating lymphocytes and prognosis in different subtypes of breast cancer: A pooled analysis of 3771 patients treated with neoadjuvant therapy. Lancet Oncol. 2017, 19, 40–50. [Google Scholar] [CrossRef]
- Siegel, R.L.; Mph, K.D.M.; Jemal, A. Cancer statistics, 2016. CA A Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef]
- Radovich, M.; Jiang, G.; Hancock, B.A.; Chitambar, C.; Nanda, R.; Falkson, C.; Lynce, F.C.; Gallagher, C.; Isaacs, C.; Blaya, M.; et al. Association of circulating tumor DNA and circulating tumor cells after neoadjuvant chemotherapy with disease recurrence in patients with triple-negative breast cancer: Preplanned secondary analysis of the BRE12-158 randomized clinical trial. JAMA Oncol. 2020. [Google Scholar] [CrossRef]
- Madic, J.; Kiialainen, A.; Bidard, F.-C.; Birzele, F.; Ramey, G.; Leroy, Q.; Frio, T.R.; Vaucher, I.; Raynal, V.; Bernard, V.; et al. Circulating tumor DNA and circulating tumor cells in metastatic triple negative breast cancer patients. Int. J. Cancer 2014, 136, 2158–2165. [Google Scholar] [CrossRef]
- Penault-Llorca, F.; Radosevic-Robin, N. Biomarkers of residual disease after neoadjuvant therapy for breast cancer. Nat. Rev. Clin. Oncol. 2016, 13, 487–503. [Google Scholar] [CrossRef] [PubMed]
- Carey, L.A.; Winer, E.P. I-SPY 2—Toward more rapid progress in breast cancer treatment. N. Engl. J. Med. 2016, 375, 83–84. [Google Scholar] [CrossRef] [PubMed]
- Zardavas, D.; Irrthum, A.; Swanton, C.; Piccart, M. Clinical management of breast cancer heterogeneity. Nat. Rev. Clin. Oncol. 2015, 12, 381–394. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, A.A.; Aranda, V.; Bardelli, A.; Blanpain, C.; Bock, C.; Borowski, C.; Caldas, C.; Califano, A.; Doherty, M.; Elsner, M.; et al. Toward understanding and exploiting tumor heterogeneity. Nat. Med. 2015, 21, 846–853. [Google Scholar] [CrossRef]
- Almendro, V.; Marusyk, A.; Polyak, K. Cellular Heterogeneity and Molecular Evolution in Cancer. Annu. Rev. Pathol. Mech. Dis. 2013, 8, 277–302. [Google Scholar] [CrossRef]
- Yap, T.A.; Gerlinger, M.; Futreal, P.A.; Pusztai, L.; Swanton, C. Intratumor heterogeneity: Seeing the wood for the trees. Sci. Transl. Med. 2012, 4, 127ps10. [Google Scholar] [CrossRef]
- Parker, J.S.; Perou, C.M. Tumor heterogeneity: Focus on the leaves, the trees, or the forest? Cancer Cell 2015, 28, 149–150. [Google Scholar] [CrossRef][Green Version]
- Swain, S.M.; Baselga, J.; Kim, S.-B.; Ro, J.; Semiglazov, V.; Campone, M.; Ciruelos, E.M.; Ferrero, J.-M.; Schneeweiss, A.; Heeson, S.; et al. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N. Engl. J. Med. 2015, 372, 724–734. [Google Scholar] [CrossRef]
- Paoletti, C.; Li, Y.; Muñiz, M.C.; Kidwell, K.; Aung, K.; Thomas, D.G.; Brown, M.E.; Abramson, V.G.; Irvin, W.J.; Lin, N.U.; et al. Significance of circulating tumor cells in metastatic triple-negative breast cancer patients within a randomized, phase II trial: TBCRC 019. Clin. Cancer Res. 2015, 21, 2771–2779. [Google Scholar] [CrossRef]
- Konner, M.; Hayes, D.F. Progress in the treatment of breast cancer. N. Engl. J. Med. 2020, 382, e4. [Google Scholar] [CrossRef]
- Van Sciver, R.E.; Lee, M.P.; Lee, C.D.; LaFever, A.C.; Svyatova, E.; Kanda, K.; Collier, A.L.; Van Reesema, L.L.S.; Tang-Tan, A.M.; Zheleva, V.; et al. A New strategy to control and eradicate “undruggable” oncogenic k-ras-driven pancreatic cancer: Molecular insights and core principles learned from developmental and evolutionary biology. Cancers 2018, 10, 142. [Google Scholar] [CrossRef] [PubMed]
- Van Sciver, R.E.; Njogu, M.M.; Isbell, A.J.; Odanga, J.J.; Bian, M.; Svyatova, E.; van Reesema, L.L.S.; Zheleva, V.; Eisner, J.L.; Bruflat, J.K.; et al. Blocking SIAH proteolysis, an important K-RAS vulnerability, to control and eradicate K-RAS-driven metastatic cancer. In Conquering RAS: From Biology to Cancer Therapy; Azmi, A.S., Ed.; Academic Press: Boston, MA, USA, 2016. [Google Scholar] [CrossRef]
- Schmid, P.; Rugo, H.S.; Adams, S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Henschel, V.; Molinero, L.; Chui, S.Y.; et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): Updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 44–59. [Google Scholar] [CrossRef]
- Adams, S.; Diéras, V.; Barrios, C.; Winer, E.; Schneeweiss, A.; Iwata, H.; Loi, S.; Patel, S.; Henschel, V.; Chui, S.; et al. Patient-reported outcomes from the phase III IMpassion130 trial of atezolizumab plus nab-paclitaxel in metastatic triple-negative breast cancer. Ann. Oncol. 2020, 31, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Woodward, W.A. Building momentum for subsets of patients with advanced triple-negative breast cancer. Lancet Oncol. 2020, 21, 3–5. [Google Scholar] [CrossRef]
- Killock, D. Chemotherapy as a TONIC to invigorate PD-1 inhibition in TNBC. Nat. Rev. Clin. Oncol. 2019, 16, 464. [Google Scholar] [CrossRef] [PubMed]
- Sidaway, P. Setting dictates efficacy of pembrolizumab in TNBC. Nat. Rev. Clin. Oncol. 2018, 16, 66. [Google Scholar] [CrossRef] [PubMed]
- Romero, D. Benefit in patients with PD-L1-positive TNBC. Nat. Rev. Clin. Oncol. 2018, 16, 6. [Google Scholar] [CrossRef]
- Brockwell, N.K.; Owen, K.L.; Zanker, D.; Spurling, A.; Rautela, J.; Duivenvoorden, H.M.; Baschuk, N.; Caramia, F.; Loi, S.; Darcy, P.K.; et al. Neoadjuvant interferons: Critical for effective PD-1–based Immunotherapy in TNBC. Cancer Immunol. Res. 2017, 5, 871–884. [Google Scholar] [CrossRef]
- Williford, J.M.; Ishihara, J.; Ishihara, A.; Mansurov, A.; Hosseinchi, P.; Marchell, T.M.; Potin, L.; Swartz, M.A.; Hubbell, J.A. Recruitment of CD103+ dendritic cells via tumor-targeted chemokine delivery enhances efficacy of checkpoint inhibitor immunotherapy. Sci. Adv. 2019, 5, eaay1357. [Google Scholar] [CrossRef]
- Tomioka, N.; Azuma, M.; Ikarashi, M.; Yamamoto, M.; Sato, M.; Watanabe, K.I.; Yamashiro, K.; Takahashi, M. The therapeutic candidate for immune checkpoint inhibitors elucidated by the status of tumor-infiltrating lymphocytes (TILs) and programmed death ligand 1 (PD-L1) expression in triple negative breast cancer (TNBC). Breast Cancer 2017, 26, 34–42. [Google Scholar] [CrossRef]
- Karn, T.; Denkert, C.; Weber, K.; Holtrich, U.; Hanusch, C.; Sinn, B.; Higgs, B.; Jank, P.; Sinn, H.; Huober, J.; et al. Tumor mutational burden and immune infiltration as independent predictors of response to neoadjuvant immune checkpoint inhibition in early TNBC in GeparNuevo. Ann. Oncol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Mittendorf, E.A.; Philips, A.V.; Meric-Bernstam, F.; Qiao, N.; Wu, Y.; Harrington, S.; Su, X.; Wang, Y.; Gonzalez-Angulo, A.M.; Akcakanat, A.; et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol. Res. 2014, 2, 361–370. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.A.; Wright, G.S.; et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef] [PubMed]
- Cetin, B.; Gumusay, O.; Schmid, P.; Dent, R.; O’Shaughnessy, J. Pembrolizumab for early triple-negative breast cancer. N. Engl. J. Med. 2020, 382, e108. [Google Scholar] [CrossRef] [PubMed]
- Salgado, R.; Denkert, C.; DeMaria, S.; Sirtaine, N.; Klauschen, F.; Pruneri, G.; Wienert, S.; Eynden, G.V.D.; Baehner, F.L.; Penault-Llorca, F.; et al. The evaluation of tumor-infiltrating lymphocytes (TILs) in breast cancer: Recommendations by an International TILs Working Group 2014. Ann. Oncol. 2015, 26, 259–271. [Google Scholar] [CrossRef]
- Beckers, R.K.; Selinger, C.I.; Vilain, R.; Madore, J.; Wilmott, J.; Harvey, K.; Holliday, A.; Cooper, C.L.; Robbins, E.; Gillett, D.; et al. Programmed death ligand 1 expression in triple-negative breast cancer is associated with tumour-infiltrating lymphocytes and improved outcome. Histopathology 2016, 69, 25–34. [Google Scholar] [CrossRef]
- Lee, J.; Kim, D.M.; Lee, A. Prognostic role and clinical association of tumor-infiltrating lymphocyte, programmed death ligand-1 expression with neutrophil-lymphocyte ratio in locally advanced triple-negative breast cancer. Cancer Res. Treat. 2018, 51, 649–663. [Google Scholar] [CrossRef]
- Cimino-Mathews, A.; Thompson, E.; Taube, J.M.; Ye, X.; Lu, Y.; Meeker, A.; Xu, H.; Sharma, R.; Lecksell, K.; Cornish, T.C.; et al. PD-L1 (B7-H1) expression and the immune tumor microenvironment in primary and metastatic breast carcinomas. Hum. Pathol. 2015, 47, 52–63. [Google Scholar] [CrossRef]
- Planes-Laine, G.; Rochigneux, P.; Bertucci, F.; Chrétien, A.-S.; Viens, P.; Sabatier, R.; Gonçalves, A.; Laine, P. PD-1/PD-L1 Targeting in breast cancer: The first clinical evidences are emerging. A literature review. Cancers 2019, 11, 1033. [Google Scholar] [CrossRef]
- Zhang, M.; Sun, H.; Zhao, S.; Wang, Y.; Pu, H.; Zhang, Q. Expression of PD-L1 and prognosis in breast cancer: A meta-analysis. Oncotarget 2017, 8, 31347–31354. [Google Scholar] [CrossRef]
- Schmid, P.; Cortes, J.; Pusztai, L.; McArthur, H.; Kümmel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; Harbeck, N.; et al. Pembrolizumab for early triple-negative breast cancer. N. Engl. J. Med. 2020, 382, 810–821. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Salgado, R.; Park, Y.; Muñoz-Couselo, E.; Kim, S.; Sohn, J.; Im, S.A.; Foukakis, T.; Kuemmel, S.; Dent, R.; et al. Pembrolizumab plus chemotherapy as neoadjuvant treatment of high-risk, early-stage triple-negative breast cancer: Results from the phase 1b open-label, multicohort KEYNOTE-173 study. Ann. Oncol. 2020, 31, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Seliger, B.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Seliger, B.; Allison, J.P. Immune checkpoint targeting in cancer therapy: Toward combination strategies with curative potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef]
- Schachter, J.; Ribas, A.; Long, G.V.; Arance, A.; Grob, J.-J.; Mortier, L.; Daud, A.I.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus ipilimumab for advanced melanoma: Final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet 2017, 390, 1853–1862. [Google Scholar] [CrossRef]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Sosman, J.A.; Atkins, M.B.; Leming, P.D.; et al. Five-year survival and correlates among patients with advanced melanoma, renal cell carcinoma, or non-small cell lung cancer treated with nivolumab. JAMA Oncol. 2019, 5, 1411–1420. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti–PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef]
- First Anti-PD-L1 Drug Approved for NSCLC. Cancer Discov. 2016, 6, OF1. [CrossRef]
- Okazaki, T.; Honjo, T. PD-1 and PD-1 ligands: From discovery to clinical application. Int. Immunol. 2007, 19, 813–824. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Larkin, J.; Sileni, V.C.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Venkitaraman, A.R. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell 2002, 108, 171–182. [Google Scholar] [CrossRef]
- Narod, S.A.; Foulkes, W.D. BRCA1 and BRCA2: 1994 and beyond. Nat. Rev. Cancer 2004, 4, 665–676. [Google Scholar] [CrossRef]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2011, 12, 68–78. [Google Scholar] [CrossRef]
- Hatano, Y.; Tamada, M.; Matsuo, M.; Hara, A. Molecular trajectory of BRCA1 and BRCA2 mutations. Front. Oncol. 2020, 10, 361. [Google Scholar] [CrossRef]
- Ashworth, A.; Lord, C.J. Synthetic lethal therapies for cancer: What’s next after PARP inhibitors? Nat. Rev. Clin. Oncol. 2018, 15, 564–576. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Guha, M. PARP inhibitors stumble in breast cancer. Nat. Biotechnol. 2011, 29, 373–374. [Google Scholar] [CrossRef]
- Mullard, A. PARP inhibitor pick-me-up. Nat. Rev. Drug Discov. 2019, 18, 814–815. [Google Scholar] [CrossRef] [PubMed]
- Underhill, C.; Toulmonde, M.; Bonnefoi, H. A review of PARP inhibitors: From bench to bedside. Ann. Oncol. 2011, 22, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Angulo, A.M.; Timms, K.M.; Liu, S.; Chen, H.; Litton, J.K.; Potter, J.; Lanchbury, J.S.; Stemke-Hale, K.; Hennessy, B.T.; Arun, B.K.; et al. Incidence and outcome of BRCA mutations in unselected patients with triple receptor-negative breast cancer. Clin. Cancer Res. 2011, 17, 1082–1089. [Google Scholar] [CrossRef] [PubMed]
- McCann, K.E.; Hurvitz, S.A. Advances in the use of PARP inhibitor therapy for breast cancer. Drugs Context 2018, 7, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Pascal, J.M. The comings and goings of PARP-1 in response to DNA damage. DNA Repair 2018, 71, 177–182. [Google Scholar] [CrossRef]
- Caron, M.C.; Sharma, A.K.; O’Sullivan, J.; Myler, L.R.; Ferreira, M.T.; Rodrigue, A.; Coulombe, Y.; Ethier, C.; Gagné, J.P.; Langelier, M.F.; et al. Poly(ADP-ribose) polymerase-1 antagonizes DNA resection at double-strand breaks. Nat. Commun. 2019, 10, 2954. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Beniey, M.; Haque, T.; Hassan, S. Translating the role of PARP inhibitors in triple-negative breast cancer. Oncoscience 2019, 6, 287–288. [Google Scholar] [CrossRef]
- Robson, M.E.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Robson, M.E.; Tung, N.; Conte, P.; Im, S.-A.; Senkus, E.; Xu, B.; Masuda, N.; Delaloge, S.; Li, W.; Armstrong, A.; et al. OlympiAD final overall survival and tolerability results: Olaparib versus chemotherapy treatment of physician’s choice in patients with a germline BRCA mutation and HER2-negative metastatic breast cancer. Ann. Oncol. 2019, 30, 558–566. [Google Scholar] [CrossRef]
- Robson, M.E.; Ruddy, K.J.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Li, W.; Tung, N.; Armstrong, A.; et al. Patient-reported outcomes in patients with a germline BRCA mutation and HER2-negative metastatic breast cancer receiving olaparib versus chemotherapy in the OlympiAD trial. Eur. J. Cancer 2019, 120, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Litton, J.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Fasching, P.A.; Loibl, S.; Hu, C.; Hart, S.N.; Shimelis, H.; Moore, R.; Schem, C.; Tesch, H.; Untch, M.; Hilfrich, J.; et al. BRCA1/2 Mutations and bevacizumab in the neoadjuvant treatment of breast cancer: Response and prognosis results in patients with triple-negative breast cancer from the geparquinto study. J. Clin. Oncol. 2018, 36, 2281–2287. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; kConFab Investigators; Geyer, F.C.; Blecua, P.; Lee, J.Y.; Selenica, P.; Brown, D.N.; Pareja, F.; Lee, S.S.K.; Kumar, R.; et al. Homologous recombination DNA repair defects in PALB2-associated breast cancers. NPJ Breast Cancer 2019, 5, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Noordermeer, S.M.; Van Attikum, H. PARP Inhibitor resistance: A tug-of-war in BRCA-mutated cells. Trends Cell Biol. 2019, 29, 820–834. [Google Scholar] [CrossRef]
- Hoyer, J.; Vasileiou, G.; Uebe, S.; Wunderle, M.; Kraus, C.; Fasching, P.A.; Thiel, C.T.; Hartmann, A.; Beckmann, M.W.; Lux, M.P.; et al. Addition of triple negativity of breast cancer as an indicator for germline mutations in predisposing genes increases sensitivity of clinical selection criteria. BMC Cancer 2018, 18, 926. [Google Scholar] [CrossRef]
- Shi, Y.; Jin, J.; Ji, W.; Guan, X. Therapeutic landscape in mutational triple negative breast cancer. Mol. Cancer 2018, 17, 99. [Google Scholar] [CrossRef]
- Hu, C.; Polley, E.C.; Yadav, S.; Lilyquist, J.; Shimelis, H.; Na, J.; Hart, S.N.; Goldgar, D.E.; Shah, S.; Pesaran, T.; et al. The contribution of germline predisposition gene mutations to clinical subtypes of invasive breast cancer from a clinical genetic testing cohort. J. Natl. Cancer Inst. 2020. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, H.; Fu, F.; Li, Z.; Feng, Q.; Wu, W.; Liu, Y.; Wang, C.; Chen, Y. Spectrum of PALB2 germline mutations and characteristics of PALB2 -related breast cancer: Screening of 16,501 unselected patients with breast cancer and 5890 controls by next-generation sequencing. Cancer 2020, 126, 3202–3208. [Google Scholar] [CrossRef]
- Ellsworth, D.L.; Turner, C.E.; Ellsworth, R.E. A review of the hereditary component of triple negative breast cancer: High- and moderate-penetrance breast cancer genes, low-penetrance loci, and the role of nontraditional genetic elements. J. Oncol. 2019, 2019, 4382606. [Google Scholar] [CrossRef]
- Goldenberg, D.M.; Stein, R.; Sharkey, R.M. The emergence of trophoblast cell-surface antigen 2 (TROP-2) as a novel cancer target. Oncotarget 2018, 9, 28989–29006. [Google Scholar] [CrossRef] [PubMed]
- Ocean, A.J.; Starodub, A.N.; Bardia, A.; Vahdat, L.T.; Isakoff, S.J.; Guarino, M.; Messersmith, W.A.; Picozzi, V.J.; Mayer, I.A.; Wegener, W.A.; et al. Sacituzumab govitecan (IMMU-132), an anti-Trop-2-SN-38 antibody-drug conjugate for the treatment of diverse epithelial cancers: Safety and pharmacokinetics. Cancer 2017, 123, 3843–3854. [Google Scholar] [CrossRef] [PubMed]
- Rugo, H.S.; Bardia, A.; Tolaney, S.M.; Arteaga, C.; Cortes, J.; Sohn, J.; Marmé, F.; Hong, Q.; Delaney, R.J.; Hafeez, A.; et al. TROPiCS-02: A Phase III study investigating sacituzumab govitecan in the treatment of HR+/HER2- metastatic breast cancer. Futur. Oncol. 2020, 16, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Bardia, A.; Mayer, I.A.; Diamond, J.R.; Moroose, R.L.; Isakoff, S.J.; Starodub, A.N.; Shah, N.C.; O’Shaughnessy, J.; Kalinsky, K.; Guarino, M.; et al. Efficacy and safety of anti-trop-2 antibody drug conjugate sacituzumab govitecan (IMMU-132) in heavily pretreated patients with metastatic triple-negative breast cancer. J. Clin. Oncol. 2017, 35, 2141–2148. [Google Scholar] [CrossRef] [PubMed]
- Bardia, A.; Mayer, I.A.; Vahdat, L.T.; Tolaney, S.M.; Isakoff, S.J.; Diamond, J.R.; O’Shaughnessy, J.; Moroose, R.L.; Santin, A.D.; Abramson, V.G.; et al. Sacituzumab govitecan-hziy in refractory metastatic triple-negative breast cancer. N. Engl. J. Med. 2019, 380, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Killock, D. AKT inhibition improves OS in TNBC. Nat. Rev. Clin. Oncol. 2020, 17, 135. [Google Scholar] [CrossRef]
- Saini, K.S.; Loi, S.; De Azambuja, E.; Metzger-Filho, O.; Saini, M.L.; Ignatiadis, M.; Dancey, J.E.; Piccart, M. Targeting the PI3K/AKT/mTOR and Raf/MEK/ERK pathways in the treatment of breast cancer. Cancer Treat. Rev. 2013, 39, 935–946. [Google Scholar] [CrossRef]
- Aggarwal, S.; Verma, S.S.; Aggarwal, S.; Gupta, S.C. Drug repurposing for breast cancer therapy: Old weapon for new battle. Semin. Cancer Biol. 2019, 13. [Google Scholar] [CrossRef]
- Jamdade, V.S.; Sethi, N.; Mundhe, N.A.; Kumar, P.; Lahkar, M.; Sinha, N. Therapeutic targets of triple-negative breast cancer: A review. Br. J. Pharmacol. 2015, 172, 4228–4237. [Google Scholar] [CrossRef]
- Nakajima, H.; Ishikawa, Y.; Furuya, M.; Sano, T.; Ohno, Y.; Horiguchi, J.; Oyama, T. Protein expression, gene amplification, and mutational analysis of EGFR in triple-negative breast cancer. Breast Cancer 2012, 21, 66–74. [Google Scholar] [CrossRef]
- Herold, C.I.; Anders, C.K. New targets for triple-negative breast cancer. Oncology 2013, 27, 846–854. [Google Scholar] [PubMed]
- Masuda, H.; Zhang, N.; Bartholomeusz, C.; Doihara, H.; Hortobagyi, G.N.; Ueno, N.T. Role of epidermal growth factor receptor in breast cancer. Breast Cancer Res. Treat. 2012, 136, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Carey, L.A.; Rugo, H.S.; Marcom, P.K.; Mayer, E.L.; Esteva, F.J.; Ma, C.X.; Liu, M.C.; Storniolo, A.M.; Rimawi, M.F.; Forero-Torres, A.; et al. TBCRC 001: Randomized phase II study of cetuximab in combination with carboplatin in Stage IV triple-negative breast cancer. J. Clin. Oncol. 2012, 30, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Nico, B.; Ruggieri, S.; Tamma, R.; Simone, G.; Mangia, A. Angiogenesis and antiangiogenesis in triple-negative breast cancer. Transl. Oncol. 2016, 9, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, L. BEATRICE bevacizumab trial—Every cloud has a silver lining. Nat. Rev. Clin. Oncol. 2013, 10, 548. [Google Scholar] [CrossRef] [PubMed]
- Cameron, D.; Brown, J.; Dent, R.; Jackisch, C.; Mackey, J.; Pivot, X.; Steger, G.G.; Suter, T.M.; Toi, M.; Parmar, M.; et al. Adjuvant bevacizumab-containing therapy in triple-negative breast cancer (BEATRICE): Primary results of a randomised, phase 3 trial. Lancet Oncol. 2013, 14, 933–942. [Google Scholar] [CrossRef]
- Bell, R.; Brown, J.; Parmar, M.; Toi, M.; Suter, T.; Steger, G.G.; Pivot, X.; Mackey, J.; Jackisch, C.; Dent, R.; et al. Final efficacy and updated safety results of the randomized phase III BEATRICE trial evaluating adjuvant bevacizumab-containing therapy in triple-negative early breast cancer. Ann. Oncol. 2017, 28, 754–760. [Google Scholar] [CrossRef]
- Miles, D.; Diéras, V.; Cortés, J.; Duenne, A.A.; Yi, J.; O’Shaughnessy, J. First-line bevacizumab in combination with chemotherapy for HER2-negative metastatic breast cancer: Pooled and subgroup analyses of data from 2447 patients. Ann. Oncol. 2013, 24, 2773–2780. [Google Scholar] [CrossRef]
- Von Minckwitz, G.; Eidtmann, H.; Rezai, M.; Fasching, P.A.; Tesch, H.; Eggemann, H.; Schrader, I.; Kittel, K.; Hanusch, C.; Kreienberg, R.; et al. Neoadjuvant chemotherapy and bevacizumab for HER2-negative breast cancer. N. Engl. J. Med. 2012, 366, 299–309. [Google Scholar] [CrossRef]
- Bear, H.D.; Tang, G.; Rastogi, P.; Geyer, C.E., Jr.; Robidoux, A.; Atkins, J.N.; Baez-Diaz, L.; Brufsky, A.M.; Mehta, R.S.; Fehrenbacher, L.; et al. Bevacizumab added to neoadjuvant chemotherapy for breast cancer. N. Engl. J. Med. 2012, 366, 310–320. [Google Scholar] [CrossRef]
- Bear, H.D.; Tang, G.; Rastogi, P.; Geyer, C.E., Jr.; Zoon, C.K.; Kidwell, K.M.; Robidoux, A.; Baez-Diaz, L.; Brufsky, A.M.; Mehta, R.S.; et al. The Effect on surgical complications of bevacizumab added to neoadjuvant chemotherapy for breast cancer: NRG Oncology/NSABP protocol B-40. Ann. Surg. Oncol. 2016, 24, 1853–1860. [Google Scholar] [CrossRef] [PubMed]
- Bossuyt, V.; Provenzano, E.; Symmans, W.F.; Boughey, J.C.; Coles, C.; Curigliano, G.; Dixon, J.M.; Esserman, L.J.; Fastner, G.; Kuehn, T.; et al. Recommendations for standardized pathological characterization of residual disease for neoadjuvant clinical trials of breast cancer by the BIG-NABCG collaboration. Ann. Oncol. 2015, 26, 1280–1291. [Google Scholar] [CrossRef] [PubMed]
- Nahleh, Z.A.; Barlow, W.E.; Hayes, D.F.; Schott, A.F.; Gralow, J.R.; Sikov, W.M.; Perez, E.A.; Chennuru, S.; Mirshahidi, H.R.; Corso, S.W.; et al. SWOG S0800 (NCI CDR0000636131): Addition of bevacizumab to neoadjuvant nab-paclitaxel with dose-dense doxorubicin and cyclophosphamide improves pathologic complete response (pCR) rates in inflammatory or locally advanced breast cancer. Breast Cancer Res. Treat. 2016, 158, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Pascual, J.; Turner, N. Targeting the PI3-kinase pathway in triple-negative breast cancer. Ann. Oncol. 2019, 30, 1051–1060. [Google Scholar] [CrossRef]
- Costa, R.L.B.; Han, H.S.; Gradishar, W.J. Targeting the PI3K/AKT/mTOR pathway in triple-negative breast cancer: A review. Breast Cancer Res. Treat. 2018, 169, 397–406. [Google Scholar] [CrossRef]
- Chan, J.J.; Tan, T.J.Y.; Dent, R. Novel therapeutic avenues in triple-negative breast cancer: PI3K/AKT inhibition, androgen receptor blockade, and beyond. Ther. Adv. Med Oncol. 2019, 11, 1758835919880429. [Google Scholar] [CrossRef]
- Rugo, H.S.; André, F.; Yamashita, T.; Cerda, H.; Toledano, I.; Stemmer, S.; Jurado, J.; Juric, D.; Mayer, I.; Ciruelos, E.; et al. Time course and management of key adverse events during the randomized phase III SOLAR-1 study of PI3K inhibitor alpelisib plus fulvestrant in patients with HR-positive advanced breast cancer. Ann. Oncol. 2020. [Google Scholar] [CrossRef]
- André, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef]
- Mosele, F.; Stefanovska, B.; Lusque, A.; Dien, A.T.; Garberis, I.; Droin, N.; Le Tourneau, C.; Sablin, M.P.; Lacroix, L.; Enrico, D.; et al. Outcome and molecular landscape of patients with PIK3CA-mutated metastatic breast cancer. Ann. Oncol. 2020, 31, 377–386. [Google Scholar] [CrossRef]
- Basho, R.K.; Gilcrease, M.; Murthy, R.K.; Helgason, T.; Karp, D.D.; Meric-Bernstam, F.; Hess, K.R.; Herbrich, S.; Valero, V.; Albarracin, C.; et al. Targeting the PI3K/AKT/mTOR pathway for the treatment of mesenchymal triple-negative breast cancer. JAMA Oncol. 2017, 3, 509–515. [Google Scholar] [CrossRef]
- Lin, J.; Sampath, D.; Nannini, M.; Lee, B.B.; Degtyarev, M.; Oeh, J.; Savage, H.; Guan, Z.; Hong, R.; Kassees, R.; et al. Targeting activated Akt with GDC-0068, a novel selective akt inhibitor that is efficacious in multiple tumor models. Clin. Cancer Res. 2013, 19, 1760–1772. [Google Scholar] [CrossRef] [PubMed]
- Davies, B.R.; Greenwood, H.; Dudley, P.; Crafter, C.; Yu, D.; Zhang, J.; Li, J.; Gao, B.; Ji, Q.; Maynard, J.; et al. Preclinical pharmacology of AZD5363, an inhibitor of AKT: Pharmacodynamics, antitumor activity, and correlation of monotherapy activity with genetic background. Mol. Cancer Ther. 2012, 11, 873–887. [Google Scholar] [CrossRef]
- Kim, S.B.; Dent, R.; Im, S.A.; Espié, M.; Blau, S.; Tan, A.R.; Isakoff, S.J.; Oliveira, M.; Saura, C.; Wong, M.J.; et al. Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2017, 18, 1360–1372. [Google Scholar] [CrossRef]
- Schmid, P.; Abraham, J.; Chan, S.; Wheatley, D.; Brunt, A.M.; Nemsadze, G.; Baird, R.D.; Park, Y.H.; Hall, P.S.; Perren, T.; et al. Capivasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer: The PAKT Trial. J. Clin. Oncol. 2020, 38, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Ricciardi, G.R.R.; Adamo, B.; Ieni, A.; Licata, L.; Cardia, R.; Ferraro, G.; Franchina, T.; Tuccari, G.; Adamo, V. Androgen receptor (AR), E-Cadherin, and Ki-67 as Emerging targets and novel prognostic markers in triple-negative breast cancer (TNBC) patients. PLoS ONE 2015, 10, e0128368. [Google Scholar] [CrossRef]
- Bhattarai, S.; Klimov, S.; Mittal, K.; Krishnamurti, U.; Li, X.B.; Oprea-Ilies, G.; Wetherilt, C.S.; Riaz, A.; Aleskandarany, M.A.; Green, A.R.; et al. Prognostic role of androgen receptor in triple negative breast cancer: A multi-institutional study. Cancers 2019, 11, 995. [Google Scholar] [CrossRef]
- Gucalp, A.; Tolaney, S.; Isakoff, S.J.; Ingle, J.N.; Liu, M.C.; Carey, L.A.; Blackwell, K.; Rugo, H.; Nabell, L.; Forero, A.; et al. Phase II trial of bicalutamide in patients with androgen receptor-positive, estrogen receptor-negative metastatic Breast Cancer. Clin. Cancer Res. 2013, 19, 5505–5512. [Google Scholar] [CrossRef]
- Bonnefoi, H.; Grellety, T.; Trédan, O.; Saghatchian, M.; Dalenc, F.; Mailliez, A.; L’Haridon, T.; Cottu, P.; Abadie-Lacourtoisie, S.; You, B.; et al. A phase II trial of abiraterone acetate plus prednisone in patients with triple-negative androgen receptor positive locally advanced or metastatic breast cancer (UCBG 12-1). Ann. Oncol. 2016, 27, 812–818. [Google Scholar] [CrossRef]
- Schwartzberg, L.S.; Yardley, D.A.; Elias, A.D.; Patel, M.; Lorusso, P.; Burris, H.A.; Gucalp, A.; Peterson, A.C.; Blaney, M.E.; Steinberg, J.L.; et al. A phase I/Ib study of enzalutamide alone and in combination with endocrine therapies in women with advanced breast cancer. Clin. Cancer Res. 2017, 23, 4046–4054. [Google Scholar] [CrossRef]
- Traina, T.A.; Miller, K.; Yardley, D.A.; Eakle, J.; Schwartzberg, L.S.; O’Shaughnessy, J.A.; Gradishar, W.J.; Schmid, P.; Winer, E.; Kelly, C.; et al. Enzalutamide for the treatment of androgen receptor–Expressing triple-negative breast cancer. J. Clin. Oncol. 2018, 36, 884–890. [Google Scholar] [CrossRef]
- Saini, G.; Bhattarai, S.; Gogineni, K.; Aneja, R. Quadruple-negative breast cancer: An uneven playing field. JCO Glob. Oncol. 2020, 6, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Speirs, V.; Skliris, G.P.; Burdall, S.E.; Carder, P.J. Distinct expression patterns of ER and ER in normal human mammary gland. J. Clin. Pathol. 2002, 55, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Lam, E.W.F.; Sunters, A.; Enmark, E.; De Bella, M.T.; Coombes, R.C.; Gustafsson, J.-Å.; Dahlman-Wright, K. Expression of estrogen receptor β isoforms in normal breast epithelial cells and breast cancer: Regulation by methylation. Oncogene 2003, 22, 7600–7606. [Google Scholar] [CrossRef] [PubMed]
- Hieken, T.J.; Carter, J.M.; Hawse, J.R.; Hoskin, T.L.; Bois, M.; Frost, M.; Hartmann, L.C.; Radisky, D.C.; Visscher, D.W.; Degnim, A.C. ERβ expression and breast cancer risk prediction for women with atypias. Cancer Prev. Res. 2015, 8, 1084–1092. [Google Scholar] [CrossRef]
- Dotzlaw, H.; Leygue, E.; Watson, P.H.; Murphy, L.C. Estrogen receptor-beta messenger RNA expression in human breast tumor biopsies: Relationship to steroid receptor status and regulation by progestins. Cancer Res. 1999, 59, 529–532. [Google Scholar]
- Roger, P.; Sahla, M.E.; Mäkelä, S.; Gustafsson, J.A.; Baldet, P.; Rochefort, H. Decreased expression of estrogen receptor beta protein in proliferative preinvasive mammary tumors. Cancer Res. 2001, 61, 2537–2541. [Google Scholar]
- Hawse, J.R.; Carter, J.M.; Aspros, K.G.M.; Bruinsma, E.S.; Koepplin, J.W.; Negron, V.; Subramaniam, M.; Ingle, J.N.; Rech, K.L.; Goetz, M.P. Optimized immunohistochemical detection of estrogen receptor beta using two validated monoclonal antibodies confirms its expression in normal and malignant breast tissues. Breast Cancer Res. Treat. 2019, 179, 241–249. [Google Scholar] [CrossRef]
- Reese, J.M.; Suman, V.J.; Subramaniam, M.; Wu, X.; Negron, V.; Gingery, A.; Pitel, K.S.; Shah, S.S.; Cunliffe, H.E.; McCullough, A.E.; et al. ERβ1: Characterization, prognosis, and evaluation of treatment strategies in ERα-positive and -negative breast cancer. BMC Cancer 2014, 14, 749. [Google Scholar] [CrossRef]
- Badve, S.S.; Gökmen-Polar, Y. TP53 status and estrogen receptor-beta in triple-negative breast cancer: Company matters. J. Natl. Cancer Inst. 2019, 111, 1118–1119. [Google Scholar] [CrossRef]
- Sellitto, A.; D’Agostino, Y.; Kovalskaya-Alexandrova, E.; Lamberti, J.; Pecoraro, G.; Memoli, D.; Rocco, D.; Coviello, E.; Giurato, G.; Nassa, G.; et al. Insights into the role of estrogen receptor β in triple-negative breast cancer. Cancers 2020, 12, 1477. [Google Scholar] [CrossRef]
- Lazennec, G.; Bresson, D.; Lucas, A.; Chauveau, C.; Vignon, F. ER beta inhibits proliferation and invasion of breast cancer cells. Endocrinology 2001, 142, 4120–4130. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Rajapaksa, G.; Nikolos, F.; Hao, R.; Katchy, A.; Mccollum, C.; Bondesson, M.; Quinlan, P.R.; Thompson, A.M.; Krishnamurthy, S.; et al. ERβ1 represses basal-like breast cancer epithelial to mesenchymal transition by destabilizing EGFR. Breast Cancer Res. 2012, 14, R148. [Google Scholar] [CrossRef] [PubMed]
- Shanle, E.K.; Zhao, Z.; Hawse, J.; Wisinski, K.; Keles, S.; Yuan, M.; Xu, W. Research resource: Global identification of estrogen receptor β target genes in triple negative breast cancer cells. Mol. Endocrinol. 2013, 27, 1762–1775. [Google Scholar] [CrossRef] [PubMed]
- Schüler-Toprak, S.; Häring, J.; Inwald, E.C.; Moehle, C.; Ortmann, O.; Treeck, O. Agonists and knockdown of estrogen receptor β differentially affect invasion of triple-negative breast cancer cells in vitro. BMC Cancer 2016, 16, 951. [Google Scholar] [CrossRef] [PubMed]
- Reese, J.M.; Bruinsma, E.S.; Monroe, D.G.; Negron, V.; Suman, V.J.; Ingle, J.N.; Goetz, M.P.; Hawse, J.R. ERβ inhibits cyclin dependent kinases 1 and 7 in triple negative breast cancer. Oncotarget 2017, 8, 96506–96521. [Google Scholar] [CrossRef] [PubMed]
- Reese, J.M.; Bruinsma, E.S.; Nelson, A.W.; Chernukhin, I.; Carroll, J.S.; Li, Y.; Subramaniam, M.; Suman, V.J.; Negron, V.; Monroe, D.G.; et al. ERβ-mediated induction of cystatins results in suppression of TGFβ signaling and inhibition of triple-negative breast cancer metastasis. Proc. Natl. Acad. Sci. USA 2018, 115, E9580–E9589. [Google Scholar] [CrossRef]
- Austin, D.; Hamilton, N.; Elshimali, Y.; Pietras, R.J.; Wu, Y.; Vadgama, J.V. Estrogen receptor-beta is a potential target for triple negative breast cancer treatment. Oncotarget 2018, 9, 33912–33930. [Google Scholar] [CrossRef]
- Wisinski, K.B.; Xu, W.; Tevaarwerk, A.J.; Saha, S.; Kim, K.; Traynor, A.; Dietrich, L.; Hegeman, R.; Patel, D.; Blank, J.; et al. Targeting estrogen receptor beta in a phase 2 study of high-dose estradiol in metastatic triple-negative breast cancer: A Wisconsin oncology network study. Clin. Breast Cancer 2016, 16, 256–261. [Google Scholar] [CrossRef]
- Jiang, W.; Wang, X.; Zhang, C.; Xue, L.; Yang, L. Expression and clinical significance of MAPK and EGFR in triple-negative breast cancer. Oncol. Lett. 2020, 19, 1842–1848. [Google Scholar] [CrossRef]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef]
- Wright, K.L.; Adams, J.R.; Liu, J.C.; Loch, A.J.; Wong, R.G.; Jo, C.E.; Beck, L.A.; Santhanam, D.R.; Weiss, L.; Mei, X.; et al. Ras signaling is a key determinant for metastatic dissemination and poor survival of luminal breast cancer patients. Cancer Res. 2015, 75, 4960–4972. [Google Scholar] [CrossRef] [PubMed]
- Tebbutt, N.; Pedersen, M.W.; Johns, T.G. Targeting the ERBB family in cancer: Couples therapy. Nat. Rev. Cancer 2013, 13, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef]
- Ahmed, A.U.; Schmidt, R.L.; Park, C.H.; Reed, N.R.; Hesse, S.E.; Thomas, C.F.; Molina, J.R.; Deschamps, C.; Yang, P.; Aubry, M.C.; et al. Effect of disrupting seven-in-absentia homolog 2 function on lung cancer cell growth. J. Natl. Cancer Inst. 2008, 100, 1606–1629. [Google Scholar] [CrossRef]
- Schmidt, R.L.; Park, C.H.; Ahmed, A.U.; Gundelach, J.H.; Reed, N.R.; Cheng, S.; Knudsen, B.E.; Tang, A.H. Inhibition of RAS-Mediated transformation and tumorigenesis by targeting the downstream E3 ubiquitin ligase seven in absentia homologue. Cancer Res. 2007, 67, 11798–11810. [Google Scholar] [CrossRef]
- Clark, G.J.; Der, C.J. Aberrant function of the Ras signal transduction pathway in human breast cancer. Breast Cancer Res. Treat. 1995, 35, 133–144. [Google Scholar] [CrossRef]
- Von Lintig, F.C.; Dreilinger, A.D.; Varki, N.M.; Wallace, A.M.; Casteel, D.E.; Boss, G.R. Ras activation in human breast cancer. Breast Cancer Res. Treat. 2000, 62, 51–62. [Google Scholar] [CrossRef]
- Eckert, L.B. Involvement of Ras activation in human breast cancer cell signaling, invasion, and anoikis. Cancer Res. 2004, 64, 4585–4592. [Google Scholar] [CrossRef]
- Malaney, S.; Daly, R. The ras signaling pathway in mammary tumorigenesis and metastasis. J. Mammary Gland. Biol. Neoplasia 2001, 6, 101–113. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Bonati, A.; Libra, M.; Stivala, F.; Martelli, A.; Franklin, R.A.; Navolanic, P.; Steelman, L.S. Combining chemo-, hormonal and targeted therapies to treat breast cancer (Review). Mol. Med. Rep. 2008, 1, 139–160. [Google Scholar] [CrossRef]
- McGlynn, L.M.; Kirkegaard, T.; Edwards, J.; Tovey, S.; Cameron, D.; Twelves, C.; Bartlett, J.M.; Cooke, T.G. Ras/Raf-1/MAPK pathway mediates response to tamoxifen but not chemotherapy in breast cancer patients. Clin. Cancer Res. 2009, 15, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Loi, S.; Dushyanthen, S.; Beavis, P.A.; Salgado, R.; Denkert, C.; Savas, P.; Combs, S.; Rimm, D.L.; Giltnane, J.M.; Estrada, M.V.; et al. RAS/MAPK Activation is associated with reduced tumor-infiltrating lymphocytes in triple-negative breast cancer: Therapeutic cooperation between MEK and PD-1/PD-L1 immune checkpoint inhibitors. Clin. Cancer Res. 2015, 22, 1499–1509. [Google Scholar] [CrossRef] [PubMed]
- Behling, K.C.; Tang, A.H.; Freydin, B.; Chervoneva, I.; Kadakia, S.; Schwartz, G.F.; Rui, H.; Witkiewicz, A.K. Increased SIAH expression predicts ductal carcinoma in situ (DCIS) progression to invasive carcinoma. Breast Cancer Res. Treat. 2010, 129, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Van Reesema, L.L.S.; Zheleva, V.; Winston, J.S.; Jansen, R.J.; O’Connor, C.F.; Isbell, A.J.; Bian, M.; Qin, R.; Bassett, P.T.; Hinson, V.J.; et al. SIAH and EGFR, two RAS pathway biomarkers, are highly prognostic in locally advanced and metastatic breast cancer. EBioMedicine 2016, 11, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Zipursky, S.L.; Rubin, G.M. Determination of neuronal cell fate: Lessons from the R7 neuron of Drosophila. Annu. Rev. Neurosci. 1994, 17, 373–397. [Google Scholar] [CrossRef]
- Tang, A.H.; Neufeld, T.P.; Kwan, E.; Rubin, G.M. PHYL acts to down-regulate TTK88, a transcriptional repressor of neuronal cell fates, by a SINA-dependent mechanism. Cell 1997, 90, 459–467. [Google Scholar] [CrossRef]
- Corkery, B.; Crown, J.; Clynes, M.; O’Donovan, N. Epidermal growth factor receptor as a potential therapeutic target in triple-negative breast cancer. Ann. Oncol. 2009, 20, 862–867. [Google Scholar] [CrossRef]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef]
- Olayioye, M.A.; Neve, R.M.; Lane, H.A.; Hynes, N.E. New Embo members’ review: The ErbB signaling network: Receptor heterodimerization in development and cancer. EMBO J. 2000, 19, 3159–3167. [Google Scholar] [CrossRef]
- Hynes, N.E.; Lane, H.A. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gupta, G.K.; Collier, A.L.; Lee, D.; Hoefer, R.A.; Zheleva, V.; Siewertsz van Reesema, L.L.; Tang-Tan, A.M.; Guye, M.L.; Chang, D.Z.; Winston, J.S.; et al. Perspectives on Triple-Negative Breast Cancer: Current Treatment Strategies, Unmet Needs, and Potential Targets for Future Therapies. Cancers 2020, 12, 2392. https://doi.org/10.3390/cancers12092392
Gupta GK, Collier AL, Lee D, Hoefer RA, Zheleva V, Siewertsz van Reesema LL, Tang-Tan AM, Guye ML, Chang DZ, Winston JS, et al. Perspectives on Triple-Negative Breast Cancer: Current Treatment Strategies, Unmet Needs, and Potential Targets for Future Therapies. Cancers. 2020; 12(9):2392. https://doi.org/10.3390/cancers12092392
Chicago/Turabian StyleGupta, Gagan K., Amber L. Collier, Dasom Lee, Richard A. Hoefer, Vasilena Zheleva, Lauren L. Siewertsz van Reesema, Angela M. Tang-Tan, Mary L. Guye, David Z. Chang, Janet S. Winston, and et al. 2020. "Perspectives on Triple-Negative Breast Cancer: Current Treatment Strategies, Unmet Needs, and Potential Targets for Future Therapies" Cancers 12, no. 9: 2392. https://doi.org/10.3390/cancers12092392
APA StyleGupta, G. K., Collier, A. L., Lee, D., Hoefer, R. A., Zheleva, V., Siewertsz van Reesema, L. L., Tang-Tan, A. M., Guye, M. L., Chang, D. Z., Winston, J. S., Samli, B., Jansen, R. J., Petricoin, E. F., Goetz, M. P., Bear, H. D., & Tang, A. H. (2020). Perspectives on Triple-Negative Breast Cancer: Current Treatment Strategies, Unmet Needs, and Potential Targets for Future Therapies. Cancers, 12(9), 2392. https://doi.org/10.3390/cancers12092392